

SUMMARY
Protein arginine methyltransferase 5 (PRMT5) is overexpressed in many cancer types and is a promising therapeutic target for several of them, including leukemia and lymphoma. However, we and others have reported that PRMT5 is essential for normal physiology. This dependence may become dose-limiting in a therapeutic setting, warranting the search for combinatorial approaches. Here we report that PRMT5 depletion or inhibition impairs homologous recombination (HR) DNA repair, leading to DNA damage accumulation, p53 activation, cell cycle arrest and cell death. PRMT5 symmetrically dimethylates histone and non-histone substrates, including several components of the RNA splicing machinery. We find that PRMT5 depletion or inhibition induces aberrant splicing of the multifunctional epigenetic and DNA repair factor TIP60/KAT5, which selectively affects its lysine acetyltransferase activity and leads to impaired HR. As HR-deficiency sensitizes cells to PARP inhibitors, we demonstrate here that PRMT5 and PARP inhibitors have synergistic effects on acute myeloid leukemia cells.
INTRODUCTION
Protein arginine N-methyltransferase 5 (PRMT5) is a type II protein arginine methyltransferase that catalyzes the symmetrical arginine dimethylation of histones and non-histone substrates, including three subunits of the Survival of Motor Neuron (SMN) complex (SmB, SmD1 and SmD3), involved in the assembly of snRNPs, essential components of the spliceosome machinery (Friesen et al., 2001; Matera and Wang, 2014; Meister et al., 2001). PRMT5 depletion can trigger aberrant splicing in the adult hematopoietic compartment (Bezzi et al., 2013; Koh et al., 2015; Liu et al., 2015), and splicing appears to play a critical role in normal hematopoiesis, as mutations in RNA splicing factors, such as SF3B1 or SRSF2, are found in myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML) patients (Makishima et al., 2012; Yoshida et al., 2011). RNA splicing factor mutations result in the mis-splicing of epigenetic regulators, such as EZH2, and impaired hematopoietic cell differentiation (Kim et al., 2015). Recent reports also suggest that this pathway is amenable to therapeutic intervention (Bonnal et al., 2012; Lee et al., 2016).
PRMT5 is overexpressed in a variety of human cancers, including several hematological malignancies, and inhibition of PRMT5 has shown anti-tumor activity in lymphomas (Chan-Penebre et al., 2015), MLL-rearranged acute leukemia models (Kaushik et al., 2017), and several other types of leukemia in vitro (Tarighat et al., 2016). However, fully inhibiting PRMT5 activity in the hematopoietic compartment might lead to substantial toxicities, as PRMT5 knockout in adult mouse hematopoietic stem and progenitor cells (HSPCs) triggers lethal pancytopenia (Liu et al., 2015). Should these toxicities become dose-limiting in the clinical setting, identifying combinatorial approaches that exploit synergistic or synthetic vulnerabilities, may be advantageous. One such vulnerability was recently identified, as cells lacking MTAP, a critical enzyme in the methionine salvage pathway that is deleted in approximately 15% of all human cancers, are more sensitive to PRMT5 depletion than MTAP wild type cells (Kryukov et al., 2016; Marjon et al., 2016; Mavrakis et al., 2016).
PRMT5 depletion can induce DNA damage and genomic instability in a variety of tissues (Table S1), and a potential mechanism was recently identified, as PRMT5 methylates RUVBL1, an interactor of the multifunctional, epigenetic and DNA repair factor TIP60/KAT5, a lysine acetyltransferase (Clarke et al., 2017). DNA double strand breaks (DSBs) are damaging to cells; they trigger a complex DNA damage response that includes the activation of several Phosphatidylinositol 3-kinase-related protein kinases (PIKKs), such as ATM, that can phosphorylate histone H2AX, also known as γH2AX. The generation of γH2AX in the surrounding regions of the DNA break site together with other histone modifications leads to the recruitment of specific proteins involved in the non-homologous end joining (NHEJ) or homologous recombination (HR) DNA repair pathways, including 53BP1 (Daley and Sung, 2014). 53BP1 stimulates the repair of DSBs via NHEJ, while inhibiting homology-dependent DNA repair. In S and G2 phases of the cell cycle, when the sister chromatids are available, the BRCA1 complex competes with 53BP1, leading to 53BP1 dissociation from the DSB sites, and the resection of the DSB ends. DSB-end resection is followed by the accumulation of other HR proteins, including RAD51, which promotes the repair of the original lesion, via DNA recombination with the sister chromatid (Symington and Gautier, 2011).
A deficiency in the HR DNA repair pathway creates a vulnerability in cells as they increasingly rely on poly ADP ribose polymerase (PARP) enzymes to repair their DNA. Olaparib is an FDA-approved PARP1/2 inhibitor that “traps” PARP1/2 on DNA and induces lethal DSBs in HR-deficient cells such as BRCA1-null cancer cells (Farmer et al., 2005). Similarly, it was recently reported that PARP inhibitors can selectively kill subsets of human AML deficient in HR (Esposito et al., 2015).
Here we show that depletion or inhibition of PRMT5 triggers the accumulation of DNA double strand breaks (DSBs) in cells, at least in part due to defective HR-based repair, which leads to activation of the p53 pathway, and subsequent cell cycle arrest and/or cell death. Our data reveals that PRMT5 deficiency or inhibition induces aberrant RNA splicing, including alternative splicing of TIP60. We find that aberrant splicing of exon 5 of the TIP60 gene in hematopoietic cells leads to impaired acetyltransferase activity and reduced histone H4 acetylation, a key event in DNA DSB repair pathway commitment (Jacquet et al., 2016). Indeed, we find that ectopic expression of properly spliced TIP60 in PRMT5-null cells can rescue the homologous recombination phenotype. Thus, PRMT5 controls HSPC homeostasis at least in part through its ability to affect the RNA splicing of key DNA repair pathway factors. Importantly, using human AML cell lines, we show that PRMT5 inhibitors and PARP inhibitors have synergistic cytotoxic effects on leukemia cells, demonstrating the advantages of combining targeted epigenetic and non-epigenetic inhibitors in the treatment of this hematologic malignancy.
RESULTS
PRMT5 inhibition and DNA damage are correlated in AML cells.
To define the dependency of AML cells on PRMT5 activity, we treated 15 AML human cell lines, and normal human cord blood (CB) CD34+ cells, with the PRMT5 inhibitor (PRMT5i) GSK3186000A (compound no.14) (Duncan et al., 2016) (Table S2). The AML cell lines exhibited diverse sensitivity to PRMT5 inhibition, with IC50s ranging from 0.25μM to over 10μM (Figure 1A). While PRMT5 mRNA has been shown to be overexpressed in several types of cancers (Wei et al., 2013), our western blot analysis showed that PRMT5 protein levels vary across AML cell lines, ranging from approximately 0.5 to 4-fold the level of PRMT5 expression in CB CD34+ cells (Figure 1B). There is a positive trend between PRMT5 protein levels in AML cell lines, and sensitivity to a PRMT5i, but this correlation did not reach significance, suggesting that while protein levels might contribute to sensitivity, other factors are likely involved (Figures 1B and C) (pearson r = 0.1584, P=0.13).
Figure 1. PRMT5 inhibition correlates with impaired DNA repair and increased DNA damage in leukemia cell lines.
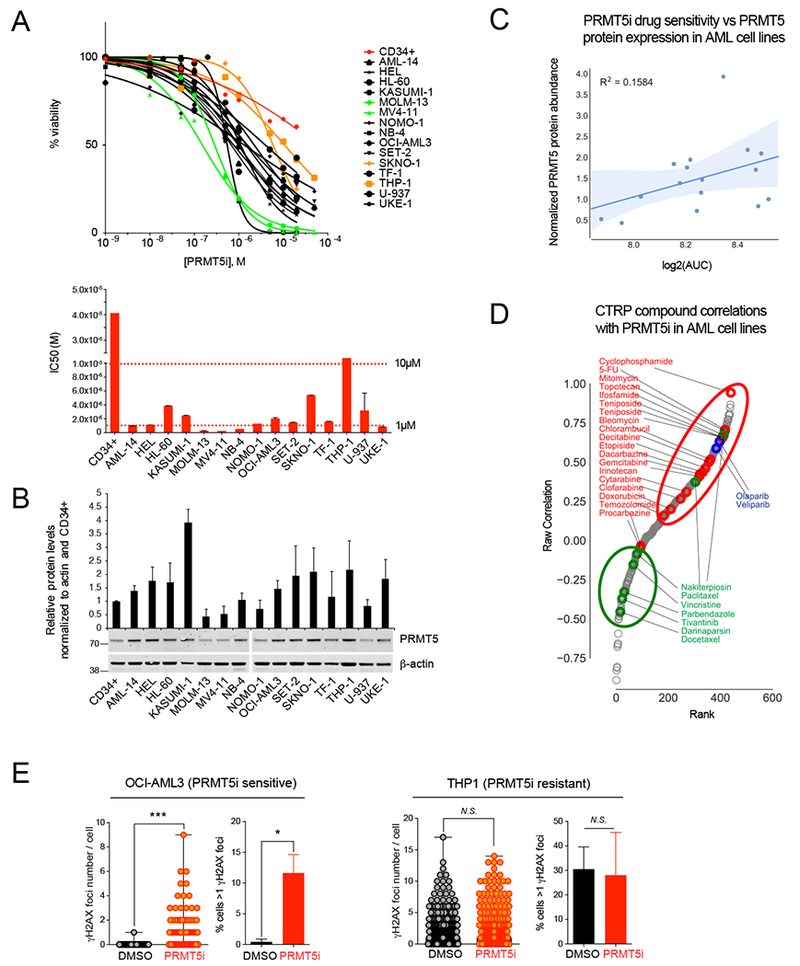
(A) Top: Viability assay at day 5 for 15 AML cells lines (the 2 most sensitive and resistant cell lines are labeled in green and in orange, respectively) and normal human cord blood derived CD34+ cells (red) treated with increasing doses of PRMT5 inhibitor (PRMT5i) GSK3186000A. Bottom: graphical depiction of the IC50 of GSK3186000A for the panel of cell lines.
(B) Immunoblot with PRMT5 and β-actin antibodies and quantification of PRMT5 relative abundance within all tested cell lines.
(C) Plot of PRMT5i drug sensitivity vs PRMT5 protein expression in AML cell lines showing no significant correlation (R2 = 0.1584, P=0.13).
(D) Distribution of PRMT5i and CTRP compound sensitivity Spearman correlations in AML cell lines showing a positive correlation between PRMT5i and genotoxic agents (red, P=0.0089) as well as PARP1/2 inhibitors (blue, P=0.016), and a negative correlation between PRMT5i and microtubule poisons (green, P=0.02).
(E) Quantification of the number of γH2AX foci per cell and the percent of cells with >1 γH2AX foci by immunofluorescence analysis performed with antibody to γH2AX in leukemia cell lines treated with GSK3186000A (IC50 dose (μM) for 4 days) showing increased DNA damage in OCI-AML3 cells (sensitive to PRMT5i) and in THP1 cells (not sensitive). Error bars indicate the SEM of 3 biological replicates. *** P<0.0001, * P<0.05.
See also Table S2.
To identify potential dependencies in an unbiased manner, we turned to the Cancer Therapeutics Response Portal dataset (CTRPv2) of the Broad Institute, which correlates the sensitivity patterns of 481 compounds with ~19,000 basal transcript levels across 823 different human cancer cell lines. We compared the effect of PRMT5 inhibition with the effect of the compounds utilized on 12 AML cell lines in the CTRPv2 dataset and found that the DNA damaging agent cyclophosphamide had the strongest correlation with PRMT5i sensitivity (Figure 1D). We then analyzed all compounds by class and found that chemotherapeutic agents were positively and significantly correlated with the effects of PRMT5i (5th out of 111 classes of compounds, Figure 1D, P=0.009, mHG Test (minimum Hypergeometric Test)). We also found that compounds affecting DNA integrity in a replication-dependent manner, such as PARP1/2 inhibitors, were among the most enriched classes (7th out of 111, Figure 1D). Conversely, microtubule poisons were anti-correlated with the effects of PRMT5i (ranked 2nd out of 111 for negative enrichment). Since microtubule inhibitors usually arrest cells in mitosis, precluding the accumulation of replication-dependent breaks, these data suggest that PRMT5 inhibitors may only show synergy with other drugs in dividing cells. We confirmed that pharmacological inhibition of PRMT5 in human AML cell lines leads to increased DNA damage as measured by γH2AX staining, in the PRMT5i sensitive cell lines but not in the resistant cell lines (Figure 1E and data not shown). Together, these findings indicate that PRMT5 inhibition correlates with DNA damage and suggest that it may affect DNA damage repair mechanisms.
PRMT5 depletion leads to DNA damage accumulation.
Given the possible role that PRMT5 may play in maintaining DNA repair capacity, and consequently genomic stability in hematopoietic cells, we first crossed Prmt5fl/fl mice with Vav1-Cre transgenic mice (Figures S1A-1C). This system triggers PRMT5 deletion just prior to the start of fetal liver (FL) hematopoiesis and we observed that loss of PRMT5 led to severe hematopoietic defects, with an ~2.5-fold decrease in the cellularity of the knockout (KO) FL on day 14.5 of embryonic development (E14.5) (Figure S1D). Ultimately PRMT5 deletion in the FL compartment was embryonic lethal and the embryos were consistently smaller, and severely anemic, compared to their normal counterparts (Figure S1E). Fetal hematopoiesis produces mainly erythropoietic cells, with up to 90% of FL cells expressing the erythrocytic marker Ter119 (Pop et al., 2010). Given the severe anemia observed in the KO animals, we assessed erythroid differentiation by flow cytometry and found a severe block in erythroid development, at the least differentiated stage (S0), with most of the cells expressing neither the CD71 nor Ter119 erythrocytic markers (Figure S1F). Both the relative and absolute number of erythroid progenitor cells were diminished in the PRMT5 KO embryos (Figures S1G and S1H) and unlike what we documented in PRMT5-null adult hematopoiesis (Liu et al., 2015), the block in cell differentiation was accompanied by an abundance of apoptotic cells (Figure S11). Performing cell cycle analysis, we found that more than 50% of the PRMT5 KO fetal hematopoietic progenitor cells (defined as Ter119 negative cells, hereafter Ter119−) were in the SubG1 phase, compared to less than 10% of the Ter119− wild-type (WT) cells, and nearly all of the remaining PRMT5 KO Ter119− cells were in the G1 phase (Figure 2A). We also observed increased caspase 3 cleavage in the PRMT5 KO E14.5 FL cells (Figure 2B), which is consistent with the increased apoptosis.
Figure 2. PRMT5 depletion leads to DNA damage accumulation.
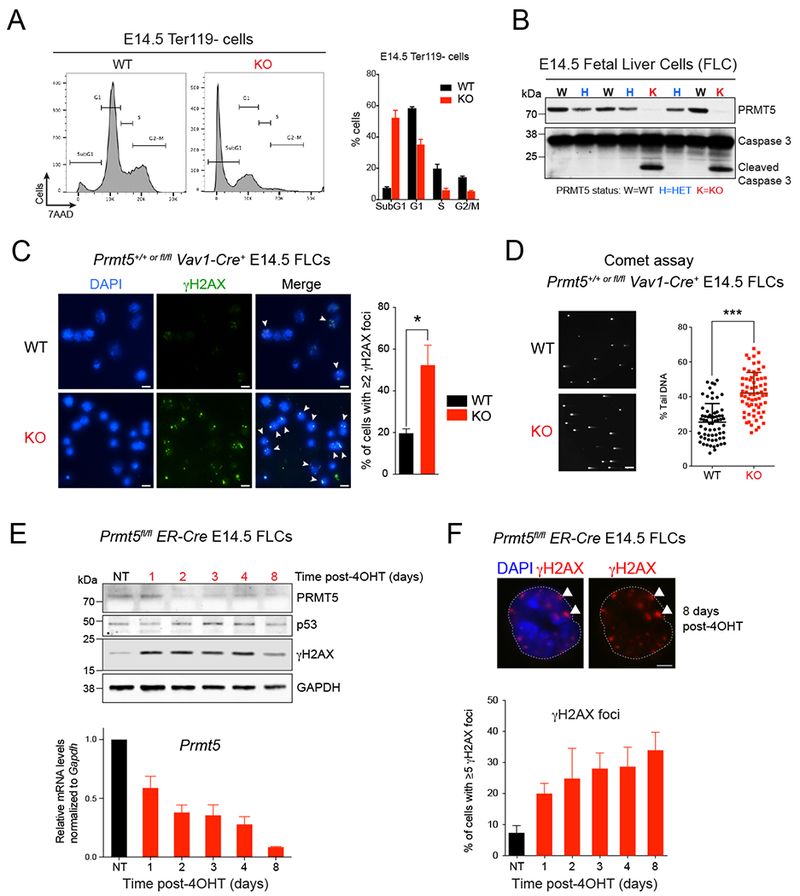
(A) Flow cytometry with fixed E14.5 fetal liver cells (FLCs) showing a substantial increase in subG1 population in KO cells compared to WT. Cells were gated on the Ter119− subset. The percent of cells in subG1, G1, S, and G2/M phases of the cell cycle were quantified and are shown on the right. Error bars indicate the SEM of 3 biological replicates.
(B) Immunoblot with PRMT5 and Caspase 3 antibodies showing increased Caspase 3 cleavage in PRMT5 KO whole FLCs at E14.5.
(C) Immunofluorescence analysis performed with antibody to γH2AX in PRMT5 WT vs KO E14.5 FLCs. Left: representative pictures. Right: quantification of the percent of cells showing γH2AX foci staining. Scale bar, 10μm. White arrows point to γH2AX foci. Error bars indicate the SEM of 3 biological replicates. * P<0.05.
(D) Neutral COMET assay performed in PRMT5 WT vs KO E14.5 FLCs. A representative field showing increased percent tail DNA in KO cells is showed on the left and the quantification for 100 cells is showed on the right. Scale bar, 20 μm. *** P<0.0001
(E) Top: Immunoblot with PRMT5, p53 and γH2AX antibodies in Prmt5fl/fl;ER-Cre E14.5 FLCs treated with 1μM 4-Hydroxytamoxifen (4-OHT) for 8 days. Bottom: Corresponding qRT-PCR analysis showing decreasing levels of Prmt5 upon 4-OHT treatment. Error bars indicate the SEM of 3 biological replicates.
(F) Immunofluorescence analysis performed with antibody against γH2AX in Prmt5fl/fl;ER-Cre E14.5 FLCs treated with 1μM 4-OHT for 8 days. Top: representative pictures. Scale bar, 5 μm. White arrows point to γH2AX foci. Bottom: quantification of the percent of cells with ≥5 γH2AX foci. Error bars indicate the SEM of 3 biological replicates.
Importantly, we found a substantial increase in DNA double strand breaks (DSBs) in the E14.5 PRMT5 KO Lin- FL cells, as measured by both γH2AX staining and the neutral COMET assay (Figures 2C and 2D). To further confirm that PRMT5 deletion triggers the accumulation of DNA lesions in hematopoietic cells, we generated an inducible Prmt5fl/fl;ER-Cre+ mouse model and showed that 4-hydroxytamoxifen (4-OHT) treatment of E14.5 FL cells also significantly increased the number of DSBs, based on γH2AX staining (as assessed by western blot and immunofluorescence assays, Figures 2E and 2F). Taken together, these results show that PRMT5 is needed to maintain genomic stability in normal HSPCs, consistent with previous studies reporting the effects of knocking out PRMT5 in different cell types (Table S1).
PRMT5 depletion leads to cell cycle defects.
To address whether the accumulation of DNA breaks in PRMT5 deficient cells affected the proliferation of normal HSPCs, we further examined the cell cycle status of the fetal hematopoietic cells, by injecting pregnant female mice with BrdU and harvesting fetal liver cells 1h post-injection. Consistent with prior reports (Pop et al., 2010), ~50% of normal E14.5 S0 cells and 70-80% of S1 cells were in S-phase (Figure S1J); in contrast only 20-50% of the PRMT5 KO S0 or S1 cells were in S-phase. This difference was even more striking at E16.5, when the majority of the S0 and S1 KO cells were held up at the G1-phase (Figure S1K). Staining of E14.5 Lin- cells with the proliferation marker Ki67 showed decreased proliferation in the KO cells, consistent with the observed cell cycle arrest (Figure S1L). Thus, similar to what we observed in the adult hematopoietic compartment, loss of PRMT5 impairs FL hematopoietic cell differentiation by blocking cell cycle progression.
Collectively, these data show that PRMT5 loss in the hematopoietic compartment leads to DSB accumulation and a subsequent G1 cell cycle arrest and apoptosis, possibly due to a defect in DNA repair mechanisms.
PRMT5 depletion triggers p53 pathway upregulation and splicing defects in hematopoietic cells.
To better understand the molecular mechanisms driving the accumulation of DNA damage observed in PRMT5-deficient hematopoietic cells, we analyzed the transcriptome of Ter119− E14.5 FL cells by RNA sequencing and identified ~650 differentially expressed genes (≥2-fold change) between the WT and the KO genotypes (Figure 3A). While gene ontology (GO) analysis did not identify erythropoietic cells differentiation as a PRMT5 regulated process (Figure 3A), GO terms associated with cell cycle progression, DNA damage or DNA repair pathways were significantly enriched in PRMT5 KO gene signatures (Figure 3A). As fetal hematopoietic progenitors cells repeatedly enter the cell cycle as they differentiate, it was proposed that mutual inhibition between specific transcription factors and S-phase progression constitute a “synchromesh” mechanism that makes the differentiation of hematopoietic progenitors indissociable from cell cycle progression (Pop et al., 2010). Our data are consistent with these findings and indicate that the block in differentiation we observed in PRMT5 KO fetal hematopoietic cells may be due to cell cycle arrest induced by DNA damage accumulation.
Figure 3. PRMT5 depletion triggers the aberrant splicing of epigenetic factors.
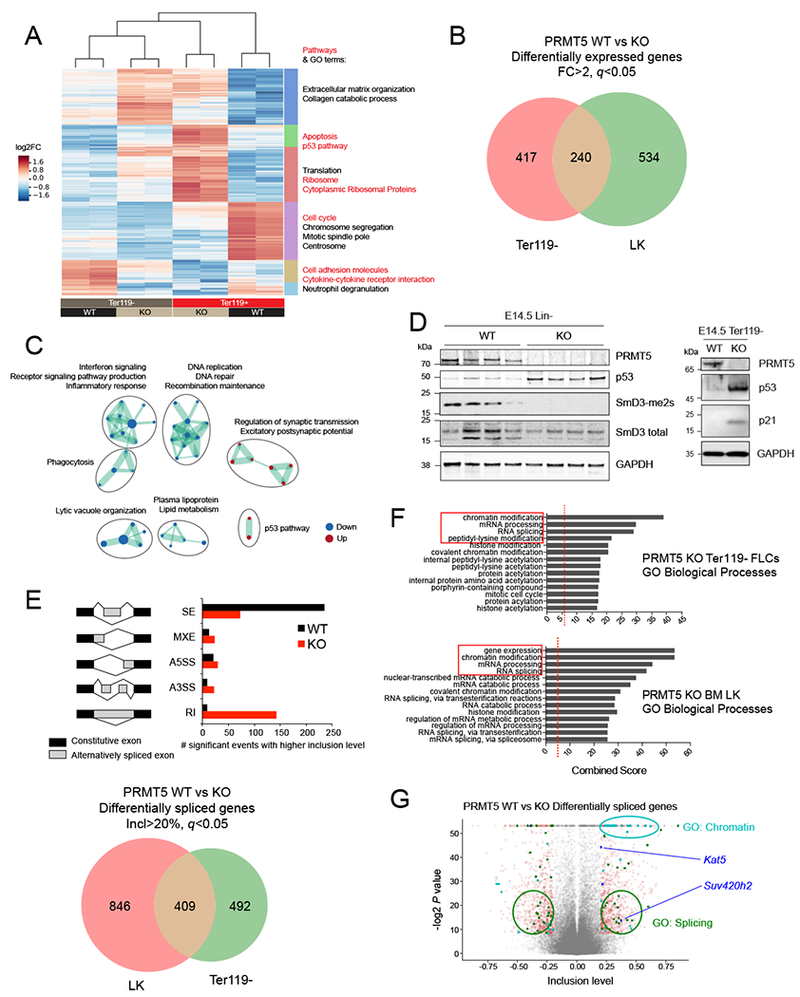
(A) Heat map showing the expression of differentially expressed protein-coding genes in PRMT5 WT (n=2) or KO (n=2) Ter119− or Ter119+ E14.5 FLCs (log2 fold change). On the right are shown the top significant pathways (red) and GO terms (black) determined for 6 clusters defined by differential expression (DE) between WT vs KO and/or Ter119− vs Ter119+ cells.
(B) Venn diagram showing the overlap of differentially expressed genes between adult (LK, lineage-, Kit+, Sea1- cells) and fetal (Ter119− cells) hematopoietic progenitors’ populations. The cutoffs were fold change (FC) >2 and FDR (q value) <0.05.
(C) Gene ontology analysis of overlapping DE genes detected upon PRMT5 depletion in Ter119− and LK cells.
(D) Left: Immunoblot with PRMT5, p53, SmD3-me2s, SmD3 total and GAPDH antibodies showing increased p53 and decreased SmD3 dimethylation in PRMT5 KO lin− E14.5 FLCs. Right: Immunoblot with PRMT5, p53, p21 and GAPDH antibodies showing increased p53 and p21 in PRMT5 KO lin− E14.5 FLCs.
(E) Top: schematic representation of the events detected and number of significant events with higher inclusion levels (inclusion level > 20%) in Ter119− E14.5 FLCs. The MATS algorithm distinguishes the following events: Skipped Exons (SE), Mutually exclusive exons (MXE), Alternative 5’ Splice site selection (A5SS), Alternative 3’ Splice site selection (A3SS), and Retained Intron (RI). The output of the analysis consists of the degree of confidence (FDR and p value) of the differences in ratios between 2 different isoforms. Bottom: Venn diagram showing the overlap of differentially spliced genes between adult (LK cells) and fetal (Ter119− cells) hematopoietic progenitors’ populations.
(F) Gene ontology analysis of all genes whose splicing is affected by PRMT5 loss in LK cells and Ter119− E14.5 FLCs (>20% inclusion level cutoff).
(G) Volcano plot representing all the splicing events detected by MATS. The significant events (p value < 0.05) with an inclusion level >0.2 or <−0.2 are shown in red. Among these, the ones affecting genes that fall in the GO categories Splicing and Chromatin are labeled in green and cyan, respectively. The chromatin modifiers involved in DNA repair are shown in blue (Kat5/Tip60 and Suv420h2).
We have combined our adult (Prmt5fl/fl;Mx1-Cre+ 7 days after poly(I:C) treatment, Lin-/Kit+ (LK) cells) and fetal hematopoietic progenitor cells (Prmt5fl/fl;Vav1-Cre+ E14.5, Ter119− cells) datasets to generate a comprehensive picture of the PRMT5 gene regulatory network in hematopoietic cells. We found ~900 genes to be significantly differentially regulated upon PRMT5 loss, with an overlap of 240 common genes between these cell types (Figure 3B). GO analyses confirmed the downregulation of gene signatures involved in DNA replication, and DNA repair, while the p53 pathway was significantly upregulated (Figures 3C and S2A). p53 was also upregulated at the protein level in both the E14.5 KO Lin- and Ter119− cells, as assessed by western blot analysis (Figure 3D). We also confirmed the upregulation of several p53 direct target genes in the KO cells using qRT-PCR, including Cdkn1a (p21), Bbc3 (Puma), Pmaip1 (Noxa), Mdm2 and Serpine1 (Figures S2B-D).
PRMT5 plays a critical role in RNA splicing by complexing with and methylating the proteins SmB, SmD1 and SmD3, three components of the so-called ‘methylosome’ (Friesen et al., 2001). PRMT5-mediated symmetric dimethylation of these three proteins facilitates the formation of the SMN complex, which is required for maturation of spliceosome-related, small nuclear RNAs (Matera and Wang, 2014). First, we confirmed that PRMT5 loss impairs global Sm protein dimethylation in HSPCs (Figure 3D). We then used an unbiased approach, performing a multivariate analysis of transcript splicing (MATS) (Shen et al., 2014), to determine how PRMT5 loss affects RNA splicing in hematopoietic progenitor cells. We identified 579 alternative splicing events (corresponding to 470 unique genes) that were differentially affected by PRMT5 deletion, the majority being retained introns (RI) and skipped exons (SE) (Figure 3E). We also confirmed the aberrant splicing of a number of genes by visualization using Sashimi plots (Figure S2E). For example, the alternative splicing of the p53 negative regulator Mdm4 in the absence of PRMT5 has been reported (Table S1, Allende-Vega et al., 2013; Bezzi et al., 2013) and we confirmed this finding in PRMT5 KO hematopoietic cells, detecting primarily the isoform of Mdm4 that lacks Exon 6 (Figures S2F, S2G, and S2H). We also found aberrant splicing of Mdm2, another major p53 negative regulator (Figure S2I), leading to an mRNA that would encode a protein lacking most of the p53 binding domain (AA 18-50); this too can contribute to the observed p53 upregulation.
Data from our ER-Cre driven PRMT5 KO mouse model suggested that the accumulation of DNA damage observed after PRMT5 knockdown precedes p53 upregulation (Figure 2E). Thus, aberrant splicing of the p53 negative regulators Mdm2 and Mdm4 may not be the primary event driving p53 activation. Indeed, we generated Prmt5fl/fl;p53−/−;Vav1-Cre+ compound mice and found that p53 KO only partially rescued PRMT5 ablation-driven phenotypes. Notably, the PRMT5 and p53 double KO embryos showed only a 15% increase in Ter119+ FL cells compared to their PRMT5 KO counterparts (from ~50% to ~65%) compared to WT animals where >90% of the cell population are Ter119+ (Figure S2J). Ultimately, no PRMT5/p53 double KO pups were born, again suggesting that PRMT5 depletion induces defects upstream of p53 induction, but also critical defects in p53-independent pathways as well.
PRMT5 loss leads to aberrant splicing of key DNA repair regulators.
Gene ontology analysis of the aberrant splicing events revealed a significant enrichment of genes involved in chromatin and histone modification, and in RNA processing, in both fetal and adult hematopoietic progenitors (Figures 3F and 3G). A number of chromatin modifiers were found to be aberrantly spliced after PRMT5 loss, including the lysine methyltransferases KMT6/EZH2, and KMT1E/SETDB1 (Table S3). Interestingly, we found that several chromatin modifier genes relevant for DSB repair (Sulli et al., 2012) were aberrantly spliced after PRMT5 loss, including the KMT5C/SUV4-20H2 lysine methyltransferase and the TIP60/KAT5 histone acetyltransferase (HAT) (Figures 3G, S3A and S3B). TIP60/KAT5 primarily catalyzes histone H4 acetylation (Bird et al., 2002), while KMT5C/SUV4-20H2 catalyzes histone H4K20 trimethylation. Both enzymes have multiple other substrates but these marks play critical roles in the DNA damage response (DDR) and in transcriptional regulation (Botuyan et al., 2006; Doyon and Côté, 2004; Squatrito et al., 2006; Sulli et al., 2012) (Figure S3B). We confirmed the aberrant splicing of both SUV4-20H2 (Figures S4A-S4C) and TIP60/KAT5 in the PRMT5 KO cells, by semi-quantitative PCR or qRT-PCR (Figures 4A, 4B and 4C) and western blot analysis (Figure 4D). The Kat5 gene generates two alternative transcripts (full-length TIP60 aka TIP60α, and TIP60ΔEx5 aka TIP60β), which vary based on the inclusion or exclusion of Exon 5 in the final mRNA (McAllister et al., 2002; Ran and Pereira-Smith, 2000). In PRMT5 KO cells, TIP60α expression is significantly decreased, both at the RNA and protein level (Figure 4B, 4C and 4D). There is no significant change in TIP60β expression, leading to an enrichment of the shorter isoform at the expense of the longer one. We also saw decreased levels of TIP60 in the PRMT5 KO and heterozygous (HET) mouse HSPCs. We also treated murine erythroleukemia (MEL) cells with the PRMT5 inhibitor and found an identical effect on TIP60/KAT5 splicing (Figure S4D). Thus, these data suggest that this splicing event is due to impaired PRMT5 activity and that loss of PRMT5 activity triggers similar splicing defects in different hematopoietic cell types.
Figure 4. Aberrant splicing of TIP60 affects its activity and DNA repair pathway choice.
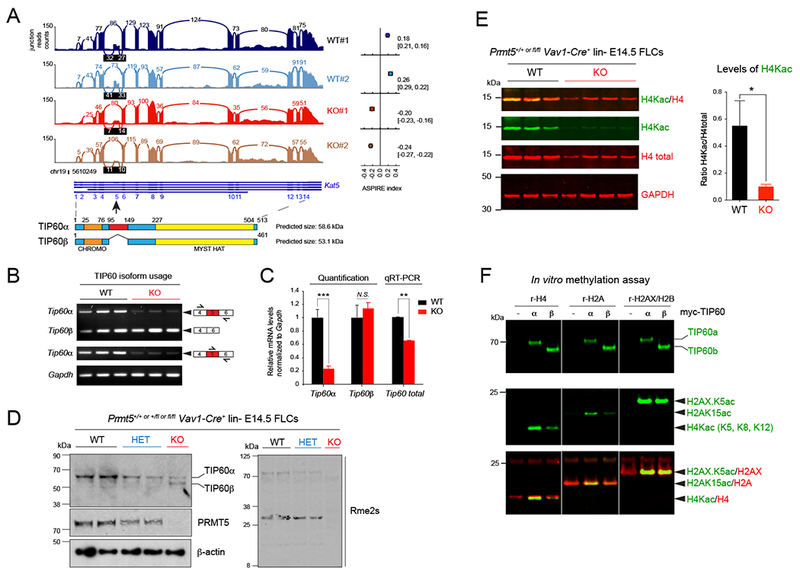
(A) Sashimi plot showing the decreased read counts between exons 4 and 5 and between exons 5 and 6 of the gene Kat5 coding for the protein TIP60. The ASPIRE index on the right indicates a decreased usage of exon 5 in PRMT5 KO Ter119− FLCs compared to WT cells and was calculated with AltAnalyze. Below the Sashimi plot, the structures of Kat5 and of TIP60 are shown.
(B) Semi-quantitative PCR of the indicated transcripts in E14.5 Ter119− PRMT5WT vs KO FLCs.
(C) Quantification of the relative abundance of Kat5/Tip60 isoforms shown in (B) and total mRNA in E14.5 Ter119− PRMT5 WT vs KO FLCs by real-time PCR using a Taqman probe that recognizes both TIP60 isoforms, relative to Gapdh. Error bars are SEM of at least 3 biological replicates. *** P<0.0001, ** P<0.01, N.S. non-significant.
(D) Immunoblot showing aberrant splicing of TIP60 in E14.5 lin− PRMT5 +/+ (WT), +/Δ (HET), and Δ/Δ (KO) FLCs, and decreasing levels of arginine methylation after PRMT5 loss.
(E) Multiplex immunoblots showing decreased H4 acetylation in PRMT5 KO E14.5 Ter119− FLCs. Quantification of H4Kac relative to H4 (n=5 biological replicates) is shown on the right. Error bars are SEM. * P<0.05.
(F) In vitro acetylation experiment and multiplex immunoblots with TIP60, H2AXK5ac, H2AK15ac, H4K5/8/12ac, and total H2A & H4 antibodies. Myc-tagged TIP60 proteins (full length or α vs shorter isoform or β) were immunoprecipitated from transiently transfected 293T cells, and incubated with recombinant histones H4, H2A or the H2AX/H2B dimer.
Aberrant splicing of TIP60 affects its acetyltransferase activity.
To identify the functional consequences of the aberrant splicing of TIP60/KAT5, and KMT5C/SUV4-20H2, on their enzymatic activity, we first assessed global histone acetylation levels in hematopoietic progenitors by western blot, comparing WT vs KO fetal liver or adult bone marrow cells. We found a striking reduction in histone H4 acetylation in the PRMT5 KO cells (Figure 4E, S5A and S5B) as well as a significant decrease in H4K20 tri-methylation, but not di-methylation (Figure S4E). To discern whether the two TIP60 isoforms have different histone acetyltransferase activities, we performed in vitro acetylation assays using myc-tagged TIP60α or TIP60β protein, purified from transfected 293T cells, and recombinant histones. While histone H2AX was acetylated equally by both TIP60 isoforms, histones H4 and H2A were acetylated two-fold more by TIP60α than TIP60β (Figure 4F), consistent with the decreased histone H4 acetylation observed in PRMT5 KO HSPCs, which primarily express TIP60β. TIP60 auto-acetylation has been shown to enhance its activity (Wang and Chen, 2010; Yang et al., 2012; Yi et al., 2014), and most auto-acetylation sites are in the domain that is absent in TIP60γ. As predicted, the level of TIP60β auto-acetylation was also much lower than the level of TIP60α autoacetylation in 293T cells engineered to express either isoform (Figure S4F). We also observed a significant decrease in H4 acetylation after PRMT5 deletion in Prmt5fl/fl;ER-Cre mouse embryonic fibroblasts (MEFs) (Figure S5C), and after exposing normal human CB cells to a PRMT5 inhibitor for 96 hours (Figure S5D). Collectively, these data indicate that PRMT5 regulates TIP60 activity at least in part through alternative splicing of TIP60/Kat5 mRNA, and that PRMT5 directly affects histone acetylation levels in multiple cell types.
PRMT5 loss leads to defective DNA repair in hematopoietic cells.
Given the increased number of DSBs after PRMT5 depletion, we examined both the NHEJ and HR DNA repair pathways, examining 53BP1 deposition at DSB sites as an indicator of NHEJ activity, and RAD51 localization to DNA, as an indicator of HR activity. Six hours after inducing DNA damage by exposure to ionizing radiation (IR), we assessed the number of IR-induced foci (IRIF) for γH2AX, 53BP1 and RAD51, as well as the number of apoptotic cells in sorted E14.5 LK and LSK cells from PRMT5 WT and KO mice. While the number of γH2AX foci was similar between genotypes, the number of 53BP1 foci was consistently higher in the E14.5 PRMT5 KO cells (Figure 5A and S6A). In contrast, the number of RAD51 IRIF was consistently lower in PRMT5 KO cells (Figure 5A and S6A). Consistent with our cell cycle profiling and caspase 3 western blot data (Figure 2A and 2B), we observed increased apoptotic cell numbers in PRMT5 KO LK cells (Figure S6B). Similar results were observed using IR-exposed Prmt5fl/fl;ER-Cre MEFs that were treated with 4-OHT for 5 days (Figure 5B). Importantly, we observed no difference in the cell cycle phase distribution after exposure to IR, as both the PRMT5 deficient and the control cells showed a similar G2/M phase arrest (Figure 5C). Together, these data suggest that PRMT5 deletion impairs HR-dependent DNA repair in hematopoietic cells, consistent with a recent report (Clarke et al., 2017). This defect likely explains the observed accumulation of DSBs in PRMT5-deficient cells, given the essential role of HR in repairing DSBs during replication.
Figure 5. PRMT5 loss leads to DNA repair defects in vivo.
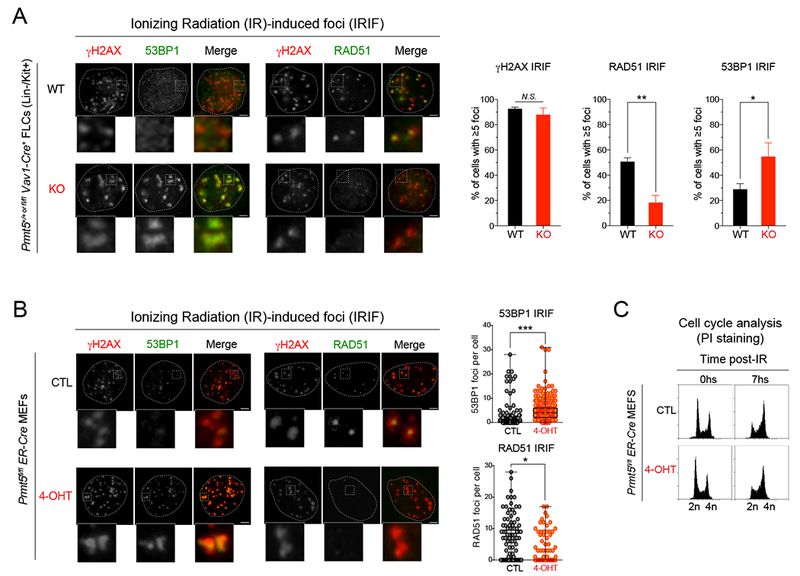
(A) Immunofluorescence analysis performed with antibodies to γH2AX, 53BP1 and RAD51 in sorted HSPCs (LK: lineage negative, Kit+) from PRMT5 WT vs KO E14.5 FLCs, 6h after γIR-induced DNA damage (10Gy). Left: representative pictures of WT vs. PRMT5 KO LK cells stained for RAD51, γH2AX and 53BP1 after IR. Right: Quantification of percent of cells with ≥5 γIR-induced foci for the indicated proteins. A minimum of 100 cells were quantified for each condition. Scale bar, 5 μm. ** P<0.001, *P<0.05, N.S. non-significant.
(B) Immunofluorescence analysis performed with antibodies against γH2AX, RAD51 and 53BP1 in Prmt5fl/fl;ER-Cre MEFs −/+ 4-OHT 1μM for 72h, 6h after γIR-induced DNA damage (4Gy). Left: representative pictures of cells stained for RAD51, γH2AX and 53BP1 before (CTL) and after (4-OHT) PRMT5 knockdown. Right: Quantification of percent of cells with ≥5 γIR-induced RAD51 and 53BP1 foci before and after PRMT5 knockdown. Scale bar, 2 γm. *** P<0.0001, *P<0.05.
(C) Flow cytometry analysis using Prmt5fl/fl;ER-Cre MEFs −/+ 4-OHT 1γM for 72h, 6h after γIR-induced DNA damage (4Gy), and quantification of cell cycle phases by DNA content (PI staining).
See also Figure S6.
PRMT5 effects on DNA repair are dependent on TIP60.
To determine the role of TIP60 isoforms in HSPCs lacking PRMT5, we conducted rescue experiments by transducing Prmt5fl/fl;ER-Cre E14.5 FL cells with lentiviruses expressing PRMT5, TIP60α or TIP60β (as described in Figures 6A and 6B), and performed colony formation assays (Figure 6C). The expression of WT PRMT5, but not PRMT5 E444Q (the enzymatically dead version of PRMT5) (Antonysamy et al., 2012; Ho et al., 2013; Liu et al., 2015) partially rescued the colony-forming potential of PRMT5 KO FL cells. However, expression of neither TIP60α or TIP60β was able to rescue the ability of PRMT5 KO progenitor cells to form colonies. This confirms that PRMT5 loss leads to multiple defects in essential pathways, and rescue of its functions on both cell differentiation and cell growth, will not be attainable by overexpressing (OE) a single PRMT5 downstream target.
Figure 6. PRMT5 effects on DNA repair are dependent on TIP60/KAT5-mediated histone acetylation.
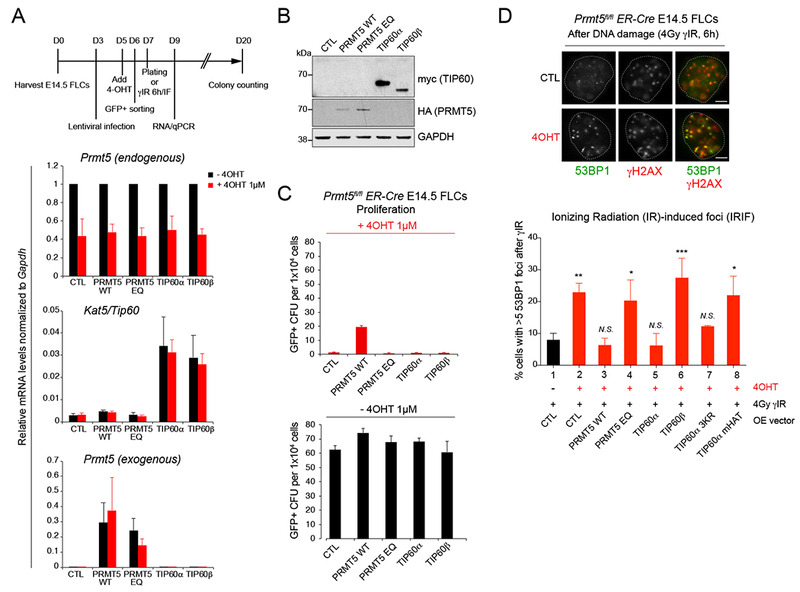
(A) Top: schematic illustration of experimental design. Bottom: qRT-PCR analysis showing relative mRNA levels of Prmt5 (endogenous and exogenous), and Kat5/TIP60 in Prmt5fl/fl ;ER-Cre E14.5 FLCs −/+ 4-OHT 1μM for 72h retrovirally infected with the corresponding overexpressing GFP+ lentiviral vectors. PRMT5 EQ: enzymatically dead mutant with a E444Q point mutation within PRMT5 methyltransferase enzymatic domain. Error bars are SEM of at least 3 biological replicates.
(B) Immunoblot with myc, HA and GAPDH antibodies in Prmt5fl/fl;ER-Cre E14.5 FLCs overexpressing HA-tagged PRMT5 or myc-tagged TIP60 before 4-OHT treatment.
(C) Quantification of GFP+ colony forming units before (bottom) and after (top) 4-OHT treatment with Prmt5fl/fl;ER-Cre E14.5 FLCs overexpressing the indicating proteins. Error bars are SEM of 3 biological replicates.
(D) Immunofluorescence analysis performed with antibodies against γH2AX and 53BP1 in Prmt5fl/fl;ER-Cre E14.5 FLCs, before (lane 1) and after (lanes 2 to 8) 4-OHT treatment (1μM for 72h), overexpressing HA-tagged PRMT5 WT (lane 3), or enzymatically dead mutant EQ (lane 4), or myc-tagged TIP60α (lane 5), TIP60β (lane 6), TIP60α 3KR (lane 7), and TIP60α enzymatically dead mutant (TIP60 mHAT, lane 8), and treated with 10Gy of gamma ionizing radiation (γIR) for 6h. Top: representative picture of 53BP1 and γH2AX foci in Prmt5fl/fl;ER-Cre E14.5 FLCs before and after treatment with 1 μM 4-OHT, 6h after γIR-induced DNA damage (4Gy). Scale bar, 5 μm. Bottom: Quantification of percent of cells with ≥5 γIR-induced 53BP1 foci from three biological replicates. *P < 0.05, **P < 0.01, ***P < 0.001, N.S., non-significant (one-way ANOVA Tukey’s multiple comparisons test, Adjusted P values).
See also Figure S6.
We also examined whether OE of TIP60α or TIP60β could rescue the DNA repair phenotype seen in PRMT5 KO cells. OE of TIP60α, but not TIP60β, reduced the number of IR-induced 53BP1 foci similar to what occured following OE of WT PRMT5 (Figure 6D). The increased number of 53BP1 foci seen in PRMT5 KO cells (see Figure 5), could reflect the decreased activity of TIP60β on histone H4 and H2A acetylation compared to TIP60α (see Figure 4), since these acetylation marks can decrease the recruitment (or retention) of 53BP1 at chromatin (Jacquet et al., 2016; Panier and Boulton, 2013; Tang et al., 2013).
To confirm that the enzymatic activity of TIP60 was essential for the DNA damage pathway rescue, we generated two mutants of TIP60α (TIP60 3KR, in which 3 lysines subject to autoacetylation and located within the domain absent in TIP60β were mutated to arginines, and TIP60 mHAT, an enzymatically-dead version of TIP60 with two point-mutations in the MYST-HAT domain, Q377E/G380E). We found that overexpressing TIP60 mHAT could not overcome the accumulation of 53BP1 foci triggered by PRMT5 knockdown, confirming the importance of TIP60 enzymatic activity in the early steps of DSB repair pathway (Figure 6D). However, TIP60α 3KR overexpression was almost as efficient as TIP60α at decreasing PRMT5 loss-dependent 53BP1 foci accumulation (Figure 6D), suggesting that TIP60 autoacetylation might not be a critical regulator of the effects of TIP60α on 53BP1 foci. Alternatively, mutation of the three lysines contained in the domain coded by exon 5 might not affect the autoacetylation levels of TIP60 sufficiently to detect a difference with the WT protein. In fact, a previous report suggested that the 3KR mutant autoacetylation levels were only ~50-60% less than the WT protein in certain cells (Yi et al., 2014). The TIP60β protein lacks a proline-rich region contained in TIP60α, which has been suggested to play a role in protein–protein interactions (Ran and Pereira-Smith, 2000). One possibility is that the TIP60 isoforms may have a differential interaction with RUVBL1, which appears to play an integral role in regulating DNA repair pathways downstream of PRMT5 in epithelial cells (Clarke et al., 2017). To assess this possibility, we looked for a detectable interaction between TIP60 and RUVBL1 but could not find one (data not shown). We also examined whether RUVBL1 regulated TIP60 function in hematopoietic cells; we knocked down RUVBL1 in IR-exposed Prmt5fl/fl;ER-Cre FLCs and examined histone H4 acetylation and the number of 53BP1 foci, before or after PRMT5 depletion (Figure S6C and S6D). We saw no effect of RUVBL1 KD on the level of histone H4 acetylation or the number of 53BP1 foci in wild type or PRMT5-null hematopoietic cells (Figure S6C and S6D), suggesting that RUVBL1 might not be a prominent player in DNA repair in hematopoietic cells. In addition, we could not detect the PRMT5- dependent methylation of RUVBL1 in fetal liver cells, indicating that this mechanism might be cell type specific (data not shown). Together, these data demonstrate a key role of PRMT5-driven TIP60 alternative splicing in regulating TIP60 enzymatic activity in hematopoietic cells, perhaps by mechanisms including altered protein-protein interactions with the TIP60 complex.
Collectively, these data support a model whereby PRMT5 loss leads to aberrant splicing of key factors involved in chromatin modifications and DNA repair, including TIP60/KAT5, which in turn leads to defective chromatin landscape at DNA damage sites, the accumulation of 53BP1 at DSBs, and blockage of the HR DNA repair pathway. This triggers accumulation of DNA damage and enhanced genomic instability, ultimately leading to p53 upregulation and either cell cycle arrest or cell death.
PRMT5 inhibition and PARP inhibition have synergistic effects on leukemia cells.
The observed defects in HR that follow PRMT5 depletion suggest that PRMT5 inhibitors and PARP inhibitors might have synergistic cytotoxic effects on AML cells. To address this possibility, we treated several AML cell lines with the PARP inhibitor Olaparib (PARPi thereafter), and the PRMT5 inhibitor GSK3186000A, alone or in combination.
In all cell lines, GSK3186000A reduced global arginine symmetric demethylation levels in a time and concentration dependent manner, with full reduction requiring at least three days of treatment (Figure 7A). Except for MV4-11 cells, the cell lines tested exhibited a low sensitivity to PARPi, with an IC50 in the micromolar range (Figure S7A), an order of magnitude higher than what is reported for other cancer cells in vitro, such as a BRCA1/2-deficient breast cancer cell line (Farmer et al., 2005). We examined cell viability after four days of treatment, and used two different methods to assess possible synergistic effects between PRMT5i and PARPi: the Bliss synergy method (He et al., 2018; Iniguez et al., 2018) (Figure 7B), and the Chou-Talalay method (Chou, 2010) (Figure S7B). We observed positive Bliss scores for all cell lines tested (Figure 7B), indicating a synergistic effect. Combination Indexes (CI) calculated by the Chou-Talalay method were less than 1 for a wide range of concentrations in all four cell lines tested (Figure S7B), also demonstrating the synergy. The synergism was evident with SKNO-1 cells, which are somewhat resistant to PRMT5 inhibition, and with THP-1 cells, which are the most resistant cell line to PRMT5i (Figure 7B and S7B).
Figure 7. PRMT5 and PARP inhibitors synergistically target leukemia cells.
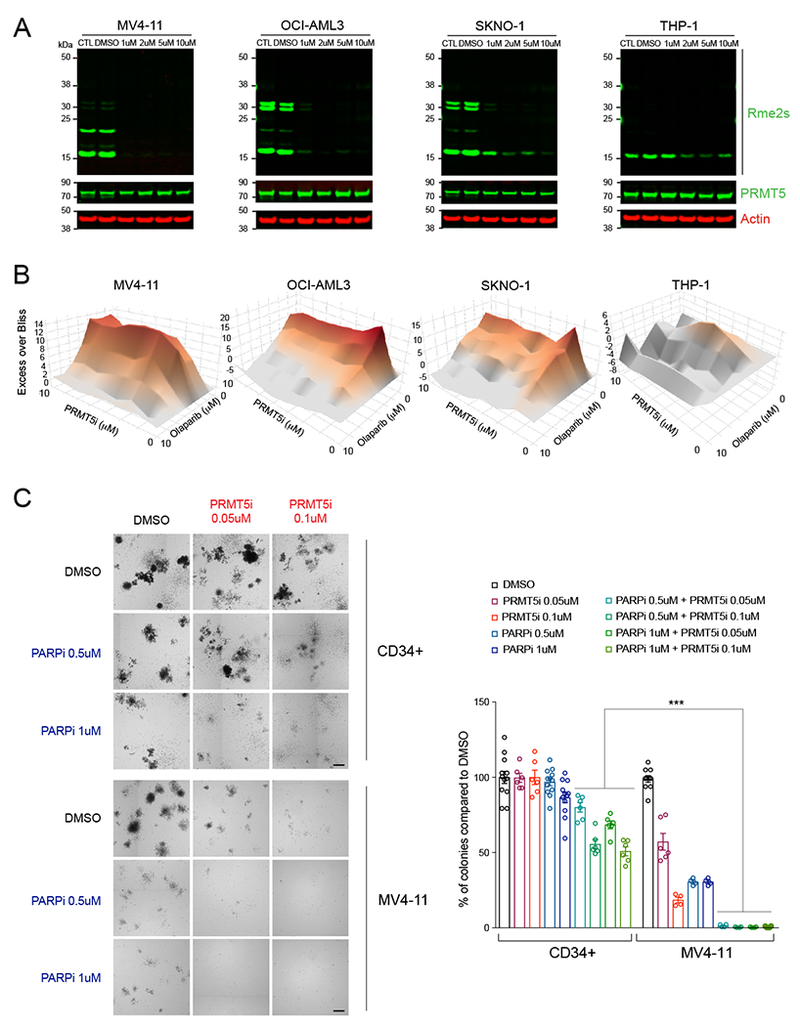
(A) Multiplex immunoblots with Rme2s, PRMT5, and GAPDH in the indicated human leukemia cell lines before and after treatment with increasing doses of GSK3186000A (PRMT5i) for 4 days, showing decreasing levels of arginine methylation.
(B) Excess Over Bliss plots (Bliss method) showing synergistic effects between PRMT5i and the PARP inhibitor Olaparib (PARPi). Cells were treated with increasing amounts of PRMT5i, PARPi, or both combined at 1:1 ratio and cell viability was assessed with the CellTiter Glo method after 4 days of treatment.
(C) Colony forming units after treatment of normal cord blood derived CD34+ cells and the AML cell line MV4-11 with DMSO (control), PRMT5i, PARPi or a combination of PRMT5i and PARPi at the indicated concentrations. Left: representative pictures of colonies on semi-solid methylcellulose media. Scale bar, 1 mm. Right: Quantification of the number of colonies as percent of colonies compared to DMSO, 7 days after plating. *** P<0.0001 (one-way ANOVA Tukey’s multiple comparisons test, Adjusted P values).
See also Figure S7.
Knowing that PRMT5 inhibition might induce genomic instability in normal cells as well, we examined the selectivity of this combinatorial approach. We performed colony formation assays using AML cell lines and normal human CB-derived CD34+ cells, treated with PRMT5i and PARPi, alone or in combination. Consistent with the cell viability assays, AML cells treated with both compounds did not form colonies in methylcellulose at drug concentrations less than the IC50 of each individual compound (Figure 7C and S7C). In contrast, at similar concentrations, CD34+ cells were still capable of forming colonies, albeit in decreased numbers and of smaller size at higher concentrations (Figure 7C and S7C). These data suggest that combining PRMT5i and PARPi has the potential to sensitize PARPi-resistant AML cells while sparing normal hematopoietic cells, possibly representing an effective therapeutic strategy for treating AML.
DISCUSSION
We have uncovered a mechanism by which PRMT5 controls the DNA repair pathway choice in fetal and adult hematopoiesis, leading to a block in differentiation and increased cell death, particularly affecting erythroid progenitors. Several other reports, using conditional knockout mouse models, also indicate that acute loss of PRMT5 increases DNA damage and cell death in vivo (Table S1). Here, we demonstrate further that PRMT5 mediates its effects on DNA repair by controlling the RNA splicing of several epigenetic regulators, especially the histone H4 acetyltransferase TIP60 (KAT5) and possibly the histone H4 methyltransferase SUV4-20H2 (KMT5C). The ensuing functional dysregulation of these proteins leads to global chromatin defects and genomic instability, ultimately engaging p53-dependent apoptosis and other pathways that trigger cell death.
PRMT5 depletion has been reported to trigger aberrant splicing in several cell types (Bezzi et al., 2013; Koh et al., 2015; Liu et al., 2015), including hematopoietic cells. Here we confirm that a significant number of genes are aberrantly spliced following PRMT5 loss, including the ones encoding the epigenetic enzymes TIP60 and SUV4-20H2.
TIP60 has been shown to acetylate several proteins involved in the cascade of events following the detection of a DSB site and leading to DNA repair, including the DNA damage sensing kinase ATM and histones H2A, H2AX and H4 (Figure S3B). Here we show that the absence of PRMT5 selectively affects TIP60 activity via its alternative spicing: while global H4 acetylation and H2AK15 acetylation were significantly decreased, γ-H2AX and H2AXK5 acetylation levels were unchanged. The 54 amino acid domain that is missing in TIP60P may be important for protein-protein interactions within the TIP60 complex, which would confer its substrate specificity, but be dispensable for its ability to acetylate ATM and H2AX. More structural studies are warranted to explore this possibility.
PRMT5 was recently reported as a critical regulator of TIP60 activity and the HR DNA repair pathway through the methylation of its cofactor RUVBL1 in epithelial cells (Clarke et al., 2017). Our RUVBL1 knockdown data in fetal liver cells suggest that this protein is not essential in mediating PRMT5’s role in DNA repair in hematopoietic cells. Moreover, we could not detect an interaction between TIP60 and RUVBL1 in hematopoietic cells (data not shown). Perhaps the deleted region in TIP60β interacts with components on the DNA repair machinery, which would explain the observed defects. Nevertheless, these findings and the present report indicate that PRMT5 can regulate TIP60 activity through multiple mechanisms in a cell-specific manner, ultimately controlling critical steps of the DNA repair pathway.
We show that PRMT5 also regulates the splicing of another epigenetic factor involved in the early steps of DNA damage recognition, the lysine methyltransferase SUV4-20H2/KMT5C, a member of the family of histone methyltransferases that specifically target histone H4K20 (Figure S3A and S3B). While SET8/KMT5A and SUV4-20H1/KMT5B primarily monomethylate and dimethylate H4K20, respectively, SUV4-20H2/KMT5C primarily trimethylates this lysine residue (Schotta et al., 2008). H4K20 dimethylation promotes the recruitment of 53BP1 to DSBs, through direct binding of 53BP1 to this mark (Botuyan et al., 2006; Fradet-Turcotte et al., 2013). Of note, H4K20me2 is the primary target of the 53BP1 Tudor domains, with H4K20me3 showing a very low ability to bind 53BP1, at least in vitro (Botuyan et al., 2006). Consistently, in the absence of PRMT5, we observed a decrease in H4K20me3 global levels and an increased retention of 53BP1 at the DSBs. Moreover, 53BP1 eviction is mediated in part by the TIP60 complex through binding of its MBTD1 subunit to H4K20me1/2, and by TIP60-dependent H2AK15 acetylation, which blocks H2AK15 ubiquitination (Jacquet et al., 2016). Here we observed that PRMT5 loss leads to decreased H4 and H2A acetylation and decreased H4K20me3, all of which favor 53BP1 retention at DSBs, thus impairing HR-mediated DNA repair.
Finally, we show that the genomic instability triggered by PRMT5 inactivation can be exploited therapeutically by combining a specific PRMT5 inhibitor and the FDA-approved PARP inhibitor Olaparib. Despite recent advances in our understanding of AML pathogenesis and the molecular events underlying leukemogenesis, the first line of treatment against this disease has barely changed over the last 50 years and still relies heavily on highly toxic chemotherapeutic approaches. AML remains one of the most difficult cancers to treat, and most AML patients have yet to benefit from more tolerated targeted therapies (Zeisig et al., 2012). Recently however, PARP inhibitors were shown to effectively target AML cells driven by certain oncogenic transcription factors, including AML1-ETO and PML-RARa, whereas MLL-fusion-driven AML were generally insensitive to PARP inhibition (Esposito et al., 2015; Wang et al., 2015). Here, we found that PRMT5 inhibition sensitizes human AML cell lines to PARP inhibition regardless of their genotype, similar to what is observed in HR-deficient models (e.g. BRCA1 deficient cells). Indeed, our data suggest that PRMT5 activity promotes efficient DNA repair in hematopoietic cells as we show that genetic ablation, or pharmacological inhibition, of this crucial enzyme impairs the homologous recombination DNA repair pathway, rendering AML cells sensitive to PARP inhibitors. This is important considering not only the potential toxicities that strong suppression of PRMT5 activity might have on the hematopoietic compartment, but also the potential testing of additional combination therapies. Combining PRMT5 inhibitors and PARP inhibitors might widen the therapeutic window, while reducing PRMT5 inhibitor-associated side effects. Future studies will define the efficacy of such combined treatments in various animal models of AML.
EXPERIMENTAL PROCEDURES
PRMT5 KO mice generation
The Prmt5 conditional knockout mice were obtained from the European Conditional Mouse Mutagenesis Program (EUCOMM, EMMA ID: 05300) and previously described (Liu et al., 2015; White et al., 2013). After crossing the mice with a Flippase transgenic mouse line to remove the LacZ-Neomycin cassette, Prmt5fl/fl mice were crossed with Vav1-Cre recombinase transgenic mice (Jackson Labs, stock #: 018968) to delete exon 7 in the Prmt5 gene and generate Prmt5 KO mice. Prmt5fl/fl mice were crossed with Cre-ERT2 recombinase transgenic mice (Jackson Labs, stock #: 008085) to generate tamoxifen-inducible Prmt5 KO mice. All studies were conducted in accordance with the Institutional Animal Care and Use Committee (IACUC) at the University of Miami.
Immunofluorescence
After two washes with 1× PBS, cells were fixed with 4% formaldehyde in 1X PBS during 10 minutes at RT, washed again twice with 1X PBS, bound to poly-L-lysine coated slides and fixed 5 minutes with 2% formaldehyde. After a 5 min wash with 1X PBS, cells were incubated with Blocking Solution (1 mg/ml BSA, 3% goat serum, 0.5% Triton X-100 in PBS) for 30 min at RT. Next, the cells were incubated with the primary antibodies in Blocking Solution for 1hr at RT, washed three times with 1X PBS, and incubated with Alexa 488 or 594 coupled secondary antibodies (ThermoFisher Scientific) for another 30 min. Finally, cells were incubated with 0.5ug/mL 4’,6-diamino-2-phenylindole (DAPI) (Sigma) in 1X PBS, washed with 1X PBS for 2 min, air dried, and mounted in SlowFade Gold antifade mounting reagent (ThermoFisher Scientific). Samples were analyzed using a Leica DMI6000B microscope with LASX software (Leica).
Drug combinations and synergy analyses
Cells were seeded on 96 well plates at 10,000 cells/well and treated for four days with the indicated doses of drugs, alone or in combination. Combination index values were generated using CalcuSyn v2.11 (http://www.biosoft.com/w/calcusyn.htm) and the Chou-Chou plots (Chou, 2010) were generated from the Dose Reduction Indexes (DRI−1) of the experimental values. Bliss synergy was calculated using the bioconductor package synergyfinder v1.0.0 using default parameters for calculating bliss independence (He et al., 2018). Excess over bliss was calculated as the difference between the observed response and the expected bliss independence (Iniguez et al., 2018).
Statistical Analysis
All data were analyzed with GraphPad Prism 6. Comparison of two groups or more than two groups was performed by unpaired t-test or ANOVA with Tukey’s multiple comparisons test, respectively. All error bars are SEM.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Bruno Amati for providing the TIP60 antibodies. We thank Luisa Luciani, Guoyan Cheng, Madhavi Tadi, Giovanni Lenguito, and Brett Schrand for technical assistance, and Delphine Prou, Daniel Bilbao and Gloria Mas Martin for scientific discussions, and their critical reading of this manuscript. We thank the Oncogenomics Shared Resource Facility and the Flow Cytometry Shared Resource Facility at Sylvester Comprehensive Cancer Center for technical help and support. This work was supported by funds from Sylvester Comprehensive Cancer Center and grant R01 CA166835 from the National Cancer Institute to S.D.N, and grant R01 GM121595 from the National Institute of General Medical Sciences to R.E.V.
Footnotes
DATA AND SOFTWARE AVAILABILITY
The accession number for the RNA-seq data reported in this paper is GEO: GSE107854.
SUPPLEMENTAL INFORMATION
Supplemental information includes Supplemental Experimental procedures, seven figures and five tables.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Allende-Vega N, Dayal S, Agarwala U, Sparks A, Bourdon J-C, and Saville MK (2013). p53 is activated in response to disruption of the pre-mRNA splicing machinery. Oncogene 32, 1–14. [DOI] [PubMed] [Google Scholar]
- Antonysamy S, Bonday Z, Campbell RM, Doyle B, Druzina Z, Gheyi T, Han B, Jungheim LN, Qian Y, Rauch C, et al. (2012). Crystal structure of the human PRMT5:MEP50 complex. Proc Natl Acad Sci U S A 109, 17960–17965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezzi M, Teo SX, Muller J, Mok WC, Sahu SK, Vardy LA, Bonday ZQ, and Guccione E (2013). Regulation of constitutive and alternative splicing by PRMT5 reveals a role for Mdm4 pre-mRNA in sensing defects in the spliceosomal machinery. Genes Dev 27, 1903–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird AW, Yu DY, Pray-Grant MG, Qiu Q, Harmon KE, Megee PC, Grant PA, Smith MM, and Christman MF (2002). Acetylation of histone H4 by Esa1 is required for DNA double-strand break repair. Nature 419, 411–415. [DOI] [PubMed] [Google Scholar]
- Bonnal S, Vigevani L, and Valcárcel J (2012). The spliceosome as a target of novel antitumour drugs. Nature Reviews Drug Discovery 11, 847–859. [DOI] [PubMed] [Google Scholar]
- Botuyan MV, Lee J, Ward IM, Kim J-E, Thompson JR, Chen J, and Mer G (2006). Structural Basis for the Methylation State-Specific Recognition of Histone H4-K20 by 53BP1 and Crb2 in DNA Repair. Cell 127, 1361–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan-Penebre E, Kuplast KG, Majer CR, Boriack-Sjodin PA, Wigle TJ, Johnston LD, Rioux N, Munchhof MJ, Jin L, Jacques SL, et al. (2015). A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat Chem Biol 11, 1–10. [DOI] [PubMed] [Google Scholar]
- Chou T-C (2010). Drug Combination Studies and Their Synergy Quantification Using the Chou-Talalay Method. Cancer Res 70, 440–446. [DOI] [PubMed] [Google Scholar]
- Clarke TL, Sanchez-Bailon MP, Chiang K, Reynolds JJ, Herrero-Ruiz J, Bandeiras TM, Matias PM, Maslen SL, Skehel JM, Stewart GS, et al. (2017). PRMT5-Dependent Methylation of the TIP60 Coactivator RUVBL1 Is a Key Regulator of Homologous Recombination. Mol Cell 65, 900–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daley JM, and Sung P (2014). 53BP1, BRCA1, and the Choice between Recombination and End Joining at DNA Double-Strand Breaks. Mol Cell Biol 34, 1380–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyon Y, and Côté J (2004). The highly conserved and multifunctional NuA4 HAT complex. Curr Opin Genet Dev 14, 147–154. [DOI] [PubMed] [Google Scholar]
- Duncan KW, Rioux N, Boriack-Sjodin PA, Munchhof MJ, Reiter LA, Majer CR, Jin L, Johnston LD, Chan-Penebre E, Kuplast KG, et al. (2016). Structure and Property Guided Design in the Identification of PRMT5 Tool Compound EPZ015666. ACS Med. Chem. Lett. 7, 162–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito MT, Zhao L, and So CW (2015). Synthetic lethal targeting of oncogenic transcription factors in acute leukemia by PARP inhibitors. Nat Med 1–34. [DOI] [PubMed] [Google Scholar]
- Farmer H, McCabe N, Lord CJ, Tutt ANJ, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. (2005). Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434, 917–921. [DOI] [PubMed] [Google Scholar]
- Fradet-Turcotte A, Canny MD, Escribano-Díaz C, Orthwein A, Leung CCY, Huang H, Landry M-C, Kitevski-LeBlanc J, Noordermeer SM, Sicheri F, et al. (2013). 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature 499, 50–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesen WJ, Paushkin S, Wyce A, Massenet S, Pesiridis GS, Van Duyne G, Rappsilber J, Mann M, and Dreyfuss G (2001). The methylosome, a 20S complex containing JBP1 and pICln, produces dimethylarginine-modified Sm proteins. Mol Cell Biol 21, 8289–8300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Kulesskiy E, Saarela J, Turunen L, Wennerberg K, Aittokallio T, and Tang J (2018). Methods for High-throughput Drug Combination Screening and Synergy Scoring In Cancer Systems Biology, (New York, NY: Springer New York; ), pp. 351–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho M-C, Wilczek C, Bonanno JB, Xing L, Seznec J, Matsui T, Carter LG, Onikubo T, Kumar PR, Chan MK, et al. (2013). Structure of the Arginine Methyltransferase PRMT5-MEP50 Reveals a Mechanism for Substrate Specificity. PLoS ONE 8, e57008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iniguez AB, Stolte B, Wang EJ, Conway AS, Alexe G, Dharia NV, Kwiatkowski N, Zhang T, Abraham BJ, Mora J, et al. (2018). EWS/FLI Confers Tumor Cell Synthetic Lethality to CDK12 Inhibition in Ewing Sarcoma. Cancer Cell 33, 202–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquet K, Fradet-Turcotte A, Avvakumov N, Lambert J-P, Roques C, Pandita RK, Paquet E, Herst P, Gingras A-C, Pandita TK, et al. (2016). The TIP60 Complex Regulates Bivalent Chromatin Recognition by 53BP1 through Direct H4K20me Binding and H2AK15 Acetylation. Mol Cell 62, 409–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik S, Liu F, Veazey K, Gao G, Das P, Neves L, Li K, Zhong Y, Lu Y, Giuliani V, et al. (2017). Genetic deletion or small molecule inhibition of the arginine methyltransferase PRMT5 exhibit anti-tumoral activity in mouse models of MLL-rearranged AML. Leukemia 32, 499–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Ilagan JO, Liang Y, Daubner GM, Lee SCW, Ramakrishnan A, Li Y, Chung YR, Micol J-B, Murphy ME, et al. (2015). SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell 27, 617–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh CM, Bezzi M, Low DHP, Ang WX, Teo SX, Gay FPH, Al-Haddawi M, Tan SY, Osato M, Sabo A, et al. (2015). MYC regulates the core pre-mRNA splicing machinery as an essential step in lymphomagenesis. Nature 523, 96–100. [DOI] [PubMed] [Google Scholar]
- Kryukov GV, Wilson FH, Ruth JR, Paulk J, Tsherniak A, Marlow SE, Vazquez F, Weir BA, Fitzgerald ME, Tanaka M, et al. (2016). MTAP deletion confers enhanced dependency on the PRMT5 arginine methyltransferase in cancer cells. Science 351, 1214–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SC-W, Dvinge H, Kim E, Cho H, Micol J-B, Chung YR, Durham BH, Yoshimi A, Kim Y-J, Thomas M, et al. (2016). Modulation of splicing catalysis for therapeutic targeting of leukemia with mutations in genes encoding spliceosomal proteins. Nat Med 22, 672–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Cheng G, Hamard P-J, Greenblatt S, Wang L, Man N, Perna F, Xu H, Tadi M, Luciani L, et al. (2015). Arginine methyltransferase PRMT5 is essential for sustaining normal adult hematopoiesis. J Clin Invest 125, 3532–3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makishima H, Visconte V, Sakaguchi H, Jankowska AM, Abu Kar S, Jerez A, Przychodzen B, Bupathi M, Guinta K, Afable MG, et al. (2012). Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis. Blood 119, 3203–3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marjon K, Cameron MJ, Quang P, Clasquin MF, Mandley E, Kunii K, McVay M, Choe S, Kernytsky A, Gross S, et al. (2016). MTAP Deletions in Cancer Create Vulnerability to Targeting of the MAT2A/PRMT5/RIOK1 Axis. Cell Reports 15, 574–587. [DOI] [PubMed] [Google Scholar]
- Matera AG, and Wang Z (2014). A day in the life of the spliceosome. Nat Rev Mol Cell Biol 15, 108–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavrakis KJ, McDonald ER, Schlabach MR, Billy E, Hoffman GR, deWeck A, Ruddy DA, Venkatesan K, Yu J, McAllister G, et al. (2016). Disordered methionine metabolism in MTAP/CDKN2A-deleted cancers leads to dependence on PRMT5. Science 351, 1208–1213. [DOI] [PubMed] [Google Scholar]
- McAllister D, Merlo X, and Lough J (2002). Characterization and expression of the mouse tat interactive protein 60 kD (TIP60) gene. Gene 289, 169–176. [DOI] [PubMed] [Google Scholar]
- Meister G, Eggert C, Bϋhler D, Brahms H, Kambach C, and Fischer U (2001). Methylation of Sm proteins by a complex containing PRMT5 and the putative U snRNP assembly factor pICln. Curr Biol 11, 1990–1994. [DOI] [PubMed] [Google Scholar]
- Panier S, and Boulton SJ (2013). Double-strand break repair: 53BP1 comes into focus. Nat Rev Mol Cell Biol 15, 7–18. [DOI] [PubMed] [Google Scholar]
- Pop R, Shearstone JR, Shen Q, Liu Y, Hallstrom K, Koulnis M, Gribnau J, and Socolovsky M (2010). A key commitment step in erythropoiesis is synchronized with the cell cycle clock through mutual inhibition between PU.1 and S-phase progression. Plos Biol 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran Q, and Pereira-Smith OM (2000). Identification of an alternatively spliced form of the Tat Interactive Protein (Tip60), Tip60(e). Gene 258, 141–146. [DOI] [PubMed] [Google Scholar]
- Schotta G, Sengupta R, Kubicek S, Malin S, Kauer M, Callen E, Celeste A, Pagani M, Opravil S, La Rosa-Velazquez, De IA, et al. (2008). A chromatin-wide transition to H4K20 monomethylation impairs genome integrity and programmed DNA rearrangements in the mouse. Genes Dev 22, 2048–2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S, Park JW, Lu Z-X, Lin L, Henry MD, Wu YN, Zhou Q, and Xing Y (2014). rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc Natl Acad Sci U S A 111, E5593–E5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squatrito M, Gorrini C, and Amati B (2006). Tip60 in DNA damage response and growth control: many tricks in one HAT. Trends Cell Biol 16, 433–442. [DOI] [PubMed] [Google Scholar]
- Sulli G, Di Micco R, and di Fagagna FD (2012). Crosstalk between chromatin state and DNA damage response in cellular senescence and cancer. Nat Rev Cancer 12, 709–720. [DOI] [PubMed] [Google Scholar]
- Symington LS, and Gautier J (2011). Double-strand break end resection and repair pathway choice. Annu Rev Genet 45, 247–271. [DOI] [PubMed] [Google Scholar]
- Tang J, Cho NW, Cui G, Manion EM, Shanbhag NM, Botuyan MV, Mer G, and Greenberg RA (2013). Acetylation limits 53BP1 association with damaged chromatin to promote homologous recombination. Nat Struct Mol Biol 20, 317–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarighat SS, Santhanam R, Lai H, Anghelina M, Alinari L, Walker A, Li L, Garzon R, Li C, and Baiocchi RA (2016). The dual epigenetic role of PRMT5 in acute myeloid leukemia: gene activation and repression via histone arginine methylation. Leukemia 30, 789–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, and Chen J (2010). SIRT1 regulates autoacetylation and histone acetyltransferase activity of TIP60. Journal of Biological Chemistry 285, 11458–11464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Hamard P-J, and Nimer SD (2015). PARP inhibitors: a treatment option for AML? Nat Med 21, 1393–1394. [DOI] [PubMed] [Google Scholar]
- Wei H, Wang B, Miyagi M, She Y, Gopalan B, Huang D-B, Ghosh G, Stark GR, and Lu T (2013). PRMT5 dimethylates R30 of the p65 subunit to activate NF-κB. Proc Natl Acad Sci U S A 110, 13516–13521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JK, Gerdin A-K, Karp NA, Ryder E, Buljan M, Bussell JN, Salisbury J, Clare S, Ingham NJ, Podrini C, et al. (2013). Genome-wide Generation and Systematic Phenotyping of Knockout Mice Reveals New Roles for Many Genes. Cell 154, 452–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Wu J, and Zheng YG (2012). Function of the Active Site Lysine Autoacetylation in Tip60 Catalysis. PLoS ONE 7, e32886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi J, Huang X, Yang Y, Zhu W-G, Gu W, and Luo J (2014). Regulation of histone acetyltransferase TIP60 function by histone deacetylase 3. Journal of Biological Chemistry 289, 33878–33886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, et al. (2011). Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 478, 64–69. [DOI] [PubMed] [Google Scholar]
- Zeisig BB, Kulasekararaj AG, Mufti GJ, and Eric So CW (2012). SnapShot: Acute Myeloid Leukemia. Cancer Cell 22, 698–698 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.