

Liquid chromatography (LC) is an incredibly successful analytical separation tool. Its versatility is unprecedented because of the many different separation modes (reversed-phase LC, ion-exchange chromatography, size-exclusion chromatography, etc.) and because almost all samples can be dissolved in some kind of solvent, ranging from water to organic solvents to strong acids or bases. Conditions (mobile and stationary phases, additives, pH, temperatures, etc.) can be found to separate almost all pairs of analytes. For example, LC is immensely successful in the separation of enantiomers. Good selectivities can be accompanied by high efficiencies in a very short time, using contemporary ultrahigh-performance liquid chromatography (UHPLC) instrumentation and (short) columns packed with sub-2-μm particles. However, LC cannot deliver very high efficiencies in a short time. Unlike other techniques, such as gas chromatography (GC) or capillary electrophoresis (CE), plate counts exceeding 100 000 are not routinely obtained in LC. As a result, LC cannot easily deal with complex mixtures that contain more than a few dozen analytes. While the selectivity between any pair of analytes can be maximized, these peaks may then start to overlap with other relevant analytes or with matrix compounds. There simply is not enough room in LC chromatograms to separate very many compounds that behave “statistically”,1 and the attainable peak capacity does not suffice to separate complex samples. As a rule of thumb, LC offers a high probability of success for separating samples containing 10 or 20 components in 1 or 2 h or up to 50 components in about 10 h.2,3
When dealing with complex samples, comprehensive two-dimensional liquid chromatography (LC × LC) is an attractive approach (Figure 1). Peak capacities of several thousands4−7 can be achieved, and 10 000 is within reasonable reach.8 For high-resolution separations, LC × LC is also much faster, with a peak-production rate (peak capacity divided by the analysis time) of about 1 peak per second, as compared to 1 peak per minute for typical high-resolution one-dimensional LC (1D-LC). The “room” in the chromatogram created by the much-enhanced peak capacity creates the possibility to fully employ two different selectivities. Groups (or “classes”) of analytes can be very efficiently separated from each other,9−11 provided that the selectivities (retention mechanisms) employed in the two dimensions are very different. In the most favorable case, in which the retention times in the two dimensions are completely independent, we speak of orthogonal separations. When separation is obtained using two very different retention mechanisms, the uncertainty of peak assignment12 can be dramatically reduced. Because of the diverse selectivities, high degrees of orthogonality can be achieved in combination with mass spectrometry (LC × LC-MS). In comparison, the combination of ion-mobility spectrometry (IMS) and MS is very fast but very much less orthogonal.13
Figure 1.

Examples of separations by 2D-LC. Top-left, HILIC × RPLC of polyphenols in apple extract;6 top-middle, RPLC × RPLC of tryptic digest of three proteins;14 top-right, HILIC × RPLC of polyether polyols;9 bottom-left, HILIC × RPLC of therapeutic antibodies subunits;15 bottom-middle, SAX × IP-RPLC of aged, synthetic dyes;5 bottom-right, RPLC × RPLC of TCM Dengzhan Shengmai.16 See respective papers for details. Top-left figure reproduced from Development of an improved online comprehensive hydrophilic interaction chromatography × reversed-phase ultrahigh-pressure liquid chromatography platform for complex multiclass polyphenolic sample analysis, Sommella, E.; Ismail, O. H.; Pagano, F.; Pepe, G.; Ostacolo, C.; Mazzoccanti, G.; Russo, M.; Novellino, E.; Gasparrini, F.; Campiglia, P. J. Sep. Sci., Vol. 40, Issue 10 (ref (6)). Copyright 2017 Wiley. Top-middle figure reprinted from J. Chromatogr. A, 1498, Sarrut, M.; Rouvière, F.; Heinisch, S., Theoretical and Experimental Comparison of One Dimensional versus Online Comprehensive Two Dimensional Liquid Chromatography for Optimized Sub-Hour Separations of Complex Peptide Samples, pp. 183–195 (ref (14)). Copyright (2017), with permission from Elsevier. Top-right figure reprinted from J. Chromatogr. A, 1569, Groeneveld, G.; Dunkle, M.N.; Rinken, M.; Gargano, A.F.G.; de Niet, A.; Pursch, M.; Mes, E.P.C.; Schoenmakers, P.J., Characterization of complex polyether polyols using comprehensive two-dimensional liquid chromatography hyphenated to high-resolution mass spectrometry, pp. 128–138 (ref (9)). Copyright (2018), with permission from Elsevier. Bottom-left figure reproduced from Stoll, D. R.; Harmes, D. C.; Staples, G. O.; Potter, O. G.; Dammann, C. T.; Guillarme, D.; Beck, A. Anal. Chem.2018, 90 (9), 5923–5929 (ref (15)). Copyright 2018 American Chemical Society. Bottom-middle figure reprinted from J. Chromatogr. A, 1436, Pirok, B. W. J.; Knip, J.; van Bommel, M. R.; Schoenmakers, P. J., Characterization of synthetic dyes by comprehensive two-dimensional liquid chromatography combining ion-exchange chromatography and fast ion-pair reversed-phase chromatography, pp. 141–146 (ref (5)). Copyright (2016), with permission from Elsevier. Bottom-right figure reprinted from J. Chromatogr. A, 1517, Sheng, N.; Zheng, H.; Xiao, Y.; Wang, Z.; Li, M.; Zhang, J., Chiral Separation and Chemical Profile of Dengzhan Shengmai by Integrating Comprehensive with Multiple Heart-Cutting Two-Dimensional Liquid Chromatography Coupled with Quadrupole Time-of-Flight Mass Spectrometry, pp. 97–107 (ref (16)). Copyright (2017), with permission from Elsevier.
Of course, there are some caveats. LC × LC separations typically take longer than 1D-LC separations (analysis times of 30 min to several hours are common). Successive dilution of the sample during two separations may result in decreased detection sensitivity and compatibility issues may arise, because without further intervention (e.g., “active modulation”, see section Modulation below) the first-dimension (1D) effluent is the second-dimension (2D) injection solvent. Also, dedicated data-analysis and visualization software is needed. Finally, method development is arguably more complex and potentially (much) more time-consuming in LC × LC than in 1D-LC. These obstacles to successful implementation and application of LC × LC techniques and possible remedies have been discussed in detail elsewhere.17
To separate a single peak from a complex matrix or to reduce assignment uncertainty for a specific peak the separation of a single fraction of the 1D effluent in a second dimension suffices. Indeed, such “heart-cut” two-dimensional LC approaches (which we denote with a hyphen, LC-LC) are very powerful tools for obtaining single, pure peaks or for establishing the purity of a specific compound.18−20 In LC × LC all fractions are subjected to two different separations to obtain a comprehensive characterization of the sample. The middle ground is occupied by multiple-heart-cut techniques, in which a number of fractions from across the chromatogram are subjected to the second dimension, and “selective-comprehensive” two-dimensional LC, in which one or more bunches of successive 1D fractions are subjected to the second dimension. The main strengths and weaknesses of heart-cut LC-LC are summarized in the SWOT analysis presented as Table 1. The corresponding analysis for LC × LC is presented as Table 2.
Table 1. SWOT Analysis of Heart-Cut Two-Dimensional Liquid Chromatography (LC-LC).
| strengths | weaknesses |
|---|---|
| • Very high resolving power | • Somewhat increased conceptual and instrumental complexity |
| • Added selectivity from second (“orthogonal”) dimension | • Analysis time is increased (especially when multiple fractions are selected for analysis in the second dimension) |
| • Choice from many different retention mechanisms | • Possibly reduced detection sensitivitya |
| • Enhanced purification or purity assessment of target analytes | • Phase-system incompatibility issuesa |
| • Preparative separations possible | • Method development is relatively straightforward |
| • Greatly reduced uncertainty of peak assignments (in comparison with 1D-LC) | |
| • Readily combined with MS and MS/MS techniques |
| opportunities | threats |
|---|---|
| • Rigorous assessment of peak purity is tantamount in (bio-) pharmaceutical industries | • For qualitative analysis high-resolution hyphenated techniques (LC-MS or LC-MS/MS) are usually preferred |
| • “Spatial” comprehensive two-dimensional (and three-dimensional) LC21 |
These issues may (largely) be addressed by incorporating active-modulation techniques (see section Modulation below).
Table 2. SWOT Analysis of Comprehensive Two-Dimensional Liquid Chromatography (LC × LC).
| strengths | weaknesses |
|---|---|
| • High peak capacities (1 000–10 000) routinely possible | • Added conceptual and instrumental complexity |
| • High peak-production rates (typically 1 peak per second) | • Rather long analysis times (typically 30 min–2 h) |
| • Choice from many different retention mechanisms | • Possibly reduced detection sensitivityb |
| • Added selectivity from second (“orthogonal”) dimension | • Phase-system incompatibility issuesb |
| • Structured, readily interpretable chromatogramsa | • Data-analysis software needed |
| • “Group-type” separations of classes of analytes | • Difficult and time-consuming method developmentc |
| • Readily combined with MS and MS/MS techniques | |
| • Greatly reduced uncertainty of peak assignments (in comparison with 1D-LC) |
| opportunities | threats |
|---|---|
| • Increased need for detailed characterization of complex samples from many fieldsd | • High-resolution hyphenated techniques (LC-MS, LC-IMS-MS, IMS-MS) may compete for certain applicationse |
| • “Spatial” comprehensive two-dimensional (and three-dimensional) LC21 |
In the case of low sample dimensionality.22
These issues may (largely) be addressed by incorporating active-modulation techniques (see section Modulation below).
This may be overcome by using advanced method-development software.
IMS = ion-mobility spectrometry.
The astounding separation power offered by LC-LC and LC × LC entices chromatographers to overcome the challenges mentioned above, and 2D-LC methods are increasingly being implemented in a variety of application areas traditionally served by 1D-LC.
In this review, we consider some 160 applications of two-dimensional LC (2D-LC) techniques that have been published in 2016, 2017, or 2018. Progress in methodology in the same period is discussed in detail. Older publications are cited when needed to provide a foundation for the discussion. While an inventory is made of all applications, we will focus on online application of 2D-LC in our discussions. Much technical progress has been focused on the interface (modulator) between the two separation dimensions. These smart modulation techniques, such as stationary-phase assisted modulation and active solvent modulation, are discussed in detail in the Modulation section. Finally, significant attention will be paid to emerging strategies for developing 2D-LC methods in the Method Development and Optimization Strategies section. For a detailed treatment of the fundamental principles and successful implementation of 2D-LC techniques, readers are referred to useful guides published elsewhere.2,23,24 Following the accepted nomenclature for comprehensive two-dimensional separations,25 we refer to the respective dimensions with a prefix, i.e., 1D refers to the first dimension and 2D to the second dimension.
Modulation
Incompatibility
At the heart of any 2D-LC setup is the modulation interface, which has the function of transferring fractions of the first-dimension (1D) effluent to the second-dimension (2D) column. The most common tool for fraction transfer is a 2-position 8- or 10-port valve equipped with two identical storage loops, which “passively” (see below) sample the 1D separation. Alternatingly, one loop samples the 1D effluent, while the contents of the other loop are injected into the 2D column.
Using the setup of Figure 2, the 1D effluent is the injection solvent of the second dimension. This may give rise to “incompatibility issues”. Regardless of the mode of 2D-LC operation (LC-LC or LC × LC), the order in which the two separation methods are combined is extremely important. The pursuit of maximal orthogonality between two separation dimensions with respect to the sample dimensions often culminates in a challenge to combine two incompatible solvent systems, with the 1D effluent detrimentally affecting the 2D separation.
Figure 2.

Generic scheme of a loop-based, passive-modulation interface for use in 2D-LC. As the 1D effluent is sampled by one loop, the contents of the other loop are, without any further modification, injected into the second dimension. In the configuration shown, the loops are filled and emptied in opposite directions (“backflush” mode). The scheme shows an 8-port valve, but a 10-port valve can also be used.26,27 In (multiple) heart-cut 2D-LC, the loops can be replaced by “decks” each containing an array of sampling loops to allow storage of more than one fraction.
An extreme case arises when the two mobile phases are completely immiscible, but more-subtle incompatibilities often occur. For example, a significant difference in viscosity between the two solvent systems may result in flow instabilities. At the interface of the two solvents, a low viscosity 2D mobile phase can penetrate a high-viscosity injection plug in finger-shaped cones as it percolates through the porous medium of the 2D column. This effect is known as viscous fingering and potentially results in peak deformation and even peak splitting.28,29 However, it seems that this is really only a serious issue in cases of extreme viscosity contrast between the mobile phases used in the two dimensions.30 In our experience, other factors such as a solvent strength mismatch between the two dimensions can be far more problematic in most applications.31
A significant difference in solvent strength may also result in peak deformation or peak splitting. If the 1D effluent is a relatively strong injection solvent in comparison with the 2D eluent, the intended retention mechanism may be disturbed as the analytes are not strongly retained by the stationary phase in the strong injection solvent. A classic example of this problem is the combination of organic size-exclusion chromatography (SEC) and reversed-phase LC (RPLC), where the fully organic 1D effluent can prevent hydrophobic analytes from being retained by the hydrophobic stationary phase. Within the injection plug, size-exclusion conditions prevail, and larger analytes will move faster than the average velocity of mobile-phase molecules. Ahead of the plug, the eluent is weak, causing analytes to slow down and be caught up by the strong-solvent plug. Conversely, analytes at the rear of the plug will disperse in the weak 2D eluent and be retained by adsorption effects in the SEC separation. This effect is known as breakthrough, with an unretained (“breakthrough”) peak eluting with the dead volume and a typically much smaller “real” peak at the normal location.32 However, the potential combination of RPLC and SEC in reverse order may also lead to problems because the aqueous RPLC effluent may induce undesired adsorption effects.33 Components of the 1D effluent matrix may potentially complicate detection after the second dimension as well. One example of such detector incompatibility is the use of salts in the 1D separation with MS or ELSD detection of the 2D, where the salt from the 1D elutes as a concentrated band into the detector. Finally, the 1D effluent may lead to column degradation in the second dimension. For example, an ion-chromatography 1D separation with a sodium-hydroxide mobile phase may not be combined with a conventional 2D reversed-phase separation because silica-based ODS columns usually degrade rapidly in basic eluents. The frequent switching of the modulation valve may also affect the lifetime of the 2D column. The modulation valve may be constructed so as to minimize the pressure pulses that accompany valve switching.34
It is not surprising that much research in the 2D-LC community is devoted to overcoming these incompatibility issues. Generally, this is addressed by making modifications to the modulation process. When some type of action is taken between the two dimensions, other than just “passively” collecting fractions of the 1D effluent as shown in Figure 2, we speak of active modulation. Chromatographers attempt to adjust the 1D effluent matrix to prevent incompatibility issues. In some cases, the application of active-modulation techniques offers opportunities to significantly improve the 2D separation, rather than affecting it negatively. Importantly, active modulation may also result in a concentration of the analyte band prior to injection in the second dimension, thus enhancing the detection sensitivity.
Active-Solvent Modulation (ASM)
In 2017, Stoll and co-workers introduced an active-modulation approach, which they refer to as Active Solvent Modulation (ASM).35 The concept behind this approach is illustrated schematically in Figure 3. In this case, two ports are added to a typical valve used for 2D-LC (see Figure 2), along with a bypass capillary. Also, two rotational positions are added to the two normally used with a conventional valve. Two of the positions (A and C) resemble those shown in Figure 2, where all of the 1D effluent passes through one of the loops and all of the 2D mobile phase passes through the other loop, displacing previously collected 1D effluent into the 2D column. In the two additional positions (B and D), the flow from the 2D pump is split into two parts, one that goes through the loop and one that bypasses the loop, such that the 2D eluent acts as a diluent for the fraction of 1D effluent. The benefit of this approach is best appreciated through chromatographic examples. In some of their most recent work, Stoll and co-workers15 have used ASM to effectively couple HILIC and RP separations of proteins in an online LC × LC format, which is otherwise quite difficult because of the solvent-strength mismatch between the conditions typically used for these two separation modes. Figure 4 shows a comparison of 2D and 2D chromatograms obtained for a partially digested monoclonal-antibody sample, with and without implementation of ASM. The left two panels (A and B) show the 2D chromatograms that are obtained with and without the use of ASM. In panel A the peaks circled in red are due to breakthrough of protein analytes. These proteins elute around the dead time of each 2D separation because of the solvent-strength mismatch. On the other hand, when ASM is used, as shown in panel B, no breakthrough is observed at all, even though the same volume of 1D effluent is ultimately injected into the 2D column in the two cases. This is because in the case of ASM the 1D effluent fraction is diluted 1:2 with water-rich diluent, which lowers the sample concentration of ACN to about 23%, below the starting point in the 2D RP gradient (about 25%). Panel C offers a more focused view of the quality of 2D separations in the two cases.
Figure 3.

Illustration of the function of Active Solvent Modulation (ASM) for coupling the two dimensions of separation in a 2D-LC system. This valve has eight ports and four positions. Postions A and C are functionally identical to those of a conventional 8- or 10-port two position valve (e.g., see Figure 2). In positions B and D, however, part of the flow from the 2D pump is split and travels through the bypass capillary. This portion of the flow joins the stream of fluid exiting the sample loop before the mixture leaves the valve and enters the 2D column. In this way, this split part of the 2D flow acts as a diluent for the 1D effluent fraction injected into the 2D column.
Figure 4.

Comparison of two 2D and 2D chromatograms for LC × LC separations of mAb fragments with (B) and without ASM (A). First dimension separations are in the HILIC mode with about 70% ACN in the eluting mobile phase for the peaks of interest, and 2D separations are in the RP mode with about 25% ACN in the starting mobile phase. In each case, 40 μL of 1D effluent is ultimately transferred to the 2D separation. In case B, the sample is diluted 1:2 with water-rich diluent, such that the total volume injected in each 2D cycle is 120 μL. The 2D chromatograms in panel C are extracted from the 2D chromatograms on the left at the position of the gray dashed line. Reproduced from Stoll, D. R.; Harmes, D. C.; Staples, G. O.; Potter, O. G.; Dammann, C. T.; Guillarme, D.; Beck, A. Anal. Chem.2018, 90 (9), 5923–5929 (ref15). Copyright 2018 American Chemical Society.
In addition to the HILIC × RP separation of proteins described above, ASM has been implemented for separations of peptides by mLC-LC with RP separations in both dimensions,35 quantitative determination of target molecules in polymer matrixes,36 and the separation of water- and fat-soluble vitamins by sLC × LC using HILIC and RP separations.37
As ASM is a relatively new approach, a lot remains to be learned about how to efficiently optimize 2D-LC separations. Some of the early papers contain guidance about the effects of different method parameters relevant to ASM. Additionally, Stoll and co-workers have developed numerical simulation methods that can be used to both make predictions of the effects of different ASM-related method parameters, such as dilution factor and injection volume, on retention and peak width and to visualize what happens inside the 2D column under these conditions.31,38
Stationary-Phase-Assisted Modulation (SPAM)
Another increasingly popular active-modulation strategy relies on the use of low-volume trapping (or “enrichment”) columns, often referred to simply as “traps”, rather than large storage loops (Figure 5). Demonstrated first in LC × LC by Vonk et al.,39 this technique has been referred to as stationary-phase-assisted modulation (SPAM)39 or as focusing modulation.40 Typically, guard columns containing a stationary phase similar to that of the2D column are used as trapping columns.
Figure 5.

Schematic of the two positions of a stationary-phase-assisted modulation (SPAM) interface. Rather than using large storage loops, analytes are effectively filtered out of the 1D effluent using low-volume trapping columns. Optionally, the 1D column effluent may be diluted using a weak eluent to facilitate retention on the traps. Moreover, the waste line may be equipped with a detector to monitor premature elution from the traps during method development. (Multiple) heart-cut 2D-LC setups are possible as long as the multiple traps are identical.
As the 1D effluent is sampled by the modulator (Figure 5), it is envisaged that the analytes are retained by the stationary phase in the traps, whereas the 1D solvent system passes unretained and leaves the chromatographic system. Upon switching of the valve, the 2D mobile phase (gradient program) elutes the trapped analytes as sharp, concentrated bands and introduces these into the second-dimension column. To facilitate sufficient retention on the traps, the 1D effluent flow may be diluted (optionally inserting a mixer) to significantly lower the elution strength of the 1D effluent prior to entering the trap.
The advantages of SPAM include (i) reducing solvent incompatibility issues, thanks to removal of most of the 1D mobile phase,2,40 (ii) improvement of detection sensitivity as a result of analyte focusing on the trapping cartridges,4,41,42 (iii) a decrease in 2D injection volumes which allows the use of short 2D columns without a loss in efficiency, significantly reducing the total analysis time.4,42
However, the use of SPAM is not without disadvantages. It is imperative that all analytes from the 1D fraction are sufficiently retained (i.e., during the full duration of the modulation) to avoid loss of analytes (incomplete recovery) and to prevent discrimination effects. This can be challenging if the various analytes have vastly different chemical properties and a dilution solvent may not always ensure complete trapping of all analytes. In addition, the trapping columns themselves potentially reduce the overall robustness of the system. It is imperative that both traps are identical (and share the same history) to avoid different (“asymmetric”) performance of alternating modulations.43
After struggling with the low loadability of nanoscale 1D and microscale 2D separations, Vonk et al. first implemented SPAM in an LC × LC workflow for the SCX × RPLC–HRMS characterization of peptides.39 The use of the active-modulation technique allowed the authors to use a high-loadability 1D column in combination with a nanoscale 2D column and splitless hyphenation with a high-resolution MS instrument. The authors noted that these developments opened the possibility of online SCX × RPLC–HRMS separations to replace the currently dominant off-line SCX prefractionation followed by LC-MS. Gargano and co-workers recently developed a nanoflow LC × LC system for the separation of intact proteins.44 The combination of silica-based weak cation exchanger run in HILIC mode and RPLC through a SPAM interface followed by HR-MS yielded a highly orthogonal system, targeting a separation based on charge and hydrophilicity in the first dimension and hydrophobicity in the second-dimension. The method allowed the authors to identify twice as many histone proteoforms compared to previous methods.
Recently, Sommella et al. combined HILIC and RPLC for the separation of polyphenols using trapping columns.6 The authors compared the performance of the SPAM approach with passive modulation and concluded that with SPAM higher peak capacities and sensitivities could be obtained. In addition, the use of trapping columns allowed the authors to circumvent the requirement to use microbore columns in the first dimension. Toro-Uribe and co-workers used SPAM in their HILIC × RPLC separation of procyanidins from green cocoa beans.40 The authors diluted the 1D effluent (20 μL min–1) with 100 μL min–1 of weak 2D eluent and observed improved resolving power and less solvent-mismatch issues as the 1D effluent was removed by the SPAM process. That the enhancement of peak intensies can be significant is visible from the comparison shown in Figure 6. This work, by Baglai and co-workers,41 utilized two identical cyano trap columns for improvement of the RPLC × RPLC-MS separation of steroids in bovine urine. As is also visible in Figure 6, a strong enhancement of peak intensities and signal-to-noise ratios was obtained by using SPAM relative to passive modulation. The authors reported an increase in S/N ratio by a factor of 7 and were able to detect 76 compounds using SPAM, relative to 36 for passive modulation.
Figure 6.
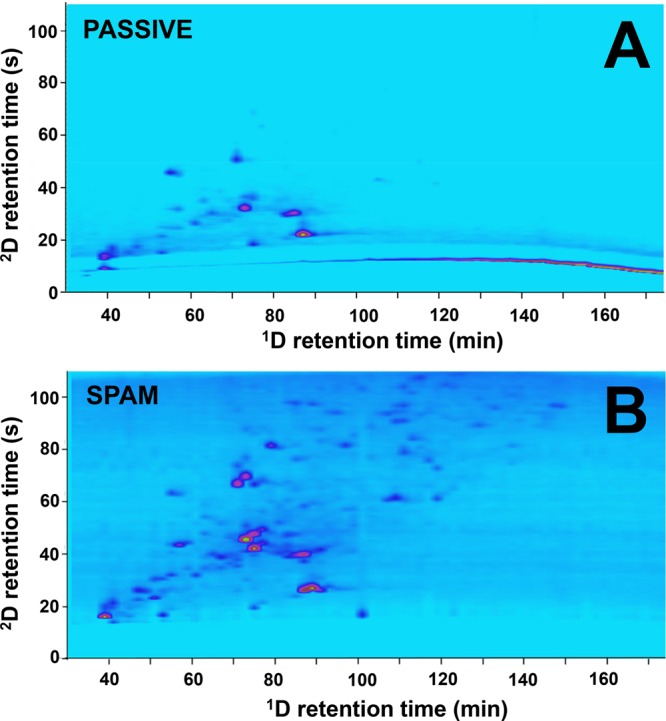
RPLC × RPLC-MS separation of steroids in bovine urine using (A) passive modulation and (B) stationary-phase assisted modulation (SPAM). 76 analytes could be detected using SPAM due to the improvement in S/N, relative to 36 using passive modulation.41 Reprinted from Anal. Chem. Acta, 1013, Baglai, A.; Blokland, M.H.; Mol, H.G.J.; Gargano, A.F.G.; van der Wal, Sj.; Schoenmakers, P.J., Enhancing detectability of anabolic-steroid residues in bovine urine by actively modulated online comprehensive two-dimensional liquid chromatography–high-resolution mass spectrometry, pp. 87–97 (ref (41)). Copyright (2018), with permission from Elsevier.
Jakobsen et al. combined trapping-based active modulation with pulsed-elution of the 1D to increase the flexibility of the 2D-LC separation.42 Pulsed elution utilizes pulses of strong eluent rather than a continuous linear gradient with increasing levels of strong eluent. Using this elution technique, the authors reported the ability to control the elution of analytes in a sufficient number of modulations to prevent loss of 1D peak capacity as a result of undersampling. As the time between pulses could be increased from 1 up to at least 10 min, the 2D analysis was not time-constrained and could be tailored toward increasing the 2D peak capacity (2nc). By combining this strategy with SPAM, the authors furthermore reported a reduction of the additional band broadening that would occur in the 2D separation in case of poor refocusing. The concept was demonstrated in practice by a separation of a fraction of vacuum gas oil.
The potential of actively modulating the 1D effluent is not necessarily restricted to overcoming solvent-compatibility issues. Modulation may also target the analytes. Pirok and co-workers combined an aqueous hydrodynamic chromatography (HDC) separation of a polymeric nanoparticle dispersion with a separation of the constituent polymers using organic SEC.43 The authors first achieved online sample transformation by diluting the aqueous 1D effluent with tetrahydrofuran in an Agilent Jet Weaver V35 mixer. The composition of THF and aqueous buffer was fine-tuned to destabilize the particles so as to obtain the constituting polymers, while preserving a sufficiently large fraction of buffer to facilitate retention on the trapping columns in the modulator. The THF used in the 2D SEC separation finally eluted the polymers for analysis. A second diode-array detector was used to monitor the modulation effluent for eventual premature elution of trapped polymers.
SPAM is also frequently applied in heart-cut 2D-LC separations.45−48 Chen et al. used SPAM in their heart-cut SCX-RPLC-MS/MS separation of tobacco-specific N-nitrosamines in cigarette smoke.46 Trapping was facilitated by diluting the 1D effluent with a weak-eluent and by adjusting the pH.
The implementation of SPAM in 2D-LC is far from optimized, and its effectiveness can be further improved by miniaturizing the trap volumes,39 enhancing the robustness of the traps,43 and by developing micromixers to combine two solvent systems rapidly and effectively.49
Other modulation strategies
Aside from ASM and SPAM, other active-modulation techniques have also been introduced, although none of these is currently widely applied. Recent examples are evaporative membrane modulation50 and longitudinal on-column thermal modulation.51 A completely different approach that was recently proposed is fractionized stacking and sampling, where the 1D effluent is split in a number of segments prior to introduction to the second-dimension column.52 The last approach that we will discuss is vacuum-evaporation modulation (VEM), which was developed by the group of Guan.53 The setup is illustrated in Figure 7. It was mainly devised to overcome incompatibility problems encountered during the combination of normal-phase LC (NPLC) and RPLC separations. Briefly, the 1D effluent is passed through a heated loop (Figure 7, left, blue) which in turn is routed to a vacuum outlet. The strong NPLC solvents are envisaged to evaporate rapidly as a result of the increased temperature and reduced pressure and the (nonvolatile) analytes are thought to be deposited on the wall of the loop. Upon switching (Figure 7, right, red) the 2D eluent is introduced, which, with the help of the heat, quickly redissolves the analytes so that they are introduced into the 2D column. Since its introduction, the VEM interface was used in a number of applications in noncomprehensive mode, including the analysis of Traditional Chinese Medicines (TCM)54 and, more recently, the analysis of lipid species in human plasma55 and mouse serum.56 While the potential of VEM has been clearly demonstrated, studies in its fundamental principles and limitations are required, including (i) the required volatility of the to-be-evaporated solvent, (ii) the extent of accidental loss of volatile analytes, and (iii) the rate of dissolution for large analytes that are difficult to dissolve. Data demonstrating to what degree different analytes are deposited on the loops during vacuum evaporation is needed to provide insight in these above fundamental limitations.
Figure 7.

Schematic of a vacuum-evaporation modulation (VEM) interface. 1D column effluent is thought to be rapidly evaporated due to the combination of supplied heat and applied vacuum, so that the analytes are deposited in the loop. Upon switching, the 2D eluent redissolves the analytes for introduction into the 2D column. Based on the work of ref (54).
Method Development and Optimization Strategies
Since the introduction of single heart-cut (LC-LC)57 and comprehensive (LC × LC)58 2D-LC separations in the late 1970s as online, automated methods, these two modes of 2D separation have been the dominant implementation of the method. However, in the past 10 years or so we have seen the development of hybrid modes of 2D separation that combine features of both heart-cutting and comprehensive 2D separations. Figure 8 illustrates the differences between the conventional single heart-cut and comprehensive modes, and the two hybrid modes multiple heart-cutting (mLC-LC)59 and selective comprehensive (sLC × LC)60 2D separation.
Figure 8.

Illustration of four different modes of 2D-LC separation. For a more thorough explanation of the similarities and differences between these different modes of 2D separation see ref61.
A thorough discussion of the similarities and differences between these different modes of 2D separation was presented recently by Stoll and co-workers61 and will not be repeated here. One of the most practically important virtues of the mLC-LC implementation as it is drawn in Figure 8B relative to the traditional single heart-cut approach is that the processes of sampling the 1D separation and separating previously collected fractions of 1D effluent in the second dimension are executed in parallel. This provides tremendous flexibility to the user during method development, particularly with respect to increasing 2D analysis times in the interest of improving the resolving power that each 2D separation contributes to the overall 2D separation. Nevertheless, one of the disadvantages of the heart-cutting approach, whether single or multiple heart-cuts are made, is that analytes that had been resolved by the 1D separation can be mixed back together if the fractions collected are larger (in time or volume) than the volume of the peaks as they exit the 1D column. This problem is referred to as “undersampling”.62 Readers interested in more detail on this issue are referred to a number of previous articles that address the problem thoroughly.24,63 The desire to avoid or at least minimize the effect of this problem gives rise to sLC × LC, as illustrated in Figure 8C. Here, regions of interest in the 1D separation are first finely sampled to avoid remixing of previously separated analytes. These fractions are temporarily stored in an array of sampling loops or traps prior to subsequent further separation in the second dimension. The best answer to the question, which mode of 2D separation should I use?, is entirely application dependent but depends strongly on the number of target analytes of interest in a particular assay. Methods with a small number of targets are well served by single heart-cut methods, whereas applications in which the entire sample is of interest, such as “-omics” types of applications, benefit most from fully comprehensive 2D separations. The mLC-LC and sLC × LC modes are typically most suitable for applications involving some 10–30 target compounds.
Method Development for Noncomprehensive Modes
When developing a single heart-cut application (i.e., Figure 8A), two very important questions encountered in method development are (1) What volume of 1D effluent will be transferred to the second dimension? and (2) What will be the dimensions of and operating conditions for the 2D column? In applications where the analyte of interest is abundant and detection sensitivity is not a primary concern, the user has a lot of flexibility. In these cases, one should minimize the volume of 1D effluent transferred to minimize the risk of undersampling the first-dimension separation and to avoid compromising the separation performance of the 2D column. However, when the analyte of interest is present at a low concentration, we must consider ways to transfer large volumes of 1D effluent to the 2D column, while minimizing the elution peak volume of the analyte from the 2D column. In this context, the active modulation strategies discussed above may be very useful because they enable transfer of relatively large volumes of 1D effluent without compromising the performance of the 2D separation.
In addition to avoiding undersampling, as already discussed above, one of the attractive aspects of the sLC × LC mode of separation is that it enables a kind of piecewise transfer of the entire volume of a 1D peak that would otherwise be too large to transfer in a single fraction. For example, suppose one wants to study an unknown peak that suddenly appears in an existing 1D separation that is carried out using a 4.6 mm i.d. column at 1 mL/min. If the peak of interest is 10-s wide at the base (i.e., 6σ level), then transferring “all” of the peak would require a sample loop of about 200 μL. However, in the sLC × LC mode the peak can be split up into a number of fractions with volumes that are more convenient. For example, the peak may be sampled five times, each time using a 40-μL loop. This kind of flexibility is valuable from a method-development point of view and enables precise quantitation, even for 1D peaks with large volumes.64 However, this line of thinking quickly leads to the question, what is the “right” number of fractions to transfer per 1D peak to use as a starting point during method development? In a series of two recent papers, Davis and Stoll3,65 addressed this question using simulations to study the effect of the number of transfers of each 1D peak on the probability of fully resolving sample mixtures of increasing complexity by mLC-LC/sLC × LC. Interestingly, they found that, from a probabilistic point of view, making a single transfer of each 1D peak maximizes the probability that the resulting 2D separation will fully resolve a random mixture of analytes. This outcome is observed even after undersampling is accounted for. Nevertheless, here are two important caveats to keep in mind here. First, the simulations used in this work did not take into account the volume of 1D peaks, and therefore splitting the 1D peak into multiple fractions may still be warranted for practical reasons, even if this is not optimal from the point of view of resolving power. Second, the conclusions of these studies are based on the outcomes of tens of thousands of simulations of 2D separations of random mixtures. Even if the simulations suggest the use of a single transfer of each 1D peak as a starting point, there undoubtedly will be specific separation scenarios where transferring multiple fractions of a 1D peak is needed to avoid undersampling and the concomitant loss of 1D resolution.
Comprehensive Mode
The step toward comprehensive 2D-LC (LC × LC) is accompanied by a number of additional constraints on the method. For the method to be truly comprehensive, it is required that all of the analytes are transferred completely from the 1D effluent to the 2D column. In the case of passive modulation, this requires that the 1D flow rate is adjusted to match the size of the loop.
One focus within the chromatographic community has been the development of faster 2D separations. Shorter modulation times imply that more fractions can be collected and transferred, thus decreasing the likelihood of undersampling of the 1D separation and/or reducing the total analysis time. In this context it is not surprising that RPLC proves to be the most popular mechanism in LC × LC (see Table 3 below). Indeed, RPLC offers many advantages when used as 2D separation mechanism2, including very fast gradient-elution separations, thanks to the swift column equilibration.66 The use of an ion-pairing reagent does not necessarily lead to impractically long gradient times. For the separation of synthetic dyes by SAX × IP-RPLC,5 Pirok and co-workers combined a programmed increase of the fraction of organic modifier in the 2D mobile phase with a proportional decrease in the ion-pair concentration.
Table 3. Overview of 2D-LC Applications from 2016 until October 2018a.
| application | mode | 1D | 2D | detection | remarks | ref | ||
|---|---|---|---|---|---|---|---|---|
| Biopharma | ||||||||
| Antibody-drug conjugates | N | - | P | SEC | RPLC | UV–vis | (93) | |
| Antibody-drug conjugates | N | - | P | RPLC | RPLC | UV–vis | (94) | |
| Antibody-drug conjugates | N | - | P | SEC | RPLC | UV–vis | (95) | |
| Antibody-drug conjugates | N | - | P | HIC | RPLC | MS | (96) | |
| Antibody-drug conjugates | N | × | P | RPLC | RPLC | MS, UV–vis | (97) | |
| N | - | P | RPLC | RPLC | ||||
| Antibody-drug conjugates | N | × | P | HIC | RPLC | MS, UV–vis | (98,99) | |
| Antibody-drug conjugates | N | × | P | HIC | SEC | MS | (100) | |
| Antibody-drug conjugates (free species) | N | - | P | SEC | RPLC | UV–vis | (93) | |
| Antibody-drug conjugate (free species) | N | - | P | MM | RPLC | MS | (101) | |
| Biopharmaceuticals (impurities) | N | - | N | RPLC | RPLC | MS | (102) | |
| Bovine insulin (degradants) | N | - | A | RPLC | RPLC | UV–vis | (35) | |
| Monoclonal antibodies | N | - | P | Affinity | SEC | UV–vis | (103) | |
| Monoclonal antibodies | N | - | P | WCX | SEC | UV–vis | mLC-LC | (104) |
| Monoclonal antibodies | N | × | P | SCX | RPLC | MS | (105) | |
| Monoclonal antibodies | N | - | P | SCX | RPLC | MS | (106) | |
| Oligonucleotides (impurities) | N | - | S | SEC | IP-RPLC | MS | Utilized a 50 mm × 4.6 mm C18 trap. | (47) |
| N | × | S | RPLC | IP-RPLC | ||||
| N | - | S | SAX | IP-RPLC | ||||
| N | × | S | IP-RPLC | IP-RPLC | ||||
| Therapeutic antibodies | N | - | P | IEX | RPLC | MS | (107) | |
| Therapeutic antibodies | N | × | A | HILIC | RPLC | MS | (15) | |
| Environmental | ||||||||
| AChE inhib. in wastewater effluent | N | × | P | RPLC | RPLC | MS, UV–vis | 2D effluent fractionated and studied. | (108) |
| Emerging contaminants | N | × | P | RPLC | RPLC | MS | Column selection by orthogonality metrics. | (84) |
| Polycyclic aromatics | N | × | P | RPLC | RPLC | UV–vis | Theoretical gradient optimization. | (109) |
| Food | ||||||||
| Black chokeberries pomace | N | × | P | HILIC | RPLC | MS, UV–vis | (110) | |
| Overripe fruits (carotenoids) | N | × | P | NPLC | RPLC | MS, UV–vis | (111,112) | |
| Hop cones, pellet extracts | N | × | P | RPLC | RPLC | MS, UV–vis | (113) | |
| N | × | P | HILIC | RPLC | ||||
| Soybean (Kunitz Trypsin Inhibitor) | F | - | M | WAX | SEC | MS | (114) | |
| Corn oil (Triacylglycerols) | N | × | P | AgLC | RPLC | MS | Compared data-analysis techniques. | (115) |
| Tea, Grape seed, red wine (extracts) | N | × | P | HILIC | RPLC | IMS-MS | Optimized LC × LC × IMS-MS separation. | (80) |
| Wine (polyphenols and contaminants) | N | × | P | RPLC | RPLC | MS, UV–vis | (116) | |
| Metabolites | ||||||||
| Acyl-Coenzyme A (mouse liver) | N | - | P | RPLC | RPLC | MS | Two parallel 2D columns. | (117) |
| Amino acids (of gramicidin, bacitracin) | N | × | P | Chiral | Chiral | UV–vis | Amino acids were derivatized. | (118) |
| Amino acids (tea, amino acids) | F | - | M | RPLC | Chiral | UV–vis | (119) | |
| Antioxidants (Malus hupehensis) | F | - | M | HILIC | RPLC | MS, UV–vis | (120) | |
| Breast milk (fluoxetine, norfluoxetine) | N | - | P | SEC | Chiral | MS | (121,122) | |
| N | - | P | SEC | RPLC | ||||
| Flavonoids | F | - | M | HILIC | RPLC | MS | (123) | |
| Flavonoids | N | × | P | RPLC | RPLC | UV–vis | Microprep. to investigate recovery. | (124) |
| Flavonoids from licorice | F | - | M | NPLC | RPLC | MS, UV–vis | Preparative scale. | (125) |
| Furanocoumarins | N | × | P | RPLC | RPLC | UV–vis | Compared multivariate curve-resolution strategies in LC × LC. | (85) |
| Lipidomics (brain tissue) | N | - | S | HILIC | RPLC | MS | (126) | |
| Lipidomics (human plasma) | N | - | V | NPLC | RPLC | MS | (55,127) | |
| Lipidomics (mice serum) | N | - | V | NPLC | RPLC | MS | (56,128) | |
| Lipidomics (rat plasma) | F | - | M | MM | RPLC | MS | (129) | |
| Lipidomics (cyanobacteria) | N | - | V | NPLC | RPLC | MS | (130) | |
| Lipidomics (human plasma) | N | × | P | RPLC | HILIC | MS | Comparison with LC-TIMS-MS. | (13) |
| Lipids (rice) | N | × | P | RPLC | HILIC | MS | (131) | |
| Metabolites (Escherichia coli) | N | × | P | RPLC | RPLC | MS, UV–vis | (132) | |
| Metabolites (green cocoa beans) | N | × | S | HILIC | RPLC | MS, UV–vis | (40) | |
| Metabolites (licorice) | N | × | P | HILIC | RPLC | MS, UV–vis | ZIC-HILIC | (133) |
| Metabolites (microbial) | N | × | S | RPLC | RPLC | UV–vis | (134) | |
| Metabolites (Panax notoginseng leaves) | F | × | M | HILIC | RPLC | MS, UV–vis | (135) | |
| Metabolites (rice plant) | N | × | P | HILIC | RPLC | MS | (136) | |
| Metabolites (Vitamin D, human serum) | N | - | S | RPLC | RPLC | MS | PFP in 1D, C18 in 2D | (137) |
| Metabolites and lipids (human plasma) | N | - | P | RPLC | RPLC | MS | Online prefractionation before 1D | (138) |
| Metabolomics (Glycyrrhiza glabra) | N | × | P | RPLC | RPLC | MS, UV–vis | Multisegment shifting gradients. | (139) |
| Phenolic acids | N | × | E | RPLC | RPLC | UV–vis | Evaporative membrane modulation. | (50) |
| Phenolic acids and flavonoids | N | × | P | MM | RPLC | UV–vis | Simultaneous HILIC and RP in 1D | (140) |
| Phenolic compounds (Grapevine canes) | N | × | P | HILIC | RPLC | MS, UV–vis | Compared a number of selectivities. | (141) |
| Plant extracts and coffee | N | - | P | RPLC | RPLC | MS | Special concept of LC+LC. | (142) |
| Polyphenols | N | × | S | HILIC | RPLC | MS | (6) | |
| Polyphenols in red raspberry fruits shoots | N | × | P | RPLC | RPLC | MS | (143) | |
| Procyanidins (cocoa) | N | × | P | HILIC | RPLC | UV–vis | Reported kinetic optimization tool. | (79) |
| Proteinogenic amino acids | N | - | P | RPLC | Chiral | UV–vis | (144) | |
| Pyrrolizidine alkaloids | N | - | P | RPLC | RPLC | MS | 1D at pH = 3, 2D at pH = 10 | (145) |
| Steroids | N | × | P | TRLC | RPLC | UV–vis | TRLC as 1D facilitates 2D focusing. | (77) |
| Testosterone (human serum) | N | - | P | RPLC | RPLC | MS | (146) | |
| Urine (bovine) | N | × | S | RPLC | RPLC | MS | (41) | |
| Urine (Steroids, Sulphonamides) | N | × | P | RPLC | RPLC | MS | CN, BEH and Phenyl studied for 1D | (147) |
| Miscellaneous | ||||||||
| Bioactives in plant extracts | N | × | S | RPLC | RPLC | ELSD | Fractionation system | (148) |
| Heavy-oil fractions | N | × | P | NA-RPLC | NA-RPLC | CAD, UV–vis | CN, PFP and BiPh studied for 1D | (149) |
| Household dust and dryer lint | N | × | P | RPLC | RPLC | MS | (150) | |
| Lignin phenols | N | × | S | RPLC | SFC | UV–vis | (151) | |
| Synthetic cannabinoids | N | × | P | RPLC | RPLC | MS | (152) | |
| Synthetic dyes | N | × | P | SAX | IP-RPLC | UV–vis | (5) | |
| Tobacco | N | - | S | SCX | RPLC | MS | (46,153) | |
| Tobacco (snus) | N | - | P | RPLC | RPLC | MS | (154) | |
| Vacuum-gas oil fraction | N | × | S | RPLC | RPLC | MS | Pulsed elution in 1D | (42) |
| Natural Medicines | ||||||||
| Phlorotannins in brown algae | N | × | P | HILIC | RPLC | MS, UV–vis | Use of Hansen solubility parameter to study extraction selectivity. | (155) |
| Brown seaweed | N | × | P | HILIC | RPLC | MS, UV–vis | (156) | |
| TCM (Additives) | N | - | S | Affinity | RPLC | UV–vis | (157,158) | |
| TCM (Dracocephalum heterophyllium) | F | - | M | RPLC | HILIC | MS, NMR | (159) | |
| TCM (U. rhynchophylla) | F | - | M | MM | MM | UV–vis | (160) | |
| TCM (Curcuma kwangsiensis) | N | × | P | RPLC | RPLC | MS | (161) | |
| TCM (Flos Carthami, dried flowers) | N | × | S | MM | RPLC | UV–vis | 1D: SEC-RPLC | (162) |
| TCM (Gardenia jasminoides Ellis) | F | × | M | RPLC | HILIC | MS | (163) | |
| TCM (Gegen-Qinlian Decoction) | N | × | P | RPLC | RPLC | MS | (164) | |
| TCM (Notoginseng total saponins) | F | × | M | HILIC | RPLC | MS | (165) | |
| TCM (Oxytropis falcata) | F | - | M | RPLC | RPLC | UV–vis | Preparative scale | (166) |
| TCM (Salvia miltiorrhiza) | F | × | M | HILIC | RPLC | MS | (167) | |
| TCM (Salvia miltiorrhiza) | N | × | P | RPLC | RPLC | MS | (168) | |
| TCM (sapinins and alkaloids) | F | - | M | RPLC | RPLC | MS | (169) | |
| TCM (saponins in Gleditsia sinensis) | N | × | P | RPLC | RPLC | MS | (170) | |
| TCM (Sphaerophysa salsula) | F | - | M | RPLC | HILIC | NMR,UV–vis | Preparative scale | (171) |
| TCM (toad skin) | F | × | M | NPLC | RPLC | MS | (172) | |
| TCM (Tropane alkaloids) | N | - | P | RPLC | SCX | UV–vis | (173) | |
| TCM (Xuebijing) | N | - | P | RPLC | RPLC | MS, UV–vis | (174) | |
| TCM (Zhibai Dihuang Granule) | F | × | M | SCX | RPLC | MS | (175) | |
| TCM (Denzhan Shenmai) | N | × | P | RPLC | RPLC | MS | Integrated data from LC × LC with that from heart-cut 2D-LC. | (16) |
| N | - | P | RPLC | Chiral | ||||
| TCM (Ginseng powder) | N | - | P | HILIC | RPLC | UV–vis | (52) | |
| TCM (Ginkgo biloba) | F | × | P | HILIC | RPLC | MS | (176) | |
| Peptides | ||||||||
| Peptide biomarkers (rat urine) | F | - | M | RPLC | RPLC | MS | (177) | |
| Peptic digests | N | - | S | RPLC | MM | MS | (178) | |
| Peptides | N | × | P | RPLC | RPLC | MS, UV–vis | (14) | |
| Protein digests (plasma) | F | - | M | RPLC | RPLC | MS | (179) | |
| Tryptic peptides | N | - | S | SCX | RPLC | MS | DCM instead of Gradient | (180) |
| Tryptic peptides (Cortical neurons) | N | × | S | SCX | RPLC | MS | (181) | |
| Tryptic peptides (Elaeis Guineensis Jacq) | N | - | S | RPLC | RPLC | MS | (182) | |
| Tryptic peptides (Flammulina velutipes) | F | - | M | RPLC | RPLC | MS | (183) | |
| Tryptic peptides (human tissue, cancer) | N | - | P | RPLC | RPLC | MS | (184) | |
| Tryptic peptides (mitochondrial proteins) | F | - | M | SCX | RPLC | MS | (185) | |
| Tryptic peptides (lung adenocarcinoma) | F | - | M | SCX | RPLC | MS | (186) | |
| Tryptic peptides (Proteus mirabilis) | F | - | M | RPLC | RPLC | MS | (187) | |
| Tryptic peptides (Salmonella bacteria) | F | - | M | SCX | RPLC | MS | (188) | |
| Tryptic peptides (serum) | F | - | M | SCX | RPLC | MS | (189) | |
| Tryptic peptides (serum) | F | - | M | SCX | RPLC | MS | (190) | |
| Peptides | N | × | P | RPLC | SEC | UV–vis | Studied stop-flow effects in 1D | (191) |
| Peptides | N | - | P | Chiral | RPLC | FLD, MS | (192) | |
| N | - | P | Chiral | Affinity | ||||
| Peptides | N | × | P | SEC | RPLC | UV–vis | 1D: Stop-flow; 1D dispersion studied. | (193) |
| Peptides | N | - | P | RPLC | RPLC | MS, UV–vis | 2D-LC used as desalting tool. | (194) |
| Pharmaceuticals | ||||||||
| Antibiotic drug (cefonicid sodium) | N | - | P | RPLC | RPLC | MS | (195) | |
| Antibiotic residues in dairy products | N | - | P | HILIC | RPLC | MS | (196) | |
| Antibiotics | N | - | P | RPLC | RPLC | MS | (197) | |
| Beta-blockers in human plasma | N | - | P | SEC | RPLC | FLD | RAM as 1D for prefractionation. | (198) |
| Desonide cream | N | - | P | RPLC | RPLC | MS | Strong salt buffer in 1D | (199) |
| Parental drug microdosing vehicle | N | - | P | RPLC | RPLC | MS, UV–vis | (200) | |
| Pharmaceutical materials | N | - | P | RPLC | RPLC | MS, UV–vis | (201) | |
| Pharmaceuticals | N | - | P | RPLC | SFC | UV–vis | sLC × SFC | (202) |
| Pharmaceuticals | N | × | P | Chiral | Chiral | UV–vis | (203) | |
| N | - | P | RPLC | Chiral | ||||
| Pharmaceuticals | N | - | S | RPLC | SFC | MS, UV–vis | (18) | |
| Pharmaceuticals | N | × | P | RPLC | RPLC | MS, UV–vis | Applied theoretical optimization. | (204) |
| Pharmaceuticals, Metabolites | N | - | S | RPLC | Chiral | MS | Chiral at SFC conditions. | (45) |
| Therapeutic drug (in human plasma) | N | - | S | RPLC | RPLC | UV–vis | SCX as trapping column. | (205) |
| Vidarabine monophosphate | N | - | S | RPLC | MM | MS | (206) | |
| Vitamins | N | - | A | HILIC | RPLC | UV–vis | sLC × LC | (37) |
| Polymers | ||||||||
| Oligomers (Oxidzed waxes) | N | × | P | NPLC | SEC | ELSD | 1D and 2D at high temperature. | (207,208) |
| Polyether polyols | N | × | P | HILIC | RPLC | MS | (9) | |
| Polymeric nanoparticles | N | × | S | HDC | SEC | UV–vis | (43) | |
| Synthetic polymers (Polystyrene/Polybutadiene Block Copolymers) | N | × | S | NA-TGIC | SEC | UV–vis | (209) | |
| N | × | S | RP-TGIC | LCCC | ||||
| Synthetic polymers (novolac) | N | × | A | SEC | RPLC | UV–vis | (36) | |
| Synthetic polymers (branched poly (bisphenol A-carbonate)) | N | × | P | LCCC | SEC | LS, RID, UV–vis, VI | MALDI used off-line. LC × LC Correlated with Monte Carlo simulations. | (210,211) |
| Synthetic polymers (HEUR) | N | × | P | SEC | RPLC | ELSD | (212) | |
| Synthetic polymers (nonlinear) | N | × | P | NPLC | SEC | LS, RID, VI | (213) | |
| Synthetic polymers (poloxamers) | N | × | P | LCCC | LCCC | ELSD | Use two 2D columns. | (214) |
| Synthetic polymers (polystyrene) | N | × | P | NA-RPLC | SEC | UV–vis | 2D-col with longitudinal porosity gradient. | (215) |
| (Intact) Proteins | ||||||||
| Intact histone proteoforms | N | × | S | MM | RPLC | MS | Nanoflow LC × LC; 1D WCX-HILIC | (44) |
| Intact proteins | N | × | P | SCX | RPLC | UV–vis | Application of multichannel detector216 | (216−218) |
| Intact proteins and protein digests | N | × | P | SCX | IP-RPLC | MS, UV–vis | Photografted monolith for SCX. Optimized various parameters. | (219) |
| Metaproteomics (soil) | N | - | S | SCX | RPLC | MS | (48) | |
| Proteins in human plasma | N | × | S | SAX | RPLC | None | Array (8) of 2D columns, Fractionation | (220) |
| Surfactants | ||||||||
| Ethoxylate phosphate surfactants | N | × | S | HILIC | RPLC | UV–vis | (4) | |
| Ionic surfactants | N | × | P | MM | RPLC | CAD | 1D: WCX-RPLC | (2) |
| Nonionic surfactants in pharmaceuticals | N | × | P | HILIC | RPLC | ELSD, MS | (221) | |
| Polymeric dispersants in detergents | N | - | P | SEC | RPLC | ELSD | (222) | |
N = Online, F = Offline, × = Comprehensive, - = Heart-cut, P = Passive modulation (empty loops), A = Active-Solvent Modulation (ASM), E = Evaporation membrane, S = Active stationary-phase assisted modulation (SPAM), V = Vacuum-evaporation modulation (VEM), M = Manual/No modulation. CAD = charged-aerosol detection, ELSD = evaporative light-scattering detection, FLD = fluorescence detection, LS = light scattering, MS = mass spectrometry, RID = refractive-index detection, VI = viscometry. LS, RID, and VI are typically combined to obtain “triple detection”. See Modulation section for a detailed discussion of the different modulation techniques.
The acceleration of size-exclusion chromatography (SEC) for 2D separations has recently also received attention. Separation in SEC is thought to be promoted by the largest possible pore volume,67 and as a result the polymer-separation community has largely ignored the introduction of core–shell (or superficially porous) particles, which are now routinely applied in LC due to their superior efficiency.68,69 Recent studies suggest that, despite the decrease in pore volume, the increase in efficiency yields similar or better resolution in a shorter time.70,71 This type of stationary phase was applied in a second-dimension separation of acrylate polymers. The authors exploited the possibility of overlapping injections, as no separation is expected to occur outside the SEC range of the column.43
In this context, the demonstration of subsecond (isocratic) HILIC, chiral, and achiral separations by Wahab et al. represent an attractive concept.72 The authors packed superficially porous particles in 5 mm-long columns. While the results were promising, it was noted that modified UHPLC hardware with minimized extra-column dispersion was required to successfully realize subsecond separations. In addition, sufficiently high detection sampling frequencies are required to accurately describe the shape of elution bands.
A completely different approach to LC × LC is based on spatial separations. In a spatial separation, analytes are not eluted from a column but separated “spatially” because they reach different locations. In a second step they may be eluted from a flat bed or a series of channels in an orthogonal direction. In the latter case there is the potential for a third dimension. The fundamental advantage of using a spatial separation in the first dimension is that all 2D separations can be performed simultaneously, rather than sequentially. As a result, spatial 2D (and 3D) separations are fundamentally very attractive, up to a point that a peak capacity approaching one million has been predicted for a spatial 3D-LC.73 However, spatial separations at elevated pressures have hardly been developed yet. Early studies illustrate that some serious obstacles need to be overcome.74,75 Most importantly, to realize successful LC × LC with a spatial 1D separation we need to find ways to confine the flow to the 1D separation channel.
Very recently Adamopoulou et al. described a potentially revolutionary two-dimensional insertable separation tool (TWIST).76 The 1D spatial separation can take place in the TWIST channel. After turning the TWIST, a series of fractions can be sent simultaneously from different locations along the 1D channel to a series of 2D channels. This approach has potential advantages, due to its simplicity and the potential for highly selective (and ultimately high-resolution) separations in a relatively short time. However, the concept is quite new and still highly immature.
Other technological advancements include the introduction of new modulation strategies to help overcome incompatibility and undersampling issues, as discussed in the previous section. Baert et al. recently advocated the use of temperature-responsive LC (TRLC) in purely aqueous environments as 1D separation, in combination with RPLC in the second dimension.77 Their concept relied on the use of temperature-responsive stationary phases, which exhibit strong changes in retention behavior upon small changes in temperature. The purely aqueous 1D mobile phases were ideally compatible with a 2D RPLC separation. Using a sample containing neutral organic analytes the authors demonstrated that undersampling constraints could be relaxed and complete analyte refocusing could be achieved.
Optimization
There are a variety of useful studies that provide general guidelines for developing LC × LC methods.2,23,78 However, genuine optimization of methods is much more complex.2,239 This is reflected in different interpretations of the word “optimization”. Chromatographers may have specific objectives, such as improving peak shapes or enhancing resolution in targeted sections of the separation space. Specific adjustments based on a trial-and-error approach may suffice to answer the analytical question at hand. In untargeted optimization, the most-efficient route toward optimal conditions is less-clearly defined. In the absence of a specific question, this type of optimization encompasses maximizing or minimizing quality descriptors (e.g., peak capacity and analysis time, respectively).
Quality descriptors are objective parameters that quantify specific performance properties, such as orthogonality, peak capacity, dilution factors, resolution, etc. Readers seeking to learn more about these and other quality descriptors are referred elsewhere.2 Often a known (theoretical) relationship exists between a method parameter and a quality descriptor. For example, increasing the particle diameter of the packing material of either dimension (while keeping all other parameters constant) results in a reduced pressure drop but also in a decreased efficiency and peak capacity. Changing one parameter typically also affects other parameters. In the case of passive modulation, changing the 1D flow rate affects the required loop volume, which in turn affects the suitable 2D column dimensions and, consequently, the 2D flow rate. Sarrut et al. tried to create a network diagram to visualize the relations between a large number of parameters.7 They certainly succeeded in illustrating the complexity of LC × LC method development. The number of possible method parameters is much larger in 2D-LC than in 1D-LC, where method development is already thought to be challenging. This sheer complexity greatly encumbers the method development, increasing the time, effort, and knowledge required to arrive at an acceptable solution. When developing a method “manually”, using a trial-and-error process as a solution is unlikely to be truly optimal. Therefore, several groups work on computer-aided method-development tools, which aim to significantly accelerate method development using multiobjective algorithms.
Vivó-Truyols et al. proposed the use of Pareto optimization for sample-independent optimization of two-dimensional chromatographic separations.8 In this approach, multiple method parameters (e.g., gradient slope, column dimensions, flow rates) are varied and, using theoretical relations, their impact on objective parameters is assessed. The Pareto-optimization strategy involves defining two or more of such objective parameters. A point is considered Pareto optimal if no other point exists that has superior values for all objective parameters. All other points are suboptimal. The collection of Pareto-optimal points forms the Pareto-optimal front. Vivó-Truyols and co-workers argued that this approach was eminently suited to deal with the trade-offs between different objectives, such as peak capacity, analysis time, and dilution factors.8 They took the loss of peak capacity due to undersampling the 1D separation and due to large injection volumes in the 2D into account. The method yielded optimal values for a large number of parameters including the 1D and 2D column particle sizes and diameters and the modulation time. Sarrut et al. applied a systematic optimization approach to a real RPLC × RPLC separation of peptides.7 More recently, Muller et al. reported a predictive kinetic optimization program to derive the optimum column combinations and chromatographic conditions for a HILIC × RPLC separation of procyanidins.79 The method was also applied in a different study where a phenolic extract of red wine was separated using the Pareto-optimized chromatographic method but now hyphenated with IMS-MS (Figure 9).80
Figure 9.
HILIC × RP-LC × IMS-MS TIC separation of a phenolic extract of red wine. The chromatographic method was optimized using predictive Pareto optimization.79 Reproduced from Venter, P.; Muller, M.; Vestner, J.; Stander, M.A.; Tredoux, A.G.J.; Pasch, H.; de Villiers, A. Anal. Chem.2018, 90, 11643–11650 (ref (80)). Copyright 2018 American Chemical Society.
The optimization of physical properties, as described above, is pivotal for the design of efficient 2D-LC separation systems. Sample-independent optimization procedures typically aim to maximize quality descriptors that are largely independent of the sample, such as the peak capacity. Resolution in chromatography is known to depend on three factors, viz., selectivity, retention, and efficiency. In sample-independent optimization, the focus is on efficiency, maximizing the plate counts in the two dimensions and ultimately the overall peak capacity. As we discussed in the introduction, the peak capacity must be very high relative to the number of analytes in the mixture if the analytes follow Poison statistics. However, very large gains are possible if the retention and selectivity are optimized. If we understand the properties of the sample, conditions can be selected such that all components are eluted with optimal k values. In LC this is often achieved by establishing suitable gradient parameters. If the number of analytes is manageable or if (group-type) selectivity is sought for a limited number of component classes, the selectivity of the LC × LC separation may be optimized.
Such sample dependent optimization is being pursued by Pirok and co-workers.81 Their PIOTR program is based on the realization that a linear relationship between a retention parameter (such as k or log k) and a composition parameter (such as the volume fraction of modifier in RPLC) can be accurately described based on two (gradient-elution or isocratic) measurements. Furthermore, Pirok et al. realized that a single LC × LC experiment in principle provides retention data in two dimensions for all analytes. They demonstrated the principle for a separation of a complex mixtures of dyes with gradient-elution IEC (where log k is expected to vary linearly with the log of the counterion concentration) and gradient-elution ion-pair LC. An approach like this, based on modeling the retention of large numbers of analytes, in principle allows a great acceleration and improvement of LC × LC method development. To achieve this, accurate retention models are needed for all popular mechanisms. While a (log-) linear model for RPLC usually suffices, models for, for example, HILIC are still under study and not necessarily linear.82,83 The principle of a program like PIOTR is hardly affected, provided that it allows establishing nonlinear retention models based on three or four experiments. To obtain correct input data for retention modeling, “peak matching” is a critical requirement.240 While it is not necessary to identify all peaks in the input (LC × LC) chromatograms, it is vital to know which peaks refer to the same analytes. Given the large numbers of analytes typically encountered in the complex samples subjected to LC × LC, another requirement is a high degree of automation of the entire optimization process.
Work has also been invested in studying some of the quality descriptors themselves. Leonhardt and co-workers investigated the performance of different orthogonality metrics.84 They noted that the convex hull and bin-counting methods do not provide information on the peak distribution, whereas the asterisk method does not work optimally when only a limited number of components are used for method development. The authors therefore introduced a new concept for peak distribution assessment across the separation space of two-dimensional separation systems in combination with clustering detection.
While, LC × LC separations typically deal less with coelution relative to 1D-LC, overlapping peaks often still occur in LC × LC data sets. Indeed, for quantification it is imperative to be able to deconvolute overlapping peaks. One frequently applied tool is multivariate curve-resolution alternating least-squares (MCR-ALS). Cook et al. recently compared several MCR-ALS strategies.85
Automated optimization programs typically need to revert to data analysis strategies. One interesting example is the work of Navarro-Reig et al., where the authors proposed an untargeted protocol that used MCR-ALS on region-of-interest (ROI) compressed data to find relevant information from the increasingly highly complex LC × LC-MS data sets. The authors applied this protocol on HILIC × RPLC-MS/MS data from separations of arsenic-exposed rice samples and found that arsenic exposure had significant effects on the rice lipidome.
Applications
Table 3 provides a comprehensive overview of recent applications of 2D-LC. We made an effort to include every relevant paper from early 2016 until now. Two-dimensional combinations of electrophoresis with liquid chromatography are not included but have been reviewed recently.86 Applications where a solid-phase extraction cartridge was used as “first-dimension separation” were considered to fall in the category online sample preparation and were not considered here. Yang and Pursch87 recently provided an overview of a number of interesting applications of 2D-LC.
The applications in Table 3 are grouped in a number of categories. Some application fields (chiral separation, biopharmaceuticals, and synthetic polymers) are discussed in more detail in later sections of this review. For each application a distinction is made between online (N) or offline (F) 2D-LC. Comprehensive two-dimensional separations are denoted with “×”; all other modes (heart-cut LC-LC, mLC-LC, multiple heart-cut and selective comprehensive, sLC × LC) are grouped under “noncomprehensive” methods and are denoted with “-”. Finally, the separation modes are classified through the type of modulation used, being either passive (P), active solvent modulation (A), evaporation-membrane modulation (E), stationary-phase assisted modulation (S), vacuum-evaporation modulation (V), and manual (M). The latter mode is typically associated with the offline mode. The retention mechanisms used, and the detection method are listed in the table for each application. In some cases, two applications are described in one publication or vice versa.
The popularity of different separation mechanisms is illustrated in the pie charts shown as Figure 10. RPLC is popular as first dimension technique, but it is used in less than 50% of all applications. Due to the pursuit of orthogonal mechanisms, other mechanisms are of considerable interest in 2D-LC and most of these are more suitable as 1D separations than as 2D separations.2 In a significant number of cases (some 35% of all online applications) RPLC is used in both dimensions, with very different mobile phases or different stationary phases. Other popular 1D techniques include HILIC, ion-exchange methods (grouped under IEX) and SEC. IEX and SEC using aqueous solvents are highly compatible with a 2D RPLC separation, thanks to inherent analyte focusing at the inlet of the 2D column. HILIC is popular as 1D technique, especially in comprehensive 2D-LC. When combining 1D HILIC with 2D RPLC some form of active modulation is highly desirable.
Figure 10.

(A) Overview of applied retention mechanisms in the first (left) and second (right) dimensions. (B) Use of modulation strategies in noncomprehensive (left) and comprehensive (right) applications (see Modulation section for a technical clarification). (C) Overview of applied detection techniques. Note that one application may use more than one detection technique so that a pie chart is less appropriate in this case. Total number of applications: 161 (online noncomprehensive, 58; online comprehensive, 76; offline, 27. CAD = charged-aerosol detection, ELSD = evaporative light-scattering detection, FLD = fluorescence detection, LS = light scattering, MS = mass spectrometry, RID = refractive-index detection, VI = viscometry. Data covers all online applications from Table 3.
RPLC is the dominant technique used in the second dimension. This is easily explained by the advantageous behavior of RPLC systems under gradient conditions. Column equilibration is much faster than in other modes of LC and gradients from high to low percentages of water allow analytes with a very broad range of polarities to be eluted under optimal conditions. One the other hand, NPLC, HIC, IEX, and HILIC are almost exclusively used as 1D techniques, because of slow equilibration and more-limited applicability. Other popular 2D techniques include SEC, which is an isocratic method, allowing simpler instrumentation and a wider selection of detectors to be used. Ion-pair (RP)LC is used as 2D separation in a substantial number of cases, probably because of the greatly different selectivity it offers for ionic analytes in comparison with RPLC. Chiral separations are used as 1D and, especially, 2D separations, mainly in heart-cut 2D-LC.
Figure 10B summarizes the use of different types of modulation systems. Passive modulation is still dominant, which may at least in part be explained by the recent emergence of some active-modulation strategies. Stationary-phase assisted modulation (SPAM) is by far the most popular active-modulation technique. Vacuum evaporation modulation has been used in some noncomprehensive applications. It may at present still be too slow to be used in LC × LC applications, while analyte recovery may also be an issue. Membrane evaporation modulation has been used in one LC × LC application. Active solvent modulation may be used more often in forthcoming years.
The dominant detection techniques in 2D-LC applications are UV spectrometry and, especially, mass spectrometry (Figure 10C). Various types of MS analyzers (quadrupole, time-of-flight, Orbitrap, and ion-trap) and hybrid systems (such as Q-TOF) are all frequently used.
In the following sections, we will discuss a number of application fields from Table 3 in more detail. Readers interested in other application areas are referred to the recent reviews on herbal medicines,88,89 food,90,91 and pharmaceuticals.92
Chiral Separations
The use of 2D-LC for pharmaceutical applications was reviewed recently by Iguiniz and co-workers,223 including the development of 2D-LC separations involving chiral separations in one or both dimensions. Here, we are focusing our discussion on developments in this area to the most recent years. Recent developments have been focused on two primary areas of research, separations of chiral amino acids, and separations of pharmaceutical materials having at least some components of the sample that are chiral.
Although the use of 2D-LC for separations of amino acids actually has a considerable history,224 the recent developments in this area are particularly exciting, because they reflect and leverage recent developments in 2D-LC technology to produce some impressive separations. Chen and Liao have demonstrated the utility of LC-LC separations, with chiral columns in one or both dimensions, for bottom-up sequencing of short peptides that yields not only the sequence but also knowledge of the enantiomeric composition of each amino acid in the peptide.192 In a different study, Wang and co-workers used offline LC × LC separations to characterize the enantiomeric composition of free amino acids in tea.119 Amino acids were derivatized with 9-fluorenylmethoxycarbonyl (FMOC) chloride prior to 2D-LC using the RP mode in the first dimension followed by the chiral mode in the second dimension. Woiwode et al. describe what they refer to as “two-dimensional correlation liquid chromatography” for characterizing the chirality of peptides and proteins by chiral × chiral separations of their constituent amino acids.118 In this work they deployed stationary phases for the 1D and 2D separations that were very similar except for the configuration of the chiral selector bonded to the stationary phase. In this way they refer to “R columns and S columns”. This is quite clever in that this particular configuration makes interpretation of the resulting 2D chromatograms quite straightforward. Molecules that are achiral all elute along a diagonal line in the 2D plot because the selectivities of the R and S columns are nominally identical for these molecules. On the other hand, all d-amino acids elute one side of this diagonal line, while the l-amino acids elute on the other side of the line. According to the authors, this clear separation of the enantiomers makes it possible to quantify the enantiomeric composition of each amino acid present in the peptide or protein that it came from. Finally, in a different paper from the same group, the authors demonstrate the ability to resolve and quantify both the d- and l-forms of all 20 proteinogenic amino acids (plus five others) using a fully automated,144 online 2D-LC separation operated in multiple heartcutting and selective comprehensive modes. In this case, the amino acids were derivatized prior to analysis using 2,4-dinitrobenzene, and the 1D and 2D columns were RP and chiral, respectively. Figure 11 is a representative chromatogram from this work that shows where each cut is made over the course of the 2D separation, and the corresponding 2D separations of the d- and l-amino acids.
Figure 11.
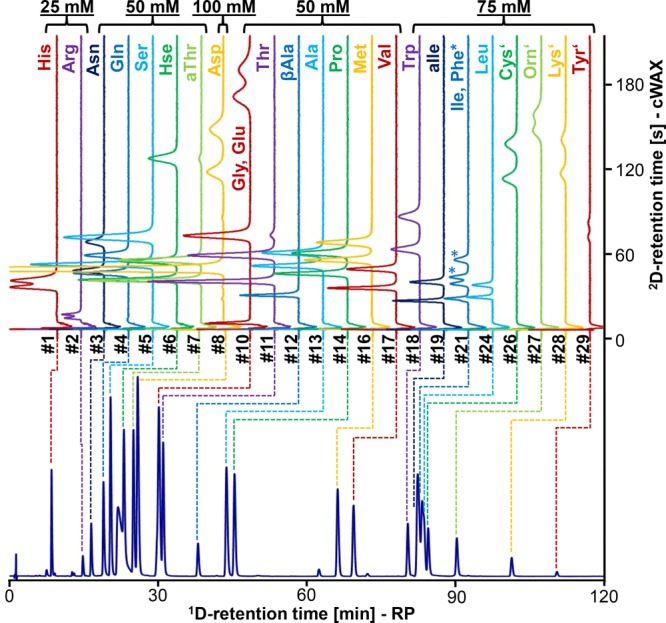
Online 2D-LC separation of 2,4-dinitrobenzene (DNP) derivatives of all 20 proteinogenic amino acids (plus allo-threonine, allo-isoleucine, homoserine, ornithing, and β-alanine). The d-enantiomer always elutes after l-, except for glutamine and ornithine. The 1D separation is carried out in the RP mode and the 2D separation in the chiral mode. Figure based on ref (144) and kindly provided by M. Lämmerhofer.
In the pharmaceutical domain, Barhate and co-workers have demonstrated a variety of 2D separations of chiral small pharmaceutical molecules that leverage very fast 2D chiral separations (e.g., 30–60 s per 2D cycle).203 These fast 2D chiral separations enable a number of possibilities for online 2D separations, including the use of chiral separations in one or both dimensions, and different 2D separation modes, ranging from simple single heart-cut all the way up to fully comprehensive separations. Furthermore, the fast 2D separations render the overall analysis time for these 2D separations much shorter than what has been possible historically.
The last three papers we will discuss here involve 2D separations with achiral RP-LC separations in the first dimension, followed by chiral SFC separations in the second dimension. Although the feasibility of LC-SFC separation was demonstrated in 1992 by Cortes co-workers,225 there has been little development in this area until recently. Venkatramani and co-workers made a significant step forward through the development of a simpler interface for the two separations that involves the use of trapping devices between the two dimensions to address the solvent incompatibility inherent to this coupling of separation modes.226 Using a multiple heart-cutting approach, they demonstrated a fast (18 min) and efficient separation of a mixture of eight stereoisomers of a single drug substance. Whereas most work involving trapping columns in the interface has used just two traps, in this work they also demonstrated the use of an array of five traps for more flexibility in the multiple heart-cutting mode.
In a follow-up study,45 the same group discussed the results of experiments aimed at systematically studying the variables that affect detection sensitivity in LC-SFC systems. Parameters studied included column diameters in both dimensions (2.1, 3.0, and 4.6 mm i.d.), volume of 1D RP effluent transferred to the 2D SFC separation, chemistry of the trapping stationary phase, dimensions of connecting tubing in the system, and detection techniques. By choosing optimal values of these variables, they established a detection limit of 10 ng/mL for the drug metabolites studied and applied the resulting system to the study of in vivo interconversion of a chiral drug.
Finally, Iguiniz and co-workers227 have described the development of 2D separations involving 1D achiral RP-LC separation and 2D chiral SFC separation that is significantly different from the work of Venkatramani et al. First, these separations were carried out in the selective comprehensive mode (sLC × SFC), which makes the simultaneous optimization of method variables more challenging. Second, they explored conditions that do not involve trapping columns in the interface. Although relatively small transfer volumes of 10 μL were used along with large 2D columns, the authors report a detection limit of 0.5% of a minor enantiomer of a drug substance relative to the major enantiomer. These are interesting developments that should promote more studies comparing the advantages and disadvantages of 2D separations coupling LC to either LC or SFC for achiral–chiral analyses.
Separations of Biopharmaceuticals
Biopharmaceuticals include therapeutic peptides, oligonucleotides, monoclonal antibodies (mAbs), antibody drug conjugates (ADCs), and other therapeutic proteins. Protein biopharmaceuticals present significant challenges to analytical techniques because of their large size and the presence of many variants that result from small changes in structure (isomers, glycosylation, etc.). Increasingly, analysts are turning to 2D separation methods, often coupled with MS detection, to help characterize these complex materials. Work in this area has been reviewed many times in recent years. In some cases the focus of the review is on biopharmaceuticals, and 2D-LC is discussed among an array of other techniques.228,229 In other reviews, the articles are organized around different 2D-LC techniques with a focus on application to biopharmaceutical problems.61,230−232 Since these reviews are pretty recent and thorough, we will not reiterate a detailed discussion of recent work in this area here. Rather, we provide an overview of recent trends in this area, with an emphasis on the most recent publications.
Over the past few years most work in this area has been focused on mAbs and ADCs. Arguably, the single most compelling demonstration of the value of 2D-LC separations for the analysis of mAbs was published recently by Sandra and co-workers.233 In this work they demonstrated multiple modes of 2D separation, ranging from single heart-cut to fully comprehensive separations. They showed examples of separations involving multiple combinations of different separation modes including Protein-A affinity chromatography (ProA) combined with SEC, IEX, or RP separation and RP combined with RP for LC × LC separations of peptide mixtures (in this case very different pH buffers are used in the two dimensions). Other groups have been exploring the potential of these new applications as well. Williams and co-workers discussed the development of an online ProA-SEC system for automated determination of mAb titer and degree of aggregation in a single 2D-LC analysis.103 Gstöttner and co-workers have described the development of a complex but powerful mLC-LC approach for characterization of mAb charge variants.106 In this work fractions from a 1D cation-exchange (CEX) separation were first reduced online with dithiothreitol (DTT) and then digested online with trypsin prior to separation and identification of the resulting peptides in a 2D RP-LC-MS analysis. The entire workflow is automated, which eliminates many of the manual sample manipulations associated with the characterization of mAb charge variants. In a very different study, Yan and co-workers used ion-exchange coupled with SEC separations in a mLC-LC system to study the degradants produced in a forced degradation of a mAb.104 Whereas all prior 2D-LC separations of mAbs at the intact or subunit level have been noncomprehensive, Stoll and co-workers demonstrated both WCX × RP-MS105 and HILIC × RP-MS15 separations of mAbs after partial digestion with the IdeS enzyme. The HILIC × RP separation is particularly interesting for heavily glycosylated mAbs, because of the exquisite selectivity of the HILIC separation mode for separating forms of the proteins with subtly different glycans.
In recent years, the most active area of development for 2D-LC separations of biopharmaceuticals has involved ADCs, perhaps because an additional level of complexity is added to the already complex mAbs when small-molecule cytotoxic drugs are conjugated to the protein. This conjugation step gives rise to a number of important questions that must be answered in the course of developing an ADC as a therapeutic agent. These include questions about how much free small-molecule drug is present in the drug formulation and the extent and localization of the small molecule conjugation to cysteine or lysine residues in the protein. To quantify the level of unconjugated (free) drug present in the protein sample, several groups have used LC-LC or mLC-LC separations.93,95,101 In the first dimension, free drug molecules are separated from protein by SEC. The fraction containing the free drug is transferred to a 2D RP separation for desalting, further separation, and eventual detection by UV or MS. In the work of Goyon and co-workers, the authors report excellent quantitative accuracy, precision, and recovery for the SEC-RP method, consistent with our expectations of conventional 1D-LC around these performance metrics.95 Since the conjugation of the small-molecule drug to the protein involves a “linker” molecule, this is an additional potential source of impurities and variation in the drug product. Venkatramani and co-workers have demonstrated the use of sLC × LC with RP separations in both dimensions to efficiently study the effect of process conditions on the impurity profile of these linker-drug intermediate molecules.94
Understanding the extent and localization of conjugated small-molecule drugs on an ADC is a challenging task. Although hydrophobic-interaction chromatography (HIC) is a powerful tool for separating ADCs according to the number of small molecule drugs that are attached (i.e., drug-to-antibody ratio, DAR), these separations typically require very high eluent concentrations of nonvolatile salts (often exceeding 1 M) and thus are not directly compatible with MS detection.234 Adding a second dimension of separation by RPLC both substantially increases the resolving power of the HIC method and provides an easy and effective desalting step to enable direct MS detection. Sarrut and co-workers have demonstrated the utility of such a HIC-RP-MS system for online, automated, and thorough mapping of the extent and localization of small-molecule drug conjugation in cysteine-linked ADCs.99,235 Ehkirch and co-workers100 have coupled HIC and SEC separations with the SEC separation carried out under nondenaturing conditions to enable determine DAR and molecular mass of ADCs in their native state. In this work they further coupled the HIC × SEC separation to IM-MS detection, thus enabling confirmation of conformational homogeneity of each detected protein. Gilroy and Eakin studied a family of related heart-cutting approaches to assemble a detailed picture of conjugation in a cysteine-linked ADC.96 This included HIC-SEC-MS, where the SEC conditions were nondenaturing, thus allowing the determination of the intact mass of each species observed in the HIC separation. They also used HIC-RP-MS separations under nonreducing or reducing conditions. In case of reducing conditions, an online reduction step (using DTT) was implemented after the HIC separation but before injection into the 2D RP column. Whereas most conjugation-mapping work has been demonstrated using cysteine-linked ADCs, Sandra and co-workers have used mLC-LC and LC × LC separations to study the conjugation of the lysine-linked ADC ado-trastuzumab emtansine.97 This is an even more challenging problem, because there are many more potential lysine conjugation sites in a typical mAb than there are cysteine conjugation sites. CEX and RP separations were used in the first and second dimensions of the mLC-LC system, respectively, to study intact and partially digested forms of the molecule, again with MS detection. Also, RP × RP-MS was used to localize sites of conjugation, following digestion of the molecule with trypsin.
To date there have not been many published reports on 2D-LC applied to the characterization of therapeutic peptides or oligonucleotides, although we expect this to change going forward. Petersson et al. have demonstrated how mLC-LC can be used to rapidly identify unknown impurities observed in 1D-RPLC separations of peptides that rely on high eluent-salt concentrations to obtain good peak shapes.102 Adding a second dimension of RP separation with a MS-friendly eluent not only enables direct MS detection, but also provides additional selectivity that is useful in cases of coelution in the 1D separation that would otherwise complicate interpretation of MS spectra. Finally, Roussis et al. have demonstrated the use of mLC-LC and LC × LC separations involving different combinations of AEX, RP, and IP-RP separation modes to identify impurities present in synthetic oligonucleotides as well as degradants observed in stability studies.47 This work provides a compelling example of the ability of 2D-LC to achieve separations “...not currently possible by mass spectrometry alone or 1D-LC”.
Separations of Synthetic Polymers
LC × LC is particularly relevant for the characterization of polymers for several reasons. The dimensionality of the sample236 is relatively low. In case of homopolymers there may just be one parameter, molecular weight, that describes the nature of each molecule. In case the same homopolymer molecules have variable end groups, two parameters suffice and the dimensionality of the sample is two. Two-dimensional samples, or samples with two dominant dimensions, may yield structured and readily interpretable chromatograms in LC × LC. The dimensions of the sample may reflect key properties. For example, if a polymer is to be used to form a network in a reacting formulation, both the functional (reactive) groups and the molecular weight (length of the cross-link) reflect essential properties of the polymer. A second reason why LC × LC is attractive for characterizing polymers is that LC-MS is usually not a good alternative. In most cases online LC-(ESI-)MS is not a viable alternative. Only for relatively small and relatively polar polymers can MS be used successfully, at least for qualitative analysis. Quantitative LC-MS is always difficult, because the response depends on the exact structure (e.g., end groups) and on the molecular weight of the molecules. For the above reasons, the characterization of polymers was one of the first domains in which LC × LC started to be regularly applied and as a result a number of applications are relatively mature and routinely performed in industry. Routine applications of LC × LC for polymer characterization often involve RPLC × SEC or NPLC × SEC separations, and these are barely published anymore.237,238 Recent publications often focus on unique (combinations of) selectivities to solve difficult problems in polymer science and industry.
Groeneveld et al. describe the separation of complex polyether polyols (polymers of ethylene oxide, EO, and propylene oxide, PO) by HILIC × RPLC.9 One of their chromatograms is shown in Figure 12. This is a good example of highly structured 2D chromatograms of samples that are complex but have a low sample dimensionality. The relatively small and relatively polar polymers allowed the use of online high-resolution MS to characterize samples in great detail. Vanhoenacker et al. demonstrated HILIC × RPLC-separations of nonionic surfactant used in pharmaceutical formulations, using mass-spectrometric detection.221 Malik et al. achieved impressive separations of triblock copolymers of EO and PO using an isocratic first-dimension with an acetonitrile/water mixture on a silica column,214 corresponding to the critical conditions (no retention) for the PO block and a RPLC separation with a methanol/water mixture that corresponded to critical conditions for the EO block.
Figure 12.

HILIC × RPLC-(+)HRMS separation of the castor oil ethoxylates. The 1D HILIC dimension (horizontal) indicates the degree of ethoxylation, while the 2D RPLC column (vertical) separates the ethoxylated species according to hydrophobicity. Various ethoxylated fatty acids as well as glycerol ethoxylated mono-, di-, tri-, tetra-, and penta-esters were identified using the obtained accurate mass and isotope distributions. Reprinted from J. Chromatogr. A, 1569, Groeneveld, G.; Dunkle, M.N.; Rinken, M.; Gargano, A.F.G.; de Niet, A.; Pursch, M.; Mes, E.P.C.; Schoenmakers, P.J., Characterization of complex polyether polyols using comprehensive two-dimensional liquid chromatography hyphenated to high-resolution mass spectrometry, pp. 128–138 (ref (9)). Copyright (2018), with permission from Elsevier.
Lee et al. described the separation of a series of polystyrene-polybutadiene block copolymers with temperature-gradient interaction chromatography (TGIC) in the first dimension.209 Normal-phase TGIC was combined with either SEC or NPLC at the critical conditions in the second dimension. Likewise, RP-TGIC was combined with either SEC or RPLC at the critical conditions.
One example of newly charted territory are high-temperature LC × LC separations. High temperatures are typically required to resolve poly olefins (including ubiquitous polymers, such as poly ethene, PE, and polypropene, PP). Ndiripo and Pasch used high-temperature LC × SEC to characterize oxidized waxes at 140 °C.207 A porous-graphitic-carbon column and a gradient from n-decane to o-dichlorobenzene were used in the first dimension. The latter solvent was also used for the second-dimension SEC separation. In a separate study, interaction LC on a silica column with a gradient from n-decane to c-hexanone was used in the first dimension,208 either with a composition gradient at a constant temperature (110 °C) or in TGIC mode, increasing the temperature at a constant mobile-phase composition.
Apel et al. performed RPLC in the first dimension with a very shallow gradient close to the critical conditions,210 followed by SEC in the second dimension to characterize branched poly (bisphenol A carbonate) structures. They combined this RPLC × SEC method with a triple-detector system (concentration detector, light scattering, and viscometry) and they tried to correlate the results with the outcome of Monte Carlo simulations.211 Yang et al. used SEC × RPLC to determine the number of hydrophobic end groups in modified ethylene-oxide-urethane polymers.212 They used an ultrahigh-performance column packed with sub-2-μm particles and a mobile phase containing 100 mM ammonium acetate in methanol in the first dimension and a gradient RPLC method in the second dimension. The authors developed a multistep second-dimension water–acetonitrile gradient to avoid breakthrough. The same group used a similar system for the separation of polymeric dispersants in detergents.222 A very wide (8 mm i.d.) 1D SEC column was used with an aqueous mobile phase and up to 60% of the effluent was diverted to waste.
Lee et al. exploited the unique selectivity of LC × LC to separate polystyrenes with interesting configurations (“figure-eight-shaped” and “cage-shaped” cyclic molecules) from branched polystyrenes using NPLC × SEC with a bare silica column (NPLC) and a n-hexane/THF gradient in the first dimension and SEC with THF as eluent in the second dimension.213 Separation is based on the fact that molecules with different conformations but the same molecular weight have a different size in solution (hydrodynamic radius) and thus different elution times in SEC, whereas the extent of interaction with the silica column is not strictly related to the hydrodynamic radius.
Pursch et al. used the full capabilities of contemporary LC × LC instrumentation, including the use of ultrahigh pressures to realize SEC × RPLC separations of Novolac-type polymers within 20 min.36 Active solvent modulation was used to successfully avoid breakthrough in the second dimension.
A unique application of LC × LC for the analysis of polymeric nanoparticles was described by Pirok et al.43 They first separated the nanoparticles by packed column hydrodynamic chromatography (HDC) using an aqueous mobile phase. At the modulation stage, the particles were dissolved in THF to yield a solution of the constituting polymer molecules. After concentrating these on a trap column, the molecular-weight distribution was determined by organic SEC in THF.
Summary and Outlook
The popularity of two-dimensional liquid chromatography (2D-LC) is on the rise, due to a range of successful applications. When two very different selectivities (retention mechanisms) are selected, heart-cut LC-LC allows target peaks to be rigorously purified and the purity of target analytes to be assessed. This is, for example, of significant interest in the emerging field of biopharmaceuticals. LC-LC also provides additional certainty of peak identification, which is relevant, for example, in forensics. Comprehensive two-dimensional liquid chromatography (LC × LC) is of general interest for analyzing complex samples that contain nonvolatile analytes or matrix compounds. Resolution for such samples can be much improved, thanks to the much higher peak capacities in comparison with conventional 1D-LC and thanks to the additional selectivity. As a result, LC × LC is potentially a much better “sample-preparation” method prior to analysis by MS or MS/MS. LC × LC-MS may thus be advantageous for the analysis of, for example, complex mixtures of peptides. When the two separation dimensions are selected judiciously, LC × LC may provide reliable fingerprints that allow rapid (often visual) and efficient comparison of samples, for example, in the field of natural medicines. For complex samples with a low dimensionality, such as synthetic polymers, LC × LC may provide highly structured, readily interpretable two-dimensional chromatograms.
The applications discussed in this review provide plenty of motivation to accelerate the development of LC-LC and LC × LC techniques. Research may be focused on a number of aspects. First and foremost, 2D-LC will thrive if the techniques (instruments, columns, modulators) are robust and reliable. Arguably, 2D-LC is a complex technique. This creates high demands on user interfaces for applications and, especially, for method development. Third-party method development (by instrument manufacturers or research groups) may accelerate the proliferation of 2D-LC techniques, provided that methods can be transferred reliably and efficiently. Technical issues that must be addressed include generic approaches to overcome compatibility issues that arise from the selection of two very different (“orthogonal”) retention mechanisms. If the first-dimension effluent is not suitable as second-dimension injection solvent active-modulation techniques must be applied. These may have additional advantages. If the analyte bands are effectively focused by the modulator detection sensitivity may be enhanced, and the second-dimension separation may be performed more efficiently and on narrower columns, reducing the amounts of solvent required. Generally, 2D-LC can benefit from any progress in LC. In this century better columns (sub-2-μm particles, core–shell particles) and ultrahigh-performance instruments have emerged. Two developments are especially relevant for 2D-LC. Very fast analysis are desirable, especially in the second dimension of LC × LC methods. All 2D-LC methods can be improved by minimizing extra-column dispersion.
In the short period covered by this review (2016–2018) we have seen great activity in the development and application of 2D-LC techniques. Both heart-cut (LC-LC) and comprehensive (LC × LC) techniques appear to have a bright future.
Acknowledgments
Bob W. J. Pirok acknowledges the MANIAC project, which is funded by The Netherlands Organisation for Scientific Research (NWO) in the framework of the Programmatic Technology Area PTA-COAST3 of the Fund New Chemical Innovations (Project 053.21.113). Dwight R. Stoll acknowledges financial support from the National Science Foundation (Grant CHE-1508159). Prof. Dr. Michael Lämmerhofer (University of Tübingen, Germany) is kindly acknowledged for providing Figure 11.
Biographies
Bob W. J. Pirok worked at Shell after obtaining his M.Sc. degree in 2014. He now is a Ph.D. student in the MANIAC (Making ANalytically Incompatible Approaches Compatible) project at the University of Amsterdam since 2015. He developed the (LC × )LC optimization tool Program for Interpretive Optimization (PIOTR) and has published on fast size-exclusion chromatography using core–shell particles, comprehensive integrated analysis of nanoparticles and the constituting macromolecules by HDC × SEC, and the application and optimization of LC × LC separations. Bob W. J. Pirok was decorated with a Shimadzu Young-Scientist Award at HPLC2015 Beijing, the Young-Scientist-Award Lecture during the SCM-8 meeting in Amsterdam in 2017, and the Csaba Horváth Young-Scientist Award at HPLC2017 Prague.
Dwight R. Stoll is Professor and Co-Chair of Chemistry at Gustavus Adolphus College in St. Peter, MN. He has authored or coauthored 57 peer-reviewed publications, four book chapters in separation science, and over 100 conference presentations. His primary research focus is on the development of two-dimensional liquid chromatography (2D-LC) for both targeted and untargeted analyses. Within this area he has made contributions on many aspects of the technique including stationary phase characterization, biopharmaceutical analysis, new 2D-LC methodologies and instrumentation, and fundamental aspects including re-equilibration in gradient elution reversed-phase liquid chromatography and analyte focusing. He is the 2011 recipient of LCGC’s Emerging Leader in Chromatography Award, the 2015 recipient of the American Chemical Society Division of Analytical Chemistry Award for Young Investigators in Separation Science, and was named to The Analytical Scientist’s list of Top Ten Separation Scientists in 2017.
Peter J. Schoenmakers is Professor of Analytical Chemistry at the University of Amsterdam. He investigates analytical separations, focussing on multidimensional liquid chromatography. He studied in Delft, The Netherlands (with Professor Leo de Galan) and in Boston, MA (with Professor Barry Karger). Thereafter, he worked for Philips in Eindhoven (The Netherlands) and for Shell in Amsterdam and in Houston, TX. He was awarded the AJP Martin Medal in 2011, the John Knox Medal in 2014, and the Csaba Horváth Award and the CASSS Award in 2015. In 2016 he obtained an Advanced Grant for Excellent Research from the European Research Council.
The authors declare no competing financial interest.
References
- Davis J. M.; Giddings J. C. Anal. Chem. 1983, 55 (3), 418–424. 10.1021/ac00254a003. [DOI] [Google Scholar]
- Pirok B. W. J.; Gargano A. F. G.; Schoenmakers P. J. J. Sep. Sci. 2018, 41 (1), 68–98. 10.1002/jssc.201700863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis J. M.; Stoll D. R. J. Chromatogr. A 2014, 1360, 128–142. 10.1016/j.chroma.2014.07.066. [DOI] [PubMed] [Google Scholar]
- Gargano A. F. G.; Duffin M.; Navarro P.; Schoenmakers P. J. Anal. Chem. 2016, 88, 1785–1793. 10.1021/acs.analchem.5b04051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirok B. W. J.; Knip J.; van Bommel M. R.; Schoenmakers P. J. J. Chromatogr. A 2016, 1436, 141–146. 10.1016/j.chroma.2016.01.070. [DOI] [PubMed] [Google Scholar]
- Sommella E.; Ismail O. H.; Pagano F.; Pepe G.; Ostacolo C.; Mazzoccanti G.; Russo M.; Novellino E.; Gasparrini F.; Campiglia P. J. Sep. Sci. 2017, 40 (10), 2188–2197. 10.1002/jssc.201700134. [DOI] [PubMed] [Google Scholar]
- Sarrut M.; D’Attoma A.; Heinisch S. J. Chromatogr. A 2015, 1421, 48–59. 10.1016/j.chroma.2015.08.052. [DOI] [PubMed] [Google Scholar]
- Vivó-Truyols G.; van der Wal S.; Schoenmakers P. J. Anal. Chem. 2010, 82 (20), 8525–8536. 10.1021/ac101420f. [DOI] [PubMed] [Google Scholar]
- Groeneveld G.; Dunkle M. N.; Rinken M.; Gargano A. F. G.; de Niet A.; Pursch M.; Mes E. P. C.; Schoenmakers P. J. J. Chromatogr. A 2018, 1569, 128–138. 10.1016/j.chroma.2018.07.054. [DOI] [PubMed] [Google Scholar]
- Mondello L.; Tranchida P. Q.; Stanek V.; Jandera P.; Dugo G.; Dugo P. J. Chromatogr. A 2005, 1086 (1–2), 91–98. 10.1016/j.chroma.2005.06.017. [DOI] [PubMed] [Google Scholar]
- Elsner V.; Laun S.; Melchior D.; Köhler M.; Schmitz O. J. J. Chromatogr. A 2012, 1268, 22–28. 10.1016/j.chroma.2012.09.072. [DOI] [PubMed] [Google Scholar]
- Ochoa C. M.; Shoenmakers P.; Mallet C. R.; Lurie I. S. Anal. Methods 2018, 10 (26), 3178–3187. 10.1039/C8AY00565F. [DOI] [Google Scholar]
- Baglai A.; Gargano A. F. G.; Jordens J.; Mengerink Y.; Honing M.; van der Wal S.; Schoenmakers P. J. J. Chromatogr. A 2017, 1530, 90–103. 10.1016/j.chroma.2017.11.014. [DOI] [PubMed] [Google Scholar]
- Sarrut M.; Rouvière F.; Heinisch S. J. Chromatogr. A 2017, 1498, 183–195. 10.1016/j.chroma.2017.01.054. [DOI] [PubMed] [Google Scholar]
- Stoll D. R.; Harmes D. C.; Staples G. O.; Potter O. G.; Dammann C. T.; Guillarme D.; Beck A. Anal. Chem. 2018, 90 (9), 5923–5929. 10.1021/acs.analchem.8b00776. [DOI] [PubMed] [Google Scholar]
- Sheng N.; Zheng H.; Xiao Y.; Wang Z.; Li M.; Zhang J. J. Chromatogr. A 2017, 1517, 97–107. 10.1016/j.chroma.2017.08.037. [DOI] [PubMed] [Google Scholar]
- Pirok B. W. J.; Schoenmakers P. J. LC-GC Eur. 2018, 31 (5), 242–249. [Google Scholar]
- Venkatramani C. J.; Al-Sayah M.; Li G.; Goel M.; Girotti J.; Zang L.; Wigman L.; Yehl P.; Chetwyn N. Talanta 2016, 148, 548–555. 10.1016/j.talanta.2015.10.054. [DOI] [PubMed] [Google Scholar]
- Stoll D. R.; Venkatramani C. J.; Rutan S. C. LC-GC N. Am. 2018, 36 (6), 356–361. [Google Scholar]
- Simpkins S. W.; Bedard J. W.; Groskreutz S. R.; Swenson M. M.; Liskutin T. E.; Stoll D. R. J. Chromatogr. A 2010, 1217 (49), 7648–7660. 10.1016/j.chroma.2010.09.023. [DOI] [PubMed] [Google Scholar]
- Wouters B.; Davydova E.; Wouters S.; Vivo-Truyols G.; Schoenmakers P. J.; Eeltink S. Lab Chip 2015, 15 (23), 4415–4422. 10.1039/C5LC01169H. [DOI] [PubMed] [Google Scholar]
- Giddings J. C. J. Chromatogr. A 1995, 703 (1–2), 3–15. 10.1016/0021-9673(95)00249-M. [DOI] [PubMed] [Google Scholar]
- Stoll D. R.; Carr P. W. Anal. Chem. 2017, 89 (1), 519–531. 10.1021/acs.analchem.6b03506. [DOI] [PubMed] [Google Scholar]
- Stoll D. R. In Handbook of Advanced Chromatography/Mass Spectrometry Techniques; Elsevier, 2017; pp 227–286. [Google Scholar]
- Marriott P. J.; Wu Z.-Y.; Schoenmakers P. LC-GC Eur. 2012, 25 (5), 266–275. [Google Scholar]
- François I.; Sandra K.; Sandra P.; Dugo P.; Mondello L.; Cacciola F.; Donato P.. Comprehensive Chromatography in Combination with Mass Spectrometry; Mondello L., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, 2011. [Google Scholar]
- Dugo P.; Cacciola F.; Kumm T.; Dugo G.; Mondello L. J. Chromatogr. A 2008, 1184 (1–2), 353–368. 10.1016/j.chroma.2007.06.074. [DOI] [PubMed] [Google Scholar]
- Mayfield K. J.; Shalliker R. A.; Catchpoole H. J.; Sweeney A. P.; Wong V.; Guiochon G. J. Chromatogr. A 2005, 1080 (2), 124–131. 10.1016/j.chroma.2005.04.093. [DOI] [PubMed] [Google Scholar]
- Samuelsson J.; Shalliker R. A.; Fornstedt T. Microchem. J. 2017, 130, 102–107. 10.1016/j.microc.2016.08.007. [DOI] [Google Scholar]
- Shalliker R. A.; Guiochon G. J. Chromatogr. A 2009, 1216 (5), 787–793. 10.1016/j.chroma.2008.11.067. [DOI] [PubMed] [Google Scholar]
- Stoll D. R.; Sajulga R. W.; Voigt B. N.; Larson E. J.; Jeong L. N.; Rutan S. C. J. Chromatogr. A 2017, 1523, 162–172. 10.1016/j.chroma.2017.07.041. [DOI] [PubMed] [Google Scholar]
- Jiang X.; Van Der Horst A.; Schoenmakers P. J. J. Chromatogr. A 2002, 982 (1), 55–68. 10.1016/S0021-9673(02)01483-8. [DOI] [PubMed] [Google Scholar]
- Reingruber E.; Jansen J. J.; Buchberger W.; Schoenmakers P. J. Chromatogr. A 2011, 1218 (8), 1147–1152. 10.1016/j.chroma.2010.12.080. [DOI] [PubMed] [Google Scholar]
- Talus E. S.; Witt K. E.; Stoll D. R. J. Chromatogr. A 2015, 1378, 50–57. 10.1016/j.chroma.2014.12.019. [DOI] [PubMed] [Google Scholar]
- Stoll D. R.; Shoykhet K.; Petersson P.; Buckenmaier S. Anal. Chem. 2017, 89 (17), 9260–9267. 10.1021/acs.analchem.7b02046. [DOI] [PubMed] [Google Scholar]
- Pursch M.; Wegener A.; Buckenmaier S. J. Chromatogr. A 2018, 1562, 78–86. 10.1016/j.chroma.2018.05.059. [DOI] [PubMed] [Google Scholar]
- Bäurer S.; Guo W.; Polnick S.; Lämmerhofer M. Chromatographia 2018, 10.1007/s10337-018-3615-0. [DOI] [Google Scholar]
- Jeong L. N.; Sajulga R.; Forte S. G.; Stoll D. R.; Rutan S. C. J. Chromatogr. A 2016, 1457, 41–49. 10.1016/j.chroma.2016.06.016. [DOI] [PubMed] [Google Scholar]
- Vonk R. J.; Gargano A. F. G.; Davydova E.; Dekker H. L.; Eeltink S.; de Koning L. J.; Schoenmakers P. J. Anal. Chem. 2015, 87 (10), 5387–5394. 10.1021/acs.analchem.5b00708. [DOI] [PubMed] [Google Scholar]
- Toro-Uribe S.; Montero L.; López-Giraldo L.; Ibáñez E.; Herrero M. Anal. Chim. Acta 2018, 1036, 204–213. 10.1016/j.aca.2018.06.068. [DOI] [PubMed] [Google Scholar]
- Baglai A.; Blokland M. H.; Mol H. G. J.; Gargano A. F. G.; van der Wal S.; Schoenmakers P. J. Anal. Chim. Acta 2018, 1013, 87–97. 10.1016/j.aca.2017.12.043. [DOI] [PubMed] [Google Scholar]
- Jakobsen S. S.; Christensen J. H.; Verdier S.; Mallet C. R.; Nielsen N. J. Anal. Chem. 2017, 89 (17), 8723–8730. 10.1021/acs.analchem.7b00758. [DOI] [PubMed] [Google Scholar]
- Pirok B. W. J.; Abdulhussain N.; Aalbers T.; Wouters B.; Peters R. A. H.; Schoenmakers P. J. Anal. Chem. 2017, 89 (17), 9167–9174. 10.1021/acs.analchem.7b01906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gargano A. F. G.; Shaw J. B.; Zhou M.; Wilkins C. S.; Fillmore T. L.; Moore R. J.; Somsen G. W.; Paša-Tolić L. J. Proteome Res. 2018, 17, 3791–3800. 10.1021/acs.jproteome.8b00458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel M.; Larson E.; Venkatramani C. J.; Al-Sayah M. A. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2018, 1084, 89–95. 10.1016/j.jchromb.2018.03.029. [DOI] [PubMed] [Google Scholar]
- Chen M.; Wang L.; Dong H.; Shao X.; Wu D.; Liu B.; Zhang X.; Chen C. J. Sep. Sci. 2017, 40 (9), 1920–1927. 10.1002/jssc.201601367. [DOI] [PubMed] [Google Scholar]
- Roussis S. G.; Cedillo I.; Rentel C. Anal. Biochem. 2018, 556, 45–52. 10.1016/j.ab.2018.06.019. [DOI] [PubMed] [Google Scholar]
- Callister S. J.; Fillmore T. L.; Nicora C. D.; Shaw J. B.; Purvine S. O.; Orton D. J.; White R. A.; Moore R. J.; Burnet M. C.; Nakayasu E. S.; Payne S. H.; Jansson J. K.; Paša-Tolić L. Soil Biol. Biochem. 2018, 125, 290–299. 10.1016/j.soilbio.2018.07.018. [DOI] [Google Scholar]
- Ianovska M. A.; Mulder P. P. M. F. A.; Verpoorte E. RSC Adv. 2017, 7 (15), 9090–9099. 10.1039/C6RA28626G. [DOI] [Google Scholar]
- Fornells E.; Barnett B.; Bailey M.; Hilder E. F.; Shellie R. A.; Breadmore M. C. Anal. Chim. Acta 2018, 1000, 303–309. 10.1016/j.aca.2017.11.053. [DOI] [PubMed] [Google Scholar]
- Creese M. E.; Creese M. J.; Foley J. P.; Cortes H. J.; Hilder E. F.; Shellie R. A.; Breadmore M. C. Anal. Chem. 2017, 89 (2), 1123–1130. 10.1021/acs.analchem.6b03279. [DOI] [PubMed] [Google Scholar]
- Ji B.; Xia B.; Liu J.; Gao Y.; Ding L.; Zhou Y. J. Chromatogr. A 2016, 1466, 199–204. 10.1016/j.chroma.2016.09.014. [DOI] [PubMed] [Google Scholar]
- Tian H.; Xu J.; Xu Y.; Guan Y. J. Chromatogr. A 2006, 1137 (1), 42–48. 10.1016/j.chroma.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Tian H.; Xu J.; Guan Y. J. Sep. Sci. 2008, 31 (10), 1677–1685. 10.1002/jssc.200700559. [DOI] [PubMed] [Google Scholar]
- Shen S.; Yang L.; Li L.; Bai Y.; Cai C.; Liu H. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2017, 1068–1069, 41–48. 10.1016/j.jchromb.2017.10.004. [DOI] [PubMed] [Google Scholar]
- Weng R.; Shen S.; Burton C.; Yang L.; Nie H.; Tian Y.; Bai Y.; Liu H. Anal. Bioanal. Chem. 2016, 408 (11), 2963–2973. 10.1007/s00216-015-9256-3. [DOI] [PubMed] [Google Scholar]
- Johnson E. L.; Gloor R.; Majors R. E. J. Chromatogr. A 1978, 149, 571–585. 10.1016/S0021-9673(00)81012-2. [DOI] [Google Scholar]
- Erni F.; Frei R. W. J. Chromatogr. A 1978, 149, 561–569. 10.1016/S0021-9673(00)81011-0. [DOI] [Google Scholar]
- Zhang K.; Li Y.; Tsang M.; Chetwyn N. P. J. Sep. Sci. 2013, 36 (18), 2986–2992. 10.1002/jssc.201300493. [DOI] [PubMed] [Google Scholar]
- Groskreutz S. R.; Swenson M. M.; Secor L. B.; Stoll D. R. J. Chromatogr. A 2012, 1228, 31–40. 10.1016/j.chroma.2011.06.035. [DOI] [PubMed] [Google Scholar]
- Stoll D. R.; Zhang K.; Staples G. O.; Beck A.. Advances in Chromatography CRC Press: Boca Raton, FL, 2018, Vol. 56. [Google Scholar]
- Murphy R. E.; Schure M. R.; Foley J. P. Anal. Chem. 1998, 70 (8), 1585–1594. 10.1021/ac971184b. [DOI] [Google Scholar]
- Davis J. M.; Stoll D. R.; Carr P. W. Anal. Chem. 2008, 80 (2), 461–473. 10.1021/ac071504j. [DOI] [PubMed] [Google Scholar]
- Pursch M.; Buckenmaier S. Anal. Chem. 2015, 87 (10), 5310–5317. 10.1021/acs.analchem.5b00492. [DOI] [PubMed] [Google Scholar]
- Davis J. M.; Stoll D. R. J. Chromatogr. A 2018, 1537, 43–57. 10.1016/j.chroma.2017.12.035. [DOI] [PubMed] [Google Scholar]
- Schellinger A. P.; Stoll D. R.; Carr P. W. J. Chromatogr. A 2005, 1064 (2), 143–156. 10.1016/j.chroma.2004.12.017. [DOI] [PubMed] [Google Scholar]
- Striegel A. M.; Yau W. W.; Kirkland J. J.; Bly D. D.. Modern Size-Exclusion Liquid Chromatography, 2nd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, 2009. [Google Scholar]
- Vanderheyden Y.; Cabooter D.; Desmet G.; Broeckhoven K. J. Chromatogr. A 2013, 1312, 80–86. 10.1016/j.chroma.2013.09.009. [DOI] [PubMed] [Google Scholar]
- Gritti F.; Leonardis I.; Abia J.; Guiochon G. J. Chromatogr. A 2010, 1217 (24), 3819–3843. 10.1016/j.chroma.2010.04.026. [DOI] [PubMed] [Google Scholar]
- Pirok B. W. J.; Breuer P.; Hoppe S. J. M.; Chitty M.; Welch E.; Farkas T.; van der Wal S.; Peters R.; Schoenmakers P. J. J. Chromatogr. A 2017, 1486, 96–102. 10.1016/j.chroma.2016.12.015. [DOI] [PubMed] [Google Scholar]
- Schure M. R.; Moran R. E. J. Chromatogr. A 2017, 1480, 11–19. 10.1016/j.chroma.2016.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahab M. F.; Wimalasinghe R. M.; Wang Y.; Barhate C. L.; Patel D. C.; Armstrong D. W. Anal. Chem. 2016, 88 (17), 8821–8826. 10.1021/acs.analchem.6b02260. [DOI] [PubMed] [Google Scholar]
- Davydova E.; Schoenmakers P. J.; Vivó-Truyols G. J. Chromatogr. A 2013, 1271 (1), 137–143. 10.1016/j.chroma.2012.11.043. [DOI] [PubMed] [Google Scholar]
- Wouters B.; Davydova E.; Wouters S.; Vivo-Truyols G.; Schoenmakers P. J.; Eeltink S. Lab Chip 2015, 15 (23), 4415–4422. 10.1039/C5LC01169H. [DOI] [PubMed] [Google Scholar]
- Davydova E.; Wouters S.; Deridder S.; Desmet G.; Eeltink S.; Schoenmakers P. J. J. Chromatogr. A 2016, 1434, 127–135. 10.1016/j.chroma.2016.01.003. [DOI] [PubMed] [Google Scholar]
- Adamopoulou T.; Deridder S.; Desmet G.; Schoenmakers P. J. J. Chromatogr. A 2018, 1577, 120. 10.1016/j.chroma.2018.09.054. [DOI] [PubMed] [Google Scholar]
- Baert M.; Martens S.; Desmet G.; de Villiers A.; Du Prez F.; Lynen F. Anal. Chem. 2018, 90 (8), 4961–4967. 10.1021/acs.analchem.7b04914. [DOI] [PubMed] [Google Scholar]
- Schoenmakers P. J.; Vivó-Truyols G.; Decrop W. M. C. J. Chromatogr. A 2006, 1120 (1–2), 282–290. 10.1016/j.chroma.2005.11.039. [DOI] [PubMed] [Google Scholar]
- Bedani F.; Schoenmakers P. J.; Janssen H.-G. Theories to Support Method Development in Comprehensive Two-Dimensional Liquid Chromatography - A Review. J. Sep. Sci. 2012, 35 (14), 1697–1711. 10.1002/jssc.201200070. [DOI] [PubMed] [Google Scholar]
- Muller M.; Tredoux A. G. J.; de Villiers A. J. Chromatogr. A 2018, 1571, 107–120. 10.1016/j.chroma.2018.08.004. [DOI] [PubMed] [Google Scholar]
- Venter P.; Muller M.; Vestner J.; Stander M. A.; Tredoux A. G. J.; Pasch H.; de Villiers A. Anal. Chem. 2018, 90 (19), 11643–11650. 10.1021/acs.analchem.8b03234. [DOI] [PubMed] [Google Scholar]
- Pirok B. W. J.; Pous-Torres S.; Ortiz-Bolsico C.; Vivó-Truyols G.; Schoenmakers P. J. J. Chromatogr. A 2016, 1450, 29–37. 10.1016/j.chroma.2016.04.061. [DOI] [PubMed] [Google Scholar]
- Pirok B. W. J.; Molenaar S. R. A.; van Outersterp R. E.; Schoenmakers P. J. J. Chromatogr. A 2017, 1530, 104. 10.1016/j.chroma.2017.11.017. [DOI] [PubMed] [Google Scholar]
- Česla P.; Vaňková N.; Křenková J.; Fischer J. J. Chromatogr. A 2016, 1438, 179–188. 10.1016/j.chroma.2016.02.032. [DOI] [PubMed] [Google Scholar]
- Pirok B. W. J.; Molenaar S. R. A.; Roca L. S.; Schoenmakers P. J.. Peak-Tracking Algorithm for Use in Automated Interpretive Method-Development Tools in Liquid Chromatography. Anal. Chem. 2018, 10.1021/acs.analchem.8b03929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonhardt J.; Teutenberg T.; Buschmann G.; Gassner O.; Schmidt T. C. Anal. Bioanal. Chem. 2016, 408 (28), 8079–8088. 10.1007/s00216-016-9911-3. [DOI] [PubMed] [Google Scholar]
- Cook D. W.; Burnham M. L.; Harmes D. C.; Stoll D. R.; Rutan S. C. Anal. Chim. Acta 2017, 961, 49–58. 10.1016/j.aca.2017.01.047. [DOI] [PubMed] [Google Scholar]
- Ranjbar L.; Foley J. P.; Breadmore M. C. Anal. Chim. Acta 2017, 950, 7–31. 10.1016/j.aca.2016.10.025. [DOI] [PubMed] [Google Scholar]
- Yang P.; Pursch M. Chromatogr. Today 2018, 10 (4), 24–29. [Google Scholar]
- Ji S.; Wang S.; Xu H.; Su Z.; Tang D.; Qiao X.; Ye M. J. Pharm. Biomed. Anal. 2018, 160, 301–313. 10.1016/j.jpba.2018.08.014. [DOI] [PubMed] [Google Scholar]
- Li Z.; Chen K.; Guo M.; Tang D. J. Sep. Sci. 2016, 39 (1), 21–37. 10.1002/jssc.201500634. [DOI] [PubMed] [Google Scholar]
- Cacciola F.; Farnetti S.; Dugo P.; Marriott P. J.; Mondello L. J. Sep. Sci. 2017, 40 (1), 7–24. 10.1002/jssc.201600704. [DOI] [PubMed] [Google Scholar]
- Cacciola F.; Dugo P.; Mondello L. TrAC, Trends Anal. Chem. 2017, 96, 116–123. 10.1016/j.trac.2017.06.009. [DOI] [PubMed] [Google Scholar]
- Iguiniz M.; Heinisch S. J. Pharm. Biomed. Anal. 2017, 145, 482–503. 10.1016/j.jpba.2017.07.009. [DOI] [PubMed] [Google Scholar]
- Li Y.; Stella C.; Zheng L.; Bechtel C.; Gruenhagen J.; Jacobson F.; Medley C. D. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2016, 1032, 112–118. 10.1016/j.jchromb.2016.05.011. [DOI] [PubMed] [Google Scholar]
- Venkatramani C. J.; Huang S. R.; Al-Sayah M.; Patel I.; Wigman L. J. Chromatogr. A 2017, 1521, 63–72. 10.1016/j.chroma.2017.09.022. [DOI] [PubMed] [Google Scholar]
- Goyon A.; Sciascera L.; Clarke A.; Guillarme D.; Pell R. J. Chromatogr. A 2018, 1539, 19–29. 10.1016/j.chroma.2018.01.039. [DOI] [PubMed] [Google Scholar]
- Gilroy J. J.; Eakin C. M. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2017, 1060, 182–189. 10.1016/j.jchromb.2017.06.005. [DOI] [PubMed] [Google Scholar]
- Sandra K.; Vanhoenacker G.; Vandenheede I.; Steenbeke M.; Joseph M.; Sandra P. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2016, 1032, 119–130. 10.1016/j.jchromb.2016.04.040. [DOI] [PubMed] [Google Scholar]
- Sarrut M.; Corgier A.; Fekete S.; Guillarme D.; Lascoux D.; Janin-Bussat M.-C.; Beck A.; Heinisch S. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2016, 1032, 103–111. 10.1016/j.jchromb.2016.06.048. [DOI] [PubMed] [Google Scholar]
- Sarrut M.; Fekete S.; Janin-Bussat M. C.; Colas O.; Guillarme D.; Beck A.; Heinisch S. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2016, 1032, 91–102. 10.1016/j.jchromb.2016.06.049. [DOI] [PubMed] [Google Scholar]
- Ehkirch A.; D’Atri V.; Rouviere F.; Hernandez-Alba O.; Goyon A.; Colas O.; Sarrut M.; Beck A.; Guillarme D.; Heinisch S.; Cianferani S. Anal. Chem. 2018, 90 (3), 1578–1586. 10.1021/acs.analchem.7b02110. [DOI] [PubMed] [Google Scholar]
- Birdsall R. E.; McCarthy S. M.; Janin-Bussat M. C.; Perez M.; Haeuw J.-F.; Chen W.; Beck A. MAbs 2016, 8 (2), 306–317. 10.1080/19420862.2015.1116659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersson P.; Haselmann K.; Buckenmaier S. J. Chromatogr. A 2016, 1468, 95–101. 10.1016/j.chroma.2016.09.023. [DOI] [PubMed] [Google Scholar]
- Williams A.; Read E. K.; Agarabi C. D.; Lute S.; Brorson K. A. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2017, 1046, 122–130. 10.1016/j.jchromb.2017.01.021. [DOI] [PubMed] [Google Scholar]
- An Y.; Verma S.; Chen Y.; Yu S.; Zhang Y.; Kelner S.; Mengisen S.; Richardson D.; Chen Z. J. Chromatogr. Sep. Tech. 2017, 8, 365. 10.4172/2157-7064.1000365. [DOI] [Google Scholar]
- Sorensen M.; Harmes D. C.; Stoll D. R.; Staples G. O.; Fekete S.; Guillarme D.; Beck A. MAbs 2016, 8 (7), 1224–1234. 10.1080/19420862.2016.1203497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gstöttner C.; Klemm D.; Haberger M.; Bathke A.; Wegele H.; Bell C.; Kopf R. Anal. Chem. 2018, 90 (3), 2119–2125. 10.1021/acs.analchem.7b04372. [DOI] [PubMed] [Google Scholar]
- Bathke A.; Klemm D.; Gstöttner C.; Bell C.; Kopf R. LC-GC Eur. 2018, 31 (1), 10–21. [Google Scholar]
- Ouyang X.; Leonards P. E. G.; Tousova Z.; Slobodnik J.; de Boer J.; Lamoree M. H. Anal. Chem. 2016, 88 (4), 2353–2360. 10.1021/acs.analchem.5b04311. [DOI] [PubMed] [Google Scholar]
- Graesbøll R.; Janssen H.-G.; Christensen J. H.; Nielsen N. J. J. Sep. Sci. 2017, 40 (18), 3612–3620. 10.1002/jssc.201700239. [DOI] [PubMed] [Google Scholar]
- Brazdauskas T.; Montero L.; Venskutonis P. R.; Ibañez E.; Herrero M. J. Chromatogr. A 2016, 1468, 126–135. 10.1016/j.chroma.2016.09.033. [DOI] [PubMed] [Google Scholar]
- Cacciola F.; Giuffrida D.; Utczas M.; Mangraviti D.; Beccaria M.; Donato P.; Bonaccorsi I.; Dugo P.; Mondello L. LC-GC Eur. 2016, 29 (5), 252–257. [Google Scholar]
- Cacciola F.; Giuffrida D.; Utczas M.; Mangraviti D.; Dugo P.; Menchaca D.; Murillo E.; Mondello L. Food Anal. Methods 2016, 9 (8), 2335–2341. 10.1007/s12161-016-0416-7. [DOI] [Google Scholar]
- Sommella E.; Pagano F.; Salviati E.; Chieppa M.; Bertamino A.; Manfra M.; Sala M.; Novellino E.; Campiglia P. J. Sep. Sci. 2018, 41 (7), 1548–1557. 10.1002/jssc.201701242. [DOI] [PubMed] [Google Scholar]
- Zhou T.; Han S.; Li Z.; He P. Food Anal. Methods 2017, 10 (10), 3350–3360. 10.1007/s12161-017-0902-6. [DOI] [Google Scholar]
- Navarro-Reig M.; Jaumot J.; van Beek T. A.; Vivó-Truyols G.; Tauler R. Talanta 2016, 160, 624–635. 10.1016/j.talanta.2016.08.005. [DOI] [PubMed] [Google Scholar]
- Donato P.; Rigano F.; Cacciola F.; Schure M.; Farnetti S.; Russo M.; Dugo P.; Mondello L. J. Chromatogr. A 2016, 1458, 54–62. 10.1016/j.chroma.2016.06.042. [DOI] [PubMed] [Google Scholar]
- Wang S.; Wang Z.; Zhou L.; Shi X.; Xu G. Anal. Chem. 2017, 89 (23), 12902–12908. 10.1021/acs.analchem.7b03659. [DOI] [PubMed] [Google Scholar]
- Woiwode U.; Reischl R. J.; Buckenmaier S.; Lindner W.; Lämmerhofer M. Anal. Chem. 2018, 90 (13), 7963–7971. 10.1021/acs.analchem.8b00676. [DOI] [PubMed] [Google Scholar]
- Wang X.; Wu H.; Luo R.; Xia D.; Jiang Z.; Han H. Anal. Methods 2017, 9 (43), 6131–6138. 10.1039/C7AY01569K. [DOI] [Google Scholar]
- Liu M.; Huang X.; Liu Q.; Chen M.; Liao S.; Zhu F.; Shi S.; Yang H.; Chen X. J. Sep. Sci. 2018, 41 (12), 2536–2543. 10.1002/jssc.201800007. [DOI] [PubMed] [Google Scholar]
- Lopes B. R.; Cassiano N. M.; Carvalho D. M.; Moisés E. C. D.; Cass Q. B. J. Pharm. Biomed. Anal. 2018, 150, 362–367. 10.1016/j.jpba.2017.12.041. [DOI] [PubMed] [Google Scholar]
- Alvim J. Jr.; Lopes B. R.; Cass Q. B. J. Chromatogr. A 2016, 1451, 120–126. 10.1016/j.chroma.2016.05.022. [DOI] [PubMed] [Google Scholar]
- Yao C.-l.; Yang W.-z.; Si W.; Shen Y.; Zhang N.-x.; Chen H.-l.; Pan H.-q.; Yang M.; Wu W.-y.; Guo D. an J. Chromatogr. A 2017, 1491, 87–97. 10.1016/j.chroma.2017.02.041. [DOI] [PubMed] [Google Scholar]
- Corgier A.; Sarrut M.; Crétier G.; Heinisch S. Chromatographia 2016, 79 (3–4), 255–260. 10.1007/s10337-015-3012-x. [DOI] [Google Scholar]
- Fan Y.; Fu Y.; Fu Q.; Cai J.; Xin H.; Dai M.; Jin Y. J. Sep. Sci. 2016, 39 (14), 2710–2719. 10.1002/jssc.201501393. [DOI] [PubMed] [Google Scholar]
- Berkecz R.; Tömösi F.; Körmöczi T.; Szegedi V.; Horváth J.; Janáky T. J. Pharm. Biomed. Anal. 2018, 149, 308–317. 10.1016/j.jpba.2017.10.043. [DOI] [PubMed] [Google Scholar]
- Yang L.; Lv P.; Ai W.; Li L.; Shen S.; Nie H.; Shan Y.; Bai Y.; Huang Y.; Liu H. Anal. Bioanal. Chem. 2017, 409 (12), 3211–3222. 10.1007/s00216-017-0261-6. [DOI] [PubMed] [Google Scholar]
- Shen S.; Yang L.; Li L.; Bai Y.; Liu H. Anal. Chim. Acta 2018, 1037, 75–86. 10.1016/j.aca.2017.11.005. [DOI] [PubMed] [Google Scholar]
- Narváez-Rivas M.; Vu N.; Chen G.-Y.; Zhang Q. Anal. Chim. Acta 2017, 954, 140–150. 10.1016/j.aca.2016.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan Y.; Liu Y.; Yang L.; Nie H.; Shen S.; Dong C.; Bai Y.; Sun Q.; Zhao J.; Liu H. J. Sep. Sci. 2016, 39 (19), 3745–3753. 10.1002/jssc.201600315. [DOI] [PubMed] [Google Scholar]
- Navarro-Reig M.; Jaumot J.; Tauler R. J. Chromatogr. A 2018, 1568, 80–90. 10.1016/j.chroma.2018.07.017. [DOI] [PubMed] [Google Scholar]
- Cacciola F.; Mangraviti D.; Rigano F.; Donato P.; Dugo P.; Mondello L.; Cortes H. J. Anal. Bioanal. Chem. 2018, 410 (15), 3473–3482. 10.1007/s00216-017-0744-5. [DOI] [PubMed] [Google Scholar]
- Montero L.; Ibáñez E.; Russo M.; di Sanzo R.; Rastrelli L.; Piccinelli A. L.; Celano R.; Cifuentes A.; Herrero M. Anal. Chim. Acta 2016, 913, 145–159. 10.1016/j.aca.2016.01.040. [DOI] [PubMed] [Google Scholar]
- Yan X.; Wang L.-J.; Wu Z.; Wu Y.-L.; Liu X.-X.; Chang F.-R.; Fang M.-J.; Qiu Y.-K. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2016, 1033–1034, 1–8. 10.1016/j.jchromb.2016.07.053. [DOI] [PubMed] [Google Scholar]
- Yao C.; Pan H.; Wang H.; Yao S.; Yang W.; Hou J.; Jin Q.; Wu W.; Guo D. J. Chromatogr. A 2018, 1538, 34–44. 10.1016/j.chroma.2018.01.040. [DOI] [PubMed] [Google Scholar]
- Navarro-Reig M.; Jaumot J.; Baglai A.; Vivó-Truyols G.; Schoenmakers P. J.; Tauler R. Anal. Chem. 2017, 89 (14), 7675–7683. 10.1021/acs.analchem.7b01648. [DOI] [PubMed] [Google Scholar]
- Mena-Bravo A.; Priego-Capote F.; Luque de Castro M. D. J. Chromatogr. A 2016, 1451, 50–57. 10.1016/j.chroma.2016.05.006. [DOI] [PubMed] [Google Scholar]
- Wang S.; Zhou L.; Wang Z.; Shi X.; Xu G. Anal. Chim. Acta 2017, 966, 34–40. 10.1016/j.aca.2017.03.004. [DOI] [PubMed] [Google Scholar]
- Wong Y. F.; Cacciola F.; Fermas S.; Riga S.; James D.; Manzin V.; Bonnet B.; Marriott P. J.; Dugo P.; Mondello L. Electrophoresis 2018, 39 (15), 1993–2000. 10.1002/elps.201700469. [DOI] [PubMed] [Google Scholar]
- Hájek T.; Jandera P.; Staňková M.; Česla P. J. Chromatogr. A 2016, 1446, 91–102. 10.1016/j.chroma.2016.04.007. [DOI] [PubMed] [Google Scholar]
- Montero L.; Sáez V.; von Baer D.; Cifuentes A.; Herrero M. J. Chromatogr. A 2018, 1536, 205–215. 10.1016/j.chroma.2017.06.013. [DOI] [PubMed] [Google Scholar]
- Stephan S.; Jakob C.; Hippler J.; Schmitz O. J. Anal. Bioanal. Chem. 2016, 408 (14), 3751–3759. 10.1007/s00216-016-9460-9. [DOI] [PubMed] [Google Scholar]
- Kula M.; Głód D.; Krauze-Baranowska M. J. Pharm. Biomed. Anal. 2016, 121, 99–106. 10.1016/j.jpba.2015.12.047. [DOI] [PubMed] [Google Scholar]
- Woiwode U.; Neubauer S.; Lindner W.; Buckenmaier S.; Lämmerhofer M. J. Chromatogr. A 2018, 1562, 69–77. 10.1016/j.chroma.2018.05.062. [DOI] [PubMed] [Google Scholar]
- van de Schans M. G. M.; Blokland M. H.; Zoontjes P. W.; Mulder P. P. J.; Nielen M. W. F. J. Chromatogr. A 2017, 1503, 38–48. 10.1016/j.chroma.2017.04.059. [DOI] [PubMed] [Google Scholar]
- Vamathevan V.; Murby E. J. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2016, 1038, 49–56. 10.1016/j.jchromb.2016.10.004. [DOI] [PubMed] [Google Scholar]
- Blokland M. H.; Zoontjes P. W.; Van Ginkel L. A.; Van De Schans M. G. M.; Sterk S. S.; Bovee T. F. H. Food Addit. Contam., Part A 2018, 35 (9), 1703–1715. 10.1080/19440049.2018.1506160. [DOI] [PubMed] [Google Scholar]
- Coulerie P.; Ratinaud Y.; Moco S.; Merminod L.; Naranjo Pinta M.; Boccard J.; Bultot L.; Deak M.; Sakamoto K.; Queiroz E. F.; Wolfender J.-L.; Barron D. J. Nat. Prod. 2016, 79 (11), 2856–2864. 10.1021/acs.jnatprod.6b00628. [DOI] [PubMed] [Google Scholar]
- van Beek F. T.; Edam R.; Pirok B. W. J.; Genuit W. J. L.; Schoenmakers P. J. J. Chromatogr. A 2018, 1564, 110–119. 10.1016/j.chroma.2018.06.001. [DOI] [PubMed] [Google Scholar]
- Ouyang X.; Weiss J. M.; de Boer J.; Lamoree M. H.; Leonards P. E. G. Chemosphere 2017, 166, 431–437. 10.1016/j.chemosphere.2016.09.107. [DOI] [PubMed] [Google Scholar]
- Sun M.; Sandahl M.; Turner C. J. Chromatogr. A 2018, 1541, 21–30. 10.1016/j.chroma.2018.02.008. [DOI] [PubMed] [Google Scholar]
- Eckberg M. N.; Arroyo-Mora L. E.; Stoll D. R.; DeCaprio A. P. J. Anal. Toxicol. 2018, bky081. 10.1093/jat/bky081. [DOI] [PubMed] [Google Scholar]
- Shao X.; Chen M.; Wu D.; Liu B.; Zhang X.; Chen C. Anal. Methods 2017, 9 (5), 761–767. 10.1039/C6AY02762H. [DOI] [Google Scholar]
- Qi D.; Fei T.; Liu H.; Yao H.; Wu D.; Liu B. J. Agric. Food Chem. 2017, 65 (45), 9923–9929. 10.1021/acs.jafc.7b04329. [DOI] [PubMed] [Google Scholar]
- Sánchez-Camargo A. P.; Montero L.; Cifuentes A.; Herrero M.; Ibáñez E. RSC Adv. 2016, 6 (97), 94884–94895. 10.1039/C6RA16862K. [DOI] [Google Scholar]
- Montero L.; Sánchez-Camargo A. P.; García-Cañas V.; Tanniou A.; Stiger-Pouvreau V.; Russo M.; Rastrelli L.; Cifuentes A.; Herrero M.; Ibáñez E. J. Chromatogr. A 2016, 1428, 115–125. 10.1016/j.chroma.2015.07.053. [DOI] [PubMed] [Google Scholar]
- Guo P.; Xu X.; Chen G.; Bashir K.; Shu H.; Ge Y.; Jing W.; Luo Z.; Chang C.; Fu Q. J. Pharm. Biomed. Anal. 2017, 146, 292–301. 10.1016/j.jpba.2017.09.008. [DOI] [PubMed] [Google Scholar]
- Guo P.; Xu X.; Xian L.; Ge Y.; Luo Z.; Du W.; Jing W.; Zeng A.; Chang C.; Fu Q. Talanta 2016, 161, 830–837. 10.1016/j.talanta.2016.09.041. [DOI] [PubMed] [Google Scholar]
- Dang J.; Zhang L.; Shao Y.; Mei L.; Liu Z.; Yue H.; Wang Q.; Tao Y. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2018, 1095, 267–274. 10.1016/j.jchromb.2018.08.005. [DOI] [PubMed] [Google Scholar]
- Wei Z.; Fu Q.; Cai J.; Huan L.; Zhao J.; Shi H.; Jin Y.; Liang X. J. Sep. Sci. 2016, 39 (12), 2221–2228. 10.1002/jssc.201600134. [DOI] [PubMed] [Google Scholar]
- Zhou W.; Guo Z.; Yu L.; Zhou H.; Shen A.; Jin Y.; Jin G.; Yan J.; Yang F.; Liu Y.; Wang C.; Feng J.; Liu Y.; Liang X. Talanta 2018, 186, 73–79. 10.1016/j.talanta.2018.04.014. [DOI] [PubMed] [Google Scholar]
- Wang Y. Q.; Tang X.; Li J. F.; Wu Y. L.; Sun Y. Y.; Fang M. J.; Wu Z.; Wang X. M.; Qiu Y. K. J. Chromatogr. A 2017, 1519, 145–151. 10.1016/j.chroma.2017.08.053. [DOI] [PubMed] [Google Scholar]
- Zhou X.; Chen C.; Ye X.; Song F.; Fan G.; Wu F. J. Chromatogr. Sci. 2017, 55 (1), 75–81. 10.1093/chromsci/bmw154. [DOI] [PubMed] [Google Scholar]
- Qiao X.; Wang Q.; Song W.; Qian Y.; Xiao Y.; An R.; Guo D.; Ye M. J. Chromatogr. A 2016, 1438, 198–204. 10.1016/j.chroma.2016.02.034. [DOI] [PubMed] [Google Scholar]
- Yang W.; Zhang J.; Yao C.; Qiu S.; Chen M.; Pan H.; Shi X.; Wu W.; Guo D. J. Pharm. Biomed. Anal. 2016, 128, 322–332. 10.1016/j.jpba.2016.05.035. [DOI] [PubMed] [Google Scholar]
- Wang W.; Tao Y.; Jiao L.; Fan M.; Shao Y.; Wang Q.; Mei L.; Dang J. J. Sep. Sci. 2017, 40 (18), 3593–3601. 10.1002/jssc.201700449. [DOI] [PubMed] [Google Scholar]
- Sun W.; Tong L.; Miao J.; Huang J.; Li D.; Li Y.; Xiao H.; Sun H.; Bi K. J. Chromatogr. A 2016, 1431, 79–88. 10.1016/j.chroma.2015.12.038. [DOI] [PubMed] [Google Scholar]
- Cao J.-L.; Wei J.-C.; Hu Y.-J.; He C.-W.; Chen M.-W.; Wan J.-B.; Li P. J. Chromatogr. A 2016, 1427, 79–89. 10.1016/j.chroma.2015.11.078. [DOI] [PubMed] [Google Scholar]
- Jin G.; Liu Y.; Yang F.; Yu D.; Yan J.; Zhou W.; Guo Z.; Zhu J.; Liang X. J. Sep. Sci. 2018, 41 (4), 856–867. 10.1002/jssc.201701125. [DOI] [PubMed] [Google Scholar]
- Wang S.; Wang Q.; Qiao X.; Song W.; Zhong L.; Guo D.; Ye M. Planta Med. 2016, 82 (18), 1558–1567. 10.1055/s-0042-110206. [DOI] [PubMed] [Google Scholar]
- Jiao L.; Tao Y.; Wang W.; Shao Y.; Mei L.; Wang Q.; Dang J. J. Sep. Sci. 2017, 40 (19), 3808–3816. 10.1002/jssc.201700675. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Jin H.; Li X.; Zhao J.; Guo X.; Wang J.; Guo Z.; Zhang X.; Tao Y.; Liu Y.; Chen D.; Liang X. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2016, 1026, 67–74. 10.1016/j.jchromb.2015.11.015. [DOI] [PubMed] [Google Scholar]
- Long Z.; Zhang Y.; Gamache P.; Guo Z.; Steiner F.; Du N.; Liu X.; Jin Y.; Liu X.; Liu L. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2018, 1072, 70–77. 10.1016/j.jchromb.2017.10.064. [DOI] [PubMed] [Google Scholar]
- Lin Y.; Wang C.; Shao X.; Yang L.; Hou Y.; He H.; Sun M. Curr. Pharm. Anal. 2018, 14 (5), 496–500. 10.2174/1573412913666170613085815. [DOI] [Google Scholar]
- Liu C.-M.; Chen J.; Yang S.; Mao L.-G.; Jiang T.-T.; Tu H.-H.; Chen Z.-L.; Hu Y.-T.; Gan L.; Li Z.-J.; Li J.-C. J. Ethnopharmacol. 2018, 225, 271–278. 10.1016/j.jep.2018.05.001. [DOI] [PubMed] [Google Scholar]
- Ji S.; He D.; Wang T.; Han J.; Li Z.; Du Y.; Zou J.; Guo M.; Tang D. J. Pharm. Biomed. Anal. 2017, 146, 68–78. 10.1016/j.jpba.2017.07.057. [DOI] [PubMed] [Google Scholar]
- Zhang F.; Ni Y.; Yuan Y.; Yin W.; Gao Y. Sci. China: Life Sci. 2018, 10.1007/s11427-017-9268-y. [DOI] [PubMed] [Google Scholar]
- Saleh A.; Edlund P.-O.; Gustafsson T. N.; Sahlin M.; Sjöberg B.-M.; Granelli I. Chromatographia 2016, 79 (7–8), 383–393. 10.1007/s10337-016-3048-6. [DOI] [Google Scholar]
- Yang L.; Zou Q.-H.; Zhang Y.; Shi Y.; Hu C.-R.; Hui C.-X.; Liu X.-F.; Fang Y.-F. Clin. Rheumatol. 2018, 37 (7), 1773–1782. 10.1007/s10067-017-3974-1. [DOI] [PubMed] [Google Scholar]
- Kwiatkowski M.; Krösser D.; Wurlitzer M.; Steffen P.; Barcaru A.; Krisp C.; Horvatovich P.; Bischoff R.; Schlüter H. Anal. Chem. 2018, 90 (16), 9951–9958. 10.1021/acs.analchem.8b02189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang C.-F.; King C.-E.; Ho B.-W.; Chien K.-Y.; Chang Y.-C. J. Proteome Res. 2018, 17 (5), 1953–1966. 10.1021/acs.jproteome.8b00069. [DOI] [PubMed] [Google Scholar]
- Nascimento S. V. do; Magalhães M. M.; Cunha R. L.; Costa P. H. de O.; Alves R. C. de O.; Oliveira G. C. de; Valadares R. B. da S. PLoS One 2018, 13 (4), e0195538. 10.1371/journal.pone.0195538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang D.; Yang W.; Deng Z.; An X.; Zhao L.; Hu Q. J. Agric. Food Chem. 2017, 65 (47), 10368–10381. 10.1021/acs.jafc.7b04393. [DOI] [PubMed] [Google Scholar]
- Lee H.; Mun D.-G.; So J. E.; Bae J.; Kim H.; Masselon C.; Lee S.-W. Anal. Chem. 2016, 88 (23), 11734–11741. 10.1021/acs.analchem.6b03366. [DOI] [PubMed] [Google Scholar]
- Li W.; Zhang W.; Deng W.; Zhong Y.; Zhang Y.; Peng Z.; Chen H.; Sun R.; Zhang X.; Yang S. Thorac. Cancer 2018, 9, 1366. 10.1111/1759-7714.12839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W.; Zheng H.; Qin H.; Liu G.; Ke L.; Li Y.; Li N.; Zhong X. Clin. Respir. J. 2018, 12 (6), 2036–2045. 10.1111/crj.12771. [DOI] [PubMed] [Google Scholar]
- Dong H.; Guo Z.; Feng W.; Zhang T.; Zhai G.; Palusiak A.; Rozalski A.; Tian S.; Bai X.; Shen L.; Chen P.; Wang Q.; Fan E.; Cheng Z.; Zhang K. Mol. Cell. Proteomics 2018, 17 (3), 482–494. 10.1074/mcp.RA117.000430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y.; Cai X.; Cao J.; Wu Z.; Pan D. Front. Microbiol. 2018, 9, 1078. 10.3389/fmicb.2018.01078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y. X.; Gao C. Y.; Wang X. Q.; Yuan Y. Q.; Liu Y. H.; Jia J. G. J. Trace Elem. Med. Biol. 2017, 44, 331–338. 10.1016/j.jtemb.2017.09.012. [DOI] [PubMed] [Google Scholar]
- Ye F.; Wang J.; Meng W.; Qian J.; Jin M. Sci. Rep. 2017, 7, 17981. 10.1038/s41598-017-18069-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J.; Zheng L.; Lin L.; Sun B.; Su G.; Zhao M. Anal. Chim. Acta 2018, 1018 (381), 119–126. 10.1016/j.aca.2018.02.025. [DOI] [PubMed] [Google Scholar]
- Cheng C.; Liao C.-F. J. Chin. Chem. Soc. 2018, 65 (6), 714–725. 10.1002/jccs.201700380. [DOI] [Google Scholar]
- Xu J.; Sun-Waterhouse D.; Qiu C.; Zhao M.; Sun B.; Lin L.; Su G. J. Chromatogr. A 2017, 1521, 80–89. 10.1016/j.chroma.2017.09.025. [DOI] [PubMed] [Google Scholar]
- Luo H.; Zhong W.; Yang J.; Zhuang P.; Meng F.; Caldwell J.; Mao B.; Welch C. J. J. Pharm. Biomed. Anal. 2017, 137, 139–145. 10.1016/j.jpba.2016.11.012. [DOI] [PubMed] [Google Scholar]
- Wang J.; Xu Y.; Zhang Y.; Wang H.; Zhong W. Rapid Commun. Mass Spectrom. 2017, 31 (18), 1541–1550. 10.1002/rcm.7934. [DOI] [PubMed] [Google Scholar]
- Wang L.; Yang B.; Zhang X.; Zheng H. Food Anal. Methods 2017, 10 (6), 2001–2010. 10.1007/s12161-016-0763-4. [DOI] [Google Scholar]
- Wang J.; Xu Y.; Wen C.; Wang Z. Anal. Chim. Acta 2017, 992, 42–54. 10.1016/j.aca.2017.08.028. [DOI] [PubMed] [Google Scholar]
- Gonçalves V. M. F.; Rodrigues P.; Ribeiro C.; Tiritan M. E. J. Pharm. Biomed. Anal. 2017, 141, 1–8. 10.1016/j.jpba.2017.03.064. [DOI] [PubMed] [Google Scholar]
- Wang J.; Zheng S.; Xu Y.; Hu H.; Shen M.; Tang L. J. Pharm. Biomed. Anal. 2018, 161, 399–406. 10.1016/j.jpba.2018.08.055. [DOI] [PubMed] [Google Scholar]
- Dai L.; Yeh G. K.; Ran Y.; Yehl P.; Zhang K. J. Pharm. Biomed. Anal. 2017, 137, 182–188. 10.1016/j.jpba.2017.01.036. [DOI] [PubMed] [Google Scholar]
- Yang S. H.; Wang J.; Zhang K. J. Chromatogr. A 2017, 1492, 89–97. 10.1016/j.chroma.2017.02.074. [DOI] [PubMed] [Google Scholar]
- Iguiniz M.; Corbel E.; Roques N.; Heinisch S. J. Pharm. Biomed. Anal. 2018, 159, 237–244. 10.1016/j.jpba.2018.06.058. [DOI] [PubMed] [Google Scholar]
- Barhate C. L.; Regalado E. L.; Contrella N. D.; Lee J.; Jo J.; Makarov A. A.; Armstrong D. W.; Welch C. J. Anal. Chem. 2017, 89 (6), 3545–3553. 10.1021/acs.analchem.6b04834. [DOI] [PubMed] [Google Scholar]
- Iguiniz M.; Rouvière F.; Corbel E.; Roques N.; Heinisch S. J. Chromatogr. A 2018, 1536, 195–204. 10.1016/j.chroma.2017.08.070. [DOI] [PubMed] [Google Scholar]
- Sheng Y.; Zhou B. J. Chromatogr. A 2017, 1499, 48–56. 10.1016/j.chroma.2017.02.061. [DOI] [PubMed] [Google Scholar]
- Wang H.; Xu T.; Yuan J. J. Sep. Sci. 2017, 40 (8), 1674–1685. 10.1002/jssc.201601320. [DOI] [PubMed] [Google Scholar]
- Ndiripo A.; Pasch H. Anal. Chim. Acta 2018, 1027, 137–148. 10.1016/j.aca.2018.03.007. [DOI] [PubMed] [Google Scholar]
- Ndiripo A.; Pasch H. Anal. Chem. 2018, 90 (12), 7626–7634. 10.1021/acs.analchem.8b01480. [DOI] [PubMed] [Google Scholar]
- Lee S.; Choi H.; Chang T.; Staal B. Anal. Chem. 2018, 90 (10), 6259–6266. 10.1021/acs.analchem.8b00913. [DOI] [PubMed] [Google Scholar]
- Apel N.; Uliyanchenko E.; Moyses S.; Rommens S.; Wold C.; Macko T.; Brüll R. Anal. Chem. 2018, 90 (8), 5422–5429. 10.1021/acs.analchem.8b00618. [DOI] [PubMed] [Google Scholar]
- Apel N.; Ramakrishnan V.; Uliyanchenko E.; Moyses S.; Wold C.; Macko T.; Brüll R. Macromolecules 2018, 51 (13), 4829–4839. 10.1021/acs.macromol.8b00667. [DOI] [Google Scholar]
- Yang P.; Bai L.; Wang W.; Rabasco J. J. Chromatogr. A 2018, 1560, 55–62. 10.1016/j.chroma.2018.05.033. [DOI] [PubMed] [Google Scholar]
- Lee T.; Oh J.; Jeong J.; Jung H.; Huh J.; Chang T.; Paik H. Macromolecules 2016, 49 (10), 3672–3680. 10.1021/acs.macromol.6b00093. [DOI] [Google Scholar]
- Malik M. I.; Lee S.; Chang T. J. Chromatogr. A 2016, 1442, 33–41. 10.1016/j.chroma.2016.03.008. [DOI] [PubMed] [Google Scholar]
- Urban J.; Hájek T.; Svec F. J. Sep. Sci. 2017, 40 (8), 1703–1709. 10.1002/jssc.201700048. [DOI] [PubMed] [Google Scholar]
- Lynch K. B.; Yang Y.; Ren J.; Liu S. Talanta 2018, 181, 416–421. 10.1016/j.talanta.2018.01.045. [DOI] [PubMed] [Google Scholar]
- Zhu Z.; Chen H.; Ren J.; Lu J. J.; Gu C.; Lynch K. B.; Wu S.; Wang Z.; Cao C.; Liu S. Talanta 2018, 179, 588–593. 10.1016/j.talanta.2017.11.060. [DOI] [PubMed] [Google Scholar]
- Ren J.; Beckner M. A.; Lynch K. B.; Chen H.; Zhu Z.; Yang Y.; Chen A.; Qiao Z.; Liu S.; Lu J. J. Talanta 2018, 182, 225–229. 10.1016/j.talanta.2018.01.072. [DOI] [PubMed] [Google Scholar]
- Vonk R. J.; Wouters S.; Barcaru A.; Vivó-Truyols G.; Eeltink S.; de Koning L. J.; Schoenmakers P. J. Anal. Bioanal. Chem. 2015, 407 (13), 3817–3829. 10.1007/s00216-015-8615-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z.; Yan G.; Gao M.; Zhang X. Anal. Chem. 2016, 88 (4), 2440–2445. 10.1021/acs.analchem.5b04553. [DOI] [PubMed] [Google Scholar]
- Vanhoenacker G.; Steenbeke M.; Sandra K.; Sandra P. LC-GC Eur. 2018, 31 (7), 360–370. [Google Scholar]
- Yang P.; Gao W.; Shulman J. E.; Chen Y. J. Chromatogr. A 2018, 1566, 111–117. 10.1016/j.chroma.2018.06.063. [DOI] [PubMed] [Google Scholar]
- Iguiniz M.; Heinisch S. J. Pharm. Biomed. Anal. 2017, 145, 482–503. 10.1016/j.jpba.2017.07.009. [DOI] [PubMed] [Google Scholar]
- Hamase K.; Morikawa A.; Ohgusu T.; Lindner W.; Zaitsu K. J. Chromatogr. A 2007, 1143 (1–2), 105–111. 10.1016/j.chroma.2006.12.078. [DOI] [PubMed] [Google Scholar]
- Cortes H. J.; Campbell R. M.; Himes R. P.; Pfeiffer C. D. J. Microcolumn Sep. 1992, 4 (3), 239–244. 10.1002/mcs.1220040310. [DOI] [Google Scholar]
- Venkatramani C. J.; Al-Sayah M.; Li G.; Goel M.; Girotti J.; Zang L.; Wigman L.; Yehl P.; Chetwyn N. Talanta 2016, 148, 548–555. 10.1016/j.talanta.2015.10.054. [DOI] [PubMed] [Google Scholar]
- Iguiniz M.; Corbel E.; Roques N.; Heinisch S. J. Pharm. Biomed. Anal. 2018, 159, 237–244. 10.1016/j.jpba.2018.06.058. [DOI] [PubMed] [Google Scholar]
- Beck A.; Terral G.; Debaene F.; Wagner-Rousset E.; Marcoux J.; Janin-Bussat M.-C.; Colas O.; Van Dorsselaer A.; Cianférani S. Expert Rev. Proteomics 2016, 13 (2), 157–183. 10.1586/14789450.2016.1132167. [DOI] [PubMed] [Google Scholar]
- Chen T.; Chen Y.; Stella C.; Medley C. D.; Gruenhagen J. A.; Zhang K. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2016, 1032, 39–50. 10.1016/j.jchromb.2016.07.023. [DOI] [PubMed] [Google Scholar]
- Stoll D. R.; Maloney T. D. LC-GC N. Am. 2017, 35 (9), 680–687. [Google Scholar]
- Wang X.; Buckenmaier S.; Stoll D. J. Appl. Bioanal. 2017, 3 (5), 120–126. 10.17145/jab.17.015. [DOI] [Google Scholar]
- Stoll D.; Danforth J.; Zhang K.; Beck A. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2016, 1032, 51–60. 10.1016/j.jchromb.2016.05.029. [DOI] [PubMed] [Google Scholar]
- Sandra K.; Steenbeke M.; Vandenheede I.; Vanhoenacker G.; Sandra P. J. Chromatogr. A 2017, 1523, 283–292. 10.1016/j.chroma.2017.06.052. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Aller M.; Guillarme D.; Beck A.; Fekete S. J. Pharm. Biomed. Anal. 2016, 118, 393–403. 10.1016/j.jpba.2015.11.011. [DOI] [PubMed] [Google Scholar]
- Sarrut M.; Corgier A.; Fekete S.; Guillarme D.; Lascoux D.; Janin-Bussat M.-C.; Beck A.; Heinisch S. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2016, 1032, 103–111. 10.1016/j.jchromb.2016.06.048. [DOI] [PubMed] [Google Scholar]
- Giddings J. C. C. J. Chromatogr. A 1995, 703 (1–2), 3–15. 10.1016/0021-9673(95)00249-M. [DOI] [PubMed] [Google Scholar]
- Uliyanchenko E. Chromatographia 2017, 80 (5), 731–750. 10.1007/s10337-016-3193-y. [DOI] [Google Scholar]
- Schoenmakers P.; Aarnoutse P. Anal. Chem. 2014, 86 (13), 6172–6179. 10.1021/ac301162b. [DOI] [PubMed] [Google Scholar]