

Abstract
BACKGROUND
Plumbing systems are an infrequent but known reservoir for opportunistic microbial pathogens that can infect hospitalized patients. In 2016, a cluster of clinical sphingomonas infections prompted an investigation.
METHODS
We performed whole-genome DNA sequencing on clinical isolates of multidrug-resistant Sphingomonas koreensis identified from 2006 through 2016 at the National Institutes of Health (NIH) Clinical Center. We cultured S. koreensis from the sinks in patient rooms and performed both whole-genome and shotgun metagenomic sequencing to identify a reservoir within the infrastructure of the hospital. These isolates were compared with clinical and environmental S. koreensis isolates obtained from other institutions.
RESULTS
The investigation showed that two isolates of S. koreensis obtained from the six patients identified in the 2016 cluster were unrelated, but four isolates shared more than 99.92% genetic similarity and were resistant to multiple antibiotic agents. Retrospective analysis of banked clinical isolates of sphingomonas from the NIH Clinical Center revealed the intermittent recovery of a clonal strain over the past decade. Unique single-nucleotide variants identified in strains of S. koreensis elucidated the existence of a reservoir in the hospital plumbing. Clinical S. koreensis isolates from other facilities were genetically distinct from the NIH isolates. Hospital remediation strategies were guided by results of microbiologic culturing and fine-scale genomic analyses.
CONCLUSIONS
This genomic and epidemiologic investigation suggests that S. koreensis is an opportunistic human pathogen that both persisted in the NIH Clinical Center infrastructure across time and space and caused health care-associated infections. (Funded by the NIH Intramural Research Programs.)
Health care–associated infections affect 2 million patients each year in the United States, and an increasing proportion is attributed to multidrug-resistant bacteria.1 Waterborne bacteria represent an important subset of health care–associated pathogens and are of increasing concern to public health authorities. In 2017, the Centers for Medicare and Medicaid Services established stringent requirements for health care facilities to reduce the transmission risk of waterborne organisms from hospital plumbing systems to patients.2
Historically, emphasis has been placed on preventing health care–associated transmission of legionella and pseudomonas species; however, other waterborne organisms including stenotrophomonas, sphingomonas, burkholderia, and nontuberculous mycobacteria3–5 also pose risks for health care–associated infections, particularly in immunocompromised patients. Exposure may occur through water droplets or water aerosols that are inhaled or that breach normal defenses through nonintact mucous membranes or invasive devices.6–8
Whole-genome sequence analyses have clarified epidemiologic chains of transmission for outbreaks of Mycobacterium tuberculosis, carbapenem-resistant Klebsiella pneumoniae, methicillin-resistant Staphylococcus aureus, and Legionella pneumophila.9–13 Underlying genetic diversity can be a confounder when single isolates from a given source are analyzed and cutoffs are applied to the number of single-nucleotide variants (SNVs) that constitute a “match.”14 However, characterizing the underlying genetic diversity can provide critical information to elucidate patient-to-patient transmission events or to identify reservoirs in the environment.
Although sphingomonas species are ubiquitous in natural and man-made aqueous environments, community-acquired infections are rarely reported.15 The most common species implicated as a human pathogen, S. paucimobilis, causes a range of infections, including pneumonia, meningitis, catheter-associated bloodstream infections, and wound infections.16,17
Although the Centers for Disease Control and Prevention (CDC) urges that a single case of health care–associated legionnaires’ disease should trigger an epidemiologic investigation,18 intermittent infections with most other waterborne pathogens most likely go unrecognized, unless they are clustered temporally. Over a 6-month period in 2016, infections with sphingomonas species developed in six inpatients at the National Institutes of Health (NIH) Clinical Center. Isolates from four of these six patients were identified as multidrug-resistant S. koreensis, a nonfermenting gram-negative bacillus previously reported in only two clinical cases.19,20 This cluster triggered an epidemiologic investigation that used both culture-based and genomics-based techniques to identify possible sources and to inform effective intervention strategies.
METHODS
IDENTIFICATION OF SPHINGOMONAS CASES
The epidemiologic investigation that was initiated when the cluster of sphingomonas infections was identified in 2016 included clinical and microbiologic record review and genomic analysis. Records identified 37 patients from 2001 through 2016 who had sphingomonas species cultured from clinical specimens (Fig. 1; and Table S1 in the Supplementary Appendix, available with the full text of this article at NEJM.org). Environmental and clinical S. koreensis isolates from geographically distant institutions were solicited for genomic and microbiologic comparisons. A waiver of informed consent was granted by the NIH Office of Human Subjects Research Protection for this study.
Figure 1. Sphingomonas Infections at the National Institutes of Health (NIH) Clinical Center, According to Year.

A new NIH Clinical Center building was constructed in 2004 and opened in 2005.
EPIDEMIOLOGIC INVESTIGATION
Patient records were reviewed for possible common sources of exposure, including hospital rooms, wards, invasive procedures, dialysis, respiratory treatments, and therapeutic baths. Because sphingomonas species reside in aqueous reservoirs, we cultured potable water (100 samples), sink faucets (56 samples), ice machines (7 samples), and other plumbing components (52 samples) from October 2016 through December 2017. Samples were obtained from water and from faucets in the rooms in which inpatients were staying when they acquired S. koreensis infections. Large-volume water samples were also collected from the main municipal intake pipe and its branches, from the heat exchanger, from down-stream of the heat exchanger, from the recirculating hot-water loop, and from pipes supplying rooms with known culture-positive sinks. With the aid of plumbers, three culture-positive sinks were disassembled into isolated components, and any visible biofilm and attached pipe segments were cultured.
From November 2016 through December 2016, water samples were collected in triplicate from 25 sinks (samples of hot and cold water were collected from 13 manual sinks, and 12 mixed-temperature samples were collected from automatic sinks) (Table S2 in the Supplementary Appendix). The water samples were tested for free and total chlorine concentrations with the use of a digital colorimetric assay (with the Hach DR900 colorimeter) (Table S2 in the Supplementary Appendix). Beginning in February 2017, the chlorine concentrations were measured serially from 13 faucets distributed throughout the hospital, and hot-water temperature was measured at the heat exchanger two to five times weekly.
CHARACTERIZATION OF THE BACTERIAL ISOLATES
Isolates were assessed by whole-genome sequencing, which was performed on an Illumina MiSeq instrument with 300-bp paired-end reads, as described previously.21,22 DNA sequence reads were assembled with the use of SPAdes and polished with the use of Pilon.23,24 On average, each MiSeq genome assembly contained 93 contigs with a mean N50 of 223,917 bp. To obtain a complete genome, one S. koreensis isolate from the NIH Clinical Center was sequenced with the use of the PacBio RS II SMRT platform (with the P6 polymerase and C4 sequencing kit, with a 240-minute data-collection time). PacBio reads were assembled and polished with the use of Canu.25 Further details on sequencing and bacterial culturing methods are described in the Supplementary Appendix.
COMPARATIVE GENOMIC ANALYSES
To assess the genetic relatedness of the sphingomonas isolates, the average nucleotide identity of each pairwise genome comparison was calculated with the use of the Mash algorithm, version 2.0, on the basis of 1000 21-bp substrings (k-mers) selected from each genome.26 Reads from each S. koreensis isolate were mapped to the reference S. koreensis PacBio genome to assess genome content. For fine mapping, SNVs were identified in genomic regions shared among all isolates (the core genome). In brief, the MiSeq reads for each isolate were aligned to the PacBio reference genome, and SNVs were identified with the use of Snippy, version 3.2 (with the BWA-MEM algorithm, version 0.7.17, and Free-Bayes software, version 1.1.0; https://github.com/tseemann/snippy), which requires a read depth of at least 10x; we specified that at least 90% of reads should support the variant nucleotide call. A recombination-corrected core genome alignment was used to construct a phylogenetic tree of the S. koreensis strains isolated from the NIH Clinical Center.
To detect the underlying genetic heterogeneity of the primary environmental cultures, DNA was prepared from an entire plate (or a quadrant if densely populated), and ensuing sequence reads were aligned individually to the reference S. koreensis PacBio genome. Nucleotide frequency at each unique SNV was assessed with the use of Bam-readcount (https://github.com/genome/bam-readcount). All data visualization was performed with the use of R Software, version 3.4.3 (R Foundation for Statistical Computing).27 An in-depth description of the bioinformatic methods is provided in the Supplementary Appendix. The sequencing data for this study are linked to National Center for Biotechnology Information BioProject number PRJNA445389.
RESULTS
CLINICAL CHARACTERISTICS OF THE PATIENTS
From 2006, a year after the opening of a new inpatient hospital building, through 2016, S. koreensis clinical isolates were identified in 12 patients at the NIH Clinical Center (Table S1 in the Supplementary Appendix). Seven isolates from patients were initially identified only at the genus level, and the species was assigned retrospectively. All the patients were hospitalized at the time of their positive cultures; 5 patients were in the intensive care unit and 7 were in three other wards. The median length of stay was 44 days (range, 0 to 374) before a positive culture was obtained. Four patients had a single positive culture, and the other patients had either persistent or recurrent positive cultures over a range of 2 to 43 days. One isolate per patient was sequenced. Among the 12 patients, 9 were recipients of stem-cell transplants. Eight of the 12 patients had S. koreensis bacteremia, including 2 who had concurrent S. koreensis pneumonia and 3 who had catheter-related bloodstream infections. In addition, 1 patient had pneumonia alone, 1 had cholecystitis that led to peritonitis, and 1 had a deep surgical-site infection. One of the 12 patients had S. koreensis cultured from urine with a low concentration in the absence of pyuria; this patient did not have a urinary tract infection, and the organism was believed to represent contamination or colonization. Of the remaining 11 patients, 8 patients with S. koreensis infections recovered, and 3 patients died (all 3 patients had S. koreensis sepsis as well as severe, unrelated infections) (Table S1 in the Supplementary Appendix).
INVESTIGATION OF CLINICAL ISOLATES OF SPHINGOMONAS FROM 2016
Of the six clinical sphingomonas cases in 2016, genomic sequencing classified four as S. koreensis, one as S. yanoikuyae, and one as S. trueperi. The four S. koreensis clinical isolates possessed an exceptionally high degree of genetic similarity (>99.92% average nucleotide identity), which suggested that they belonged to the same clonal strain. These S. koreensis isolates exhibited resistance to multiple classes of antibiotic agents, including aminoglycosides (amikacin and gentamicin), beta-lactams (aztreonam, piperacillin-tazobactam, cefepime, ceftazidime, ceftriaxone, and meropenem), and fluoroquinolones (levo-floxacin) (Fig. 2) — a finding that reflects the difficulty in treating these infections. The isolates were susceptible to trimethoprim-sulfamethoxazole and ciprofloxacin, and therefore these agents were used for treatment. Other sphingomonas clinical isolates were resistant to fewer classes of antibiotics. Aside from a partially conserved chloramphenicol acetyltransferase (catB) gene (96% coverage, 77% identity), no canonical resistance genes could be identified within the S. koreensis genome, a finding that is consistent with intrinsic resistance or undiscovered mechanisms of antibiotic resistance.
Figure 2. Genome Comparisons of the Sphingomonas Isolates Obtained from Patients at the NIH Clinical Center.

Panel A shows antibiotic susceptibility patterns for the sphingomonas isolates obtained from patients. Panel B shows the genetic relatedness of the isolates on the basis of average nucleotide identity, with purple squares indicating the greatest similarity. Isolates are named according to species (Sphingomonas koreensis [SK] or other sphingomonas species [S]), location (NIH), patient number (Pt), and date (month [MM] and year [YY]). Sphingomonas isolates from 2016 are highlighted in red.
EPIDEMIOLOGIC INVESTIGATION TO IDENTIFY THE SOURCE OF S. KOREENSIS
S. koreensis grew from 22 of 56 faucets (39%) and from 9 of 17 water samples (53%) collected from faucets in patient rooms. Three positive water samples were collected from faucets with culture-negative swabs.
Environmental S. koreensis isolates showed resistance to numerous antibiotics, similar to the isolates obtained from patients (Fig. S1 in the Supplementary Appendix). Isolates derived from sink components and water samples were genetically related to the S. koreensis isolates obtained from patients in 2016 (>99.7% average nucleotide identity) (Table 1, and Fig. S1 in the Supplementary Appendix), which implicated sinks or water as the most likely source of nosocomial S. koreensis infections.
Table 1.
Sphingomonas koreensis Genome Sequences Compared with the Reference Genome.*
| Isolate Source and Sequence | No. of Isolates | Mean Percent Aligned (Range)† | Average No. of SNVs (Range)‡ |
|---|---|---|---|
| Patients at the NIH Clinical Center | 12 | ||
| Chromosome | 99.11 (98.76–99.26) | 18.91 (11–41) | |
| Plasmid | 95.86 (93.94–96.98) | 60.18 (0–661)§ | |
| Sinks at the NIH Clinical Center | 55 | ||
| Chromosome | 98.94 (97.87–99.19) | 20.13 (12–29) | |
| Plasmid | 96.41 (90.98–97.05) | 6.53 (0–109)¶ | |
| CDC | 1 | ||
| Chromosome | 84.82 | 33,754 | |
| Plasmid | 34.12 | 1949 | |
| Wadsworth Center | 1 | ||
| Chromosome | 97.09 | 145 | |
| Plasmid | 72.42 | 1658 | |
| Korean environmental samples∥ | 2 | ||
| Chromosome | 84.66 (84.63–84.69) | 36,188 (36,188–36,188) | |
| Plasmid | 2.61 (2.10–3.13) | 86 (81–92) |
The S. koreensis isolate SK-NIH.Pt5_1016, obtained from a patient at the NIH Clinical Center, was sequenced with the use of the PacBio system and was used as the reference genome (GenBank accession number, PRJNA354050). External clinical isolates were obtained from the Centers for Disease Control and Prevention (CDC) and from the Wadsworth Center, New York State Department of Health (Albany, NY), and environmental isolates were obtained from Korea.
The percent aligned indicates the percentage of nucleotides in the reference sequence that have at least one read aligned with the sequenced isolate.
Single-nucleotide variants (SNVs) were called with the use of Snippy (https://github.com/tseemann/snippy).
One plasmid sequence from an isolate obtained from a patient contained 661 SNVs. The remaining 11 plasmid sequences contained 0 to 1 SNV.
Four plasmid sequences obtained from sinks contained 27 to 109 SNVs. The remaining 51 plasmid sequences contained 0 to 3 SNVs.
Isolates of S. koreensis from Korea (KCTC_2882 and KCTC_2883) were obtained from the Korean Collection for Type Cultures.
The free chlorine concentration and water temperature of the samples were evaluated. Cold-water samples contained adequate free chlorine concentrations (≥0.5 mg per liter, as recommended by the CDC28), but chlorine concentrations of hot-water samples were well below the recommended threshold. Starting in December 2016, hot water returning from the recirculating loops was flushed continuously at a rate that was calibrated to maintain circulating free chlorine levels above 0.5 mg per liter, effectively replacing water that had declining concentrations of free chlorine with more freshly chlorinated municipal water. Concentrations of free chlorine in hot water rose continuously from December 2016 through July 2017, reaching concentrations of approximately 1.0 mg per liter. Water temperature was determined at the heat exchanger and was adjusted from a temperature of 46 to 49°C to a temperature of 60°C or higher in accordance with the hospital standard of 51°C or higher.29
HISTORICAL INVESTIGATION OF S. KOREENSIS AT THE NIH CLINICAL CENTER
Eight additional S. koreensis clinical isolates from the NIH Clinical Center were identified that dated back to 2006, a year after the opening of a new hospital building (Fig. S2 in the Supplementary Appendix). These isolates shared similar levels of nucleotide identity with one another and with isolates from 2016 (>99.8% average nucleotide identity), which supports the persistence of a reservoir (Fig. 2). When the isolates obtained from patients were compared with the reference S. koreensis genome, they were found to have 11 to 41 chromosomal SNVs and 0 to 1 SNV on the plasmid, except for one variant plasmid that had 661 SNVs (Table 1). Over the course of a decade, this represents a very low mutation rate, which is consistent with the ability of sphingomonas to survive in stasis in low-nutrient environments.30,31
CHARACTERIZATION OF EXTERNAL S. KOREENSIS ISOLATES
To determine whether these genetically related S. koreensis isolates were unique to the NIH Clinical Center, four external S. koreensis isolates were obtained and sequenced: two isolates from the original type strains from environmental samples from Korea; a clinical blood isolate from the Wadsworth Center, New York State Department of Health (Albany, NY); and a clinical isolate from the CDC (Atlanta). Although they were genetically distinct, the isolate from the Wadsworth Center was the most similar to the isolates from the NIH Clinical Center, with 97% chromosomal alignment and 145 SNVs. The isolates from Korea and the CDC were notably less similar to isolates from the NIH Clinical Center, with approximately 84% chromosomal alignment with the reference genome and more than 33,000 SNVs (Table 1). These external isolates were resistant to multiple classes of antibiotics, as were the isolates from the NIH Clinical Center (Fig. S1 in the Supplementary Appendix), which underscores the intrinsic resistance of S. koreensis to antibiotics.
GENOMIC ANALYSIS TO IDENTIFY THE RESERVOIR
The phylogenetic tree, constructed on the basis of chromosomal SNVs in the core genome for S. koreensis isolates obtained from patients, revealed no clear structure and did not reflect the timing of the identification of the isolates (Fig. S3 in the Supplementary Appendix). Environmental S. koreensis isolates from the NIH Clinical Center yielded few additional connections, which suggested a widely distributed reservoir within the hospital (Fig. 3). Only the final isolate obtained from a patient (SK-NIH.Pt6_1016) was genetically linked to the corresponding sink isolate, which was collected 15 days later (five shared SNVs) (Fig. S4 in the Supplementary Appendix) — a finding consistent with a direct transmission event.
Figure 3. Phylogenetic Tree of the S. koreensis Isolates from the NIH Clinical Center.

The phylogenetic tree was constructed on the basis of single-nucleotide variants (SNVs) across the core genome. Circles of the same color at the end of the nodes indicate environmental isolates (Env) from the same sink. Black circles indicate that no other isolates were cultured from a given sink. The circles at the end of the nodes for isolates obtained from patients are colored to match the isolates from the sink in the room in which the patient was staying. The labels of isolates obtained from patients are highlighted in red, and isolates obtained from sinks are labeled in black.
S. koreensis was cultured from water in patient rooms but was not detected in the municipal water entering the hospital, in water sampled from the large pipes branching off the main intake pipe, or in ice machines. To devise a remediation strategy, we performed metagenomic sequencing of primary environmental cultures grown from the components of sinks and water pipes behind the walls of patient rooms (Fig. S5 in the Supplementary Appendix). We leveraged core genome SNVs to identify individual S. koreensis strains and their relative abundance within each sample. Sinks were found to be colonized by multiple S. koreensis strains — a discovery that would not have been made with conventional single-colony isolation methods (Fig. 4). Metagenomic analysis identified three distinct isolates from a single sink faucet; this faucet was replaced and resampled monthly (Fig. 4A and B). Four months after replacement, two of the three original S. koreensis strains were detected again, which suggested recolonization of the new faucet by a proximal reservoir. The third strain was not observed after the faucet was replaced, which suggested a localized colonization within the removed faucet. A single sink was found to be colonized by two strains; one had been isolated previously from this same sink, and the other had been isolated previously from the adjoining room (Fig. 4C and D). The two rooms shared a water pipe, which is consistent with a common reservoir. Nine faucets that were positive for S. koreensis were replaced. No further replacements were made after serial cultures showed that six of the new faucets (67%) became recolonized with S. koreensis within 5 to 90 days after replacement.
Figure 4. Metagenomic Sequencing of Multiple S. koreensis Strains Cultured from Sinks in Patient Rooms at the NIH Clinical Center.
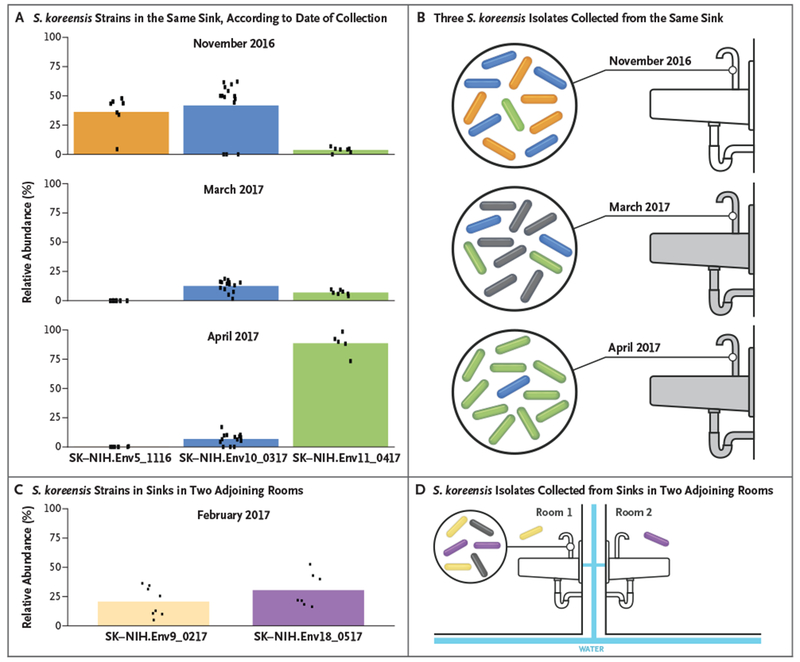
Panels A and B show the relative abundance of three S. koreensis strains (SK-NIH.Env5_1116, SK-NIH.Env10_0317, and SK-NIH. Env11_0417) collected from the same sink in one patient room in November 2016, March 2017, and April 2017. The relative abundance of each isolate is represented by the colored bars in Panel A. Relative abundance was estimated by calculation of the mean allele frequency of SNVs that were unique to each isolate (dots). The sink was replaced after the first positive sample was obtained in November 2016 (indicated by the gray sink in Panel B), but S. koreensis was cultured again from this sink in March 2017 and in April 2017. The colored rods in Panel B correspond to the three isolates in Panel A. Panels C and D show the relative abundance of two other S. koreensis strains (SK-NIH.Env9_0217 and SK-NIH.Env18_0517) that were detected in the sinks of two adjoining patient rooms. The colored rods in Panel D correspond to the two isolates in Panel C. The rods that appear outside the circle indicate individually cultured isolates. The gray rods in Panels B and D represent isolates with unknown SNV profiles. The water in the two rooms in Panel D was supplied by the same pipes (blue bars).
INVESTIGATION OF S. KOREENSIS COLONIZATION WITHIN PLUMBING FIXTURES
To assess the extent of colonization in sink components and to explore possible remediation strategies, we disassembled and cultured three sinks that showed S. koreensis colonization in faucets. Nine components grew S. koreensis, including horizontal sections of hot-water and cold-water pipes immediately proximal to the sink, mixing valves, aerators, faucets, and other plumbing fixtures. Components from two sinks were immersed in 71°C water baths for 20 minutes after culturing. Subsequent cultures of heated components were negative. Aerators were removed from affected sinks. Additional remediation strategies are under consideration. No further S. koreensis infections have occurred since the augmentation of free chlorine concentrations and the adjustment of hot-water temperature in December 2016.
DISCUSSION
In 2016, a cluster of sphingomonas infections sparked an epidemiologic investigation that identified 12 patients over 11 years who had been infected with genetically similar strains of S. koreensis, a rarely reported pathogen. Sink faucets and water from numerous patient rooms were positive for S. koreensis, which implicated hospital plumbing infrastructure as a possible reservoir. In this study, genomic and metagenomic techniques provided a higher-resolution understanding of this intermittent cluster and revealed a pervasive reservoir in the water system of the NIH Clinical Center.
Whole-genome sequencing of 68 S. koreensis isolates from the NIH Clinical Center (obtained from patients and the plumbing system) revealed a genetically diverse population, a phenomenon increasingly observed in microbial outbreaks.11,13,32,33 Understanding genetic diversity in an outbreak is of value and can be leveraged to dissect complex issues, such as identification of the point source and patient-to-patient transmission.34 One limitation of this study is that we could not perfectly match any isolate from a sink in a patient room to a clinical isolate. Matching isolates would have strongly supported S. koreensis transmission, though we would still have been unable to identify the precise modes of transmission from sinks to patients. In addition, we lack metagenomic data for the initial isolates from patients and sinks because we were not aware of strain diversity in early samples. Despite these limitations, a link was found between isolates from one patient and the corresponding room faucet, which were collected 15 days apart. In contrast, other paired patient-sink samples were acquired months and years apart. The heterogeneity among S. koreensis isolates provided a valuable “genetic barcode” to parse subsequent metagenomic samples and to enable the inferenc of a reservoir at the peripheries of the NIH Clinical Center water-distribution system.
Outbreaks of sphingomonas have been reported previously.35–37 The ubiquity of this genus in ostensibly clean water makes it a possible hazard to immunocompromised patients. Previous studies have underscored the threat that in-room sinks may pose to patients, from both potable water and splashback from the drain.38–43 The steps taken in this study to prevent further S. koreensis infections within the NIH Clinical Center are applicable to many opportunistic waterborne pathogens.
Externally collected S. koreensis isolates and banked NIH Clinical Center clinical isolates allowed us to address two important questions: has the 2016 strain caused infections before, and is the strain unique to our hospital? Our results suggest that a single S. koreensis strain entered the water system soon after construction of the new NIH Clinical Center hospital building in 2004. Reports describe colonization of pipes with waterborne bacteria in newly constructed, unused facilities in which water has stagnated.44,45 The expansive genetic diversity within a small sampling window supports our hypothesis that this strain disseminated throughout the hospital and diversified at multiple distinct locations.46
This study was a systematic genomic investigation to understand the dynamics of an indolent outbreak within a single hospital, and it assessed more than 80 sequenced isolates and 49 metagenomic samples. Whole-genome sequencing allowed us to reach back more than a decade to a time shortly after a new hospital building was occupied by patients and to identify the onset of a sporadic clonal outbreak.
Supplementary Material
Acknowledgments
Supported by the NIH Intramural Research Programs, including the National Human Genome Research Institute, the NIH Clinical Center, and the National Institute of Allergy and Infectious Diseases.
Footnotes
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
We thank the clinical laboratory scientists of the NIH Microbiology Service, Department of Laboratory Medicine and Clinical Center for their contributions to this study; and Dr. Daniel Kastner and Dr. Matthew J. Arduino for their helpful discussions and expert review of previous versions of the manuscript. The computational resources of the NIH High-Performing Computation Biowulf Cluster (http://hpc.nih.gov) were used for this study.
References
- 1.Antibiotic/antimicrobial resistance: biggest threats and data. Atlanta: Centers for Disease Control and Prevention, 2013. (http://www.cdc.gov/drugresistance/threat-report-2013). [Google Scholar]
- 2.Requirement to reduce Legionella risk in healthcare facility water systems to prevent cases and outbreaks of legionnaires’ disease (LD). Baltimore: Centers for Medicare & Medicaid Services, June 2, 2017. (https://www.cms.gov/Medicare/Provider-Enrollment-and-Certification/SurveyCertificationGenInfo/Downloads/Survey-and-Cert-Letter-17-30.pdf). [Google Scholar]
- 3.Conger NG, O’Connell RJ, Laurel VL, et al. Mycobacterium simae outbreak associated with a hospital water supply. Infect Control Hosp Epidemiol 2004;25:1050–5. [DOI] [PubMed] [Google Scholar]
- 4.Williams MM, Armbruster CR, Arduino MJ. Plumbing of hospital premises is a reservoir for opportunistically pathogenic microorganisms: a review. Biofouling 2013;29:147–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Osawa K, Shigemura K, Abe Y, et al. A case of nosocomial Legionella pneumonia associated with a contaminated hospital cooling tower. J Infect Chemother 2014;20:68–70. [DOI] [PubMed] [Google Scholar]
- 6.Aumeran C, Paillard C, Robin F, et al. Pseudomonas aeruginosa and Pseudomonas putida outbreak associated with contaminated water outlets in an oncohaematology paediatric unit. J Hosp Infect 2007; 65:47–53. [DOI] [PubMed] [Google Scholar]
- 7.Chand M, Lamagni T, Kranzer K, et al. Insidious risk of severe Mycobacterium chimaera infection in cardiac surgery patients. Clin Infect Dis 2017;64:335–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thomson R, Tolson C, Carter R, Coulter C, Huygens F, Hargreaves M. Isolation of nontuberculous mycobacteria (NTM) from household water and shower aerosols in patients with pulmonary disease caused by NTM. J Clin Microbiol 2013;51:3006–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Snitkin ES, Zelazny AM, Thomas PJ, et al. Tracking a hospital outbreak of carbapenem-resistant Klebsiella pneumonia with whole-genome sequencing. Sci Transl Med 2012;4:148ra116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Snitkin ES, Won S, Pirani A, et al. Integrated genomic and interfacility patient-transfer data reveal the transmission pathways of multidrug-resistant Klebsiella pneumoniae in a regional outbreak. Sci Transl Med 2017; 9(417): pii:eaan0093. [DOI] [PubMed] [Google Scholar]
- 11.Coll F, Harrison EM, Toleman MS, et al. Longitudinal genomic surveillance of MRSA in the UK reveals transmission patterns in hospitals and the community. Sci Transl Med 2017;9(413):pii:eaak9745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burckhardt F, Brion A, Lahm J, et al. Confirming Legionnaires’ disease outbreak by genome-based method, Germany, 2012. Emerg Infect Dis 2016;22:1303–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gardy JL, Johnston JC, Ho Sui SJ, et al. Whole-genome sequencing and social-network analysis of a tuberculosis outbreak. N Engl J Med 2011;364:730–9. [DOI] [PubMed] [Google Scholar]
- 14.Worby CJ, Lipsitch M, Hanage WP. Within-host bacterial diversity hinders accurate reconstruction of transmission networks from genomic distance data. PLoS Comput Biol 2014;10(3):e1003549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nandy S, Dudeja M, Das AK, Tiwari R. Community acquired bacteremia by Sphingomonas paucimobilis: two rare case reports. J Clin Diagn Res 2013;7:2947–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheong HS, Wi YM, Moon SY, et al. Clinical features and treatment outcomes of infections caused by Sphingomonas paucimobilis. Infect Control Hosp Epidemiol 2008;29:990–2. [DOI] [PubMed] [Google Scholar]
- 17.Ryan MP, Adley CC. Sphingomonas paucimobilis: a persistent Gram-negative nosocomial infectious organism. J Hosp Infect 2010;75:153–7. [DOI] [PubMed] [Google Scholar]
- 18.Tablan OC, Anderson LJ, Besser R, et al. Guidelines for preventing health-care–associated pneumonia, 2003: recommendations of CDC and the Healthcare Infection Control Practices Advisory Committee. MMWR Recomm Rep 2004;53 (RR-3):1–36. [PubMed] [Google Scholar]
- 19.Marbjerg LH, Gaini S, Justesen US. First report of Sphingomonas koreensis as a human pathogen in a patient with meningitis. J Clin Microbiol 2015;53:1028–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wallner J, Frei R, Burkhalter F. A rare case of peritoneal dialysis-associated peritonitis with Sphingomonas koreensis. Perit Dial Int 2016;36:224–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dotson GA NISC Comparative Sequencing Program, Dekker JP, et al. Draft genome sequence of a Klebsiella pneumonia carbapenemase-positive sequence type 111 Pseudomonas aeruginosa strain. Genome Announc 2016;4(1): pii:e01663–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Conlan S, Lau AF, NISC Comparative Sequencing Program, et al. Complete genome sequence of a Klebsiella pneumonia strain carrying blaNDM-1 on a multidrug resistance plasmid. Genome Announc 2016;4(4): pii:e00664–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bankevich A, Nurk S, Antipov D, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 2012;19:455–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Walker BJ, Abeel T, Shea T, et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 2014;9(11):e112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res 2017;27:722–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ondov BD, Treangen TJ, Melsted P, et al. Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol 2016;17:132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.R: a language and environment for statistical computing. Vienna: R Foundation for Statistical Computing, 2017. [Google Scholar]
- 28.Free chlorine testing. Atlanta: Centers for Disease Control and Prevention; (https://www.cdc.gov/safewater/chlorine–residual-testing.html). [Google Scholar]
- 29.Sehulster L, Chinn RY, CDC, HICPAC. Guidelines for environmental infection control in health-care facilities: recommendations of CDC and the Healthcare Infection Control Practices Advisory Committee (HICPAC). MMWR Recomm Rep 2003;52(RR-10):1–42. [PubMed] [Google Scholar]
- 30.La Duc MT, Dekas A, Osman S, Moissl C, Newcombe D, Venkateswaran K. Isolation and characterization of bacteria capable of tolerating the extreme conditions of clean room environments. Appl Environ Microbiol 2007;73:2600–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Novikova N, De Boever P, Poddubko S, et al. Survey of environmental biocontamination on board the International Space Station. Res Microbiol 2006;157:5–12. [DOI] [PubMed] [Google Scholar]
- 32.Stoesser N, Sheppard AE, Moore CE, et al. Extensive within-host diversity in fecally carried extended-spectrum-beta-lactamase-producing Escherichia coli isolates: implications for transmission analyses. J Clin Microbiol 2015;53:2122–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paterson GK, Harrison EM, Murray GG, et al. Capturing the cloud of diversity reveals complexity and heterogeneity of MRSA carriage, infection and transmission. Nat Commun 2015;6:6560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Correia AM, Ferreira JS, Borges V, et al. Probable person-to-person transmission of Legionnaires’ disease. N Engl J Med 2016;374:497–8. [DOI] [PubMed] [Google Scholar]
- 35.Kilic A, Senses Z, Kurekci AE, et al. Nosocomial outbreak of Sphingomonas paucimobilis bacteremia in a hemato/oncology unit. Jpn J Infect Dis 2007;60:394–6. [PubMed] [Google Scholar]
- 36.Meric M, Willke A, Kolayli F, Yavuz S, Vahaboglu H. Water-borne Sphingomonas paucimobilis epidemic in an intensive care unit. J Infect 2009;58:253–5. [DOI] [PubMed] [Google Scholar]
- 37.Mutlu M, Bayramoglu G, Yilmaz G, Saygin B, Aslan Y. Outbreak of Sphingomonas paucimobilis septicemia in a neonatal intensive care unit. Indian Pediatr 2011;48:723–5. [DOI] [PubMed] [Google Scholar]
- 38.Hota S, Hirji Z, Stockton K, et al. Outbreak of multidrug-resistant Pseudomonas aeruginosa colonization and infection secondary to imperfect intensive care unit room design. Infect Control Hosp Epidemiol 2009;30:25–33. [DOI] [PubMed] [Google Scholar]
- 39.Lowe C, Willey B, O’Shaughnessy A, et al. Outbreak of extended-spectrum β-lactamase-producing Klebsiella oxytoca infections associated with contaminated handwashing sinks. Emerg Infect Dis 2012;18:1242–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leitner E, Zarfel G, Luxner J, et al. Contaminated handwashing sinks as the source of a clonal outbreak of KPC-2-producing Klebsiella oxytoca on a hematology ward. Antimicrob Agents Chemother 2015;59:714–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Umezawa K, Asai S, Ohshima T, et al. Outbreak of drug-resistant Acinetobacter baumannii ST219 caused by oral care using tap water from contaminated hand hygiene sinks as a reservoir. Am J Infect Control 2015;43:1249–51. [DOI] [PubMed] [Google Scholar]
- 42.Kotay S, Chai W, Guilford W, Barry K, Mathers AJ. Spread from the sink to the patient: in situ study using green fluorescent protein (GFP)-expressing Escherichia coli to model bacterial dispersion from hand-washing sink-trap reservoirs Appl Environ Microbiol 2017;83(8): pii:e03327–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.De Geyter D, Blommaert L, Verbraeken N, et al. The sink as a potential source of transmission of carbapenemase-producing Enterobacteriaceae in the intensive care unit. Antimicrob Resist Infect Control 2017;6:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Francois Watkins LK, Toews KE, Harris AM, et al. Lessons from an outbreak of Legionnaires’ disease on a hematology-oncology unit. Infect Control Hosp Epidemiol 2017;38:306–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stout JE, Brennen C, Muder RR. Legionnaires’ disease in a newly constructed long-term care facility. J Am Geriatr Soc 2000;48:1589–92. [DOI] [PubMed] [Google Scholar]
- 46.McElroy KE, Hui JG, Woo JK, et al. Strain-specific parallel evolution drives short-term diversification during Pseudomonas aeruginosa biofilm formation. Proc Natl Acad Sci U S A 2014;111:E1419–E1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.