

Abstract
Isomers of chiral drugs can exhibit marked differences in biological activities. We studied the binding and inhibitory activities of 12 compounds against KDM5A. Among them are two pairs of enantiomers representing two distinct inhibitor chemotypes, namely, (R)- and (S)-2-((2-chlorophenyl)(2-(piperidin-1-yl)ethoxy)methyl)-1H-pyrrolo[3,2-b]pyridine-7-carboxylic acid (compounds N51 and N52) and (R)- and (S)-N-(1-(3-isopropyl-1H-pyrazole-5-carbonyl)pyrrolidin-3-yl)cyclopropanecarboxamide (compounds N54 and N55). In vitro, the S enantiomer of the N51/N52 pair (N52) and the R enantiomer of the N54/N55 pair (N54) exhibited about 4- to 5-fold greater binding affinity. The more potent enzyme inhibition of KDM5A by the R-isoform for the cell-permeable N54/N55 pair translated to differences in growth inhibitory activity. We determined structures of the KDM5A catalytic domain in complex with all 12 inhibitors, which revealed the interactions (or lack thereof) responsible for the differences in binding affinity. These results provide insights to guide improvements in binding potency and avenues for development of cell permeable inhibitors of the KDM5 family.
Graphical Abstract
INTRODUCTION
Histone lysine methylation is involved in a wide range of biological processes including gene expression, chromatin organization, dosage compensation, and epigenetic memory and is often compromised in cancers (reviewed in ref 1). Histone methylation can be transcriptionally repressive or activating, depending on the position and identity (lysine or arginine) of the methylated residue and the extent of its methylation (i.e., monomethylated, dimethylated, or trimethylated).2 Methylation of lysine residue 4, 36, or 79 of histone H3 is typically associated with active transcription, while methylation of lysine 9 or 27 of histone H3 and lysine 20 of histone H4 contributes to repression.3 Along with other histone modifications such as acetylation, phosphorylation, and ubiquitination, other regulatory factors, and DNA cytosine modifications, these modifications ultimately determine chromatin conformation and the local potential for gene expression.
To enable changes in chromatin structure and gene expression, histone modifications have to be dynamic. Although protein extracts containing lysine demethylase activity were identified and partially purified as early as the 1960s (reviewed in ref 4), the first protein lysine-specific demethylase 1 (LSD1, also known as KDM1A), a flavin-dependent amine oxidase, was identified nearly a half century later.5 This was immediately followed by the discovery of a second and larger class of Jumonji-domain containing demethylases:6 a family of >20 Fe(II) and α-ketoglutarate (αKG)-dependent dioxygenases (KDM2–8). Histone demethylases have since become important therapeutic targets, particularly in human cancers where these enzymes are frequently mutated and/or misregulated.7,8
Histone H3 lysine 4 (H3K4) methylation is typically associated with regions of accessible chromatin, including gene promoters and enhancers. There are two different enzyme families that act on methylated H3K4; LSD1/KDM1 removes methyl groups from low-degree (mono- or di-) methylated H3K45 (reviewed in ref 9), whereas JARID1/KDM5 removes methyl groups from the higher-degree (tri- or di-) methylated H3K4 forms10 (reviewed in ref11). The KDM5 family is unique among histone demethylases in that each member contains an atypical split catalytic domain wherein the insertion of a DNA-binding ARID and a histone-interacting PHD1 domain separates the Jumonji domain into two segments, JmjN and JmjC (Figure S1A in Supporting Information). Although all four members of the KDM5 family catalyze the demethylation of the same histone mark, they have different tissue expression profiles and are present in distinct protein complexes, suggesting different cellular functions.11,12
The KDM5 family has been implicated in the pathogenesis of several types of cancer and plays a role in drug resistance and in metastasis.11–13 For example, KDM5A is amplified or overexpressed in breast cancer,14 lung cancer,15,16 hepatocellular carcinoma,17 and gastric cancers.18,19 KDM5A has been linked to the control of proliferation and senescence by antagonizing the functions of retinoblastoma protein20–22 and through direct repression of cyclin-dependent kinase inhibitor genes.15,17,18 Knockout of KDM5A significantly prolonged survival in genetically engineered mouse tumor models.22,23 Moreover, KDM5A plays a role in the epithelial–mesenchymal transition16,24 and the suppression of invasion15,23 and metastasis.23,25 Increased levels of KDM5A have been shown to confer anticancer drug resistance in lung cancer,26 breast cancer,14 and glioblastoma.27 KDM5B is similarly overexpressed in a variety of cancers, including breast cancer and others,28,29 and is associated with a poor prognosis, chemo-resistance, and metastasis.30–35
Given the mounting evidence from human tumors and model systems supporting a role for the KDM5A and KDM5B as oncogenic drivers, extensive efforts have been devoted to develop KDM5 family specific inhibitors. Several cell-permeable inhibitors have been identified (KDM5-C70 (patent WO2014053491), CPI-455,36 and KDOAM-2537) and have been shown to alter global levels of H3K4me3 and to inhibit cancer cell growth, thus serving as the basis for further development of therapeutic agents that combat KDM5 oncogenic potential. We and others have studied the binding modes of chemically diverse KDM5 inhibitors in complex with KDM5A and KDM5B demethylases.36–39 We noted that some of the compounds that inhibit enzyme activity in vitro were racemic mixtures, and thus we were intrigued by the opportunity to identify stereospecific inhibitors of KDM5 and to understand the structural basis for that selectivity.
The individual enantiomers of chiral drugs can exhibit marked differences in biological activity, and thus detailed characterization of the individual isomers is critical.40,41 With regard to αKG-dependent dioxygenases in particular, cancer-associated mutations in the isocitrate dehydrogenases IDH1 and IDH2 alter the function of these enzymes such that instead of catalyzing the oxidative decarboxylation of isocitrate to αKG, they go on to produce the (R)-enantiomer of 2-hydroxyglutarate (2HG),42,43 a weak inhibitor of many αKG-dependent dioxygenases.44–46 Interestingly, whereas the (S)-isomer is a more potent in vitro inhibitor of most of the αKG-dependent enzymes tested, only (R)-2HG exhibits cellular activity, inducing cytokine independence and blocking differentiation of hematopoietic cells47 and targeting FTO N6-methyladenine RNA demethylase in leukemic cells,48 suggesting that enantiomeric inhibitors of the broader class of αKG-dependent dioxygenases may have distinct in vitro versus in vivo activities.
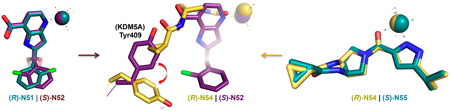
Here we study the structural features and growth inhibitory properties of two pairs of enantiomeric KDM5 inhibitors, namely, (R)- and (S)-2-((2-chlorophenyl)(2-(piperidin-1-yl)-ethoxy)methyl)-1H-pyrrolo[3,2-b]pyridine-7-carboxylic acid (compounds N51 and N52) and (R)- and (S)-N-(1-(3-isopropyl-1H-pyrazole-5-carbonyl)pyrrolidin-3-yl)-cyclopropanecarboxamide (compounds N54 and N55). We find that, in vitro, the S isoform of the N51/N52 pair (N52) and the R isoform of the N54/N55 pair (N54) exhibited greater binding and more potent inhibition of KDM5A. For the cell-permeable N54/N55 pair, this difference in potency translated to a difference in cells wherein the (R)-N54 showed enhanced accumulation of H3K4me3 and greater growth inhibition.
RESULTS
We previously defined the minimal requirements for in vitro enzymatic activity of KDM5 family to be the linked JmjN-JmjC domain coupled with the immediate C-terminal helical Zn-binding domain.49 Deletion of internal ARID and PHD1 domains (ΔAP) between JmjN and JmjC as well as the region C-terminal to the Zn-binding domain generated KDM5A(1–739)ΔAP, which was used for enzyme activity studies (Figure S1C). The protein produced without the Zn-binding domain, KDM5A(1–588)ΔAP, was used for compound binding assays and structural studies (Figure S1D). Recombinant proteins were affinity-purified from Escherichia coli using an N-terminal His-SUMO-tag, which was subsequently removed as described.49 Structural comparisons with a longer construct of KDM5A,36 which comprises the entire N-terminal half (including ARID, PHD1, and the C-terminal Zn helical domain) (Figure SlB), showed that the deletion of the ARID-PHD1 domains does not interfere with either the folding of the linked JmjN-JmjC domain or the conformation of its active site. The chemically related compounds CPI-455 and N8 bind in the active site of the two KDM5A constructs in exactly the same manner (Figure S1E). The shorter construct with the linked JmjN-JmjC domain provides an opportunity for studying numerous KDM5A demethylase inhibitors at near atomic resolution by X-ray crystallography.
Structure–Function Relationships among KDM5-Directed Inhibitors.
Among the 12 compounds we examined, 9 contain a 1H-pyrrolo[3,2-b]pyridine-7-carboxylic acid (pyridine-7-carboxylic acid) moiety and 2-chlorophenyl moiety connected to a variable length of alkoxyether (Figure 1A,B). Compounds N9, N40, N41, N42, N46, N47, and N48 are racemic mixtures containing the same chiral secondary carbon atom connected by the three different substituents and a hydrogen. The racemic mixture of compound N40 was separated by chiral chromatography (Supporting Information methods), yielding two enantiomers N51 and N52 (the assignment of R- and S-enantiomers is based on the observed electron densities in the final refined structures). Using the purified minimal catalytic domain KDM5A(1–588)ΔAP, we measured the dissociation constants (KD) by isothermal titration calorimetry (ITC) (Figures 1C and S2). Among the mixtures, N9 (patent WO2014164708), which lacks a piperidine ring, has an approximately 3-fold weaker binding affinity relative to N40. Compounds with piperidine (N40 or N48), difluoropiperidine (N41), or pyrrolidine (N46) substitutions have similar binding affinities in the range of 0.15–0.25 μM. This indicates that the identity of the basic substituents at the end of alkoxyether and the length of alkyl linker has a relatively small impact on binding affinity and is tolerant of structural modification. The substitution of pyrrolidine ring in N46 with an imidazole ring in N47 reduced the binding affinity by ~3-fold. Shortening the alkoxyether extension from two-carbon to one-carbon reduced the binding affinity by ~2-fold (N41 and N42), whereas an increase in the alkyl chain length to three carbons did not affect binding affinity (N40 and N48). Importantly, among the compounds examined, the pure S-enantiomer N52 exhibited the highest binding affinity with a KD of 60 nM, approximately 4-fold better than that of the R-enantiomer N51.
Figure 1.

Binding of KDM5A by compound N9 and its derivatives. (A) Chemical structures of N9 related compounds containing a constant pyridine-7-carboxylic acid moiety and 2-chlorophenyl moiety connected to a variable hydroxymethyl extension. (B) Chemical structures of (R)- and (S)-2-((2-chlorophenyl)(2-(piperidin-1-yl)ethoxy)methyl)-1H-pyrrolo[3,2-b]pyridine-7-carboxylic acid (compounds N51 and N52). (C) Summary of isothermal titration calorimetry measurement of dissociation constants (KD) of various compounds to KDM5A(1–588)ΔAP (see Figure S2 for original binding data). (D) Summary of the IC50 values of inhibition of demethylation for KDM5A (residues 1–1090) by AlphaLISA (see Figure S3 for original inhibition data). (E) Summary of inhibitor-bound X-ray structures of KDM5A(1–588)ΔAP (see Table S1 for detailed statistics of X-ray diffraction and refinement).
We noted that the N9-based compounds (with a pyridine-7-carboxylate moiety) are intrinsically fluorescent which interferes with the fluorescence signal detected in the formaldehyde dehydrogenase (FDH)-coupled demethylase assay. We thus employed an AlphaLISA method to assess potency of these compounds on enzyme inhibition. This assay utilizes an antibody against methylated H3K4me3 to measure the conversion (disappearance) of trimethylated lysine 4 of histone H3 peptide substrate (biotinylated H3(1–21)K4me3) and a commercially available recombinant human KDM5A large fragment (residues 1–1090) with a C-terminal FLAG-tag (BPS Bioscience). The inhibitory activities in this assay (IC50 values) correlated reasonably well with the relative binding affinities for the KDM5A linked Jumonji domain construct, as revealed by the pairwise comparisons between N9 and N40, N41 and N42, N46 and N47, and N54 and N55 (Figure 1D). However, there are some significant differences between the two assays. For example, the ~4-fold difference in dissociation constants between the two enantiomers N51 and N52 was not reflected in the enzyme inhibitory assays, where the two enantiomers displayed similar IC50 values (Figure 1D). The similar KD values observed among N40 (KD = 0.18 μM), N51 (0.22), N41 (0.21), N46 (0.15), and N48 (0.25) corresponded well with the nearly equivalent IC50 values with the exception of N41, which demonstrated reduced inhibitory activity (by approximately 6-to 15-fold) compared to other compounds: N40 (IC50 = 0.69 μM), N51 (0.67), N41 (4.5), N46 (0.29), and N48 (0.43). The inconsistency between the binding affinity and inhibitory constants might be due to the different KDM5A constructs used in the respective assays. The deleted internal domains (ARID, PHD1, and C-terminal Zn helical domain) might contribute to the interaction with the inhibitors in the active site that were not observed in the binding assays, although alternative explanations exist (see Discussion). In addition, potent compounds (with IC50 values of <1 μM) against KDM5A exhibited significant selectivity against the KDM4A catalytic domain by a factor of 20–50 (Figure S3B), whereas compound N42, with the least inhibition against KDM5A (IC50 = 22 μM), demonstrated no selectivity between the two enzymes. We note that more comprehensive selectivity screening against other histone demethylases (and the larger family of dioxygenases) will be performed in the near future.
Structures of KDM5A Linked Jumonji Domain in Complex with Nine Inhibitors Containing Pyridine-carboxylate Scaffold.
We determined the structures of nine different inhibitors in ternary complex with KDM5A(1–588)ΔAP and Mn(II) (instead of Fe(II)) to the resolution range of 1.22–1.99 Å (Figure 1E and Table S1). Compounds were soaked into preformed crystals of KDM5A complexed with αKG and thus had to displace bound αKG to be visible in the active site. The inhibitor-bound KDM5A Jumonji domains are similar in overall structure to the αKG-bound form; for example, there was a root-mean-squared deviation of 0.2 Å across 289 pairs of Cα atoms between the N46-bound structure of KDM5A (at the highest resolution of 1.22 Å) and that of αKG-bound at 1.39 Å resolution (PDB code 5IVB). Like other structurally characterized αKG-dependent dioxygenases,50 the metal ion was bound by six ligands in an octahedral coordination. The side chains of His483, Glu485, and His571 occupy three of the coordination sites (Figure 2A), while the pyridine ring nitrogen atom and two water molecules provide the other three ligands (Figure 2A).
Figure 2.

Structure of ternary complex of N46-KDM5A-Mn(II) at 1.22 Å resolution (PDB code 6BGY). (A) Six metal ligands involving three protein residues, two water molecules, and the ring nitrogen atom of pyridine. The omit electron densities, contoured at 10σ and 5σ above the mean, are shown for Mn(II) (magenta mesh) and compound N46 (gray mesh), respectively. (B) The carboxylate group of pyridine-7-carboxylic acid moiety forms ionic and hydrogen bonding interactions with Lys501 and Tyr409. (C) Extensive van der Waals contacts formed between pyridine ring and 2-chlorophenyl ring of N46 and aromatic residues of KDM5A in the active site. (D) The chlorine atom of 2-chlorophenyl ring binds in a hydrophobic pocket formed by Ala411, Tyr409, and Tyr472.
The pyridinecarboxylic acid portion of N46 occupies the αKG binding site (Figure 2B), with the carboxylate group forming an extensive H-bond network with Lys501, Tyr409, and water-mediated interactions similar to those normally engaged by αKG.38 The carboxylic acids of the other eight pyridine-based compounds (N9, N40, N51, N52, N41, N42, N47, and N48) exhibited nearly identical interactions with the protein residues (Tyr409, Asn575, Lys501, Asn493, and Ser491) involved in direct and indirect contacts (Figure 3A–I). This finding underscores the critical nature of these interactions for defining the binding mode for this class of inhibitors and clearly illustrates why any modification of the carboxylic acid reduces inhibitory potency significantly, as was observed previously when comparing the structures of the catalytic domain bound by cofactor αKG or inhibitors containing an isonicotinic acid moiety.38 In addition to these polar interactions with Lys501 and Tyr409, the aromatic pyridine ring is engaged extensively in face-to-face π–π interactions with Tyr472, Phe480 and an edge-to-edge interaction with Trp503 (Figure 2C).
Figure 3.

Structural snapshots of N9 related compounds bound into KDM5A active site. (A–I) The pyridine-7-carboxylic acid moiety involves in conserved interactions with Lys501, Asn575, and Tyr409 (top left corner of each panel). The omit electron densities, contoured at 5σ above the mean, are shown for each inhibitor (gray mesh). (A) In compound N9 (PDB code 6BGU), the relative positions of 2-chlorophenyl moiety and the propoxymethyl group are switched in comparison with other compounds containing larger chemical part attached to the end of hydroxymethyl extension. (B) In the racemic mixture of compound N40 (PDB code 6BGV), two possible conformations of 2-chlorophenyl moiety can be modeled into the electron density. (C, D) In the purified enantiomers (R)-N51 (PDB code 6BH0) and (S)-N52 (PDB code 6BH1), the chlorine atom of the chlorophenyl ring points to opposite directions. (E–I) For the other five racemic mixtures of compounds, N41 (PDB code 6BGW), N42 (PDB code 6BGX), N46 (PDB code 6BGY), N47 (PDB code 6BGZ), and N48 (PDB code 6BH5), the (R)-N52-like conformation dominates. (J) Superimposition of compounds N9 and N46. (K) Superimposition of compounds N41 and N42. Note the inhibitor-specific interaction with Trp470 (compound N41) and Asp486 (compound N42). (L) Superimposition of compounds N46 and N47. Note the conformational change of Asp412.
Moving away from the αKG binding region (occupied by the pyridinecarboxylic acid), the 2-chlorophenyl in compound N9 (in R conformation) points to an open solvent channel with the phenyl ring apparently not well-ordered (as indicated by the poor electron density). Meanwhile the propoxyl group forms van der Waals contacts (interatomic distance varying between 3.4 and 3.8 Å) with the guanidine group of Arg73, Tyr409, and Ala411 (Figure 3A). This binding site appears relatively tight, suggesting that additional substitutions on the propoxyl would not be tolerated, and explains why the remaining eight pyridine-based analogs, all of which have a larger alkoxyether extension, adopt a different orientation in this binding site. For these compounds, we observe that these two substituents on the chiral carbon (the 2-chlorophenyl and the alkoxyether extension) have switched positions (Figure 3B–J).
Knowing that compound N40 is a racemic mixture, we modeled the 2-chlorophenyl into the electron density in two conformations (Figure 3B). With the structures of purified enantiomers N51 and N52, the electron density is well-defined, allowing us to unambiguously position the chlorine atom of the chlorophenyl ring pointing either to solvent (N51) or into the protein (N52) (Figure 3C,D). The chlorine atom makes extensive van der Waals contacts with Ala411, Tyr409, and Tyr472 (Figure 2D). These Cl interactions with the protein presumably contribute to the 4-fold stronger binding by N52 (Figure 1D). Superimposition of the three structures (N40, N51, and N52) shows the density for N40 is consistent with being a mixture of the two enantiomers (Figure 4A). And the dissociation constant of N40 is between that measured for each of the individual enantiomers (Figure 1C). For the other five racemic mixtures of chlorophenyl-containing compounds (N41, N42, N46, N47, and N48), the electron density mapped well with the (S)-N52-like conformation with the chlorine atom interacting with Ala411 (Figure 3F–J), suggesting the preferential binding of this enantiomer to the protein.
Figure 4.

Chiral pairs of (R)-N51 and (S)-N52 bind in the KDM5A active site. (A) Superimposition of racemic mixture of compound N40 (magenta; PDB code 6BGV) and the purified enantiomers (R)-N51 (cyan; PDB code 6BH0) and (S)-N52 (gold; PDB code 6BH1). Note the pyridine-7-carboxylate moiety is superimposable. (B, C) In (R)-N51, Y472 forms two hydrogen bonds with the compound. In (S)-N52, Asp412 forms an ionic interaction with the compound. The 2Fo – Fc electron densities, contoured at 1.2σ, are shown for the inhibitor and immediate neighboring residues. (D) Superimposition of (R)-N51 and (S)-N52. Note the conformational change of Asp412.
Furthermore, we observed two different side-chain conformations of Asp412, as well as a shift of ~2 Å in the main-chain Cα atom when comparing the N46-bound structure with that of N47 (Figure 3L). In the pyrrolidine-containing N46 structure, Asp412 bridges between Arg73 and Tyr472, whereas in the imidazole-containing N47 structure, this Asp412-Tyr472 interaction breaks down. These observations may explain the different KD values measured for N46 and N47 (Figure 1C).
In summary, each of the three substitutions on the chiral carbon has a different impact on the interactions with KDM5A; the planar ring of 1H-pyrrolopyridine-7-carboxylic acid occupies the highly conserved, αKG-binding region of the catalytic site, linking metal binding to the Lys501-Asn575-Tyr409 network. These interactions are essential for the potency of the inhibitors and are likely the primary determinants that anchor the compound’s orientation. The 2-chlorophenyl moiety, particularly the chlorine atom, makes extensive van der Waals contacts with Ala411, Tyr409, and Tyr472. Finally, the alkoxyether extension allows a variety of different interactions specific for each inhibitor, with Asp412 being a particularly interesting site.
Structures of Chiral Pair (R)-N51 and (S)-N52.
We overlaid the two structures of N51 and N52 in the KDM5A binding pocket (Figure 4A). While the 2-chlorophenyl moiety and ethoxypiperidine branch are nonsuperimposable, the 1H-pyrrolopyridine-7-carboxylate moiety is. We find that, besides the 1H-pyrrolopyridine-7-carboxylate moiety, the other two components of the enantiomers have different interactions with KDM5A. As noted above, there are extensive hydrophobic interactions with the chlorine atom in N52 and that are absent in N51 (Figure 3C,D). In addition, the nitrogen atom of the piperidine ring in N51 is within H-bond distance to the hydroxyl oxygen atom of Tyr472 (Figure 4B). In contrast, the corresponding nitrogen atom in N52 interacts with one of the carboxylate oxygen atoms of Asp412 (Figure 4C). The six-membered piperidine ring in N52 could carry a formal charge if the ring nitrogen is protonated such that the close contact with the negatively charged Asp412 may enhance the binding affinity of N52 with KDM5A (Figure 4C). These differences in the protein–ligand interaction are likely responsible for the ~4-fold difference in binding affinity between N51 and N52. Interestingly, we observed a conformational change of Asp412 (involving both main-chain and side-chain atoms) upon binding of N51 versus N52 (Figure 4D), which is analogous to the difference observed between compounds N47 and N46 (Figure 3L). In both cases, interaction with Asp412 (Asp412-N52 or Asp412-N46) seems to correlate with stronger binding affinity (lower KD values).
The Methyl Ester Derivatives of Pyridinecarboxylic Acid Are Cell Inactive.
Some previously identified KDM inhibitors, including GSK-J151 and KDM5-C49 (patent WO2014053491), are acidic, displayed a competitive mechanism of inhibition relative to cofactor αKG in vitro, and are cell impermeable.38,39 Here we observe a similar binding mode for the compounds containing the 1H-pyrrolopyridine-7-carboxylic acid, wherein the distal carboxylic acid displays strong ionic and hydrogen bonding interactions with Lys501 and Tyr409 in the active site (Figure 2B). In the cases of GSK-J1 and KDM5-C49, the respective ethyl ester derivatives, GSK-J4 and KDM5-C70, were designed as cell-permeable prodrugs that are hydrolyzed by unknown esterase(s) within the cell to generate active GSK-J1 and KDM5-C49, respectively. By analogy, we generated methyl ester derivatives of N40, N41, and N46 to test their activity in cells. However, exposure of BT474 breast cancer cells to any of these at concentrations up to 38 μM had no impact on cellular H3K4me3 methylation levels, whereas KDM5-C70 did (Figure S4). The lack of cellular activity is probably a result of the combination of potentially reduced cellular penetration and less efficient hydrolysis of these methyl ester derivatives in the cell.
Enantiomers (R)-N54 and (S)-N55 Adopt Similar Binding Modes.
Compounds N54 and N55 represent a second pair of chiral KDM5 inhibitors based on a completely different molecular scaffold. The compounds are notable for being uncharged, without a carboxylate group (patent WO2016057924).52 The chiral center at the C3-position on the pyrrolidine ring is connected to the nitrogen of a cyclopropylamide (C3–N bond in Figure 5A). ITC data showed that the dissociation constant (KD) for binding to KDM5A is more than 5-fold lower for N54 than N55 (Figures 1D and S2). The difference in binding affinity between the two enantiomers correlated well with the relative inhibitory activity (IC50 values), and the potency of inhibition was dependent on αKG concentration (Figures 5B and S5A), suggesting that the inhibitors compete with αKG for binding. We note that the approximately 5- to 7-fold difference in IC50 values between N54 and N55 against KDM5A from two different sources, i.e., our purified KDM5A(1—739)ΔAP and the commercially available KDM5A(1—1090)-Flag, was similar and independent of the method used to determine inhibitory activity (FDH-coupled and AlphaLISA) (Figures 1D and 5B).
Figure 5.

(R)-N54 and (S)-N55 bind highly similar in the KDM5A active site. (A) Chemical structures of (R)-N54 and (S)-N55. (B) Inhibition of demethylation of KDM5A(1–739)ΔAP by (R)-N54 via FDH-coupled assay at three concentrations of αKG (see Figure S5A for inhibition data by (S)-N55). (C) Structure of N54-KDM5A-Mn(II) showing the binding of N54 compound in the active site involving two metal ligands (upper right corner), a direct hydrogen bond with Asn575 (left), and aromatic interactions via Phe480 (top), Tyr472 (bottom), and Trp503 (in the background away from the viewer). See Figure S5B for structure of (S)-N55 bound in the active site of KDM5A (D) The terminal cyclopropyl group forms a stacking interaction with Tyr409 (see Figure S5C for corresponding interaction in (S)-N55). (E) The terminal isopropyl methyl groups form van der Waals interactions with Trp470, Asn585, and Val584 (see Figure S5C for corresponding interaction in (S)-N55). (F) Superimposition of (R)-N54 and (S)-N55. (G, H) Superimposition of compounds N52 (dark magenta) and N54 (yellow) illustrates conformational changes of Tyr409 (G) and Trp780 (H). (I) An ordered glycerol molecule was observed near the inhibitor, forming direct H-bond interaction with the inhibitor. (J) A surface representation of KDM5A active site showing the binding of N54 compound together with a glycerol molecule. (K) A surface representation of KDM5A showing the binding of compound N52 occupying a much larger space in the active site.
We determined structures of N54- and N55-bound KDM5A, at resolutions of 1.45 and 1.70 Å, respectively (Table S1). The unambiguous electron density helped define the absolute stereochemistry of the two enantiomers. Both compounds occupy the αKG-binding site as expected and provide two ligands to the metal via a pyrazole ring nitrogen and an adjacent amide carbonyl oxygen atom (Figures 5C,D and S5B,C). The second pyrazole ring nitrogen (NH group) forms a H-bond with Glu485 (one of the three protein metal ligands). Like a peptide bond, the partial double bond character makes the amide carbonyl and pyrazole ring coplanar. The terminal isopropyl groups form van der Waals interactions with side chains of Trp470 and Asn585 and main-chain carbon of Val584 (Figures 5E and S5D). On the other end of the molecule, the carbonyl oxygen of the cyclopropylcarboxamide forms a H-bond network with Lys501 and Asn575 (Figures 5D and S5C). The cyclopropyl group forms a stacking interaction with Tyr409. Again, the six atoms associated with the peptide-like bond (amide nitrogen –NH and carbonyl oxygen –C=O) are planar. The central, five-membered pyrrolidine ring containing the chiral center at C3 is surrounded by aromatic residues forming a three-sided cage, with Phe480 on the ceiling, Tyr472 on the floor, and Trp503 on the back wall (Figures 5C and S5B).
Superimposition of the bound conformations of the two enantiomers revealed that the pyrrolidine ring rotates so that the chiral carbon C3 of the second enantiomer now occupies the position of ring carbon C4 in the structure of the first enantiomer (Figure 5F). During the course of our study, Liang et al.52 published a structure of (R)-N54 bound with KDM5A(12–800) (PDB code 5V9T), in which the entire N-terminal half of KDM5A was used for cocrystallization (Figure S6A). Superimposition of the two (R)-N54-bound KDM5A structures demonstrates that positioning of (R)-N54 is nearly identically in the two structures, being unaffected by the absence or presence of the internal ARID-PHD1 (PHD1 is disordered in the structure) or the helical Zn-domain (Figure S6B,C); no additional interactions with residues outside of the linked JmjN-JmjC domain were observed.
Two key interacting aromatic residues (Tyr409 and Trp780) were found to undergo substantial inhibitor-induced conformational changes. In the 1H-pyrrolopyridine-7-carboxylate based inhibitors, the side chain Asn575 bridges between Lys501 and Tyr409, the latter forming a hydrogen bond with the inhibitor carboxylate (Figure 3A–I). By contrast, in the N54 complex, the side chain of Tyr409 is pushed away by the cyclopropyl group, rotating nearly 90° as compared to its position in the N52-bound structure (Figure 5G), and forms a van der Waals contact with the cyclopropyl group (Figure 5D). Similarly, the indole ring of Trp780 rotates approximately 90° from the piperidine-interacting conformation (N52) to adopt an orientation that enables interaction with the isopropyl group (N54) (Figure 5H). In both cases, the alternative positioning involves the π-stacking forces, which could be exploited to design inhibitor specific interactions.
A highly ordered glycerol molecule, used as a cryoprotectant during crystal freezing, is observed near the inhibitor in both the N54 and N55 structures (Figure 5I). The glycerol molecule forms a direct H-bond interaction with the amide nitrogen that is bound to the chiral C3 carbon and side chain of Asp412, as well as an indirect interaction with Arg73 and a network of water-mediated interactions with Tyr409 (Figure 5I). Design of new inhibitors that expand to occupy this solvent-filled space, and pick up increased protein–ligand interactions, is an avenue for potentially increasing the potency of KDM5 inhibitors (Figure 5J). Indeed, the most potent 1H-pyrrolopyridine-7-carboxylate based compound N52 occupies a greater volume (Figure 5K) and exhibited stronger binding than N54 by a factor of 40 (Figure 1D). Superimposition of N52-bound and N54-bound structures revealed that compound N52 occupies one-third larger volume in the active site (390 Å3) than that of N54 (290 Å3).
Cellular Effect of Compounds (R)-N54 and (S)-N55.
We next examined whether the difference in biochemical activities of enantiomers N54 and N55 is also reflected in a cellular setting. We treated several cell lines representing different subtypes of breast cancer, including BT474 and SKBR3 (HER2+), MCF7 (ER+), MDA-MB231 (triple negative for ER, PR, and HER2) cells with N54 and N55 at concentrations ranging from 0.1 to 10 μM. Global levels of trimethylated lysine 4 of histone H3 (H3K4me3), the KDM5 substrate, were elevated to varying degrees across the cell lines in response to both N54 and N55, with N54 being ~10-fold more potent than N55 at driving similar levels of H3K4me3 accumulation (Figure 6A,B). Consistent with the idea that N54 and N55 are specific for KDM5,52 N54 or N55 treatment showed minimal effects on other histone methylation marks, including H3K9me3 (substrates for the KDM4 family) and H3K27me3 (substrate for the KDM6 family) (Figure 6A,B). To further compare the effects of N54 and N55 on the growth of breast cancer cells, we performed colony formation assays using MCF7 cells. Consistent with our previous studies,38 KDM5-C70 at 1 μM significantly inhibited the growth of MCF7 cells (Figure 6C,D). N54 at 10 μM showed similar growth inhibitory activity to 1 μM KDM5-C70, whereas N55 was ineffective even at 10 μM, consistent with a relative difference in potency between N54 and N55 in inducing global H4K4me3 levels.
Figure 6.

(R)-Enantiomer N54 is more active than (S)-enantiomer N55 in cellular assays. (A, B) Western blot analyses of (A) SKBR3 and BT474 cells, (B) MCF7 and MDA-MB231 cells treated with the indicated concentration of N54, N55, KDM5-C70, or DMSO control for 3 days. Representative data from biological duplicate experiments performed in duplicate are shown. (C, D) Colony-formation assays of MCF-7 cells treated with DMSO, KDM5-C70 (1 μM), and N54 and N55 (1 or 10 μM). Shown are (C) representative images and (D) quantification of biological triplicate experiments. “Intensity %” indicates the measured intensity of cell confluency using ImageJ. Error bars denote SEM: (***) p < 0.001.
The impact of KDM5 inhibitors on breast cancer cell proliferation was further assessed by live-cell imaging in the presence of 0.1–50 μM compound. Both N54 and N55 inhibited BT474 growth but had no effect on MCF7, SKBR3, or MDA-MB231 (Figure 7A–D). Notably, the growth rates for the four lines were variable, with doubling times ranging from 32 h (MDA-MB231) to 149 h (BT474). Interestingly, N54/N55 showed the greatest effects on the growth of BT474 cells with N54 being somewhat (~2-fold) more potent than N55 Figure 7E,F). Consistent with our previous studies,38 the ability of a KDM5 inhibitor to affect global H3K4me3 levels correlated poorly with its growth inhibitory activity; treatment with both N54, and to a lesser degree N55, resulted in an increase in H3K4me3 levels in all cell lines while the impact on growth varied considerably. The discordance in MCF7 growth inhibitory response as measured in bulk cultures over 4 days (Figure 7) versus in colony formation assays (Figure 6) may arise from the latter being a more sensitive measure of inhibition, requiring that cells survive plating at low density and then proliferate from a single cell over 12 days (Figure 6C). Taken together, these data are consistent with the in vitro biochemical data showing that (R)-N54 is the more potent than its enantiomer (S)-N55 in cells and further underscore the necessity for both biochemical and cell-based assays in the development of KDM5 inhibitors.
Figure 7.

Proliferation of (R)-N54 and (S)-N55 treated breast cancer lines. N54 and N55 inhibit proliferation of (A) BT474 but not (B) MCF7, (C) SKBR3, or (D) MDA- MB231. Growth curves represent percent confluence (mean ± SD, n = 3). (E) N54 is a more potent inhibitor of BT474 proliferation than N55. AUC for growth (% confluence over 288 h) is plotted versus inhibitor concentration. (F) Micrographs at 240 h posttreatment, with the confluence mask shown in yellow. Scale bar is 300 μm.
DISCUSSION
KDM5 family enzymes (particularly KDM5A and KDM5B) have been studied in several types of cancer and cancer-related processes including cell proliferation, drug resistance, and metastasis.11,12,53 Knockout of KDM5A significantly prolonged survival in genetically engineered mouse tumor models.22,23 These studies set the foundation that KDM5A inhibition could be exploited for cancer treatment. Our extensive efforts to characterize the molecular interactions of KDM5A with structurally diverse inhibitors provide important insights into structure–activity relationships for KDM5 inhibitors38,49 (this study).
Here we present two pairs of enantiomers representing two distinct inhibitor chemotypes, compounds N51 and N52 [(R)-and (S)-2-((2-chlorophenyl)(2-(piperidin-1-yl)ethoxy)-methyl)-1H-pyrrolo[3,2-b]pyridine-7-carboxylic acid] and compounds N54 and N55 [(R)- and (S)-N-(1-(3-isopropyl-1H-pyrazole-5-carbonyl)pyrrolidin-3-yl)cyclopropanecarboxamide]. In vitro, the S enantiomer of N51/N52 and the R isoform of N54/N55 exhibited more potent binding by approximately a factor of 4–5. As noted above, there was a discrepancy for compounds N51 and N52 between the binding affinity KD values measured by using the linked JmjN-JmjC domain of KDM5A(1–588)ΔAP wherein there was a ~4-fold difference in the binding affinities (Figure 1C), yet the enzyme inhibitory activities (IC50 values) measured using the commercially available KDM5A(1–1090)-Flag were comparable (Figure 1D). To further investigate, we examined the crystal structures of a larger fragment of KDM5A(12–800) published by Vinogradova et al.36 and Liang et al.52 for additional interactions that might explain the observed difference. Like (R)-N54 (Figure S6), two related compounds, CPI-455, in complex with KDM5A(12–800),36 and N8, in complex with KDM5A(1–588)ΔAP,38 showed no additional interactions with residues outside of the linked JmjN-JmjC domain (Figure S1E). Another explanation may be variation between batches of commercial enzymes used in AlphaLISA, as we noted in our previous study38 (comparing Flag-KDM5B-FL in Figures 2D and S4B in Horton et al., 2016).
For the cell-permeable N54/N55 pair the differences observed in binding affinity and in vitro enzyme inhibitory activity translated to a difference in cellular potency, with the R-isoform (N54) exhibiting a greater ability to inhibit KDM5, as indicated by the accumulation of intracellular H3K4me3 levels, and a somewhat enhanced growth inhibitory activity relative to the S-isoform (N55). Interestingly, while there was some concordance between the relative ability of the two isoforms to influence intracellular H3K4me3 levels and to impact cell growth within individual cell lines (e.g., N54 more potent than N55), there appeared to be little relationship between treatment-induced H3K4me3 accumulation and growth inhibitory effects of the two compounds across cell lines. Indeed, consistent with our previous studies on KDM5-C70,38 treatment with N54, or to a lesser extent N55, resulted in the accumulation of H3K4me3 across all cell lines, an indication that it reaches its intracellular target, but the dose-dependence of this effect had little relationship to growth inhibitory activity. These data suggest that there are additional cellular factors beyond H3K4me3 levels that contribute to differential effects of KDM5 inhibition on cell growth.
While our study should aid in the successful design of selective and potent epigenetic inhibitors of KDM5 demethylases in vitro (for example, (S)-N52 exhibited approximately 40 fold stronger binding than (R)-N54), a remaining challenge is to make these compounds cell permeable. As mentioned above, KDM5-C70 was designed as a cell-permeable prodrug that is hydrolyzed by intracellular esterase(s) to generate KDM5-C49, which contains an isonicotinic acid moiety with a carboxylic acid (PCT WO 2014053491).38,39 Like (S)-N52 and the related compounds examined in this study, the terminal carboxylic acid group forms an extensive hydrogen bonding network with Lys501, Tyr409, and Asn454, similar to those normally engaged by αKG (Figure 8A–C). The hydroxyl of Tyr409 forms two hydrogen bonds, accepting a proton from the amide group of Asn454 and donating a proton to the carboxylate group of the inhibitor. The low permeability is likely to be due to the strong acidic nature of the carboxylate moiety, or overall compound polarity, and certain modifications (for example, an ethyl ester derivative) were needed to improve cell permeability of these compounds. Subsequently, several cell-permeable compounds were discovered that lack a carboxylate moiety, including CPI-455,36 (R)-N54,52 and KDOAM-2537 which simply replaces carboxylate of KDM5-C49 with a carboxyamide (Figure 8D–F). We also crystallized the KDM5A-linked Jumonji domain in complex with KDOAM-25 (Table S1 and Figure 8E), which has a similar dissociation constant (KD = 2.7 μM) and half maximal inhibitory concentration at 1 mM αKG (IC50 =18 μM) as that of (R)-N54 (Figure S7). The common feature of these neutral, carboxylate-absent compounds, like (R)-N54, is that Tyr409 undergoes a series of conformational changes upon the binding of inhibitor (Figure 8G,H). For the carboxylic acid containing inhibitors or αKG, the side chain Asn575 bridges between Lys501 and Tyr409, which form hydrogen bonds with the carboxylic group. By contrast, in the KDOAM-25 complex, the side chain of Tyr409 is rotated away approximately 30° from that of the KDM5-C49-bound form (Figure 8G). An ordered water molecule occupies the position of the hydroxyl oxygen atom of Tyr409, thus maintaining H-bond interaction between two amide groups of Asn575 and KDOAM-25, both of which function as proton donors. In the CPI-455 and (R)-N54 bound conformation, the side chain of Tyr409 is rotated nearly 90°, resulting in a van der Waals contact with the isopropyl moiety (CPI-455) or the cyclopropyl group (N54), respectively. It seems that the conformation of Tyr409 is the largest difference observed in KDM5A after binding of these two inhibitor scaffolds, likely explained by the presence or absence of the carboxylate group, and thus, a common feature among these three cell permeable compounds that lack a carboxylate moiety. These results suggest that further development of KDM5 inhibitors of less acidity could benefit from taking advantage of the inhibitor-induced flexibility of Tyr409. Furthermore, to make more cell penetrant compounds, one could potentially take advantage of adding moieties such as a furyl ring or thienyl moiety.54
Figure 8.

Inhibitor specific Tyr409 conformations. (A–C) Tyr409 adopts a nearly identical conformation via the H-bond interaction with the carboxylate moiety of (S)-N52 (panel A; PDB code 6BH1), KDM5-C49 (panel B; PDB code 5ISL), and αKG (panel C; PDB code 5IVB). (D–F) Tyr409 adopts different conformations in the complex with (R)-N54 (panel D; PDB code 6BH2), KDOAM-25 (panel E; PDB code 5IWF), and CPI-455 (panel F; PDB code 5CEH). (G) Superimposition of KDM5-C49 and KDOAM-25 bound KDM5A linked JmjN-JmjC domain structures. (H) Superimposition of (R)-N54, KDM5-C49, and KDOAM-25 bound KDM5A linked JmjN-JmjC domain structures, illustrating conformational changes of Tyr409 upon inhibitor binding.
CONCLUSIONS
Individual enantiomers of chiral drugs are known to exhibit marked differences in biological activities. For example, the (R)- and (S)-enantiomers of 2HG are structurally similar to αKG but demonstrate distinct differences in potency and inhibitory activity against various αKG-dependent dioxygenase enzymes. Here we show that binding of N54/N55 induced conformational changes in KDM5A, particularly Tyr409, which interacts with the acidic carboxylate moiety of N51/N52, as well as the cofactor αKG. In addition, Asp412 makes specific interactions with some inhibitors but not with others. These results provide insights to guide improvements in binding potency and avenues for development of cell permeable inhibitors of the KDM5 family.
EXPERIMENTAL SECTION
Compound Synthesis.
Compounds were synthesized in the National Center for Advancing Translational Sciences (Supporting Information Table S2). Purity determination was performed using an Agilent diode array detector for both method 1 and method 2 (below). Mass determination was performed using an Agilent 6130 mass spectrometer with electrospray ionization in the positive mode. Chiral separations were performed by normal phase chromatography using a CHIRALPAKAD column (5 cm × 50 cm, 20 μm) on an Agilent 1200 HPLC. 1H NMR spectra were recorded on Varian 400 MHz spectrometers. Chemical shifts are reported in ppm with undeuterated solvent (DMSO-d6 at 2.49 ppm) as internal standard for DMSO-d6 solutions. All of the analogs tested in the biological assays have purity greater than 95%, based on both analytical methods. High resolution mass spectrometry was recorded on Agilent 6210 time-of-flight LC/MS system. Confirmation of molecular formula was accomplished using electrospray ionization in the positive mode with the Agilent Masshunter software (version B.02).
Method 1.
A 7 min gradient of 4–100% acetonitrile (containing 0.025% trifluoroacetic acid) in water (containing 0.05% trifluoroacetic acid) was used with an 8 min run time at a flow rate of 1 mL/min. A Phenomenex Luna C18 column (3 μm, 3 mm × 75 mm) was used at a temperature of 50 °C.
Method 2.
A 3 min gradient of 4–100% acetonitrile (containing 0.025% trifluoroacetic acid) in water (containing 0.05% trifluoroacetic acid) was used with a 4.5 min run time at a flow rate of 1 mL/min. A Phenomenex Gemini Phenyl column (3 μm, 3 mm × 100 mm) was used at a temperature of 50 °C.
All air or moisture sensitive reactions were performed under positive pressure of nitrogen or argon with oven-dried glassware. Anhydrous solvents and bases such as dichloromethane, N,N-dimethylforamide (DMF), acetonitrile, ethanol, DMSO, dioxane, Hunig’s base, and triethylamine were purchased from Sigma-Aldrich. Palladium catalysts were purchased from Strem chemicals and used as such. SiliaCat heterogeneous catalyst DPP-Pd (catalog no. R390-100) was purchased from SiliCycle Inc. Preparative purification was performed on a Waters semipreparative HPLC system using a Phenomenex Luna C18 column (5 μm, 30 mm × 75 mm) at a flow rate of 45 mL/min. The mobile phase consisted of acetonitrile and water (each containing 0.1% trifluoroacetic acid). A gradient of 10–50% acetonitrile over 8 min was used during the purification. Fraction collection was triggered by UV detection (220 nm). Analytical analysis was performed on an Agilent LC/MS (Agilent Technologies, Santa Clara, CA). General procedure for the syntheses of analogs is shown below:
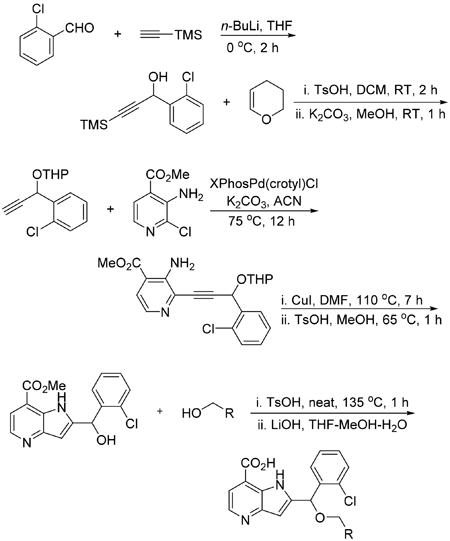
Synthesis of 1-(2-Chlorophenyl)-3-(trimethylsilyl)prop-2-yn-1-ol.
To a solution of trimethylsilylacetylene (13.07 mL, 92 mmol, 1.3 equiv) in THF (50 mL) was added 2.5 M solution of n-butyllithium in THF (37.0 mL, 92 mmol, 1.3 equiv) dropwise upon cooling in an ice bath at –5 °C. The reaction was stirred for 1 h at 0 °C, and then 2-chlorobenzaldehyde (8.01 mL, 71.1 mmol, 1 equiv) was added in dropwise manner upon cooling in ice bath. After stirring the reaction for 1 h at room temperature, it was quenched with ammonium chloride upon cooling. The reaction was concentrated and extracted with diethyl ether. The organic layer was subsequently washed with water and brine, dried over sodium sulfate. The crude product after evaporating the ether was purified on a Isco normal phase flash system using a 120 g gold column eluting with 0–30% ethyl acetate in hexanes over 20 column volumes to obtain 16.9 g (99% yield) of colorless oil.
Synthesis of 2-((1-(2-Chlorophenyl)prop-2-yn-1-yl)oxy)-tetrahydro-2H-pyran.
To a mixture of 1-(2-chlorophenyl)-3-(trimethylsilyl)prop-2-yn-1-ol (17 g, 71.2 mmol, 1 equiv) and dihydropyran (8.12 mL, 89 mmol, 1.25 equiv) in DCM (150 mL) was added p-toluenesulfonic acid monohydrate (0.677 g, 3.56 mmol, 5 mol %) upon cooling in an ice bath. After stirring the reaction for 2 h at room temperature, the reaction mixture was diluted with DCM and subsequently washed with saturated bicarbonate, water, and brine. The organic layer was dried over sodium sulfate and concentrated to obtain crude product which was taken in 100 mL of methanol and then treated with K2CO3 (9.84 g, 71.2 mmol, 1 equiv). After stirring for 1 h at room temperature excess methanol was removed. The residue obtained was suspended in DCM and filtered through a pad of silica. The filtrate was concentrated and the crude product was purified on an Isco normal phase flash system (an automated chromatography instrument) using a 330 g silica column eluting with 0–20% ethyl acetate in hexanes over 16 column volumes to obtain 14 g (78% yield) of oil.
Synthesis of Methyl 3-Amino-2-(3-(2-chlorophenyl)-3-((tetrahydro-2H-pyran-2-yl)oxy)prop-1-yn-1-yl)isonicotinate.
A mixture of methyl 3-amino-2-chloroisonicotinate (1.202 g, 6.44 mmol, 1 equiv), K2CO3 (2.226 g, 16.11 mmol, 2.5 equiv), XPhosPd(crotyl)Cl (http://jmcct.com/products-services/product_p578.html, catalog no. Pd-170) (0.217 g, 0.322 mmol, 5 mol %), and methyl 3-amino-2-(3-(2-chlorophenyl)-3-((tetrahydro-2H-pyran-2-yl)oxy)prop-1-yn-1-yl)isonicotinate (2.21 g, 5.51 mmol, 1.3 equiv) in acetonitrile (12 mL) was bubbled with argon for 5 min and then capped and stirred at 75 °C overnight. The reaction was stirred with silica thiol for 15 min and then diluted with ethyl acetate and filtered through a pad of silica. The crude product obtained after removing the solvent was purified on an Isco flash system using a 120 g gold column eluting with 0–40% ethyl acetate (contains 0.1% TEA) in hexanes over 25 column volumes to obtain a yellow solid in 86% yield (2.21 g).
Synthesis of Methyl 2-((2-chlorophenyl)(hydroxy)methyl)-1H-pyrrolo[3,2-b]pyridine-7-carboxylate (U.S. Patent 9,738,637; Example 71).
A solution of methyl 3-amino-2-(3-(2-chlorophenyl)-3-((tetrahydro-2H-pyran-2-yl)oxy)prop-1-yn-1-yl)-isonicotinate (5 g, 12.47 mmol, 1 equiv) in DMF (12 mL) was bubbled with argon 15 min, then copper(I) iodide (0.475 g, 2.495 mmol, 0.2 equiv) was added and the mixture bubbled again for 15 min. The reaction was capped and stirred at 110 °C for 7 h. The reaction was cooled and stirred with thiol resin and again stirred for 1 h at room temperature. The reaction mixture was diluted with ethyl acetate and filtered through a pad of silica. Further, the filtrate was subsequently washed with water, brine and dried over MgSO4. The crude product obtained after evaporating the solvent was purified on an Isco flash system using a 120 g silica column eluting with 10–60% ethyl acetate in hexanes over 25 column volumes to obtain pure product 72% yield (3.6 g). A mixture of the pure methyl 2-((2-chlorophenyl)((tetrahydro-2H-pyran-2-yl)oxy)methyl)-1H-pyrrolo-[3,2-b]pyridine-7-carboxylate (0.7 g, 1.746 mmol, 1 equiv) and p-toluenesulfonic acid monohydrate (0.332 g, 1.746 mmol, 5 mol %, 1 equiv) in methanol (25 mL) was stirred at 65 °C until the starting material completely disappeared. The solvent was removed and the crude product was washed with water and dried to get pure solid product.
General Procedure for the Syntheses of Analogs.
A mixture of chlorophenyl)(hydroxy)methyl)-1H-pyrrolo[3,2-b]pyridine-7-carboxylate (0.1 g, 0.316 mmol, 1 equiv), p-toluenesulfonic acid monohydrate (0.180 g, 0.947 mmol, 3 equiv), and appropriate alcohol (1.5–2 equiv) in an open vial was stirred neat at 130–135 °C for 1 h. The product formed was a mixture of acid, ester, and trans ester. The crude reaction mass was taken in water and directly injected to an Isco reverse phase column to purify on a flash system. Eluting the column with a gradient of acetonitrile in water (0.1% TFA) over 16 column volumes provided fractions containing a mixture of all three products. The residue obtained after removing the solvent was treated with 1.5 M LiOH in THF/MeOH/water. The solvent was removed and the residue was taken in DMSO and purified on a preparative HPLC to obtain pure products.
2-((2-Chlorophenyl)(propoxy)methyl)-1H-pyrrolo[3,2-b]-pyridine-7-carboxylic Acid (N9).
LC–MS retention time: t1 = 4.822 min and t2 = 3.185 min. NMR data were consistent with previously reported data for this compound in patent WO2014164708 A1 (PCT/US2014/023273; Histone demethylase inhibitors; Quanticel Pharmaceuticals, Inc.).
2-((2-Chlorophenyl)(2-(piperidin-1-yl)ethoxy)methyl)-1H-pyrrolo[3,2-b]pyridine-7-carboxylic Acid·TFA (N40).
LC–MS retention time: t1 = 3.049 min and t2 = 2.448 min. 1H NMR (400 MHz, DMSO-d6) δ 11.52 (s, 1H), 9.07 (s, 1H), 8.53–8.44 (m, 1H), 7.67–7.47 (m, 3H), 7.49–7.27 (m, 3H), 6.46 (s, 1H), 6.22 (s, 1H), 3.92–3.35 (m, 6H), 2.92 (q, J =11.2 Hz, 2H), 1.78 (d, J = 14.2 Hz, 2H), 1.66 (d, J = 12.8 Hz, 4H); HRMS (ESI) m/z (M + H)+ calculated for C22H25ClN3O3; 414.1579 found 414.1587.
2-((2-Chlorophenyl)(2-(4,4-difluoropiperidin-1-yl)ethoxy)-methyl)-1H-pyrrolo[3,2-b]pyridine-7-carboxylic Acid (N41).
LC–MS retention time: t1 = 3.069 min and t2 = 2.421 min. 1H NMR (400 MHz, DMSO-d6) δ 11.49 (s, 1H), 9.75 (s, 1H), 8.47 (d, J = 4.8 Hz, 1H), 7.59 (dd, J = 4.9, 1.3 Hz, 1H), 7.54 (m, 2H), 7.50–7.42 (m, 2H), 6.28 (s, 1H), 6.24 (s, 1H), 3.93 (m, 1H), 3.83 (m, 1H), 3.72–3.24 (m, 4H), 2.77–2.63 (m, 1H), 2.56–2.48 (m, 1H), 2.42–2.18 (m, 4H). HRMS (ESI) m/z (M + H)+ calculated for C22H23ClF2N3O3; 450.1391 found 450.1393.
2-((2-Chlorophenyl)((4,4-difluorocyclohexyl)methoxy)-methyl)-1H-pyrrolo[3,2-b]pyridine-7-carboxylic Acid (N42).
LC–MS retention time: t1 = 4.742 min and t2 = 3.148 min. 1H NMR (400 MHz, DMSO-d6) δ 11.89 (s, 1H), 8.57 (d, J = 5.3 Hz, 1H), 7.72 (d, J = 5.3 Hz, 1H), 7.51 (tt, J = 4.3, 2.7 Hz, 1H), 7.41 (q, J = 3.0 Hz, 3H), 6.38 (d, J = 1.9 Hz, 1H), 6.16 (s, 1H), 3.53 (dd, J = 8.8, 5.9 Hz, 1H), 3.41 (dd, J = 8.8, 6.2 Hz, 1H), 2.05–1.92 (m, 2H), 1.79 (tdd, J = 17.3, 12.8, 6.5 Hz, 5H), 1.24 (t, J = 10.5 Hz, 2H); HRMS (ESI) m/z (M + H)+ calculated for C22H22ClF2N2O3; 435.1282 found 435.1273.
2-((2-Chlorophenyl)(2-(1-methylpyrrolidin-2-yl)ethoxy)-methyl)-1H-pyrrolo[3,2-b]pyridine-7-carboxylic Acid (N46).
LC–MS retention time: t1 = 2.935 min and t2 = 2.468 min. 1H NMR (400 MHz, DMSO-d6) δ 11.11 (s, 1H), 8.31 (dd, J = 4.8, 3.5 Hz, 1H), 7.58 (dd, J = 7.5, 1.9 Hz, 1H), 7.53–7.32 (m, 4H), 6.17 (s, 1H), 6.11 (d, J = 4.1 Hz, 1H), 3.86–3.33 (m, 4H), 2.60–2.56 (m, 3H), 2.25–1.53 (m, 4H); HRMS (ESI) m/z (M + H)+ calculated for C22H25ClN3O3; 414.1579 found 414.1570.
2-((2-Chlorophenyl)(2-(1-methyl-1H-imidazol-2-yl)ethoxy)-methyl)-1H-pyrrolo[3,2-b]pyridine-7-carboxylic Acid (N47).
LC–MS retention time: t1 = 2.807 min and t2 = 2.338 min. 1H NMR (400 MHz, DMSO-d6) δ 11.70 (s, 1H), 8.41 (d, J = 4.9 Hz, 1H), 7.55–7.46 (m, 2H), 7.46–7.35 (m, 4H), 7.05 (d, J = 1.3 Hz, 1H), 6.86 (d, J = 1.3 Hz, 1H), 6.17 (s, 1H), 6.11 (d, J = 0.6 Hz, 1H), 3.85 (ddt, J = 32.8, 9.2, 6.6 Hz, 2H), 3.55 (s, 3H), 3.01 (td, J = 6.7, 2.1 Hz, 2H); HRMS (ESI) m/z (M + H)+ calculated for C21H20ClN4O3; 411.1218 found 411.1218.
2-((2-Chlorophenyl)(3-(piperidin-1-yl)propoxy)methyl)-1H-pyrrolo[3,2-b]pyridine-7-carboxylic Acid (N48).
LC–MS retention time: t1 = 3.009 min and t2 = 2.351 min. 1H NMR (400 MHz, DMSO-d6) δ 11.95 (s, 1H), 8.28 (d, J = 4.8 Hz, 1H), 7.59–7.52 (m, 1H), 7.49 (d, J = 4.7 Hz, 1H), 7.45–7.36 (m, 1H), 7.40–7.30 (m, 2H), 5.85 (s, 1H), 5.75 (d, J = 9.2 Hz, 1H), 3.87–3.41 (m, 4H), 3.3–3.13 (m, 4H), 2.15–1.31 (m, 8H); HRMS (ESI) m/z (M + H)+ calculated for C23H27ClN3O3; 428.1735 found 428.1741.
(R)- and (S)-2-((2-Chlorophenyl)(2-(piperidin-1-yl)ethoxy)-methyl)-1H-pyrrolo[3,2-b]pyridine-7-carboxylic Acid (N51 and N52).
Racemic mixture of ester precursor was separated by normal phase chromatography using a CHIRALPAKAD column (5 cm × 50 cm, 20 μm) on an Agilent 1200 HPLC with a hexane/isopropanol/diethylamine (85:15:0.1) isocratic mobile phase eluting at 35 mL/min and detecting at 230 nm. Separation of 70 mg of the racemic mixture provided 20 mg of enantiomer 1 (with negative rotation), with enantiomeric excess greater than 98%, and 20 mg of enantiomer 2 (with positive rotation) with enantiomeric excess 95.9%. Absolute configuration was determined by cocrystallization with KDM5A enzyme.
(R)- and (S)-N-(1-(3-Isopropyl-1H-pyrazole-5-carbonyl)-pyrrolidin-3-yl)cyclopropanecarboxamide (N54 and N55).
LC–MS retention time: t1 = 3.530 min and t2 = 2.590 min. The title compounds were prepared as individual enantiomers from enantiopure starting materials according to previously reported protocol, and NMR data were consistent with previously reported data for this compound in patent WO2016057924 A1 (PCT/US2015/ 054949; Pyrrolidine amide compounds as histone demethylase inhibitors; Genetech, Inc., Constellation Pharmaceuticals, Inc.). Absolute configuration was determined by cocrystallization with KDM5A enzyme.
Crystallography.
The N-terminal fragments of human KDM5A and the internal deletion constructs (deleting ARID and PhD1 domains (ΔAP)) were prepared in a pET28 plasmid containing an N-terminal His-SUMO tag sequence and purified by three-column chromatography utilizing affinity, anion exchange, and sizing exclusion as previously described in detail.49 The KDM5A(1–588)ΔAP, in 20 mM Hepes, pH 8.0, 300 mM NaCl, 5% glycerol, 0.5 mM tris(2-carboxyethyl)phosphine (TCEP), was mixed with MnCl2 and αKG at an approximate molar ratio of 1:5 and concentrated to ~50 mg/mL (~ 1.4 mM) and then diluted to ~ 10 mg/mL (280 μM) for cocrystallization. Experiments utilized the sitting drop technique and were conducted at 16 °C by mixing 0.2 μL of the complex with an equal volume of a well solution. Typically, clusters of physically twinned crystals grew with αKG with a well solution containing 1.5 M ammonium sulfate, 12% glycerol, and 0.1 M Tris-HCl (pH 8.5). However, these crystals could be crushed to produce microseeds and utilized to produce some large and single crystals from a 96-well screen containing 1.2–1.35 M (NH4)2SO4, 0.1 M Tris-HCl (pH 8.6–9.2), 0–20% glycerol, and 25 mM (Na/K) dibasic/monobasic phosphate. Inhibitors are soaked into these preformed crystals of KDM5A-αKG-Mn(II) complexes by transferring a crystal into a new drop containing mother liquor and an inhibitor (~500 μM), allowing the crystal to remain in this drop for a few hours to 1 day for the inhibitor to exchange with αKG. These were then mounted into nylon cryoloops (Hampton Research, Inc.) and frozen in liquid nitrogen after the addition of more glycerol (up to ~30% total) to the mother liquor as a cryoprotectant. X-ray diffraction data were collected on either the SER-CAT beamline 22-ID or 22-BM at the Advanced Photon Source at Argonne National Laboratory at 100 K with 1° oscillation images and were processed and merged with HKL200055 (Table S1). The structures were determined by molecular replacement using our initial KDM5A structure (PDB code 5E6H). Repeated rounds of manual refitting and crystallographic refinement were performed using COOT56,57 and PHENIX.58,59
Isothermal Titration Calorimetry (ITC).
ITC experiments were carried out as described.38 Briefly, enzyme was dialyzed extensively against 20 mM HEPES, pH 8.0, 300 mM NaCl, 5% glycerol, 0.5 mM tris(2-carboxyethyl)phosphine) supplemented with 500 μM MnCl2 and 10% (v/v) DMSO, and the dialysate was used to prepare fresh working solutions of various inhibitors. The manganese ion was utilized instead of iron since it would not oxidize during the time frame of these experiments. Titration was carried out at 0.42–1.1 mM compound [I] concentration and enzyme [E] concentration of 25–80 μM. Compounds were injected into protein or buffer (to measure the heat of dilution) in 25 steps of 4 μL volume using a MicroCal Auto-iTC200. Binding affinity (KD), stoichiometry (N), and binding enthalpy (ΔH) were determined by fitting the data using the ITC data analysis module of Origin version 7.0 (OriginLab Corp.).
For each ITC experiment, we performed a test run to figure out the optimal enzyme and inhibitor concentrations to be used and range of titrations. Each curve is fitted for 20 data points. Due to the small differences in the KD values of very similar chemical compounds, the data presented resulted from experiments done at the same time using the same batch of enzymes, which is essential to avoid the variation from batches of enzyme preparation.
Formaldehyde Dehydrogenase (FDH)-Coupled Demethylase Assay.
FDH assays were performed as described.38,49 In brief, inhibitors N54 and N55 with the concentrations ranging from 1.25 mM to 10 nM in a half-log serial dilution were preincubated with 0.5 μM enzyme [E] at room temperature (21 °C) in the reaction buffer, containing 50 mM MES (pH 6.8), 50 μM (NH4)2Fe(SO4)2, 2 mM ascorbic acid, 10 μg of FDH, and 10% DMSO. After 15 min, 15 μM histone peptide H3(1–24)K4me3 [S] and 0.6 mM APAD+ (3-acetylpyridine adenine dinucleotide, a more stable analogue of NAD+; Sigma-Aldrich A5251) were added to initiate the reaction for 15 min, with the fluorescence signal recorded in a BioTek Synergy 4 hybrid microplate reader using 380 nm (20 nm bandwidth) and 460 nM (40 nm bandwidth) as the excitation and emission filters, respectively. The initial velocity was calculated, and the dose–response curves were fitted by a four-parameter logistic equation using GraphPad Prism 5.0: activity (%) = B + (M – B)/[1 + 10((logIC50-log[I]·Hill slope)], where B is the baseline (usually 0 for no activity or maximum inhibition), M is maximum activity or no inhibition (usually 1), [I] is inhibitor concentration, and the Hill slope refers to the steepness of the curve (usually – 1).
AlphaLISA Assay.
Alpha streptavidin donor beads (catalog no. 6760002), anti-methyl-histone H3 lysine 4 (H3K4me2/1) AlphaLISA acceptor beads (catalog AL116), AlphaLISA 5× epigenetics buffer (catalog AL008-C1), and 30× epigenetics buffer supplement (AL008-C2) were purchased from PerkinElmer (Waltham, MA). C-terminal Flag-tagged KDM5A (residues 1–1090; catalog 50110) was purchased from BPS Bioscience (San Diego, CA). The biotinylated H3(1–21)K4me3 (catalog AS-64192) peptide was purchased from AnaSpec (Freemont, CA) and bovine serum albumin (BSA; catalog A8412) was purchased from Sigma (St. Louis, MO).
All assays were performed in 1536-well, white, solid-bottom assay plates (catalog no. 789175) obtained from Greiner Bio-One (Monroe, NC) and read using an EnVision multilabel plate reader from PerkinElmer (Waltham, MA). Reactions were performed in 50 mM HEPES, pH 7.25, containing 0.01% BSA and 0.01% Tween-20. To each well, 3 μL of enzyme mix, containing KDM5A(1– 1090)-Flag (final 10 nM), was added using a BioRAPTR flying reagent dispenser (FRD; Beckman Coulter, Fullerton, CA) while buffer alone served as a negative control. A Kalypsys pin-tool was then employed to transfer 23 nL of compound solutions in DMSO to each well, yielding a final concentration range up to 57 μM. Following a 10 min incubation at room temperature, 1 μL of 4× substrate mix containing biotinylated-H3K4(me3) peptide substrate (final 100 nM), αKG (final 25 μM), (NH4)2Fe(SO4)2 (final 50 μM), and ascorbic acid (final 10 μM) was added to initiate the reaction. Reactions were allowed to proceed at room temperature for 45 min prior to detection. To detect demethylation of biotinylated-H3K4(me3) peptide product, reactions were incubated with 20 μg/mL anti-methyl-histone H3K4 (me2/1) acceptor beads in 1× epigenetics buffer for 1 h, then followed with a 30 min incubation with 20 μg/mL streptavidin-coated donor AlphaScreen beads. Beads were added in 1.5 and 3 μL of FRD dispense, respectively, for a final volume of 7.5 μL, and plates were protected from the light at room temperature during donor bead incubation. Microplates were read on an EnVision multilabel plate reader using the 1536-well plate HTS AlphaScreen aperture (excitation time 80 ms, measurement time 240 ms). Data were normalized to the mean of the no-enzyme and uninhibited controls, and IC50 values were determined by nonlinear regression fits using GraphPad Prism4 software.
Cell Culture.
MCF7, BT474, and MDA-MB231 cells were cultured in RPMI1640 supplemented with 10% fetal bovine serum and 1% penicillin and streptomycin. SKBR3 cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and 1% penicillin and streptomycin. All cells were grown at 37 °C with 5% CO2.
Western Blot Analysis.
SKBR3, BT474, MDA-MB231, and MCF7 cells were treated with DMSO, KDM5-C70, N54, or N55 for 3 days. Histone extraction and Western blot analysis were performed as described previously.60 The following antibodies were obtained commercially: rabbit anti-histone H3 (ab1791) (Abcam); rabbit anti-H3K4me3 (C42D8), H3K9me3 (D4W1U), H3K27me3 (C36B11) (Cell Signaling Technology).
Colony Formation Assay.
MCF7 cells were seeded at 2000 cells/well and treated with 0.1% DMSO, 1 or 10 μM N54, 1 or 10 μM N55, or 1 μM KDM5-C70 the following day. Cells were treated for 12 days with media replaced every 3 days. Cells were fixed for 5 min in 4% paraformaldehyde in phosphate-buffered saline, stained in 0.05% crystal violet in double-distilled water for 45 min, gently washed with water, and allowed to dry overnight before photographing. ImageJ plug-in ColonyArea was used to quantify crystal violet staining as described previously.38
IncuCyte Experiments.
Cells were seeded in 384-well plates (Cell Carrier Ultra, PerkinElmer) at 2.5 × 103 cells per well (50 μL volume) using a Multidrop Combi Reagent Dispenser (Thermo Fisher) and returned to a humidified incubator (5% CO2) overnight. Culture medium was RPMI (Life Technologies no. 11835) + 10% fetal bovine serum + 50 U/mL penicillin + 50 μg/mL streptomycin. Compounds (190 nL in DMSO) were transferred using a pin tool. Plates were covered with a gas permeable sealing membrane (Breathe Easy, Diversified Biotech) and imaged every 4 h using an IncuCyte Zoom (Essen Bioscience) equipped with a 10× objective (Nikon). Cell growth (phase object confluence) was calculated using IncuCyte ZOOM software.
Immunocytochemistry.
BT474 cells were seeded in 384-well plates (Cell Carrier Ultra, PerkinElmer) at 2.5 × 103 cells per well (50 μL volume). After overnight incubation, 190 nL of compound (in DMSO) was added by pin transfer ([final] = 62 nM to 63 μM). After 48 h, the cells were fixed (fresh 4% paraformaldehyde in 1× PBS, pH 7.4), permeabilized (PBS + 0.1% Triton X-100 + 0.2% BSA), and blocked (PBS + 0.1% Triton X-100 + 2% BSA + 5% normal goat serum). Primary antibodies against H3K4me3 (Abcam 1012) and H3 (Cell Signaling CS4499) were incubated overnight at 4 °C. Secondary antibodies conjugated to AlexaFluor488 and 568 dyes were added, and nuclei were stained with Hoechst. Cells were imaged on an IN Cell Analyzer 2200. H3K4Me3 and total H3 intensity was measured on a per cell basis (in the nuclear region, defined as overlapping with Hoechst staining), and H3K4me3/total H3 ratio was calculated.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part with Federal funds from the National Cancer Institute (NCI), National Institutes of Health (NIH), under NCI Chemical Biology Consortium Contract HHSN261200800001E (to H.F.), NIH Grant GM114306 and Cancer Prevention Research Institute of Texas Grant RR160029 (to X.C.), American Cancer Society Research Scholar Grant RSG-13-384-01-DMC and DoD Breast Cancer Research Program Award W81XWH-14-1-0308 (to Q.Y.), and Leslie H. Warner Postdoctoral Fellowship (to L.W.). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. The Department of Biochemistry of Emory University School of Medicine supported the use of the Southeast Regional Collaborative Access Team (SERCAT) synchrotron beamlines at the Advanced Photon Source of Argonne National Laboratory. We thank Dr. Paul E. Brennan of Structural Genomics Consortium for KDOAM-25. We thank Anthony Welch, Andrew Flint, and Dane Liston of Frederick National Laboratory for Cancer Research for administrative support, and Andrew Flint for extensive comments and editing the manuscript.
ABBREVIATIONS USED
- αKG
α-ketoglutarate
- 2HG
2-hydroxyglutarate
- KDM5A
histone lysine demethylase 5A
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.8b00261.
Figures S1–S7, and Tables S1–S2 (PDF)
Molecular formula strings (CSV)
Accession Codes
The atomic coordinates and structure factors of KDM5A in complex with N9 (code 6BGU), N40 (code 6BGV), N41 (6BGW), N42 (code 6BGX), N46 (code 6BGY), N47 (code 6BGZ), N48 (code 6BH5), N51 (code 6BH0), N52 (code 6BH1), N54 (code 6BH2), N55 (code 6BH3), and KDOAM-25 (code 5IWF) have been deposited in the Protein Data Bank, and authors will release the atomic coordinates and experimental data upon article publication.
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Gasser SM, Li E, Eds. Epigenetics and Disease: Pharmaceutical Opportunities; Progress in Drug Research, Vol. 67; Springer; Basel AG, 2011; DOI: 10.1007/978-3-7643-8989-5. [DOI] [PubMed] [Google Scholar]
- (2).Bannister AJ; Kouzarides T Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Herz HM; Garruss A; Shilatifard A SET for life: biochemical activities and biological functions of SET domain-containing proteins. Trends Biochem. Sci 2013, 38, 621–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Paik WK; Paik DC; Kim S Historical review: the field of protein methylation. Trends Biochem. Sci 2007, 32, 146–152. [DOI] [PubMed] [Google Scholar]
- (5).Shi Y; Lan F; Matson C; Mulligan P; Whetstine JR; Cole PA; Casero RA; Shi Y Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [DOI] [PubMed] [Google Scholar]
- (6).Tsukada Y; Fang J; Erdjument-Bromage H; Warren ME; Borchers CH; Tempst P; Zhang Y Histone demethylation by a family of JmjC domain-containing proteins. Nature 2006, 439, 811–816. [DOI] [PubMed] [Google Scholar]
- (7).Shi YG; Tsukada Y The discovery of histone demethylases. Cold Spring Harbor Perspect. Biol 2013, 5, a017947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Jambhekar A; Anastas JN; Shi Y Histone lysine demethylase inhibitors. Cold Spring Harbor Perspect. Med 2017, 7, a026484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Zheng YC; Ma J; Wang Z; Li J; Jiang B; Zhou W; Shi X; Wang X; Zhao W; Liu HM A systematic review of histone lysine-specific demethylase 1 and its inhibitors. Med. Res. Rev 2015, 35, 1032–1071. [DOI] [PubMed] [Google Scholar]
- (10).Christensen J; Agger K; Cloos PA; Pasini D; Rose S; Sennels L; Rappsilber J; Hansen KH; Salcini AE; Helin K RBP2 belongs to a family of demethylases, specific for tri- and dimethylated lysine 4 on histone 3. Cell 2007, 128, 1063–1076. [DOI] [PubMed] [Google Scholar]
- (11).Blair LP; Cao J; Zou MR; Sayegh J; Yan Q Epigenetic regulation by lysine demethylase 5 (KDM5) enzymes in cancer. Cancers 2011, 3, 1383–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Rasmussen PB; Staller P The KDM5 family of histone demethylases as targets in oncology drug discovery. Epigenomics 2014, 6, 277–286. [DOI] [PubMed] [Google Scholar]
- (13).Han M; Xu W; Cheng P; Jin H; Wang X Histone demethylase lysine demethylase 5B in development and cancer. Oncotarget 2017, 8, 8980–8991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Hou J; Wu J; Dombkowski A; Zhang K; Holowatyj A; Boerner JL; Yang ZQ Genomic amplification and a role in drug-resistance for the KDM5A histone demethylase in breast cancer. Am. J. Transl. Res 2012, 4, 247–256. [PMC free article] [PubMed] [Google Scholar]
- (15).Teng YC; Lee CF; Li YS; Chen YR; Hsiao PW; Chan MY; Lin FM; Huang HD; Chen YT; Jeng YM; Hsu CH; Yan Q; Tsai MD; Juan LJ Histone demethylase RBP2 promotes lung tumorigenesis and cancer metastasis. Cancer Res. 2013, 73, 4711–4721. [DOI] [PubMed] [Google Scholar]
- (16).Wang S; Wang Y; Wu H; Hu L RBP2 induces epithelial-mesenchymal transition in non-small cell lung cancer. PLoS One 2013, 8, e84735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Liang X; Zeng J; Wang L; Fang M; Wang Q; Zhao M; Xu X ; Liu Z; Li W; Liu S; Yu H; Jia J; Chen C Histone demethylase retinoblastoma binding protein 2 is overexpressed in hepatocellular carcinoma and negatively regulated by hsa-miR-212. PLoS One 2013, 8, e69784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Zeng J; Ge Z; Wang L; Li Q; Wang N; Bjorkholm M; Jia J; Xu D The histone demethylase RBP2 Is overexpressed in gastric cancer and its inhibition triggers senescence of cancer cells. Gastroenterology 2010, 138, 981–992. [DOI] [PubMed] [Google Scholar]
- (19).Jiping Z; Ming F; Lixiang W; Xiuming L; Yuqun S; Han Y; Zhifang L; Yundong S; Shili L; Chunyan C; Jihui J MicroRNA-212 inhibits proliferation of gastric cancer by directly repressing retinoblastoma binding protein 2. J. Cell. Biochem 2013, 114, 2666–2672. [DOI] [PubMed] [Google Scholar]
- (20).Fattaey AR; Helin K; Dembski MS; Dyson N; Harlow E; Vuocolo GA; Hanobik MG; Haskell KM; Oliff A; Defeo-Jones D Characterization of the retinoblastoma binding proteins RBP1 and RBP2. Oncogene 1993, 8, 3149–3156. [PubMed] [Google Scholar]
- (21).Benevolenskaya EV; Murray HL; Branton P; Young RA; Kaelin WG Jr Binding of pRB to the PHD protein RBP2 promotes cellular differentiation. Mol. Cell 2005, 18, 623–635. [DOI] [PubMed] [Google Scholar]
- (22).Lin W; Cao J; Liu J; Beshiri ML; Fujiwara Y; Francis J; Cherniack AD; Geisen C; Blair LP; Zou MR; Shen X; Kawamori D; Liu Z; Grisanzio C; Watanabe H; Minamishima YA; Zhang Q; Kulkarni RN; Signoretti S; Rodig SJ; Bronson RT; Orkin SH; Tuck DP; Benevolenskaya EV; Meyerson M; Kaelin WG Jr.; Yan Q Loss of the retinoblastoma binding protein 2 (RBP2) histone demethylase suppresses tumorigenesis in mice lacking Rb1 or Men1. Proc. Natl. Acad. Sci U. S. A 2011, 108, 13379–13386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Cao J; Liu Z; Cheung WK; Zhao M; Chen SY; Chan SW; Booth CJ; Nguyen DX; Yan Q Histone demethylase RBP2 is critical for breast cancer progression and metastasis. Cell Rep. 2014, 6, 868–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Zhou D; Kannappan V; Chen X; Li J; Leng X; Zhang J; Xuan S RBP2 induces stem-like cancer cells by promoting EMT and is a prognostic marker for renal cell carcinoma. Exp. Mol. Med 2016, 48, e238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Liang X; Zeng J; Wang L; Shen L; Ma X; Li S; Wu Y; Ma L; Ci X; Guo Q; Jia M; Shen H; Sun Y; Liu Z; Liu S; Li W; Yu H; Chen C; Jia J Histone demethylase RBP2 promotes malignant progression of gastric cancer through TGF-beta1-(p- Smad3)-RBP2-E-cadherin-Smad3 feedback circuit. Oncotarget 2015, 6, 17661–17674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Sharma SV; Lee DY; Li B; Quinlan MP; Takahashi F; Maheswaran S; McDermott U; Azizian N; Zou L; Fischbach MA; Wong KK; Brandstetter K; Wittner B; Ramaswamy S; Classon M; Settleman J A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010, 141, 69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Banelli B; Carra E; Barbieri F; Wurth R; Parodi F; Pattarozzi A; Carosio R; Forlani A; Allemanni G; Marubbi D; Florio T; Daga A; Romani M The histone demethylase KDM5A is a key factor for the resistance to Temozolomide in glioblastoma. Cell Cycle 2015, 14, 3418–3429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Yamane K; Tateishi K; Klose RJ; Fang J; Fabrizio LA; Erdjument-Bromage H; Taylor-Papadimitriou J; Tempst P; Zhang Y PLU-1 is an H3K4 demethylase involved in transcriptional repression and breast cancer cell proliferation. Mol. Cell 2007, 25, 801–812. [DOI] [PubMed] [Google Scholar]
- (29).Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Wang L; Mao Y; Du G; He C; Han S Overexpression of JARID1B is associated with poor prognosis and chemotherapy resistance in epithelial ovarian cancer. Tumor Biol. 2015, 36, 2465–2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Tang B; Qi G; Tang F; Yuan S; Wang Z; Liang X; Li B; Yu S; Liu J; Huang Q; Wei Y; Zhai R; Lei B; Yu H; Jiao X; He S JARID1B promotes metastasis and epithelial-mesenchymal transition via PTEN/AKT signaling in hepatocellular carcinoma cells. Oncotarget 2015, 6, 12723–12739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Wang Z; Tang F; Qi G; Yuan S; Zhang G; Tang B; He S KDM5B is overexpressed in gastric cancer and is required for gastric cancer cell proliferation and metastasis. Am. J. Cancer Res 2015, 5, 87–100. [PMC free article] [PubMed] [Google Scholar]
- (33).Lin CS; Lin YC; Adebayo BO; Wu A; Chen JH; Peng YJ; Cheng MF; Lee WH; Hsiao M; Chao TY; Yeh CT Silencing JARID1B suppresses oncogenicity, stemness and increases radiation sensitivity in human oral carcinoma. Cancer Lett. 2015, 368, 36–45. [DOI] [PubMed] [Google Scholar]
- (34).Kuo YT; Liu YL; Adebayo BO; Shih PH; Lee WH; Wang LS; Liao YF; Hsu WM; Yeh CT; Lin CM JARID1B expression plays a critical role in chemoresistance and stem cell-like phenotype of neuroblastoma cells. PLoS One 2015, 10, e0125343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Bamodu OA; Huang WC; Lee WH; Wu A; Wang LS; Hsiao M; Yeh CT; Chao TY Aberrant KDM5B expression promotes aggressive breast cancer through MALAT1 overexpression and downregulation of hsa-miR-448. BMC Cancer 2016, 16, 160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Vinogradova M; Gehling VS; Gustafson A; Arora S; Tindell CA; Wilson C; Williamson KE; Guler GD; Gangurde P; Manieri W; Busby J; Flynn EM; Lan F; Kim HJ; Odate S; Cochran AG; Liu Y; Wongchenko M; Yang Y; Cheung TK; Maile TM; Lau T; Costa M; Hegde GV; Jackson E; Pitti R; Arnott D; Bailey C; Bellon S; Cummings RT; Albrecht BK; Harmange JC; Kiefer JR; Trojer P; Classon M An inhibitor of KDM5 demethylases reduces survival of drug-tolerant cancer cells. Nat. Chem. Biol 2016, 12, 531–538. [DOI] [PubMed] [Google Scholar]
- (37).Tumber A; Nuzzi A; Hookway ES; Hatch SB; Velupillai S; Johansson C; Kawamura A; Savitsky P; Yapp C; Szykowska A; Wu N; Bountra C; Strain-Damerell C; Burgess-Brown NA; Ruda GF; Fedorov O; Munro S; England KS; Nowak RP; Schofield CJ; La Thangue NB; Pawlyn C; Davies F; Morgan G; Athanasou N; Muller S; Oppermann U; Brennan PE Potent and selective KDM5 inhibitor stops cellular demethylation of H3K4me3 at transcription start sites and proliferation of mm1s myeloma cells. Cell Chem. Biol 2017, 24, 371–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Horton JR; Liu X; Gale M; Wu L; Shanks JR; Zhang X; Webber PJ; Bell JS; Kales SC; Mott BT; Rai G; Jansen DJ; Henderson MJ; Urban DJ; Hall MD; Simeonov A; Maloney DJ; Johns MA; Fu H; Jadhav A; Vertino PM; Yan Q; Cheng X Structural basis for KDM5A histone lysine demethylase inhibition by diverse compounds. Cell Chem. Biol 2016, 23, 769–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Johansson C; Velupillai S; Tumber A; Szykowska A; Hookway ES; Nowak RP; Strain-Damerell C; Gileadi C; Philpott M; Burgess-Brown N; Wu N; Kopec J; Nuzzi A; Steuber H; Egner U; Badock V; Munro S; LaThangue NB; Westaway S; Brown J; Athanasou N; Prinjha R; Brennan PE; Oppermann U Structural analysis of human KDM5B guides histone demethylase inhibitor development. Nat. Chem. Biol 2016, 12, 539–545. [DOI] [PubMed] [Google Scholar]
- (40).Nguyen LA; He H; Pham-Huy C Chiral drugs: an overview. Int. J. Biomed. Sci 2006, 2, 85–100. [PMC free article] [PubMed] [Google Scholar]
- (41).Chhabra N; Aseri ML; Padmanabhan D A review of drug isomerism and its significance. Int. J. Appl. Basic. Med. Res 2013, 3, 16–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Dang L; White DW; Gross S; Bennett BD; Bittinger MA; Driggers EM; Fantin VR; Jang HG; Jin S; Keenan MC; Marks KM; Prins RM; Ward PS; Yen KE; Liau LM; Rabinowitz JD; Cantley LC; Thompson CB; Vander Heiden MG; Su SM Cancer-associated IDH1 mutations produce 2- hydroxyglutarate. Nature 2009, 462, 739–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Ward PS; Patel J; Wise DR; Abdel-Wahab O; Bennett BD; Coller HA; Cross JR; Fantin VR; Hedvat CV; Perl AE; Rabinowitz JD; Carroll M; Su SM; Sharp KA; Levine RL; Thompson CB The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010, 17, 225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Xu W; Yang H; Liu Y; Yang Y; Wang P; Kim SH; Ito S; Yang C; Wang P; Xiao MT; Liu LX; Jiang WQ; Liu J; Zhang JY; Wang B; Frye S; Zhang Y; Xu YH; Lei QY; Guan KL; Zhao SM; Xiong Y Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Chowdhury R; Yeoh KK; Tian YM; Hillringhaus L; Bagg EA; Rose NR; Leung IK; Li XS; Woon EC; Yang M; McDonough MA; King ON; Clifton IJ; Klose RJ; Claridge TD; Ratcliffe PJ; Schofield CJ; Kawamura A The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011, 12, 463–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Koivunen P; Lee S; Duncan CG; Lopez G; Lu G; Ramkissoon S; Losman JA; Joensuu P; Bergmann U; Gross S; Travins J; Weiss S; Looper R; Ligon KL; Verhaak RG; Yan H; Kaelin WG Jr Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature 2012, 483, 484–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Losman JA; Looper RE; Koivunen P; Lee S; Schneider RK; McMahon C; Cowley GS; Root DE; Ebert BL; Kaelin WG Jr (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science 2013, 339, 1621–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Su R; Dong L; Li C; Nachtergaele S; Wunderlich M; Qing Y; Deng X; Wang Y; Weng X; Hu C; Yu M; Skibbe J; Dai Q; Zou D; Wu T; Yu K; Weng H; Huang H; Ferchen K; Qin X; Zhang B; Qi J; Sasaki AT; Plas DR; Bradner JE; Wei M; Marcucci G; Jiang X; Mulloy JC; Jin J; He C; Chen J R-2HG exhibits anti-tumor activity by targeting FTO/m(6)A/MYC/CEBPA signaling. Cell 2018, 172, 90–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Horton JR; Engstrom A; Zoeller EL; Liu X; Shanks JR; Zhang X; Johns MA; Vertino PM; Fu H; Cheng X Characterization of a linked jumonji domain of the KDM5/JARID1 family of histone H3 lysine 4 demethylases. J. Biol. Chem 2016, 291, 2631–2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Martinez S; Hausinger RP Catalytic mechanisms of Fe(II)- and 2-oxoglutarate-dependent oxygenases. J. Biol. Chem 2015, 290, 20702–20711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Heinemann B; Nielsen JM; Hudlebusch HR; Lees MJ; Larsen DV; Boesen T; Labelle M; Gerlach LO; Birk P; Helin K Inhibition of demethylases by GSK-J1/J4. Nature 2014, 514, E1–2. [DOI] [PubMed] [Google Scholar]
- (52).Liang J; Labadie S; Zhang B; Ortwine DF; Patel S; Vinogradova M; Kiefer JR; Mauer T; Gehling VS; Harmange JC; Cummings R; Lai T; Liao J; Zheng X; Liu Y; Gustafson A; Van der Porten E; Mao W; Liederer BM; Deshmukh G; An L; Ran Y; Classon M; Trojer P; Dragovich PS; Murray L From a novel HTS hit to potent, selective, and orally bioavailable KDM5 inhibitors. Bioorg. Med. Chem. Lett 2017, 27, 2974–2981. [DOI] [PubMed] [Google Scholar]
- (53).Horton JR; Gale M; Yan Q; Cheng X The molecular basis of histone demethylation In DNA and Histone Methylation As Cancer Targets; Kaneda A, Tsukada Y.-i., Eds.; Cancer Drug Discovery and Development; Springer, 2017; pp 151–219, DOI: 10.1007/978-3-319-59786-7_7. [DOI] [Google Scholar]
- (54).Westaway SM; Preston AG; Barker MD; Brown F; Brown JA; Campbell M; Chung CW; Diallo H; Douault C; Drewes G; Eagle R; Gordon L; Haslam C; Hayhow TG; Humphreys PG; Joberty G; Katso R; Kruidenier L; Leveridge M; Liddle J; Mosley J; Muelbaier M; Randle R; Rioja I; Rueger A; Seal GA; Sheppard RJ; Singh O; Taylor J; Thomas P; Thomson D; Wilson DM; Lee K; Prinjha RK Cell penetrant inhibitors of the KDM4 and KDM5 families of histone lysine demethylases. 1. 3-amino-4-pyridine carboxylate derivatives. J. Med. Chem 2016, 59, 1357–1369. [DOI] [PubMed] [Google Scholar]
- (55).Otwinowski Z; Borek D; Majewski W; Minor W Multiparametric scaling of diffraction intensities. Acta Crystallogr., Sect. A: Found. Crystallogr. 2003, 59, 228–234. [DOI] [PubMed] [Google Scholar]
- (56).Emsley P; Lohkamp B; Scott WG; Cowtan K Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Emsley P; Cowtan K Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr 2004, 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- (58).Adams PD; Grosse-Kunstleve RW; Hung LW; Ioerger TR; McCoy AJ; Moriarty NW; Read RJ; Sacchettini JC; Sauter NK; Terwilliger TC PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr., Sect. D: Biol. Crystallogr 2002, 58, 1948–1954. [DOI] [PubMed] [Google Scholar]
- (59).Adams PD; Afonine PV; Bunkoczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung LW; Kapral GJ; Grosse-Kunstleve RW; McCoy AJ; Moriarty NW; Oeffner R; Read RJ; Richardson DC; Richardson JS; Terwilliger TC; Zwart PH PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Sayegh J; Cao J; Zou MR; Morales A; Blair LP; Norcia M; Hoyer D; Tackett AJ; Merkel JS; Yan Q Identification of small molecule inhibitors of Jumonji AT-rich interactive domain 1B (JARID1B) histone demethylase by a sensitive high throughput screen. J. Biol. Chem 2013, 288, 9408–9417. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.