

Abstract
Histone variants such as H3.3, macroH2A, H2A.Z, and CENP-A are important epigenetic modifiers of the chromatin state in eukaryotic genomes. The centromeric histone H3 variant CENP-A/CENH3 epigenetically marks centromeres and is required for assembly of the kinetochore complex, a region of the chromosome that is responsible for proper genome segregation during mitosis. Several diverse techniques using biochemical, cell biology, and biophysical approaches have been utilized to study the nature of the CENP-A nucleosome across the cell cycle. In this chapter, we describe methods for CENP-A nucleosome purification and separation of CENP-A from other core histones using traditional SDS-PAGE and more resolving techniques such as Triton acid urea (TAU) and two-dimensional gels. We also discuss methods for observation of CENP-A on chromatin fibers using immunofluorescence. Finally, we provide a detailed description of analysis of chromatin structures using atomic force microscopy.
Keywords: CENP-A, Histones, Cell cycle, SDS-PAGE, TAU, Western blotting, Chromatin fiber, Immunofluorescence, Atomic force microscopy, AFM
1. Introduction
Histone proteins are pivotal to the higher order organization of chromatin. Histones H3, H2A, H2B, and H4 form octameric nucleosomes that wrap around DNA [1]. This higher order organization regulates DNA compaction into fibers [2] and into chromosomes during cell division. Cell division is an important step that requires equal distribution of the genome amongst the daughter cells. For this to be accomplished, a region of the chromosome, known as the centromere, is the most important domain that is targeted by the microtubules for chromosomal segregation. This centromeric domain is epigenetically marked by a variant of histone H3 found in all eukaryotes, known collectively as CENP-A/CenH3 [3, 4]. Understanding the structural composition and dynamics of all the nucleosomes across the cell cycle, including the CENP-A nucleosome, is a goal of many researchers. Although the structural composition of the CENP-A nucleosome is still under experimental investigation, the techniques we present in this work provide a comprehensive foundation to study chromatin across the cell cycle and are applicable to all histones and nucleosomes.
In a recent study by our group, CENP-A nucleosome structure and dynamics were determined over the cell cycle using human cells that were synchronized using a double thymidine block. Cells from the various cell cycle stages were harvested, and CENP-A proteins were enriched by immunoprecipitation using a CENP-A-specific antibody [5]. CENP-A dynamics were observed, including the notable depletion of its own chaperone called Holliday junction recognition protein (HJURP) from the centromeres during S-phase or DNA replication. Atomic force microscopy (AFM) studies performed on CENP-A nucleosomes derived from different stages of the cell cycle have also revealed that HJURP depletion marked a critical point of transition of CENP-A stable tetramers to stable octamers, in vivo [5].
With the exception of a few histone variants, CENP-A and the other histones are small, highly positively charged proteins, ranging from 14 to 18 kDa. They are found associated with each other on chromatin, and can be purified and studied using traditional biochemical and cell biology techniques. Such methods include SDS-PAGE or, for higher separation of the different histone species and modified forms, Triton acid urea (TAU) gel electrophoresis approach. This form of electrophoresis is capable of separating low to medium weight histones based on their charge and hydrophobicity, because the Triton X-100 detergent is capable of binding to the hydrophobic regions of proteins, allowing further retardation and separation [6–8]. This approach is also useful for studying multiacetylated forms of histones [9], such as histone H3, and is a powerful tool for downstream applications such as Western blotting (WB) and mass spectrometric (MS) analyses.
Biochemical approaches are important for understanding the nature of a protein, as well as the composition of a complex when the protein associates with other partners. However, a different approach is required to assess protein localization and to achieve further insight into its function in a cellular context. Traditional cell biology techniques use fluorescently tagged proteins or antibodies (hence immunofluorescence or IF) to visualize where the protein is localized. Certain drawback with fluorescently tagged histones is that the tags are often much larger than the histones themselves, which could result in aberrant histone localization affecting its function. One study shows that GFP-tagged CENP-A mutants were more sterile than their untagged counterparts, suggesting the large fluorescent tag may contribute to chromatin instability [10]. To alleviate that issue, specific antibodies have been produced to target the protein of interest, and small but unique peptide tags can also be cloned in frame and covalently attached to the protein to facilitate detection. These materials have provided cell biologists with useful tools to study protein localization, and allowed for such methods as DNA fluorescent in situ hybridization (FISH). Similar to SDS-PAGE gels, which later evolved to TAU gels, wherein proteins and their modifications could be definitively resolved, combined IF/FISH methods allow specific protein or histone recognition on chromatin fibers. Indeed, using high resolution imaging of chromatin fibers coupled to IF techniques, it was shown that within centromeres, domains of CENP-A alternate with domains of H3 di-methylated at lysine (H3K4Me2) [11, 12].
Another tool for chromatin and nucleosome imaging involves the use of an AFM. This technology dates back to the 1980s, and is commonly used in the fields of physics and materials chemistry, but has slowly gained acceptance over the past two decades as a robust method to study chromatin and nucleosome structure. AFM is a form of scanning probe microscopy (SPM) where the sample’s surface is probed by an extremely small tip moving over it in a scanning fashion [13]. The tip, mounted on the end of a flexible cantilever and most often composed of silicon or silicon nitride, is approximately 2–10 nm in diameter and interacts with the sample within a short range of repulsive (Coulombic) and attractive (van der Waals) atomic forces. Over the last 30 years, AFM has been used for a broad variety of physical and electronic applications, but it has proven particularly useful for observation and analysis of biological molecules, including chromatin. The main advantage of AFM is that it allows for sample imaging with resolution akin to electron microscopy but with minimal processing, making it both an excellent analytical (high precision measurements of nucleosome dimensions) and preparative (quick determination of chromatin quality) technique [5, 14, 15].
Here, we provided detailed protocols on how to purify quantitative amounts of native nucleoprotein complexes from human cells for analysis on an SDS-PAGE or high-resolution protein gels for WB and MS, how to visualize such nucleosomes bound to other protein components on extracted chromatin fibers by IF, and how to analyze nucleosome dimensions and chromatin fiber folding using AFM. These methods have been used successfully by our lab for the study of low abundance (1–5 % of total) histone variants such as CENP-A and macroH2A from mouse, Drosophila, and human cells in culture, as well as from small amounts of tumor tissues. Consequently, the collection of techniques described herein should generally be applicable to any nucleoprotein complexes present as a small fraction of the total genome.
2. Materials
2.1. Nuclei Preparation, Chromatin Digestion, Extraction, and Immunoprecipitation
1× PBS: 137 mM NaCl, 2.7 mM KCl, 1.5 mM KH2PO4, 8.1 mM Na2HPO4 pH 7.2.
Cold 1× PBS-T: PBS supplemented with 0.1 % Tween-20.
Cold TM2 buffer: 20 mM Tris–HCl pH 8.0, 2 mM MgCl2.
Nonidet P40 (NP40) or Nonidet P40 Substitute (Fluka Analytical).
0.1 M TE: 10 mM Tris–HCl pH 8.0, 0.2 mM EDTA, 100 mM NaCl.
Micrococcal Nuclease (MNase, Sigma Aldrich).
100 mM CaCl2.
MNase quenching solution: 500 mM EGTA pH 8.0.
Low-salt extraction buffer (LSEB): 0.5× PBS, 5 mM EGTA.
100 mM phenylmethanesulfonylfluoride (PMSF): dissolved in ethanol.
Anti CENP-A antibody (Santa Cruz Biotechnology, Inc., Cat #sc-22787).
Protein A/G PLUS-agarose beads (Santa Cruz Biotechnology).
0.2 M glycine pH 6.5.
2× Laemmli sample buffer: 65.8 mM Tris–HCl pH 6.8, 2.1 % SDS, 26.3 % (w/v) glycerol, 0.01 % bromophenol blue.
2.2. Histone Extraction with Hydroxylapatite
50-mL conical tubes with small stir bars.
0.35 M NaCl PBS, 0.2 mM EDTA: 1× PBS containing a total of 0.35 M NaCl and 0.2 mM EDTA.
Hydroxylapatite.
100 mM stock solution of phenylmethanesulfonylfluoride (PMSF) in ethanol.
2 M NaCl PBS, 0.2 mM EDTA: 1× PBS containing a total of 2 M NaCl and 0.2 mM EDTA.
15-mL conical tube.
2.8 M NaCl PBS, 0.2 mM EDTA: 1× PBS containing a TOTAL of 2.8 M NaCl and 0.2 mM EDTA.
Amicon Ultra-4 Centrifugal Filter Unit with Ultracel-3 membrane (Millipore).
Slide-A-Lyzer Dialysis Cassette, 7K MWCO(Thermo Scientific).
Low-salt extraction buffer (LSEB): 0.5× PBS, 5 mM EGTA.
2.3. Casting Long TAU (L-TAU) Gels
250-mL vacuum filter flask.
Urea.
40 % acrylamide solution: acrylamide/bis-acrylamide (37.5:1).
Glacial acetic acid.
N,N,N,N′-tetramethyl-ethylenediamine (TEMED).
0.3 M Triton X-100: dissolved in dH2O.
PROTEAN II xi Cell electrophoresis setup (BioRad) with proper combs and accessories.
10 % Ammonium persulfate (APS); dissolved in dH2O.
Butanol.
2.4. Running Long TAU (L-TAU) Gels and Immunoblotting
Triton X-100.
TAU Running Buffer: 65 mL glacial acetic acid, 1.3 mL Triton X-100, add water to a total of 1,300 mL.
Cysteamine Pre-run Solution: 3.84 g urea, 0.57 g cysteamine or β-mercaptoethanol, 430 μL glacial acetic acid, 160 μL 0.3 M Triton X-100, a pinch of pyronin Y (for tracking), add water to a total of 8 mL. Store at room temperature.
2-L graduated cylinder.
2× Sample Running Dye: 9.6 g urea, 750 μL β-mercaptoethanol, 750 μL glacial acetic acid, a pinch of pyronin Y (for tracking), add water to a total of 15 mL. Store at room temperature.
TAU Equilibrating Solution: 50 mM glacial acetic acid, 0.5 % SDS.
Transfer Buffer in 20 % ethanol (1× Tris-glycine transfer buffer): 25 mM Tris–HCl pH 8.0, 192 mM glycine, 20 % ethanol.
Transfer-Blot Turbo Transfer Pack: midi format, 0.2 μm nitrocellulose (BioRad).
Transfer-Blot Turbo Transfer System (BioRad).
1× PBS: 137 mM NaCl, 2.7 mM KCl, 1.5 mM KH2PO4, 8.1 mM Na2HPO4 pH 7.2.
PBS-T: 1× PBS supplemented with 0.1 % Tween-20.
Membrane Blocking Buffer: 5 % nonfat dry milk solubilized in 1× PBS.
TAU Hybridization Buffer: 3 % BSA solubilized in PBS-T.
Antibodies: CENP-A antibody—ChIP grade (Abcam, Cat #ab13939), H2A (Abcam, Cat #ab18255), and H4 (Abcam, Cat #10158).
Plastic hybridization bag.
Heat bag sealer.
IRDye secondary antibodies (LI-COR Biosciences) or other secondary antibodies.
LiCor Odyssey Imaging System (LI-COR Biosciences) or other imaging/developing system.
2.5. Preparation of Chromatin Fibers
1× PBS: 137 mM NaCl, 2.7 mM KCl, 1.5 mM KH2PO4, 8.1 mM Na2HPO4 pH 7.4.
Hemocytometer.
Hypotonic solution: 75 mM KCl diluted in sterilized distilled water.
CytoSpin 4 centrifuge (Thermo Scientific).
Shandon Single Cytofunnels with White Filter Cards (Thermo Scientific).
Shandon Cytoclips Stainless-Steel Slide Clip (Thermo Scientific).
Microscope slides.
Coplin jars.
Fiber lysis buffer: 2.5 mM Tris–HCl pH 7.5, 0.5 M NaCl, 1 % Triton X-100, 0.4 M urea.
Fixation buffer: 4 % formaldehyde, 1× PBS.
Permeabilization buffer: 1× PBS, 0.1 % Triton X-100.
2.6. Chromatin Fiber Immunofluorescence
Blocking buffer: 1× PBS, 0.5 % Bovine Serum Albumin (BSA), Triton X-100.
Wash solution: 1× PBS, 0.05 % Tween-20.
CENP-A primary antibody: mouse monoclonal [3–19] CENP-A antibody—ChIP grade (Abcam, Cat #ab13939) dilution 1:200 in blocking solution complemented with 1 % normal goat serum.
Secondary antibody: goat anti-mouse conjugated to Alexa Fluor 488 or Alexa Fluor 568 (Life Technologies, Cat #A11001 and Cat #A11004, respectively) dilution 1:500 in blocking solution complemented with 1 % normal goat serum.
4’,6-Diamidino-2-phenylindole (DAPI) solution: dissolve 1 μg/μL DAPI in 1× PBS, 50 % glycerol. Store at −20 °C. Dilute 1:5,000 in 1× PBS to prepare a working solution.
1× PBS.
Mounting Mowiol solution: add 2.4 g of anti-fading reagent MOWIOL 4–88 (Sigma-Aldrich, #81381) to 6 g of glycerol, stir to mix. Add 6 mL of distilled water and mix at room temperature for minimum 2 h. Add 12 mL of 0.2 M Tris pH 8.5, and heat to 50 °C for 30 min with occasional mixing. Clarify by centrifugation at 2,800 × g for 20 min. Add to 2.5 % (w/v) 1,4,-diazobicyclo-[2.2.2]-octane (DABCO 33-LV, Sigma-Aldrich, #290734). Aliquot and store at −20 °C.
2.7. DNA FISH with an α-Satellite Probe
Coplin jars.
Crosslinking buffer: 8 % formaldehyde diluted in distilled water.
1× PBS containing 0.5 % Tween-20.
Salmon sperm DNA.
Biotin-labeled DNA probe against centromeric α-satellite sequence: This labeling is performed by nick translation using a homemade plasmid containing repeats of centromeric α-satellite sequence (see Note 1). Mix 2 μg of DNA with 10 μL 10× nick translation buffer (0.5 M Tris–HCl pH 8.0, 50 mM MgCl2, 0.5 mg/mL BSA), 10 μL of a solution containing 0.5 mM dATP, 0.5 mM dCTP, 0.5 mM dGTP and 0.1 mM dTTP, 10 μL 0.1 M β-mercaptoethanol, 4 μL 1 mM biotin-16-dUTP for a total volume of 100 μL after addition of DNA Polymerase I and DNase I. Vortex, centrifuge, and place on ice. Add 2 μL DNA Polymerase I (Invitrogen Cat #18010–025) and 5 μL DNase I (diluted 100× in sterile water, Invitrogen Cat #AM2235), flick gently and incubate for 90 min at 15 °C. Using 5 μL of the sample, check on 2 % agarose gel the length of the DNA, it should be between 300 and 600 bp. If DNA fragments are too long, add more DNase I and incubate for additional 30 min. Stop the reaction with 1 μL 0.5 M EDTA, incubate 10 min at 55 °C, then store at −20 °C.
3 M sodium acetate pH 5.2.
100 % ethanol.
70 % ethanol.
Hybridization buffer: 20 % dextran sulfate, 4× SSC.
Denaturing solution: 70 % formamide, 2× SSC.
12 × 12 round coverslips.
Rubber cement.
Wash solution 1: 50 % formamide, 2× SSC solution.
Wash solution 2: 2× SSC, 0.05 % Tween-20.
Blocking solution: 4× SSC, 0.1 % Tween-20, 3 % BSA.
Secondary antibody: dilution 1:300 diluted in blocking buffer.
Wash solution 3: 4× SSC, 0.1 % Tween-20.
DAPI solution (see Subheading 2.6, item 5).
1× PBS.
Mounting Mowiol solution (see Subheading 2.6, item 7).
2.8. AFM
All buffers and washes should be prepared using ultrapure MilliQ filtered water. All materials listed in Subheading 2.1 are also required. For AFM, muscovite mica, grade V-1 or V-2, is recommended. This can be purchased in large sheets or pre-cut to desired dimension (discs, squares) and is available from several suppliers, including Structure Probe, Inc. and Electron Microscopy Sciences. However, untreated mica surface has a negative charge; therefore, to improve protein binding to the mica surface, a fresh, positively charged mica surface is required. Mica preparation methods have been described previously for (3-Aminopropyl)triethoxysilane (APTES)-mica (AP-mica) [16] and 1-(3-aminopropyl)silatrane (APS)-treated mica (APS-mica) [16–18].
AP-mica.
APS-mica.
Protease inhibitor cocktail.
10× PBS pH 7.4, filtered, sterile.
1 M MgCl2 filtered, sterile.
AFM scanning system (Agilent AFM 5500, Bruker MultiMode 8, or equivalent).
Gwyddion software: free and open source software for SPM analysis (other analysis software is also acceptable, provided it has the capability to export raw data as ASCII text file; however, instructions in this chapter will be provided for Gwyddion only). More information at: www.gwyddion.net.
ImageJ software: free and open source software for image processing and analysis in Java (for automated analysis of nucleosomes). More information at: rsbweb.nih.gov/ij.
3. Methods
3.1. Nuclei Preparation, Chromatin Digestion, Extraction, and Immunoprecipitation
Human cells (e.g., HeLa) are synchronized with a double thymidine block and each cell cycle stage confirmed by FACS analysis [5]. It is important to determine if synchronization protocol results in DNA damage, especially if the complexes being studied are normally involved in such mechanisms. In this scenario, it may be preferable to isolate cells at different phases of the cell cycle using flow cytometry [19] or centrifugal elutriation [20] instead. Cells are then trypsinized, washed once with 1× PBS, once with cold 1× PBS-T, and kept on ice until ready to proceed onto nuclear preparation and chromatin extraction. Make sure centrifuges are cooled to 4 °C ahead of time.
Prepare 10 mL of TM2 solution supplemented with 0.5 % NP40/NP40 substitute (see Note 2) for each sample and add the solution to the broken up cell pellet (see Note 3). Incubate on ice for 2 min and centrifuge at 230 × g for 5 min at 4 °C.
Carefully remove solution/supernatant, leaving the pellet containing nuclei behind. Gently loosen the released nuclear pellet (see Note 3) and wash with cold TM2 (NP40 free). Centrifuge at 230 × g for 5 min at 4 °C.
Carefully remove solution/supernatant, and loosen the nuclear pellet and add 2 mL of 0.1 M TE. Incubate tube(s) in the 37 °C water bath for about 5 min (see Note 4).
Add 0.4 U of MNase (see Note 5) to the top and side wall of the tube (see Note 6) and 30 μL of 100 mM CaCl2 (to achieve final concentration of 1.5 mM) to the opposite wall of the tube. Gently rotate the tube to mix the enzyme and catalyst (CaCl2) and start timer.
At the end of the chromatin digestion period, quench the reaction by adding 40 μL of 500 mM EGTA (to achieve final concentration of 10 mM). Gently rotate the tube to mix the quenching agent and place tube on ice until the next step.
Centrifuge samples at 230 × g for 5 min at 4 °C and carefully remove supernatant not to disrupt the digested nuclear pellet. Gently loosen the pellet and add 1 mL of LSEB solution to the tube. Gently break up the pellet in the LSEB solution by flicking the tube. Cut pipette tip to widen the point of entry (see Note 7) and transfer nuclear pellet/LSEB slurry to a microcentrifuge tube.
Add 5 μL of 100 mM PMSF (final concentration of 0.5 mM) (see Note 8) and rotate samples for at least 6 h to overnight at 4 °C to extract chromatin.
Cut the pipette tip before any of the following steps (see Note 7). After extraction, save 5 % for input samples (store at −20 °C until use) and/or AFM analysis (see Note 9). Transfer the rest of the extract to a new tube.
Add 12 μL CENP-A antibody (or preferred target antibody) and rotate for at least 4 h to overnight at 4 °C.
Add 30 μL Protein A/G PLUS-agarose beads and let sample(s) rotate for another 2 h at 4 °C.
Centrifuge to pellet the beads, antibody, and immunoprecipitated target protein complex at 1,503 × g for 5 min in a refrigerated microfuge. Save a small portion of the supernatant (5 %) as unbound sample, and discard the rest.
Gently wash the beads with the 1 mL LSEM buffer, and rotate for 5 min.
Centrifuge to pellet the beads, antibody, and immunoprecipitated target protein complex at 1,503 × g for 5 min in a refrigerated microfuge.
Discard the supernatant and add fresh 1 mL LSEM buffer to wash the beads again, and rotate for 5 min. A third wash is also recommended.
Transfer the wash and bead/antibody/target protein complex to a new, clean tube using a cut tip.
Centrifuge to pellet the beads, antibody, and immunoprecipitated target protein complex at 1,503 × g for 5 min in a refrigerated microfuge.
Discard the wash solution, leaving just the beads behind and very little residual LSEM buffer (if any). If the ChIP’ed samples will be imaged by AFM, perform a gentle elution by adding 10 volumes of 0.2 M glycine pH 6.5 with end-over-end rotator for 1 h at room temperature. Otherwise, for gel electrophoresis, proceed to the next step.
Add the 2× Laemmli sample buffer directly to the beads and boil sample(s) if performing traditional SDS-PAGE analysis. If sample(s) will be analyzed by TAU gel electrophoresis, dissolve in 2× Sample Running Dye (see Subheading 3.4, step 8), and proceed with the TAU gel protocol.
3.2. Histone Extraction with Hydroxylapatite
Five to ten confluent flasks of human cells are synchronized with a double thymidine block as above [5]. Cells are then trypsinized and washed at least once with 1× PBS, cold 1× PBS-T, and kept on ice until ready to proceed onto histone extraction in 50-mL conical tubes.
Perform steps 1–2 as outlined in Subheading 3.1.
Carefully remove supernatant not to disturb the nuclear pellet, and then flick tube to disperse the nuclei. To the 50-mL conical tubes, add 10 mL of the 0.35 M NaCl PBS, 0.2 mM EDTA (see Note 10), 1 g of hydroxylapatite (to achieve final concentration of 10 %), 0.5 mM PMSF, and a small stir bar that can stir freely in the tube.
Place tube(s) upside down in a thin wall plastic container, and stir overnight with a magnetic stirrer. The hydroxylapatite slurry will attach to DNA while the 0.35 M NaCl PBS, 0.2 mM EDTA will release the soluble proteins.
The next day, centrifuge the hydroxylapatite/chromatin and soluble protein mixture at 500 × g for 5 min at 4 °C and wash it twice with fresh 0.35 M NaCl PBS, 0.2 mM EDTA. This removes the excess soluble proteins, leaving the chromatin bound to the hydroxilapatite behind. Add 6 mL of 2 M NaCl PBS, 0.2 mM EDTA to the slurry (see Note 10), and magnetically stir for at least another 2–4 h. At 2 M NaCl, the majority of the histones or nucleosomes should be detached from DNA (see Note 11).
Centrifuge slurry, collect and transfer supernatant to a new 15-mL conical tube. Store samples at 4 °C until use.
To the hydroxylapatite slurry, add 4 mL of 2.8 M NaCl PBS, 0.2 mM EDTA and magnetically stir for another 2–4 h (see Note 12).
Centrifuge the slurry and pool the supernatants from the 2.8 M NaCl and 2.0 M NaCl PBS-containing solutions. Centrifuge at 900 × g for 5 min at 4 °C to sediment residual hydroxylapatite.
Transfer as much clear supernatant as possible to an Amicon Ultra Centrifugal Filter unit, and centrifuge at 2,000 × g at 4 °C until sample has been concentrated to ~0.5 mL (see Note 13).
Once samples are concentrated, dialyze sample(s) overnight at 4 °C in a beaker filled with LSEB, covered with plastic wrap, and stirred with a magnetic bar.
The following day, dialyzed samples are recovered and can be saved at −20 °C until ready to be run on a Long TAU gel and for WB.
3.3. Casting Long TAU (L-TAU) Gels
The following conditions are optimized for a 20 cm long gel that is 0.5–0.75 mm thick. For gels that are of a different size, scale the components up or down, as needed.
To a 250-mL vacuum filter flask, add the following to make the running gel: 12 g Urea, 5.6 mL 40 % acrylamide, 1.25 mL Glacial Acetic Acid, 125 μL TEMED, 0.5 mL 0.3 M Triton X-100, 7.6 mL dH2O (to 25 mL total).
Gently stir without creating bubbles until urea is completely dissolved (see Note 14).
Degas for 10 min with a vacuum hose attached to the flask.
Properly assemble glass plates and clamps that will house the TAU gel according to BioRad’s PROTEAN II xi Cell recommendations (http://www.bio-rad.com/webroot/web/pdf/lsr/literature/M1651801.pdf) or your manufacturer’s recommendations.
Add 300 μL 10 % APS to the flask containing the contents in step 1, gently swirl to mix and pipette in between the glass plates, leaving about 3 cm at the top to allow room for the stacking gel and comb.
Cover the top of the gel by adding butanol or dH2O to one side of the gel, which will slowly migrate to the opposite side, forming a uniformed monolayer that prevents the gel from over-drying.
Allow the gel to set for at least 30 min.
Prepare the stacking gel by adding the following to another 250 vacuum filter flask: 4.8 g Urea, 1.2 mL 40 % acrylamide, 500 μL Glacial Acetic Acid, 100 μL TEMED, 200 μL 0.3 M Triton X-100, 4.5 mL dH2O.
Gently stir without creating bubbles until urea is completely dissolved (see Note 14).
Degas for 10 min with a vacuum hose attached to the flask.
Take assembled glass plates with solidified TAU gel to the sink, and wash off butanol and any unpolymerized acrylamide with dH2O (see Note 15).
Assemble solidified gel/glass plate back on the gel stand, making sure the gel is leveled.
Add 250 μL 10 % APS to stacking gel mixture in flask, swirl to mix, and pipette into the top layer until it fills the lower glass plate.
Gently push comb of desired lane width in between the glass plates and allow stacking gel to set for at least 30 min to an hour (see Note 16).
3.4. Running L-TAU Gels and Western Blot
Assemble L-TAU gel apparatus in the electrophoretic unit according to BioRad’s PROTEAN II xi Cell recommendations or your manufacturer’s recommendations.
Fill the upper chamber with the TAU Running Buffer, allowing it to spill over into the outer chamber and cover the bottom of the assembled glass plates.
Pipette each lane of the gel several times to remove any unpolymerized material and debris (see Note 17).
Perform a Blank Pre-run for at least an hour without anything loaded into the wells, at a constant 15 mA. Make sure to reverse polarity of the cables (see Note 18).
Pipette each lane of the gel several times again to remove any unpolymerized material and debris (see Note 17).
Pipette 20–30 μL of the Cysteamine Pre-run Solution into each lane and electrophorese at a constant 15 mA for 16–22 h (see Note 19).
Make a fresh batch of the TAU Running Buffer, allowing it to stir with a magnetic stir bar in a 2 L graduated cylinder at room temperature overnight. This ensures the buffer is well mixed prior to next day’s sample run.
The next day, dissolve the sample(s) in the 2× Sample Running Dye, making it 1× (see Note 20). Pipette up and down a few times to mix the sample with Sample Running Dye.
The dye from the Cysteamine Pre-run Solution should have run out into the outer chamber, giving it a slight pink hue. Pipette each lane of the gel several times to remove any unpolymerized material and debris (see Note 17).
Load the appropriate amount of each sample that was dissolved in the 2× Sample Running Dye (see step 8) into their respective lanes and run at 15 mA for 4–5 h (see Note 21).
Once sample(s) are done migrating, disassemble the L-TAU gel and perform a quick wash (less than 5 min) with dH2O in a clean container (see Note 22).
For Coomassie staining, fix the gel and proceed with Coomassie stain (see Fig. 1a). For WB, proceed as described in the next step.
Cut the gel to size, and equilibrate the L-TAU gel twice for 20 min each time with TAU Equilibrating Solution, under gentle shaking conditions (see Note 23).
Equilibrate and wash the L-TAU gel twice for 20 min each with Transfer Buffer, with gentle shaking (see Note 23).
Assemble the filter paper, membrane, and gel sandwich with the midi-format Transfer-Blot Turbo Transfer Pack.
Transfer with the preset BioRad’s High Molecular Weight (MW) setting (2.5 A, 25 V) for 20 min total using Transfer-Blot Turbo Transfer System.
Disassemble the sandwich and wash the membrane with 1× PBS with gentle shaking to remove residual ethanol.
Block membrane for 30 min with Membrane Blocking Buffer at room temperature (RT).
Dilute the primary antibody to the recommended concentration with the TAU Hybridization Buffer (see Note 24).
Perform a quick rinse of the blocked membrane with PBS-T and assemble into a hybridization bag. Seal three sides of the bag with a heat bag sealer, leaving one side unsealed.
Add the antibody diluted in the TAU Hybridization Buffer to the unsealed side, and gently massage out the bubbles (see Note 25).
Seal the last side and gently rock the membrane overnight at 4 °C.
The next day, pour out the primary antibody solution, gently place membrane into a clean container, and perform two washes 5 min each with PBS-T.
Dilute the secondary antibody to the manufacturer’s recommendations with the TAU Hybridization Buffer (see Note 26).
Repeats steps 20–22 in Subheading 3.4 with the secondary antibody, and incubate with gentle rocking for an hour.
Wash the membrane twice with PBS-T with gentle rocking and image with LiCor Odyssey’s supplied imaging software (see Fig. 1b) or with the preferred method of detection.
Fig. 1.
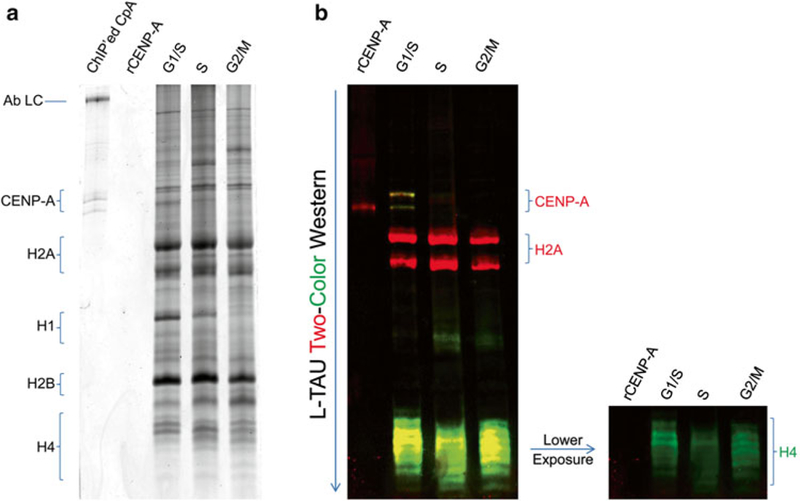
L-TAU gel and Western Blot of histones purified from human cells across the cell cycle. (a) Coomassie stained L-TAU gel. (b) Two-Color Western Blot of L-TAU gel transferred onto nitrocellulose membrane. CENP-A (red), H2A (red), and H4 (green) histones were probed. Ab LC antibody light chain, rCENP-A recombinant CENP-A
3.5. Preparation of Chromatin Fibers
This protocol was adapted from Sullivan [12] for HeLa cells, normal colon EpiCM cells, and colon cancer SW480 cells.
Trypsinize cells and wash once with 1× PBS in 50-mL conical tubes.
Count cells using a hemocytometer and resuspend in hypotonic buffer (final concentration 300,000 cells per mL). Incubate at RT for 10 min for HeLa cells and 30 min for normal colon EpiCM or tumor colon SW480 cells.
Using Cytofunnels, spin 200 μL of cells per slide for 10 min at 400 rpm in Cytospin 4. Mark with a permanent pen the circular area containing cells.
Remove slide(s) from the cytoclip and immerse them in Coplin jars filled with freshly prepared fiber lysis buffer for 15 min at RT. Do not move or agitate the slide(s) during this and the following incubation to avoid the formation of a bundle or tangle of fibers (see Fig. 2 and Note 27).
Slowly and gently pull slide(s) up and out, remaining vertical, and transfer to fixation buffer. Fix for 10 min at RT (see Note 27).
Gently transfer slide(s) from fixation buffer to permeabilization buffer to extract fibers for 7 min at RT (see Note 27).
If FISH or IF cannot be performed the same day, store slide(s) in 1× PBS at 4 ºC for a maximum of 2 weeks.
Fig. 2.
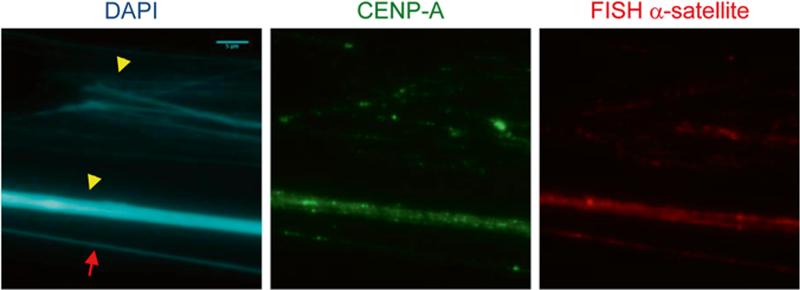
Chromatin fiber stained with CENP-A antibody and FISH α-satellite probe on HeLa cells. Three types of fibers are visible. Fiber indicated with arrow represents a homogeneous diameter and nice shape, revealing the co-localization of CENP-A and α-satellite DNA probe. Fibers designated by arrowheads are either a bundle or a tangle of fibers obstructing a clear view of the co-localization of CENP-A onto the centromeric DNA
3.6. Chromatin Fiber Immunofluorescence
Block slide(s) for 30 min in Blocking buffer at RT or overnight at 4 °C.
Incubate with primary antibody diluted in blocking solution complemented with 1 % normal goat serum overnight at 4 °C in a humidified chamber.
Wash slide(s) three times for 5 min each in wash solution. Do not agitate slide(s) (see Note 27).
Incubate with secondary antibody diluted in blocking solution complemented with 1 % serum for 1–2 h at RT in a humidified chamber in the dark.
Wash slide(s) three times for 5 min in wash solution.
Perform two more washes in 1× PBS for 2 min each at RT. Make sure slide(s) has limited exposure to light from this point onward (see Note 28).
For co-staining, repeat immunofluorescence from Subheading 3.6, step 1 using a different primary and secondary antibody. For DNA FISH, go to Subheading 3.7. Otherwise, continue with the next steps.
Incubate with DAPI solution for 15 min at RT in the dark.
Wash slide(s) three times for 5 min in 1× PBS and mount coverslip with Mowiol solution. Make sure slide(s) is kept in the dark (see Note 28).
3.7. DNA FISH with an α-Satellite Probe
Crosslink antibody-protein complexes by immersing slide(s) in a Coplin jar containing crosslinking buffer for 10 min at RT.
Store slide(s) in 1× PBS, 0.5 % Tween-20 until FISH is performed. Slide(s) may be stored at 4 °C for up to 1 week.
Precipitate 100–150 ng of biotin-labeled DNA from Subheading 2.7, item 5 with the addition of 1 μg of salmon sperm DNA, 1/10 volume of 3 M sodium acetate, and 3 volumes of 100 % ethanol per slide.
Centrifuge 30 min at 18,407 × g in a refrigerated microfuge. Wash the pellet with 70 % ethanol and centrifuge again for 5 min at 9,391 × g in a refrigerated microfuge. Resuspend the dry pellet with 5–6 μL of hybridization buffer.
Pre-warm denaturing solution in Coplin jar by successive incubations in 37 and 42 °C water baths for 10–15 min, before finally placing it into a 80 °C water bath (see Note 29).
Remove slide(s) from jar containing 1× PBS, 0.5 % Tween-20. Quickly wipe the slide(s) to remove excess solution using folded delicate task wipes or lint-free tissues, but avoid wiping the circular area containing the fibers (see Subheading 3.5, step 3).
Place slide(s) in denaturing solution for 8–10 min. During this time, place a tube containing biotin-labeled probe dissolved in hybridization buffer in 78 °C water bath to denature DNA for 9 min, and then place it on ice.
Remove the slide(s) from denaturing solution, wipe it, and add the entire denatured probe dissolved in hybridization buffer from steps 3 to 4 above to the circular area containing fibers.
Carefully and quickly cover each circular area with a 12 × 12 round coverslip, seal with Rubber cement, and incubate slide(s) in humidified chamber at 37 °C incubator for 16–48 h in the dark (see Note 28).
Wash slide(s) three times for 5 min each, at 45 °C with wash solution 1.
Wash slide(s) four times for 5 min each, at 45 °C with wash solution 2.
Block for 30 min at RT with blocking solution.
Incubate slide(s) with the secondary antibody at 37 °C for 2 h.
Wash slide(s) four times for 5 min each, at 45 °C with wash solution 3.
Incubate with DAPI solution for 15 min at RT in the dark (see Note 28).
Wash slide(s) four times for 3 min with 1× PBS, and mount coverslip with Mowiol solution. Store slide(s) in the dark at 4 °C until ready for imaging (see Note 28).
3.8. AFM
AFM can operate in several modes, each resulting in different type of information being collected. The most suitable modes for chromatin imaging are tapping (frequency slightly lower than cantilever resonance) and noncontact mode (frequency slightly higher than cantilever resonance), which generate a detailed three-dimensional image of the sample surface. Each of these modes relies on the tip rapidly oscillating up-and-down within a close proximity to the sample surface but with little to no direct contact, thereby minimizing the risk of damage or disruption of the chromatin.
Any substrate could theoretically be used for sample deposition for the AFM, as long as it could provide a very flat and uniform surface. Freshly cleaved mica has been traditionally used as it has an atomically flat sheet surface, creating a perfect background for AFM imaging. Untreated, the surface of mica has a negative charge [16, 21] and can effectively bind DNA molecules, provided that salt concentration is low and divalent cations are added to the buffer to bridge the electrostatic interaction with negatively charged phosphate backbone of the DNA. However, under most physiological conditions it does not bind proteins very well. In order to improve protein deposition, mica can be treated with APTES {(3-Aminopropyl)triethoxysilane} or APS {1-(3-aminopropyl)silatrane} silica compounds that give it a positive surface charge by functionalizing with amino groups [16, 21]. The choice between these two reagents should be determined experimentally, as well as based on feasibility at one’s facility. APTES mica is generally more consistent from batch to batch, but it also has the propensity to bind soluble proteins better, resulting in higher background from samples of lesser purity (see Fig. 3a). APS tends to have higher variability from one preparation to another (and therefore each batch needs to be checked prior to sample deposition), but when the surface is sufficiently flat the images are generally much cleaner and better looking (see Fig. 3b).
Fig. 3.
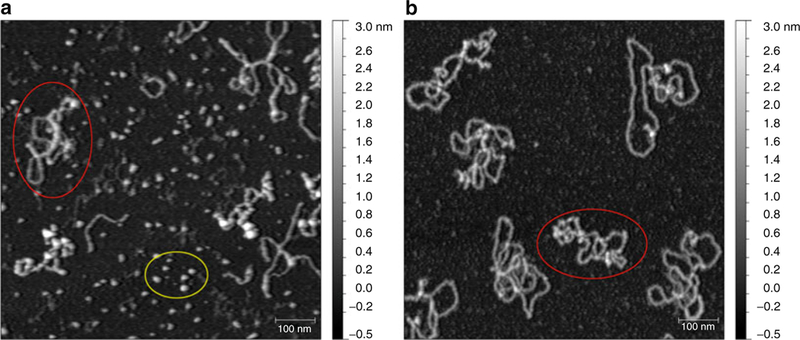
Comparison of chromatin deposited on either APTES or APS surface. (a) Partial chromatin reconstitution was deposited on a fresh APTES surface in the presence of 2 mM MgCl2. Notice the unincorporated histone proteins (bottom oval ) bound all over the sample surface in between chromatin arrays (left oval ). (b) Same sample as in (a) but deposited on freshly prepared APS surface in the presence of 2 mM MgCl2. Unincorporated histones no longer contaminate the background between chromatin arrays (oval ). Grayscale on the right represents approximate heights in nm; scale bar represents 100 nm
Another important consideration is the tip selection, as it will impact the quality of data acquired. Super sharp tips with diameters of ~2 nm (such as Bruker TESP-SS or Nanosensors SSS-SEIH) provide superior resolution and data quality, but are also costly and fragile during repeated use. Good quality, lower cost tips (such as Bruker OTESPA/Olympus AC160TS-10) can be a very good compromise, as they are much more affordable and robust while still being sharp enough to acquire good quality data.
Finally, a method of data analysis must be carefully considered. Automated analysis of surface features can be tempting, as it guarantees large number of data points with good statistical significance. However, unless the sample is exquisitely pure and all chromatin arrays are flat and evenly distributed, automated analysis can result in counting background particles in no way associated with DNA arrays (Fig. 4c). Furthermore, APS surface can degrade over time, resulting in formation of a maze of hills and valleys on a previously uniform background. This does not affect the values for nucleosome height or diameter but may interfere with the automated algorithm. Therefore, in most cases, manual evaluation of particles of interest is recommended to assure accuracy. We will discuss both analysis methods below, along with sample preparation and common scanning conditions.
Fig. 4.
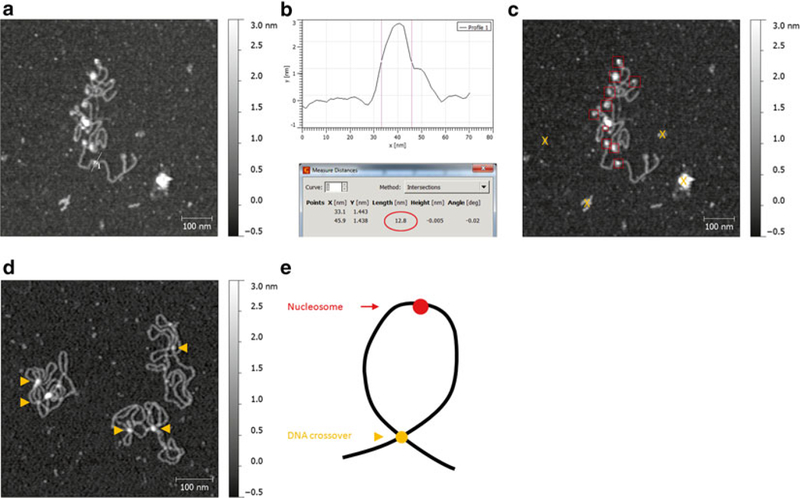
Accurate identification of nucleosome features in AFM images. (a) Immunoprecipitated chromatin from cancer cells were deposited on APS-mica in the presence of 1 mM MgCl2. Nucleosome arrays as well as features not associated with DNA are visible. Straight line represents a profile shown in (b). (b) Top: profile extraction graph of a nucleosome from (a); vertical lines denote the diameter of the nucleosome measured at half-height. Bottom: readout of the distance between the lines with the nucleosome diameter highlighted with the oval. (c) Same as in (a), but with boxes indicating the positively identified nucleosomes and “X” marking irrelevant back-ground features that would still be included in the automated analysis. (d) Failed nucleosome reconstitution. The arrowheads indicate the spots where plasmid DNA crosses on top of itself resulting in a feature that resembles a nucleosome, but should not be counted as one. Notice lack of nucleosomes on free stretches of DNA and “false-nucleosome” features only occurring at DNA crossovers. (e) A cartoon indicating how the position of the feature on the DNA can aid in distinguishing between nucleosome and DNA crossover
3.8.1. AFM Sample Preparation
Prepare a fresh, positively charged mica surface (see Subheading 2.8).
Follow the chromatin extraction protocol as outlined in Subheading 3.1, steps 1–8. Use protease inhibitor cocktail in addition to PMSF (see Note 8).
Dilute the extracted chromatin sample in the appropriate buffer to the final concentration of ~5–10 μg/mL (the dilution buffer is the same as the buffer used for chromatin extraction but supplemented with 1–2 mM MgCl2, e.g., 0.5× PBS 1 mM MgCl2 for LSEB extracted chromatin) (see Note 30).
Deposit 5–10 μL of the diluted chromatin in the center of the AP(S)-mica surface. Cover sample with a petri dish lid to protect it from dust, and incubate for 10 min at RT.
Rinse the sample with 400 μL of ultrapure water, dripping 3–4 drops at a time and gently shaking it while holding the sample with tweezers.
Dry the sample under light vacuum (see Note 31).
3.8.2. AFM Scanning Conditions
Scan the sample either in tapping or noncontact mode (see Note 32). Contact mode should not be used as the tip drag ging across the sample will easily disrupt chromatin complexes.
Use low force for scanning by setting a low free amplitude using the drive control (3–6 V on Agilent systems, 200– 400 mV on Veeco/Bruker microscopes).
Adjust the setpoint as light as possible, but low enough to avoid the “comet tail” artifact (see Fig. 5a, b). This usually means a setpoint ~85–90 % of free amplitude for the tapping mode or 75–80 % for noncontact mode.
Scan with low speed (0.5–1 line/s) and capture a large (5 μm × 5 μm), high resolution (4096 × 4096) image. This will make the analysis easier and assure that a large number of nucleosomes can be measured under the same conditions.
Fig. 5.
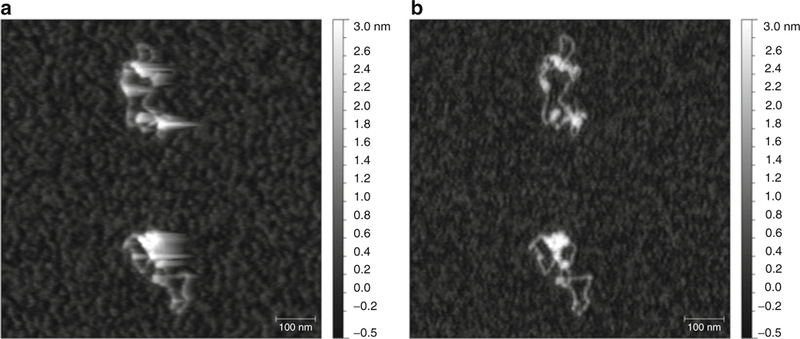
Example of a “comet tail” artifact in AFM images. (a) One week-old chromatin was scanned in tapping mode with a set point of ~98 % free amplitude, resulting in elongated tracks in the direction of the trace scan (left-to-right) resembling a tail of a comet. (b) Same sample as in (a) was scanned with a lower set point of ~90 % free amplitude, resulting in clear shape outlines and no “comet tail” artifact
3.8.3. AFM Data Analysis
Data analysis consists of two major steps: preparing the image for analysis and performing the actual analysis.
Open the image in Gwyddion.
Click “Level data by mean plane subtraction.”
Click “Correct lines by matching height median.”
Click “Correct horizontal scars (strokes).”
Click “Remove polynomial background”; verify that both horizontal and vertical polynomial degree is set to “3.” If large background structures are present in the image they may result in incorrect leveling. In that case, mask them out before subtracting polynomial background using “Mark with mask” option (see Note 33).
-
Verify that the image is properly leveled. Using the “Extract profiles” function, draw two long, intersecting, and roughly perpendicular lines spanning the entire sample. The extracted profiles should show a flat baseline, centered at the “zero” mark. If the baseline is curved then masking was not done correctly and some of the tall and/or deep features were included in the background subtraction calculation. If the base-line is flat but not centered at “zero,” adjust it using the “Read value under mouse cursor” function (see Note 34).
At this point the sample is ready for analysis. To analyze it manually continue reading from step 7. For automated analysis skip to step 10.
To analyze the sample manually, use the “Crop” function in Gwyddion to zoom in on each feature and study it carefully for presence of artifacts or contaminants. Identify nucleosomes by their characteristic round shape and association with DNA. Make sure not to confuse nucleosomes with DNA crossovers, which appear similar in height but only occur where two or more DNA strands intersect (see Fig. 4d).
To measure the exact height of each nucleosome, use the “Statistical quantities” function. Draw a square around the nucleosome and read the value corresponding to “Maximum” height.
To measure the diameter of each nucleosome, use the “Extract profiles” function. Draw a line dissecting the nucleosome at its highest point and click “Apply.” Click “Measure distances in graph” and, knowing the height of the nucleosome, mark two positions intersecting the graph on both sides of the nucleosome at exactly half of its maximum height. Read the value of “Length (nm)” as the diameter of the nucleosome (see Fig. 4a, b).
To analyze the sample automatically, it must first be exported in a numerical (ASCII) format. To save the flattened image as ASCII, click “File” → “Save As.” Type in the desired file name with “.txt” extension, select “File type” as “ASCII data matrix”, and click “Save.” In the dialog box uncheck “Add informational comment header” and select desired precision (default setting is “5”).
Exported file contains the height data as units of meters, and needs to be converted to nanometers before it can be processed with ImageJ. Open the ASCII file in MS Excel, create a new sheet, and type in “1e9” (or 1,000,000,000) in the first cell. Select that cell and click “Copy.” Go back to the sheet with ASCII data and select all cells (Ctrl+ A). Open the “Paste” dialog box, select “Paste special” followed by “Multiply.” This should result in all the values converting from meters to nanometers. Delete the created sheet (containing “1e9” in the first cell) and save the converted file.
Open the ASCII file in ImageJ using the following set of commands: “File” → “Import” → “Text Image.” Perform the operation twice resulting in two identical images being open (this is important for the analysis step 16 later). Keep track of which image was opened first (Image 1) and which was second (Image 2).
Click “Analyze” → “Set Scale.” Input the correct dimensions for the image. For example, if the image acquired was 5 μm × 5 μm and the resolution was 4096 × 4096, input “4096” for “Distance in pixels,” “5000” for “Known distance,” and “nm” as “Unit of length.” Check “Global” to apply the settings to both open pictures.
Brightness and contrast can be adjusted to change the image appearance. However, it will have no effect on the underlying data.
Set the threshold on Image 1 to average half-height of the nucleosomes in that image. Click “Image” → “Adjust” → “Threshold.” Set the upper slider (“minimum”) to the half-height value for the image (usually 1.2–1.3 nm) and the lower slider (“maximum”) all the way to the right (see Note 35).
Click “Analyze” → “Set Measurements” and check the following options: “Area,” “Min & max gray value,” “Shape descriptors,” “Integrated density,” “Mean gray value,” and “Perimeter.” Also, in the “Redirect to” field select the Image 2 as the target.
To analyze the image open “Analyze” → “Analyze particles.” Set “Size (pixel^2)” at “100–400” (the “perfect” nucleosome shouldbe~11 nm × 11 nm = 121 nm2, or~ π*(6nm)2 = 113 nm2) and “Circularity” at “0.8–1” (for less-than-perfect spheres set “0.7–1”). Also, select “Show: Outlines” and check “Display results,” “Summarize,” “Exclude on edges,” and “Include holes.”
Using the generated “Outlines” image verify that the structures analyzed are indeed nucleosomes.
Save the results as an “.xls” file and analyze it in MS Excel or any other statistical analysis software. “Max” column represents nucleosome height and “IntDen” represents volume of hypothetical sphere. Cylinder volume can be calculated using “Area” and “Max” values, and the nucleosome diameter can be calculated using the “Area” value.
Acknowledgements
We thank Dr. Rajbir Gill for expert advice on chromatin extraction, TAU gel preparation, and FISH protocols, and Dr. Emilios Dimitriadis for helpful advice on automated AFM image analysis.
Footnotes
Notes
Centromeric α-satellite sequence was based on a consensus sequence from human α-satellite DNA derived by Waye and Willard based on 130 independent monomers from at least 14 different human chromosomes [22].
NP40/NP40 substitute is difficult to get into solution. It is recommended that the pipette tip be cut to accommodate the viscous solution, and pipette around the edge of the TM2 solution, prior to vortexing to properly mix. While vortexing the TM2 + NP40 solution, bubbles will form. Continue vortexing until the NP40 is completely dissolved and not floating in the TM2, which can take several minutes.
Once the TM2/NP40 mixture is added to the cells, the cell membrane will lyse, leaving the nuclei behind. At this stage, the nuclei should be treated gently. Make sure to break up the pellet by flicking the tubes. DO NOT vortex from this point forward.
During the 37 °C incubation period, set up all necessary materials such as pipettes, tips, MNase enzyme, CaCl2, and EGTA solutions. Have a timer set to the preferred amount of time required for digestion. If long arrays are preferred (such as for AFM analysis), a 1–2 min MNase digestion should be sufficient. For chromatin of monomeric length, >8 min incubation is recommended.
Variability between the different brands and batches of MNase is likely to be expected. Thus, it is suggested that each batch of enzyme be optimized and quality control be maintained throughout the batches to maintain consistency in chromatin length.
The MNase and CaCl2 catalyst should be added towards the top and opposing sides of the round bottom tube. When ready, the tube is gently rotated to mix the two and the timer is started at that time.
It is strongly recommended that the pipette tips be cut to widen the point of entry to prevent shearing of the nuclei and maintain chromatin integrity.
PMSF should be sufficient to prevent protein degradation. However, a protease inhibitor cocktail may be the preferred choice, especially for overnight chromatin extraction and downstream applications such as AFM analysis.
For AFM samples, cut the tip of a 10 μL pipette tip and transfer to a new tube. Store at 4 °C, but samples should be imaged within a day to prevent protein degradation.
1× PBS already contains 137 mM NaCl; therefore, make sure one accounts for this NaCl concentration when preparing the 1× PBS with varying concentrations of NaCl.
The 0.35 M NaCl PBS, 0.2 mM EDTA washes will remove excess soluble proteins that are not bound to DNA, like histones. Additional washes may be necessary, especially for larger nuclei pellets. The 2 M NaCl PBS, 0.2 mM EDTA solution will release the majority of bound histones.
The extra 2.8 M NaCl PBS, 0.2 mM EDTA step with the very high salt concentration will release any leftover histones that were still bound to the chromatin after the previous 2 M NaCl PBS, 0.2 mM EDTA step. This step is optional but it does significantly increase the yield of released histone proteins.
If samples become over-concentrated, histones will start to precipitate during the process. To avoid over-concentrating, periodically check the sample with UV-spectrophotometer set at 280 nm to assure the OD is no more than 1.
Urea is difficult to get into solution. It is recommended that the flask be gently stirred while slightly immersed in a 37–45 °C water bath. The heat will help speed up the dissolution of the urea.
After washing the residual butanol and unpolymerized material, it is recommended that any excess water be removed by aspiration, prior to addition of the stacking gel.
If made properly, the bottom of each lane should be uniform and straight. The bottom should not be scraggly or dented, which could cause the bands to appear dented or scraggly as well.
It is important to remove any residual unpolymerized material from the lanes to prevent poor running and banding pattern aberrations that may make the gel look unsightly.
Because of the addition of acetic acid, the polarity of the protein migration will be reversed compared to traditional SDS-PAGE gel electrophoresis.
The BioRad PROTEAN II xi Cell apparatus is composed of large vertical electrophoresis cell that can be filled with water. This allows the glass plates and gels to remain slightly cooled. It is recommended that this chamber be filled for these long runs at room temperature, which could cause some overheating.
Unlike the traditional SDS-PAGE loading buffer like Laemmli sample buffer, the 2× Sample Running Dye for TAU gels DOES NOT need to be boiled. Pipetting up and down to mix and then directly loading into the wells is sufficient due to the presence of urea.
The sample run time of 4–5 h is generally sufficient to resolve all the histones for almost the entire length of the 20 cm L-TAU gel. However, it is recommended that run times and conditions be optimized for other protein running setups or for cases where the smaller histones (e.g., histone H4) are not needed and better resolution is required for the larger hydrophobic proteins (e.g., histone H3 or CENP-A).
This water wash will remove some residual 2× Sample Running Dye. The wash will appear slightly pink, which is normal. After all the subsequent washes, the pink hue on the gel should be mostly cleared. It is suggested that a corner of the gel be cut or marked so researcher knows which end is left or right.
Equilibration of the L-TAU gel with TAU Equilibrating and Transfer Buffer Solutions is essential for the removal of the Triton X-100, which will interfere with the transfer onto the membrane. These are the minimum steps and times, but more washes are advised, especially if the gel is still bright pink.
For CENP-A WB, a dilution of 1:2,000 is suggested. For detection of histones H4 and H2A, a dilution of 1:3,000 is suggested.
Gently massage the bubbles out of the top of the hybridization bag and seal it with a heat bag sealer. To ensure that remaining micro-bubbles do not interfere with the hybridization, it is best to orient the membrane with the transferred protein side facing down so that the bubbles float to the back of the membrane while on the rocker.
LiCor Odyssey’s IRDye secondary antibodies are recommended at the dilution of 1:20,000. For other detection systems and secondary antibodies (e.g., horseradish peroxidase), secondary antibody concentrations must be optimized by the individual.
Care must be taken to not agitate the slides and when transferring from one buffer to the next. A slow pulling up motion is critical for proper fiber formation.
Keeping slides in the dark will prevent photo-bleaching of the secondary signal.
Check the temperature of denaturing solution inside the Coplin jar to ensure that it is 78 °C before proceeding with denaturation of chromatin fibers.
Prepare the dilution carefully. Dilute in small steps and gently flick the tube to mix content (e.g., 1:400 dilution should be broken down into two 1:20 dilutions). DO NOT pipette the solution up-and-down as this will shear the chromatin and disrupt nucleosome arrays.
Small desiccator or a petri dish lid over the sample with vacuum line connected to the lid work best. The sample can still dry without the vacuum, but will take significantly longer.
In tapping mode, when the frequency is set to slightly below resonance (“left of the peak”) the tip makes very intermittent contact with the sample (“tap”). This results in sharper images but also usually more scars/background and shorter tip life. In noncontact mode, when the frequency is set to slightly above resonance (“right of the peak”) the tip relies solely on atomic repulsion and does not contact the sample. This generates a cleaner image with less interference, but the features can have less sharp and slightly blurry outlines.
If large particles and/or deep strokes are present in the image to be analyzed, it is best to mask them out before subtracting the polynomial background. To do so, click “Data Process” → “Mask” → “Mark With.” Select “Add mask” and “Data: Topography.” Adjust “Minimum” to exclude any deep scars and trenches. Adjust “Maximum” to exclude any features above surface (including objects of interest, such as DNA). What should remain marked is only the surface of the substrate to be leveled. Remove polynomial background as described but make sure that “Include only masked region” option is checked.
Sometimes leveling will result in a flat background surface but centered above or below zero. In that case read from the scale where the baseline is centered (e.g., −0.04 nm) and using “Read value under mouse cursor” option find a spot reading the opposite “Z value” of 0.04 nm and set it at zero by clicking “Set Zero.” Verify on the extracted profiles that the baseline now centers around the “zero” mark.
It is important to select the threshold value that will be as close to the nucleosome half-height as possible, but that will also allow for a clear separation between nucleosomes. Thresholded nucleosomes that are “touching” will not be recognized as independent circular structures and will be excluded from measurement. Only single nucleosomes with clear outline will be measured.
References
- 1.Thomas JO, Kornberg RD (1975) An octamer of histones in chromatin and free in solution. Proc Natl Acad Sci U S A 72(7):2626–2630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Woodcock CL, Frado LL, Rattner JB (1984) The higher-order structure of chromatin: evidence for a helical ribbon arrangement. J Cell Biol 99(1 Pt 1):42–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Palmer DK, O’Day K, Wener MH, Andrews BS, Margolis RL (1987) A 17-kD centromere protein (CENP-A) copurifies with nucleosome core particles and with histones. J Cell Biol 104(4):805–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Earnshaw WC, Migeon BR (1985) Three related centromere proteins are absent from the inactive centromere of a stable isodicentric chromosome. Chromosoma 92(4):290–296 [DOI] [PubMed] [Google Scholar]
- 5.Bui M, Dimitriadis EK, Hoischen C, An E, Quenet D, Giebe S, Nita-Lazar A, Diekmann S, Dalal Y (2012) Cell-cycle-dependent structural transitions in the human CENP-A nucleosome in vivo. Cell 150(2):317–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shechter D, Dormann HL, Allis CD, Hake SB (2007) Extraction, purification and analysis of histones. Nat Protoc 2(6):1445–1457 [DOI] [PubMed] [Google Scholar]
- 7.Zweidler A (1978) Resolution of histones by polyacrylamide gel electrophoresis in presence of nonionic detergents. Methods Cell Biol 17:223–233 [PubMed] [Google Scholar]
- 8.Waterborg JH (2002) Acid-urea-triton polyacrylamide gel electrophoresis of histones. In: Walker JM (ed) The protein protocols handbook Springer, New York, pp 113–123 [Google Scholar]
- 9.Earley KW, Shook MS, Brower-Toland B, Hicks L, Pikaard CS (2007) In vitro specificities of Arabidopsis co-activator histone acetyltransferases: implications for histone hyperacetylation in gene activation. Plant J 52(4):615–626 [DOI] [PubMed] [Google Scholar]
- 10.Ravi M, Shibata F, Ramahi JS, Nagaki K, Chen C, Murata M, Chan SW (2011) Meiosis-specific loading of the centromere-specific histone CENH3 in Arabidopsis thaliana. PLoS Genet 7(6):e1002121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blower MD, Sullivan BA, Karpen GH (2002) Conserved organization of centromeric chromatin in flies and humans. Dev Cell 2(3):319–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sullivan BA (2010) Optical mapping of protein-DNA complexes on chromatin fibers. Methods Mol Biol 659:99–115 [DOI] [PubMed] [Google Scholar]
- 13.Binnig G, Quate CF, Gerber C (1986) Atomic force microscope. Phys Rev Lett 56(9): 930–933 [DOI] [PubMed] [Google Scholar]
- 14.Bui M, Walkiewicz MP, Dimitriadis EK, Dalal Y (2013) The CENP-A nucleosome: a battle between Dr. Jekyll and Mr. Hyde. Nucleus 4(1):37–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dimitriadis EK, Weber C, Gill RK, Diekmann S, Dalal Y (2010) Tetrameric organization of vertebrate centromeric nucleosomes. Proc Natl Acad Sci U S A 107(47):20317–20322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lyubchenko YL, Gall AA, Shlyakhtenko LS (2001) Atomic force microscopy of DNA and protein-DNA complexes using functionalized mica substrates. Methods Mol Biol 148: 569–578 [DOI] [PubMed] [Google Scholar]
- 17.Shlyakhtenko LS, Gall AA, Lyubchenko YL (2013) Mica functionalization for imaging of DNA and protein-DNA complexes with atomic force microscopy. Methods Mol Biol 931:295–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shlyakhtenko LS, Gall AA, Filonov A, Cerovac Z, Lushnikov A, Lyubchenko YL (2003) Silatrane-based surface chemistry for immobilization of DNA, protein-DNA complexes and other biological materials. Ultramicroscopy 97(1–4):279–287 [DOI] [PubMed] [Google Scholar]
- 19.Juan G, Hernando E, Cordon-Cardo C (2002) Separation of live cells in different phases of the cell cycle for gene expression analysis. Cytometry 49(4):170–175 [DOI] [PubMed] [Google Scholar]
- 20.Pretlow TG II, Pretlow TP (1979) Centrifugal elutriation (counterstreaming centrifugation) of cells. Cell Biophys 1(2):195–210 [DOI] [PubMed] [Google Scholar]
- 21.Quenet D, Dimitriadis EK, Dalal Y (2012) Atomic force microscopy of chromatin. In: Atomic force microscopy investigations into biology—from cell to protein Rijeka, Croatia: InTech; 195–218 [Google Scholar]
- 22.Willard HF, Waye JS (1987) Hierarchical order in chromosome-specific human alpha satellite DNA. Trends Genet 3(7):192–198 [Google Scholar]