

Abstract
Caspases are cysteine–aspartic proteases involved in the regulation of programmed cell death (apoptosis) and a number of other biological processes. Despite overall similarities in structure and active-site composition, caspases show striking selectivity for particular protein substrates. Exosites are emerging as one of the mechanisms by which caspases can recruit, engage, and orient these substrates for proper hydrolysis. Following computational analyses and database searches for candidate exosites, we utilized site-directed mutagenesis to identify a new exosite in caspase-6 at the hinge between the disordered N-terminal domain (NTD), residues 23–45, and core of the caspase-6 structure. We observed that substitutions of the tri-arginine patch Arg-42–Arg-44 or the R44K cancer-associated mutation in caspase-6 markedly alter its rates of protein substrate hydrolysis. Notably, turnover of protein substrates but not of short peptide substrates was affected by these exosite alterations, underscoring the importance of this region for protein substrate recruitment. Hydrogen–deuterium exchange MS–mediated interrogation of the intrinsic dynamics of these enzymes suggested the presence of a substrate-binding platform encompassed by the NTD and the 240's region (containing residues 236–246), which serves as a general exosite for caspase-6–specific substrate recruitment. In summary, we have identified an exosite on caspase-6 that is critical for protein substrate recognition and turnover and therefore highly relevant for diseases such as cancer in which caspase-6–mediated apoptosis is often disrupted, and in neurodegeneration in which caspase-6 plays a central role.
Keywords: cysteine protease, allosteric regulation, apoptosis, neurodegeneration, hydrogen–deuterium exchange, mass spectrometry, protein dynamic, substrate specificity, cancer, caspase-6, substrate recognition, tri-arginine exosite
Introduction
Caspases are a class of cysteine–aspartic proteases intertwined in the regulation of programmed cell death, or apoptosis. Apoptosis is an essential homeostatic pathway mediating morphogenesis, development, and organismal protection against damaged or dangerous cells (for review, see Elmore (1)). Mis-regulation of apoptosis has been implicated in the progression of numerous multifactorial diseases, including cancer (low apoptotic activity) (2–5) or neurodegenerative disorders like Alzheimer's, Huntington's, and Parkinson's diseases during instances of heightened cell death (6–9). Briefly, caspases are divided into inflammatory (caspase-1, -4, and -5) or apoptotic classes. The apoptotic caspases are further classified as the initiator caspases (-2, -8, -9, and -10) or executioner caspases (-3, -6, and -7). Caspases are synthesized and stored in their latent-state as inactive zymogen forms or procaspases. During apoptosis, either extrinsic or intrinsic signals activate the zymogens procaspase-8 or -9. Once activated, the initiator caspases cleave the downstream zymogen-state executioners at their inter-subunit linkers, leading to mature enzymes capable of substrate hydrolysis. Upon activation, the dimeric executioners recruit and process key substrates involved in cellular homeostasis, gene transcription, and cellular structure. Cleavage of these key substrates ushers the cell to a point of no return proceeding to apoptotic cell death (10). Because of the involvement of apoptosis in disease progression, caspases have been a target for therapeutic intervention. However, the high degree of similarity of both the overall fold and the active-site geometry (Fig. 1A) of these enzymes has hindered the development of effective yet selective modulators. Therefore, understanding of the unique dynamics and structural nuances of the dozen various human caspases is essential for exogenous control over apoptosis and its involvement in various disease states.
Figure 1.

Caspases show conserved folds but different surface charges. The overall caspase fold is largely conserved across the family with minimal divergent structural differences concentrated largely to the mobile loops. A, surface topology of caspase-6 (blue, PDB code 3OD5 (80)) and caspase-7 (green, PDB code 1F1J (87)). B, calculated electrostatics highlight key differences in positive (blue) and negative (red) regions distal from the active site. Unique surface characteristics may tune caspase substrate recognition.
Numerous efforts have been made through the better part of the last two decades to understand how and why the 12 human caspases recognize their individual substrates. These studies have begun to reveal the nuanced nature of the caspases, providing new layers of unique regulation that are subject to selective modification of these enzymes. Additionally, the identification of caspase substrates has grown immensely (11–14). It has become evident that the apoptotic caspases share some substrates, but each caspase cleaves its own unique set of substrates (15, 16). In fact, each caspase performs nonredundant functions and recognizes these substrates with different affinities, occasionally in different positions, altering the downstream effects (17–19). These biological differences cannot be fully explained based solely on the specificity of each caspase for peptide substrates; thus, it became evident that there are other features in the caspase structure that influence its substrate specificity outside of the active site. An exosite, broadly defined, is a site distal from the substrate-binding groove of a protein that influences structure or function. The identification of the first exosites in caspases (Jabaiah et al. (20) and Boucher et al. (22)) has suggested that individual caspases might utilize their vastly different surface topologies (Fig. 1B) to help recruit particular protein substrates, thus tuning the overall specificity profile of these enzymes. For instance, caspase-7 utilizes an exosite in the N-terminal domain (NTD),5 a disordered region that lies just after the prodomain, which is proteolytically removed during activation. The patch of caspase-7 consists of a highly positive tetra-lysine 38KKKK41 stretch, which helps to recruit protein substrates PARP-1 and p23 (21, 22). This exosite enables caspase-7 to cleave these substrates 30-fold more efficiently than caspase-3, despite the fact these two enzymes share the same overall specificity for peptide substrates (23, 24). This difference is even more striking because caspase-3 is the most catalytically active member of the family and cleaves PARP-1 and p23 at the same positions. Moreover, changing the NTD of caspase-3 to that of caspase-7 resulted in more effective protein substrate turnover of PARP-1 by caspase-3, underscoring the idea that exosites play a role in caspase substrate recognition.
Although exosites are not a novel entity in the world of protease regulation, they are a new and exciting layer of caspase function, providing a wealth of untapped differences between these enzymes that may be utilized for exogenous therapeutic control. Exosite modulation for activity tuning of proteases has already begun to be utilized in cases where active-site inhibition was not capable of providing desired clinical outcomes. For example, cathepsin K (CatK) is a target for treatment of osteoporosis (25). One active-site CatK inhibitor, Odanacatib, was considered a promising, highly potent, and selective molecule (26, 27), but it showed increased risk of stroke and mortality in long-term studies and was subsequently halted in phase III clinical trials (28). It was hypothesized that fully inhibiting the active site of CatK would result in deleterious effects because its function in other pathways that require its proteolytic activity would be blocked. This has been circumvented by utilizing an exosite inhibitor of CatK, DHT1, which can block the collagenase activity of CatK with similar efficacy to Odanacatib, while leaving the active site free to cleave other substrates like TGF-1β (29). Therefore, utilizing exosites has allowed development of protease inhibitors that prevent unwanted off-target effects by tailoring enzymatic activity through exosite engagement.
The identification of an exosite on caspase-7 suggested that exosites on other caspases might also exist. We had previously shown, using an evolved caspase-6/-7 hybrid, that certain caspase-6 substrates, such as lamin A/C, appeared to require an exosite for recognition (38). Caspase-6 is unique among the family of apoptotic caspases for its structural transitions and its roles in neurodegeneration. Specifically, caspase-6 can interconvert between an inactive helical state and the canonically-active strand state (30). Functionally, in the context of neurodegeneration, caspase-6 cleaves amyloid precursor protein (31), Tau (32), Huntingtin protein (33), as well as Parkinson's disease protein 7 (PARK7 or DJ-1) (34, 35). Recently, the inhibition of caspase-6 in an Alzheimer's mouse model led to clinical improvements in memory deficits (36). Thus, the discovery of exosites might provide alternatives for the selective inhibition of only caspase-6. Here, we describe the discovery of an evolutionarily conserved tri-arginine patch, 42RRR44, located within the disordered NTD of caspase-6 that serves as an exosite involved in the recruitment of protein substrates. Importantly, removal of this site does not hinder the intrinsic catalytic capability of the enzyme. We also investigate the effects of a previously annotated cancer mutation R44K (37) that resides within this exosite patch and find that this seemingly silent variant has similar deleterious effects on the ability of caspase-6 to recruit protein substrates. Furthermore, we investigate the changes in dynamics that alterations within this exosite have on the caspase-6 protein and find that these changes unhinge a putative protein-binding interface. This work ultimately provides new perspectives into how caspase-6 can bind, recruit, and hydrolyze protein substrates and uncovers a scaffold that may be utilized for the selective modulation of caspase-6.
Results
Evolutionary conservation analysis identified putative caspase-6 exosite
Exosites, which are functional sites distal from the catalytic site, have been shown to play a significant role in the function of caspase-6 and -7 (21, 22, 38, 39). Based on the observation that caspase-3 and -7, which show virtually identical substrate specificity for peptide substrates, utilize an exosite to distinguish native protein substrates (22), we hypothesized that other caspases also utilize exosites to differentiate substrate pools. In particular, unique patches of solvent-exposed hydrophobic and charged residues are likely candidates for guiding substrate interactions as exosites on caspase-6. Using CaspBase (40), which contains 2879 hand-curated caspase sequences covering a wide range of phyla, classes, and taxa, caspase orthologues were aligned to the relevant human caspase (e.g. casp-3, -4, -5, etc.) using ClustalOmega (41) and then analyzed for evolutionary conservation between sequences for an individual caspase homologue with ConSurf (Fig. 2, A and C) (42–46). We identified a conserved tri-arginine patch in caspase-6, 42RRR44, which is similar in charge and location to the identified exosite in caspase-7 38KKKK41. Two of the arginine residues, Arg-43 and Arg-44, are largely conserved within caspase-6 orthologues but not conserved across the caspase family (Fig. 2, B and D), suggesting that this site may play an important role in caspase-6 function.
Figure 2.

Conservation analysis of arginine patch in caspase-6 orthologues. A, human caspases 1–14 in the N-terminal domain, the region post prodomain, curated from CaspBase (40), aligned by Clustal Omega, and colored to depict the degree of conservation calculated by ConSurf (42–46) across all caspase homologues and species. Conservation ranges from least conserved (blue) to most conserved (magenta). Arg-44 is conserved within the 205 caspase-6 sequences from a wide collection of phyla, classes, and taxa. B, sequence conservation across the 12 human caspases analyzed by ConSurf mapped onto the human caspase-6 sequence. The tri-arginine patch is not conserved across human caspases. C, conservation results of 205 caspase-6 sequences aligned and analyzed as in A and mapped onto the human caspase-6 structure. D, conservation of residues across the 12 human caspases analyzed as in B and mapped onto the human caspase-6 structure. Caspase-6 PDB code is 3OD5. ‡ denotes the caspase-7 exosite; * denotes the 42RRR44 patch in caspase-6.
Distal amino acids are predicted to be important for enzymatic function
The computational method Partial Order Optimum Likelihood (POOL) (47, 48) relies on the evaluation of theoretical titration curves of the ionizable residues as well as analysis of surface topology and phylogenetic tree information in known protein structures. For this analysis, we used a version of POOL composed of only the THEMATICS and ConCavity algorithms. This approach has been useful in identifying important functional regions in proteins (49–51). We applied POOL to a wide dataset of reported human caspase-6 structures in the PDB, in both the helical or strand conformations (Fig. 3). This method allowed us to corroborate reported structural and functional features of caspase-6, as well as to identify new sites that are most likely functional in either the helical or strand conformation or both. Notably, clustering of the predicted amino acid residues is consistent with known biological features of caspase-6. First, caspase-6 can transition between the inactive helical and active strand conformations; the region essential for this dynamic interconversion is in the 130's helix (30, 52). POOL accurately predicted that the 130's region is important only in the helical conformation but not in the strand conformation. Second, POOL accurately identifies both members of the active-site dyad, His-121 and Cys-163, regardless of the activation state of the enzyme and can differentiate the inactive structures (4IYR and 3NR2), in which the catalytic residues have been replaced (H121A and C163A), from the active structures. Third, many active-site residues are not predicted in the naturally inactive helical state. Together these findings suggest that POOL robustly identifies functionally relevant sites.
Figure 3.

POOL results show distal sites are important for biochemical function. POOL analysis performed on an ensemble of caspase-6 crystal structures in various activation states and conformations indicated by PDB codes in the figure. The top 12% of residues identified and predicted to be important for enzymatic function for each structure are highlighted in gray. Residues further clustered based on their conformational state: helical or strand, as well as their proximity to the caspase-6 active-site, 130's region, or distal positions.
In addition to known functional sites, four new residues distal from the active site were also predicted to be functionally important: Arg-44, Cys-113, Cys-116, and Lys-156. These residues are only predicted to be important in structures in the strand or the substrate binding competent conformation of caspase-6, potentially suggesting that these distal residues could be functionally important. For multiple other proteins, POOL predictions of important distal residues have been verified experimentally by site-directed mutagenesis and kinetics assays. Notably, one of the predicted distal residues for caspase-6, Arg-44, was also identified through conservation analysis (Fig. 3), and thus it became a focus in our investigation.
Catalytic parameters of exosite replacements are maintained for peptide substrates but altered for protein substrates
To investigate the functional role of the 42RRR44 patch, these residues were replaced by alanine (R42A/R43A/R44A, named R42–44A). R44K, which was previously identified as a caspase-6 mutation observed in colorectal carcinomas (37), was also investigated. When expressed from the full-length caspase-6 gene, R42–44A was unable to fully hydrolyze itself to the mature form, whereas WT caspase-6 was fully hydrolyzed under the same conditions (Fig. S1), suggesting that 42RRR44 is critical for caspase-6 self-activation. To accurately assess the catalytic function of the R42–44A variant, a constitutive two-chain (CT) construct was generated that expresses the caspase-6 large and small subunits independently (ΔN D179CT) (52). We first assayed the activity of the caspase-6 CT variants with a small fluorogenic tetrapeptide substrate. In these studies, we expected that substitution of substrate-binding exosites would maintain the same catalytic efficiency as WT caspase-6 as the inherent small size of the fluorogenic tetrapeptides are unable to engage the exosite residues. At the same time, we expected a loss in activity of the caspase-6 variants in an exosite against protein substrates due to their decreased ability to facilitate formation of the enzyme–substrate complex.
The fluorogenic peptide substrate Ac-VEID-amc was cleaved by WT caspase-6, R42–44A, and R44K variants with nearly superimposable kinetics curves (Fig. 4A). The catalytic efficiencies of R42–44A and R44K were also nearly identical to that of WT caspase-6 for Ac-VEID-amc (Fig. 4B), indicating that these substitutions did not negatively impact the function or geometry of the catalytic dyad. In contrast, when tested against full-length (FL) caspase-6 C163S, which is catalytically inactive and incapable of undergoing self-proteolysis, as a substrate, the R42–44A and R44K variants showed obvious cleavage defects. Although WT caspase-6 rapidly hydrolyzed C163S FL substrate, the R42–44A variant showed markedly decreased hydrolysis of the zymogen, with a relative 3.6-fold change (Fig. 4, C and D, and Table S1). Even the conservative R44K variant showed a decreased rate of processing, at a relative 1.8-fold change (Fig. 4, C and D, and Table S1), suggesting that the 42RRR44 patch is important for recognizing a second molecule of caspase-6. To determine whether the 42RRR44 patch is playing a role in recruitment of other substrates, the caspase-6 exosite variants R42–44A and R44K were tested for their ability to hydrolyze the canonical caspase-6 substrate lamin C. Caspase-6 has a long connection to lamin C as a substrate. Caspase-6 was identified initially as being a human laminase, and the canonical tetrapeptide substrate used to characterize caspase-6 function, Ac-VEID-amc, is derived from the caspase-6 cleavage site in lamin A/C (Asp-230) (53). In addition, it has been suggested that caspase-6 may utilize an exosite for the recognition of lamin A/C as they are only cleaved by caspase-6 and not by other caspases (54). The rate of hydrolysis of lamin C was quantified by assessing the disappearance of the full-length lamin C, 66 kDa, to the 38- and 28-kDa lamin C fragments as a function of caspase-6 concentration (Fig. 4, E–G) and fitting to a single-phase decay (Fig. 4H). WT caspase-6 hydrolyzed lamin C more rapidly (95 ± 15 m−1 s−1) than R42–44A (28 ± 6.1 m−1 s−1; Fig. 4, F and H) or R44K (41 ± 15 m−1 s−1; Fig. 4, G and H). These 3.3- and 2.3-fold decreases in the hydrolysis rates of lamin C by the R42–44A and R44K variants suggest that the 42RRR44 exosite patch may impact recognition of key caspase-6 substrates.
Figure 4.

Disruptions in the 42RRR44 patch retain activity for peptide substrates but alter kinetics for protein substrates. A, representative Michaelis-Menten kinetics of caspase-6 exosite variants compared with the WT enzyme. B, catalytic parameters of caspase-6 and exosite knockout variants R42–44A and R44K demonstrate that the active-site affinity is not altered by exosite modifications. C, cleavage of purified substrate caspase-6 C163S FL by the R42–44A or R44K variants over a time course of 0, 5, 15, 30, 45, and 60 min shows the rate of protein hydrolysis is severely delayed compared with the WT. D, quantification of caspase-6 C163S FL cleavage by WT or caspase-6 variants. E–G, hydrolysis of a constant amount of purified substrate lamin C from FL 66-kDa protein into 38/28-kDa fragments by caspase-6 (E), R42–44A (F), or R44K (G) in a 2-fold serial dilution from 25 to 0 μm for 4 h. H, quantification of the cleaved caspase-6 C163S FL band relative to the 0 μm point fit to a single-phase decay to calculate the CF50 used to calculate the hydrolysis rate of caspase-6 or variants R42–44A and R44K. Experiments performed in triplicate. *, ** denotes 90 or 95% confidence interval by unpaired two-tailed t test of the variants individually compared with the WT. In all cases, the caspase-6 variants are in the ΔN D179CT form. Only caspase-6 C163S as a substrate was in the full-length form.
Removal of tri-arginine patch significantly decreases hydrolysis of key protein substrates
The 42RRR44 exosite patch on caspase-6 clearly engages both procaspase-6 and lamin A/C and facilitates their cleavage by caspase-6. The 38KKKK41 exosite in caspase-7 engages both PARP and p23 substrates (21, 22), and it is possible that additional substrates that engage the site may soon be discovered. To distinguish whether the 42RRR44 exosite is used for many substrates or only certain substrates, we analyzed hydrolysis in whole-cell lysates isolated from SK-N-AS neuroblastoma cells. Cell lysates were incubated with 100 nm caspase-6 or the respective variants. Rates of cleavage of PARP, lamin A/C, and DJ-1 (PARK7) were analyzed. PARP was chosen for its role in DNA repair and modulating gene transcription and its known interaction with the similarly charged exosite of caspase-7. The rate of PARP hydrolysis was affected by both the removal of the exosite R42–44A, a relative 2.4-fold change, and the presence of the cancer mutation R44K, a relative 2.3-fold change, compared to the WT caspase-6 (Fig. 5, A and D, and Table S1). Similar to PARP, the rate of the A-type lamin cleavage was statistically lower (relative 1.3-fold) for both the R42–44A and R44K variants than for WT caspase-6 (Fig. 5, B and E, and Table S1). DJ-1 or Parkinson's Disease Protein 7 (PARK7) has been identified as a substrate of caspase-6. Cleavage results in an N-terminal fragment being ushered into the nucleus, increasing susceptibility to reactive oxygen species and apoptosis. The Parkinson's disease mutation D149A, which appears at the caspase-6 cleavage site, suggests that cleavage of DJ-1 may play a role in protecting the cell from the Parkinson's disease phenotype (34, 35). Similar to other substrates, the cleavage of DJ-1 by WT caspase-6 was also significantly faster than the exosite knockout counterparts, R42–44A (relative 2.3-fold) and R44K (relative 1.5-fold) (Fig. 5, C and F, and Table S1).
Figure 5.
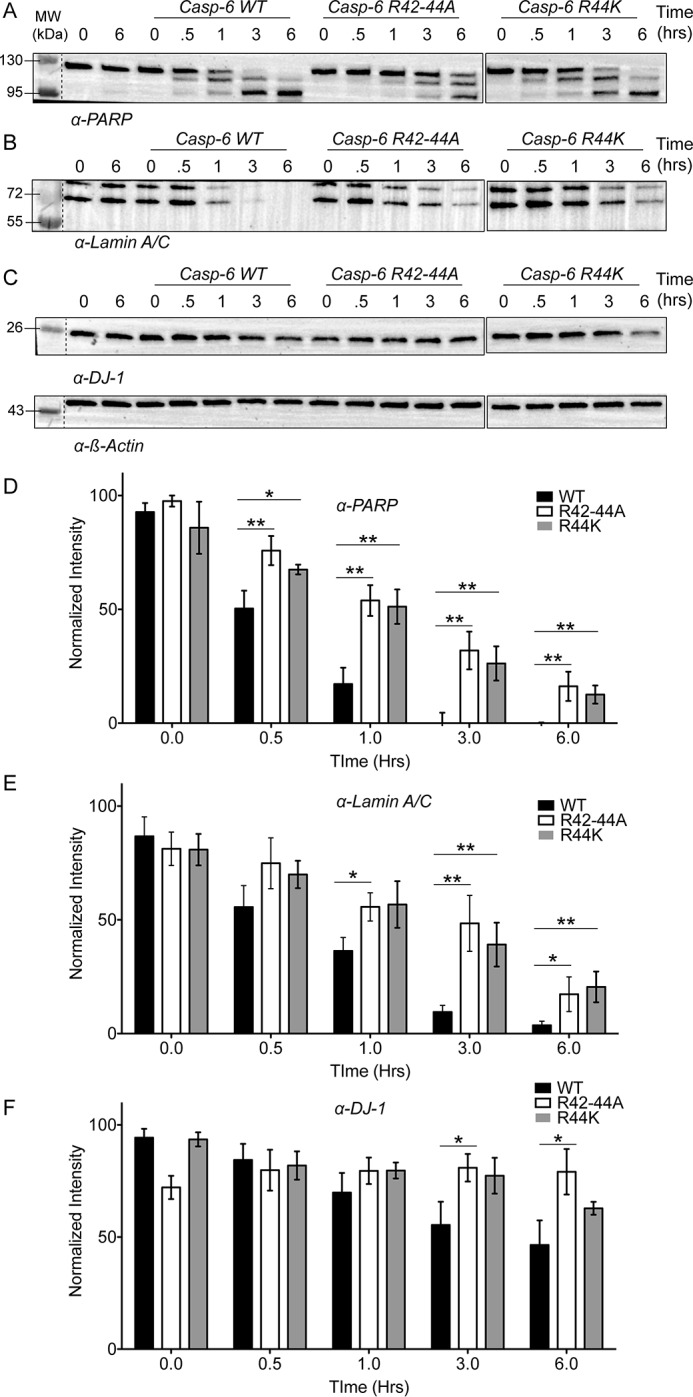
42RRR44 patch is required for general protein hydrolysis in cell lysates. A–C, lysates generated from SK-N-AS neuroblastoma cells were incubated with 100 nm of either WT caspase-6, R42–44A, or R44K to assess hydrolysis over time by Western blotting of substrate proteins. A, PARP; B, lamin A/C; or C, DJ-1 normalized to loading control β-actin. D–F, quantification of the data from WT (black), R42–44A (white), or R44K (gray) normalized to the loading control for protein substrates PARP (D), lamin A/C (E), or DJ-1 (F) demonstrates that the hydrolysis of several biologically relevant substrates of caspase-6 are impacted by the removal of the 42RRR44 exosite. Changes in substrate processing of multiple proteins suggest they are engaging caspase-6 through a similar mechanism. Experiments were performed in triplicate. *, ** denotes 90 or 95% confidence interval by unpaired two-tailed t test of the variants individually compared with the WT.
Segment polarity protein disheveled homolog DVL-3 (DVL3) was identified as being one of the substrates cleaved most quickly by caspase-6, at a rate of 2.2 × 105 m−1 s−1 (15). Once hyper-phosphorylated, DVL3 accumulates in the nucleus, where it alters the WnT pathway signal transduction and transcription. We performed DVL3 hydrolysis assays using WT caspase-6 and exosite variants. The rate of DVL3 hydrolysis by WT caspase-6 (Fig. 6A) was 3.8 × 104 ± 3.8 × 103 m−1 s−1 (Fig. 6D), a similar rate to the previous proteomic study (15). The R42–44A variant cleaved DVL3 5-fold more slowly (8.3 × 103 ± 2.7 × 103 m−1 s−1) than WT caspase-6 (Fig. 6, B and D). Finally, the cancer mutation R44K cleaved DVL3 2-fold more slowly (1.7 × 104 ± 6.7 × 103 m−1 s−1) (Fig. 6, C and D). Consistent with all substrates tested, the disruption of this exosite negatively impacts the ability of caspase-6 to proteolyze this protein substrate. Together these data suggest that the location of the 42RRR44 patch at the hinge of the NTD is participating in a global exosite for caspase-6 recognition of substrates.
Figure 6.

Cleavage of key caspase-6 substrates occurs using the 42RRR44 exosite. A–C, hydrolysis of DVL3, which is one of the substrates most quickly cleaved after caspase-6 activation, was assessed by added caspase-6 variants in the ΔN D179CT form. A 2-fold serial dilution of caspase-6 (A), R42–44A (B), or R44K (C) from 250 to 0 nm incubated with a constant amount of SK-N-AS cell lysate over a 1-h time course at 37 °C imaged by Western blotting and normalized to the loading control β-actin. D, quantification of bands fit to a single-phase decay to find the CF50 used to calculate the hydrolysis rate of DVL3 by either caspase-6 (black), R42–44A (white), R44K (gray). Experiments were performed in triplicate. *, ** denotes 90% or 95% confidence interval by unpaired two-tailed t test of the variants individually compared with the WT.
42RRR44 exosite patch influences caspase-6 dynamics
It is clear the 42RRR44 patch plays a role in caspase-6 recognition of protein substrates, but the mechanism remained unclear. To investigate the influence of the 42RRR44 exosite patch on caspase-6 dynamics, we performed hydrogen–deuterium exchange MS (H/Dx-MS) using our previously established protocol (30, 55) on a solution containing WT caspase-6 and lamin C. We were not able to detect any differences in the exchange of any peptides, probably due to technical complications (Fig. S2). WT and the R42–44A and R44K variants were likewise subjected to H/Dx-MS. Sequence coverage for the three variants was excellent, with 94.2–96.0% of all residues observed in peptic peptides (Figs. S3–S5). As we have previously observed for WT caspase-6, the cores of each variant, which are formed of buried anti-parallel β-sheets, were highly protected, whereas the solvent-exposed active-site loops showed a high level of exchange (Figs. S6–S8). Importantly, the similarity of these regions suggests that the core features of the caspase-6 variants are unchanged, and the variant enzymes are not critically perturbed.
The region spanning residues 33–48 showed a large change in the deuterium uptake in both the R42–44A and R44K variants (Fig. 7, A and B). Of the total possible deuterium uptake by their region, WT achieves only 11% deuterium uptake, suggesting that this region is shielded from exchange (Fig. S6). In R44K, this region is much more highly exchanging, reaching a maximal uptake of ∼31% (Fig. S7). This suggests that the removal of this key evolutionarily conserved arginine allows for the increased dynamic flexibility of this region consistent with playing a stabilizing role in the caspase-6 structure. Moreover, when the tri-arginines 42RRR44 are mutated to alanine (R42–44A), we find an even more increased rate of deuterium uptake, 41% (Fig. S8). This suggests that all three arginine residues play key roles in mediating the solvent exposure of this region; however, the degree of exchangeable properties suggests that Arg-44 is the key residue involved in this interaction.
Figure 7.

Disruption of the 42RRR44 exosite increases dynamics of caspase-6 variants in NTD, 240's regions. A and B, caspase-6, R42–44A, or R44K incubated in D2O and monitored for exchange over time. Difference plots generated by subtracting the deuterium uptake profiles of common peptides of WT caspase-6 from peptides from either R42–44A (A) or R44K (B) variants. Regions depicted in blue (negative) are less exchanging in the variant than in the WT, whereas regions in red (positive) were more exchanging in the variant than in WT based on the error of the data sets ±0.42 or ±0.73 for the R42–44A or R44K based on the 98 and 95% confidence interval, respectively. C and D, exchange differences mapped to the structure of caspase-6 (predicted with MODELLER to incorporate all amino acids not visible in known structures (55)) of the R42–44A (C) or R44K (D) variants. E, relative deuterium uptake difference scale. F–L, important peptides identified as significantly different upon exchange compared with the WT peptides. Experiments were repeated on 2 different days. Δ indicates the difference, wherein the data from WT is always subtracted from the data from the indicated variant. In all cases, the caspase-6 variants are the ΔN D179CT form.
To gauge the statistical significance of the differences between the variants and WT, difference plots were generated by subtracting the D2O uptake parameters of the common peptic peptides of WT from the R42–44A or R44K variants (Fig. 7, A and B) and mapped onto the caspase-6 structure (Fig. 7, C and D). Any peptides that were found to exchange in excess of error of the data sets (0.42 and 0.73 Da for R42–44A and R44K, respectively) were colored either blue (negative) or red (positive). The blue represents regions that exchange less in the variants than in the WT, whereas regions that appear in red exchange more in the variants. Consistent with observations from the uptake heatmaps, the region from 33 to 48 significantly exchanges more in the two variants (Fig. 7, F–J). Increases in deuterium uptake suggest that this region is less protected, which further suggests that the protein has undergone a change in conformation or become more dynamic (56).
The 240's region (236–246) is also more highly exchanging in the R42–44A or R44K variants than in WT (Fig. 7L). When the difference plots are mapped to the caspase-6 structure (Fig. 7, C and D), it becomes clear that the 42RRR44 exosite patch rests at the hinge point between the disordered NTD and the core of caspase-6. Moreover, the NTD lies across the caspase-6 dimer interface making clear contacts with the 240's region on the opposite half of the dimer. Altering the 42RRR44 patch leads to a dramatic increase in deuterium uptake in the 240's region. Given that the loop containing the 42RRR44 patch is more dynamic, a potential loss of contacts with the 240's region and increased exposure in that region is not surprising. Interestingly, the 240's region is moderately exchanging in WT caspase-6, indicating some degree of solvent exposure natively (Fig. S5). This observation suggests that the NTD may function as a lid, dynamically populating an open and a closed state, thus impacting the overall dynamics of the 240's region. The increased uptake upon substitution at the hinge of this lid (R42–44) increases the rate of opening and closing of the lid leading to a high rate of deuterium exposure both within the NTD and the peptides spanning from 236 to 246 (Fig. 7, A, B, and L).
Finally, only in the R42–44A variant (Fig. 7, A and K) is the 130's region also more highly exchanging. The 130's region undergoes a helix-to-strand interconversion only observed in caspase-6 (30, 57). Notably, these changes were not observed in the R44K variant (Fig. 7B). The further unhinging of the NTD by concurrent removal of all three arginines within the 42RRR44 patch led to increased dynamics of the 130's region, suggesting the motions of the NTD influence the dynamic interconversion between the helix and strand. Overall, we find that the motions of the NTD may be key to uncovering how caspase-6 can bind and recognize its various protein substrates.
Charged tri-arginine 42RRR44 patch stabilizes caspase-6 with multidentate interactions
Based on available crystal structures and H/Dx-MS data, we hypothesized that alteration of the tri-arginine patch may play a role in the stability of caspase-6. To investigate the role of 42RRR44 in the stability of caspase-6, the three variants were analyzed for changes in thermal melting temperature by differential scanning fluorimetry in the presence or absence of the covalent active-site ligand Ac-VEID-cho. WT caspase-6 has a melting temperature of 73.8 ± 0.1 °C that increases to 79.3 ± 0.06 °C, a 5.5 °C stabilization upon binding ligand (Fig. 8A). R44K showed a melting temperature of 69.9 ± 0.1 °C, 3.9 °C lower than relative to WT. Incubation with the Ac-VEID-cho inhibitor resulted in a 3.1 °C stabilization to 73.0 ± 0.07 °C relative to the unliganded form (Fig. 8B). Consistent with the H/Dx-MS, which showed greater exchange and therefore greater dynamics, R42–44A showed a larger destabilization in the unbound state, 65.8 ± 0.4 °C, an 8.0 °C decrease in stability relative to WT. Nevertheless, R42–44A was stabilized by 4.5 °C stabilization upon binding active-site inhibitor (Fig. 8C). The stabilization from 3 to 5 °C upon binding the active-site ligand suggests that all variants can form the canonical substrate-bound structure at the active site, which is consistent with observed core conservation through H/Dx-MS analysis (Fig. 7) and the superimposable kinetics for cleavage of peptide substrates (Fig. 4, A and B).
Figure 8.

42RRR44 exosite functions as a hinge between NTD and core, stabilizing caspase-6. A–C, differential scanning fluorimetry was used to observe how substitutions in the 42RRR44 hinge impact the stability of caspase-6 (A), R44K (B), or R42–44A (C) in the unliganded state or liganded to the caspase-6 cognate inhibitor Ac-VEID-cho state. D–F, changes in the observed thermal stability may be attributed to sequential loss of interactions as side chains are removed. D, WT; E, 42RRK44 (R44K), and F, 42AAA44 (R42–44A). Models (E and F) were produced using the PyMOL mutagenesis wizard to demonstrate potential loss in contacts. In all cases the caspase-6 variants are the ΔN D179CT form.
Close inspection of the caspase-6 structure reveals the critical role of Arg-44 in stabilization by making numerous contacts with residues near the N-terminal loop (Leu-81 main chain, and Asp-112 side chain) and the C-terminal loop (Phe-289 main chain) (Fig. 8D). Even in the conservative mutation R44K, replacing a guanidinium group with a primary amine ablates several points of potential contact (Fig. 8E). In this model, we find Lys-44 in position to bind to only the main chain of Phe-289 (Fig. 8E). In addition, Arg-42 and Arg-43 are positioned to form multiple coulombic and H-bonding interactions with Asp-110 of the hinge region of caspase-6 (Fig. 8D). Substitution by alanine disrupts these contacts, likely leading to the large destabilization observed (Fig. 8F). Together, the H/Dx-MS and differential scanning fluorimetry data suggest that the stabilization of the N- and C-terminal loops by the tri-arginine patch, most importantly Arg-44, is critical for the dynamics of caspase-6; moreover, this change in dynamics plays a key role in the mechanism by which the enzyme recognizes protein substrates.
Removal of the tri-arginine hinge disrupts caspase-6 protein-binding interface
Based on the crystallographic observation that the NTD crosses the dimer interface to interact with the 240's region (Fig. 9, A and B), and evidence of a similar role for the caspase-7 NTD (21), we hypothesize that caspase-6 might utilize the NTD as bait to fish protein substrates out of the cellular milieu. This interaction could orient substrates for hydrolysis by the active site on the opposite side of the caspase-6 dimer. The fact that the exact regions engaged in contacts within the crystal structure are also represented by peptides in which the H/Dx-MS was dynamically altered by substitutions in the 42RRR44 patch no longer appear to be just a coincidence but evidence of the same phenomenon. Prior to this, the 240's region has been presumed to be simply crystal packing contact in some crystallographic space groups, not a biologically important interface. Close inspection reveals several buried hydrophobic interactions spanning the interface with a bulk of the interactions centered around the NTD (Asp-32, Pro-33, Ala-34, Lys-36, and Arg-44) and CTD (Phe-289, Pro-290, and Lys-291) of one dimer interacting with the far NTD and 240's regions (Lys-238, Tyr-239, Ser-241, and Ser-242) of another caspase-6 dimer in the tetramer (Fig. 9C). Arg-44 appears to act as the base of the interaction, orienting the far CTD for the proper positioning to interact with the substrate. Distinguishing biological interfaces from crystal contacts is challenging, but computational resources such as PISA (58) often facilitate disambiguation. The identified caspase-6 interface generates a dimer of dimers, with a buried surface area of 3894 Å2 and a total calculated ΔG of −15 kcal/mol. Biologically relevant interfaces generally have a total buried surface area greater than 1500 Å2 with ΔG values less than −10 kcal/mol (59). Thus, the peptides spanning from the NTD to the 240's region could represent a biologically relevant interface driving interactions of caspase-6 with substrates.
Figure 9.

Exosite is critical for stabilization of putative protein-binding interface. A, regions identified by H/Dx-MS are displayed on the caspase-6 structure 3K7E. The NTD (green) containing the exosite patch (blue) and 240's regions (red) make contacts with the CTD (orange) on the reverse face of caspase-6. B, rotating caspase-6 90° reveals a potential binding interface that occurs in the tetrameric helical structures of caspase-6, incorporating all regions identified by H/Dx. C, putative binding interface analyzed by PISA (58), containing large portions of buried surface area primarily consisting of buried hydrophobics. Residue label subscripts correspond to the chain of the crystal structure. D, fluorescence polarization assay of FITC-labeled caspase-6 C163S FL constructs of either the WT (black), R42–44A (white), or R44K (gray) variants incubated with the WT unlabeled protein as a substrate to assess KD apparent. E as in D except incubated with their respective unlabeled R42–44A C163S FL or R44K C163S FL variants. F, KD values of all labeled proteins incubated with the unlabeled WT substrate are not statistically significantly different, as compared to those incubated with R42–44A or R44K unlabeled proteins, which have a weaker KD value. In all cases, the caspase-6 variants are the ΔN D179CT form.
We designed a two-phased fluorescence polarization assay to determine the impact that disruption of the 42RRR44 patch has on the ability of caspase-6 to bind substrate proteins. It is important to note that when an enzyme uses an exosite, by definition its substrate must necessarily contain a corresponding exosite. In this particular experiment, the enzyme exosite and the substrate exosite comprise the same region (Fig. 9, A and B). In the first phase of the experiment, we used WT caspase-6 as the substrate as it would present a properly folded substrate exosite. FITC-labeled caspase-6 FL (active-site–inactivated C163S to prevent cleavage) incubated with increasing concentrations of unlabeled caspase-6 FL showed a KD of 379 nm (Fig. 9, D and F). Similarly, when the labeled R42–44A or R44K FL variants were incubated with increasing concentrations of the unlabeled WT caspase-6 FL, they showed statistically similar KD values of 999 and 420 nm, suggesting that one intact 42RRR44 patch may be sufficient to mediate a strong interaction. In the second phase of the experiment, both the enzyme and substrate exosites have been replaced. Unlabeled R42–44A or R44K were titrated into labeled R42–44A or R44K (Fig. 9E) and showed dramatic increases of KD at 2153 or 6344 nm for R42–44A or R44K, respectively (Fig. 9F). Perturbation of all the exosite patches in the reaction profoundly disrupts caspase-6 protein binding, demonstrating the importance of the 42RRR44 region for substrate recruitment. These analyses suggest that interface mediated by the NTD and the 240's region identified through H/Dx-MS and observed in a number of crystal structures is critical for protein–protein interactions between caspase-6 and its substrates. Overall, the 42RRR44 patch plays a key role in mediating the dynamic contacts between the NTD, the CTD, and the 240's region, which serves as an extended exosite face for protein substrate recognition (Fig. 9A).
Discussion
Precise and accurate control of each step during apoptosis could result in many therapeutic advantages. Understanding the global regulatory network and functions of the caspases, the final arbiters of apoptosis, is key to this goal. Why some substrates are more efficiently recognized by individual caspases and how caspases distinguish their unique pools of substrates is an active area of investigation. The discovery of enzyme exosites (21, 22), as well as substrate exosites (39), opens the door to altering caspase activity by targeting either the enzyme or the substrate at an exosite.
Given the need for potent therapies for neurodegenerative disorders, we focused our efforts on the study of caspase-6, aiming to uncover an exosite on caspase-6 that has been predicted previously (38, 54). We identified the 42RRR44 patch lying at the hinge between the caspase-6 core and disordered NTD, which functions as an exosite in recognition of many substrates. H/Dx-MS analysis uncovered a network connecting the NTD and 240's regions, which appears to function as a protein recognition interface. The fact that binding affinities for protein substrate were severely weakened when the 42RRR44 patch was compromised, while the intrinsic catalytic activity against peptide substrates and the H/Dx-MS rates for the core were not impacted, is consistent with the function of this region being an exosite.
The exosite centered at 42RRR44 shares characteristics with exosites identified in proteases, including thrombin (60, 61), the cathepsin family (62, 63), and β-secretase (BACE) (64), as well as in kinases (65) that enable recruitment of protein substrates. Exosite compositions in various enzymes differ based on their intended substrates. Although some exosites are described primarily as being composed of hydrophobic interactions, others critically involve charged patches. The level of hydrophobicity in cathepsin exosites has been described as the distinguishing factor in the altered specificities between family members (63). Thrombin uses two separate electropositive exosite patches. Exosite 1 (residues Phe-34, Ser-36, Leu-65, Tyr-76, Arg-77, Phe-82, and Lys-110) is less charged, has more hydrophobic residues, and influences binding to substrates like fibrinogen (66) and PAR-1 (67). Na+ binding to exosite 1 enhances catalytic activity (60, 68, 69). Thrombin exosite 2 (Arg-93, Arg-97, Arg-101, Arg-233, Lys-236, and Gln-239) is highly positively charged and is used to engage substrates like heparin (60, 70). The exosite discovered on caspase-6 features residues within the NTD and spans the dimer interface interacting with the 240's region (Asp-23, Pro-33, Ala-34, Lys-36, Arg-44, Lys-238, Tyr-239, Ser-241, Ser-242, Phe-289, Pro-290, and Lys-291). These residues comprising the exosite on caspase-6 share a similar composition to exosites in other proteases. Caspase-6 also contains a stretch of amino acids 24AFYKREMF31, which is unresolved in all caspase-6 structures, and which we previously identified as being intrinsically disordered and thus likely to participate in protein–protein interactions (55). Intrinsically disordered domains often bind partners with high specificity but low affinity due to the transient and reversible nature of their folding (71–73). These characteristics are consistent with the binding and release properties required by the caspase family for rapid substrate processing.
Together, these data suggest a model for how substrate is initially recognized by caspase-6. Prior to substrate binding, caspase-6 exists in a dynamic equilibrium between the helical and strand conformations in the 130's region. Our data suggest that the dynamics of the NTD surrounding the 42RRR44 patch is coupled to the conformation of the 130's region. Early interactions between substrate and the 42RRR44 exosite promote rapid, proximity-mediated formation of the ES complex that locks caspase-6 into the canonical strand conformation (Fig. 10). When the 42RRR44 exosite is disrupted, we observe a weakening of Km due to the loss of early interactions with the 42RRR44 exosite. Additionally, it is intriguing that POOL predicts distal residues Arg-44, Cys-113, Cys-116, and Lys-156 for the strand conformation but not for the helical conformation. This suggests that the distal region has strong couplings between protonation events, a hallmark of biochemical activity, but only in the strand conformation. These interactions modulate reversible substrate-binding and/or dynamical processes that contribute to substrate recognition and catalysis. In the helical conformation, these interactions are disrupted (74, 75). Substitution of the NTD exosite in caspase-7 (22) showed similar impacts on Km. Notably, the least conserved region of caspases is the N-terminal region. Based on our work and the work of others, it appears that differences in sequence and length of the NTD in caspases is responsible for tuning interactions with protein substrates, both substrates that are common to multiple caspases and those unique to a single caspase.
Figure 10.

Model of caspase-6 protein substrate recognition. In the mature state, caspase-6 dynamically interconverts between the unique helical conformation and the canonical strand conformations (blue) (30). Herein, we propose, consistent with other models, that the intrinsically disordered NTD of caspase-6 fishes for protein substrates within the context of the cell, utilizing the unique sequences and charges of the domain to tune protein substrate recognition (green). The intrinsically disordered domain of caspase-6 is likely binding to the disordered domains of the substrate proteins (21, 55). The NTD orients the protein substrate, facilitating formation of the enzyme–substrate complex, by docking with the protein-binding interface of caspase-6. This step also coincides with the transition to the obligate strand state, which is required for enzymatic processing (red). Once assembled, chemistry proceeds producing cleaved product (yellow), wherein the remaining substrate is released allowing transition back to the dynamic resting state of mature caspase-6 (blue). Substitutions of the exosite at 42RRR44 are not proposed to affect the dynamic resting state of caspase-6, as it will still undergo helix-to-strand transitions, albeit at a different rate (blue) (Fig. 7A). However, upon substrate recruitment (green) and transition into the ES complex (red), the substitutions at the exosite of caspase-6 impact the dynamics of the NTD (Fig. 7), unhinging it, leading to perturbations of the protein docking face of the enzyme (Fig. 9, B and C) and the decreased rate of full substrate engagement, observed indirectly through the increased KD apparent of substrate binding (orange) (Fig. 9, D–F). Once the ES complex is successfully assembled (red), chemistry will proceed as expected (Fig. 4, A and B) leading to product formation (yellow) and back to the resting state of caspase-6 (blue).
In addition to its role as an exosite, the caspase-6 NTD contributes to the mechanism of zinc-mediated caspase-6 inhibition through a binding site composed of Lys-36, Glu-244, and His-287 (76). Although it was clear that zinc binding locked caspase-6 into the helical conformation, which only caspase-6 can attain (52, 57), the molecular details for how zinc promoted the helical state was unclear. Prior to now, we could only hypothesize that changes in the dynamics upon zinc binding lead to the helical state. In this work, we observed that alterations to the 42RRR44 patch led to an increase in the dynamics of the 130's region in caspase-6 suggesting that perhaps the unique helical state is coupled to the dynamics of the NTD, thus offering an explanation as to how distal zinc binding near our identified interface is able to mediate the forced helical state of caspase-6.
These data on caspase-6 also shed light on two long-standing mysteries surrounding an alternatively spliced variant, caspase-6B. Caspase-6B lacks residues 14–103 spanning the NTD. Although caspase-6B has an intact catalytic dyad (His-144 and Cys-163), it is intrinsically inactive and can bind and directly inhibit the activation of procaspase-6 to its mature and active state (77). Our data suggest that the dynamics of the NTD are coupled to the 130's region, which governs the transition to and from the natively inactive helical conformation. As caspase-6B lacks the NTD, this could result in a strictly helical and thus inactive enzyme. The binding interface between caspase-6 and caspase-6B has not been identified. Given that the NTD is missing from caspase-6B, it seems likely that the interaction is centered in the small subunits of caspase-6 and caspase-6B. Our data show that the 240's region is critical for protein binding, a region wholly maintained within the small subunit of caspase-6B. Therefore, it is tempting to speculate that this region of the protein-binding exosite we have identified (Fig. 9A) is also the platform mediating the interactions of caspase-6 with caspase-6B.
In summary, our study has identified a bona fide exosite on caspase-6. The importance of the exosite to caspase-6 function cannot be understated, as clearly emphasized by the investigation into the cancer mutation R44K (37). Minor alterations in charge critically disrupt caspase-6 recognition and turnover of all protein substrates, which could ultimately contribute to progression in disease. The tri-arginine patch plays a key role in stabilizing and mediating the dynamics of the NTD, which was directly observed to impact the extended exosite of caspase-6. Further investigation of the disordered NTD outside of the tri-arginine hinge may reveal that different proteins engage different subsections of the full exosite (Fig. 9A), similar to the way substrates interact with enzymes such as thrombin or cathepsin. Moreover, identification of this protein-binding interface provides a whole new region distal from the active site to tune the activity of this enzyme against select substrates while maintaining its crucial enzymatic activity (Fig. 4, A and B). This study furthers the narrative that caspases are utilizing distal, exosite-based interactions to recruit their protein substrates and provides evidence in favor of an intra-dimer protein substrate recruitment model. With this discovery of an exosite on a new caspase, it seems clear that other caspases engage in similar interactions to tune and promote protein substrate recognition. Further investigation into the nonconserved NTD of this family of enzymes is certainly warranted.
Experimental procedures
DNA expression constructs
The WT caspase-6 C-terminally His6-tagged human Escherichia coli optimized gene was synthesized (Celtek Bioscience) by ligation into the NdeI/BamHI sites of pET11a (52). The construct was used in both the FL C163S active-site knockouts and the constitutive CT construct (52). The exosite knockout (R42–44A) and cancer mutation (R44K) were made in both the FL active-site knockout and CT constructs using QuikChange mutagenesis (Agilent Technologies).
Caspase evolutionary conservation assessment
All caspase sequences (caspase-1–10 and caspase-14) were curated from the database CaspBase.org (40). The database contains 2879 verified sequences spanning 10 phyla, 26 classes, and 353 taxa. Each individual caspase, covering all verified sequences ranging from 10's to 100's of sequences of that specific enzyme, was aligned using Clustal Omega. The resulting multiple sequence alignments were analyzed using ConSurf. As recommended, greater than 10 verified sequences were used to maximize the quality of the corresponding conservation results. First, the conservation scores were generated and overlaid onto the previously generated multiple sequence alignments to provide perspective of the site-specific conservation between each enzyme. Second, all verified human caspase sequences were aligned using Clustal Omega and then analyzed for conservation with ConSurf. The resultant data were also mapped to the caspase-6 sequence to provide perspective of site-specific conservation between caspase-6 and all human caspases.
POOL
The computational method POOL was used to predict the residues in various caspase-6 structures that are important for biochemical function. Caspase-6 structures were picked based on the observed conformational state, either the helical state: PDB code 2WDP (78), PDB codes 3K7E and 3QNW (79), and PDB code 4FXO (76) or the strand state: PDB codes 3P45 and 3NR2 (80), PDB code 4IYR (81), PDB code 3OD5 (80), and PDB code 3P4U. POOL is a machine learning, monotonicity-constrained multidimensional isotonic regression method that predicts the residues of all types in a given protein structure that are catalytically important (47, 48). This method can accept multiple types of input features. For this analysis, THEMATICS (82) metrics and the structure-only version of ConCavity (83) were used. THEMATICS (THEoretical Microscopic Anomalous TItration Curve Shapes) is a functional site prediction method that identifies only the ionizable side chains of amino acid residues in a protein 3D structure that exhibit unusual electrostatic properties. The method generates theoretical titration curves by calculating the electrostatic potential function and the average charge of each ionizable residue as a function of pH. If these titration curve shapes are perturbed from the normal Henderson-Hasselbalch sigmoid form (84), the corresponding residue is scored as being likely to be catalytically important. The conceptual basis for THEMATICS is that local sites involved in catalysis and/or reversible binding are characterized by strong coupling between protonation events. Because THEMATICS makes predictions based on only ionizable residues, POOL uses electrostatic data from THEMATICS to calculate environment variables for amino acids of all 20 types in the protein structure; these environment variables correlate with functional importance. In addition, POOL can incorporate input features from ConCavity, which looks at the overall topology of a protein structure and scores pocket areas where a substrate/ligand could bind using structure-based geometric features. POOL takes the structure of a given protein as its input and generates a rank-ordered list of residues important for catalysis in that protein based on the combination of these methods. From this list, the top 12% of predicted residues were used in further analysis.
Caspase cleavage of peptide substrates
Caspase-6, R42–44A, and R44K CT were assayed as follows: each enzyme was diluted into a 125 nm stock buffer containing 1× Caspase Cleavage Buffer (50 mm HEPES, pH 7.5, 100 mm NaCl, 0.5% CHAPS, 1 mm EDTA, and 10 mm DTT). Substrate titrations were performed with a final concentration of 100 nm enzyme and 0–400 μm N-acetyl-Val-Glu-Ile-Asp-amc (Ac-VEID-amc; 7-amino-4-methylcoumarin) (Enzo Life Sciences). Fluorescence of cleaved coumarin was measured in a Spectramax M5 spectrometer (Molecular Devices) at excitation 365/emission 495 nm over a 7-min time course sampling every 13 s. Assays were performed in duplicate on 3 separate days in 100-μl volumes in a 96-well microtiter plate. Resultant data were analyzed using Prism 5 (GraphPad Prism) to fit initial velocities versus the concentration to determine kinetic parameters of Km and Vmax values. Concentrations of caspase constructs were accurately determined utilizing an active-site titration with the quantitatively-reacting, covalent peptide inhibitor mimetic N-acetyl-Val-Glu-Ile-Asp-cho (Enzo Life Sciences). Inhibitors were diluted and incubated with 90 μl of enzyme in a series from 0 to 300 nm inhibitor and 100 nm expected final concentrations for 1 h at room temperature in 1× Caspase Cleavage Buffer. After 1 h, 90 μl of the labeled enzyme was assayed with 10 μl of a final concentration of 100 μm N-acetyl-Val-Glu-Ile-Asp-amc in a Spectramax M5 spectrophotometer. The plot of relative fluorescence units/s versus enzyme concentration allows assessment of the lowest concentration of inhibitor capable of full inhibition of the enzyme, where Y = 0. This value is taken as the total enzyme concentration. We find this active-site titration to be more accurate than the estimation of caspase concentration by A280, which may under-/overestimate the caspase concentration due to contamination of other proteins in the preparation. These values are used to calculate concentrations of enzyme in stock solution used for future enzymatic assays and for calculation of kcat.
Caspase cleavage of protein substrates
Assessment of caspase activity was performed in two ways, as a time course and as a dilution series. For the time-course assessment, caspase-6, R42–44A, and R44K were diluted into assay conditions with a final concentration of 1× Caspase Cleavage Buffer and 100 nm enzyme. Full-length uncleaved caspase-6 as a substrate was incubated at 5 μm at 37 °C and sampled at times 0, 5, 15, 30, 45, and 60 min. Samples were diluted in SDS-PAGE loading dye, Laemmli buffer (New England Biolabs), and boiled at 95 °C for 10 min to stop the cleavage reaction at each time point. Samples were loaded into a 4–20% gradient gel (Bio-Rad) and electrophoresed at 170 V for 50 min. Gels were quantified using Image Lab software (Bio-Rad) and plotted with Prism 5 (GraphPad Prism).
Analysis by dilution series was also used. Hydrolysis of lamin C as a substrate was performed by titrating each caspase variant from 25 to 0 μm using a 2-fold serial dilution of enzyme. The concentration of lamin C was held constant at 2.5 μm. The mixtures were incubated at 37 °C for 4 h, upon which the samples were diluted with SDS-PAGE loading dye, Laemmli buffer, and boiled at 95 °C for 10 min to stop the cleavage reaction. Samples were loaded into a 4–20% gradient gel and electrophoresed at 170 V for 50 min. Gels were quantified using Image Lab software, and the data were analyzed using Prism 5. Data were fit to a one-phase decay to find the CF50 (cleavage fraction 50, where 50% of the substrate protein was cleaved). CF50 was used to calculate the hydrolysis rate in m−1 s−1, from the equation k = ((−ln(P))/(E·t), where k is the hydrolysis rate; P is the portion of the portion or the fraction cleaved (50%, 0.5); E is the concentration at which the CF50 is achieved in m, and t is time in seconds. Resultant data were compared with the WT caspase-6 and analyzed with an unpaired two-tailed t test to the 90 and 95% confidence intervals to determine statistical significance. Assays were performed in triplicate on 3 separate days.
Caspase kinetic cleavage of protein substrates in human whole-cell lysates
Whole-cell lysates were isolated from SK-N-AS neuroblastoma cells (ATCC). Briefly, cells were grown to ∼70–90% confluency in a T175 cm2 flask in Dulbecco's modified Eagle's medium (Gibco, ThermoFisher Scientific) supplemented with 10% fetal bovine serum (HyClone), 1% Glutamax (Gibco, ThermoFisher Scientific), 1% minimum Eagle's medium non-essential amino acids (Gibco, ThermoFisher Scientific), 1% sodium pyruvate (Gibco, ThermoFisher Scientific), and 1% penicillin/streptomycin (Gibco, ThermoFisher Scientific). Upon reaching confluency, cells were washed two times with 1× PBS (Gibco, ThermoFisher Scientific) and incubated with 3 ml of sterile filtered 1× Triton X-100 lysis buffer (50 mm Tris, pH 8.0, 150 mm NaCl, 1% Triton X-100) and 1× Halt Protease/Phosphatase inhibitor mixture (Pierce, ThermoFisher Scientific) with constant rotation at 4 °C for 25 min. Lysates were extracted and clarified for 30 min at 16,000 × g at 4 °C then snap-frozen in liquid nitrogen and stored until use. For use, lysates were thawed on ice, and the total concentration of protein in the cell lysates was measured using a bicinchoninic acid assay (BCA) (Pierce, ThermoFisher Scientific). A standard curve of BSA was diluted in 1× Triton X-100 lysis buffer, and the assay was performed by using the manufacturer's protocol. Assay absorbance was measured at 562 nm using a Spectramax M5 spectrophotometer in a 96-well clear bottom plate (Corning Inc.). 300 μg of total protein was diluted into 1× Caspase Cleavage Buffer with a final concentration of 10 mm DTT and 100 nm caspase and brought to a final volume of 500 μl. Samples were incubated at 37 °C and sampled at times 0, 0.5, 1, 3, and 6 h. Samples were diluted with SDS-PAGE loading dye and Laemmli buffer (New England Biolabs) and boiled at 95 °C for 10 min to stop the reaction. Samples were stored at room temperature for no longer than 24 h prior to analysis by Western blotting. Independent assays were repeated on 3 separate days. Blots were quantified using the Image Lab software (Bio-Rad) and normalized to the loading control β-actin. Loading control normalized values from all assays were set to a 1–100 scale using Prism 5 (GraphPad Prism). Resultant data were compared with the WT caspase-6 data set using an unpaired two-tailed t test at the 90 or 95% confidence intervals.
Caspase titration for cleavage of protein substrates in human whole-cell lysates
For DVL3 hydrolysis, rather than performing a kinetic assay, a titration of caspase was performed with whole-cell lysates prepared as described above. Caspases were diluted in a 2-fold serial dilution from 250 to 0 nm in a final concentration of 1× Caspase Cleavage Buffer. 30 μg of total protein was added to the serial dilution to bring the final volume of the reactions to 15 μl. Samples were incubated at 37 °C for 1 h and then immediately boiled at 95 °C for 10 min with 5 μl of SDS-PAGE loading dye and Laemmli buffer (New England Biolabs) to stop the reaction. Samples were stored for no longer than 24 h prior to analysis by Western blotting. Independent assays were repeated on 3 separate days. Blots were quantified using the Image Lab software (Bio-Rad) and normalized to the loading control β-actin. DLV3 data were further analyzed by a one-phase decay as described above to calculate the hydrolysis rate of the three enzymes. Data for caspase-6 variants were compared with WT caspase-6 to determine statistical significance using an unpaired two-tailed t test at the 90 or 95% confidence intervals.
Immunoblotting
Samples were loaded so that the same total volume of lysate was added into each well of a 4–20% gradient gel (Bio-Rad) and run at 170 V for 50 min. Samples were transferred to a polyvinylidene difluoride membrane (Millipore). After transfer, blots were sliced into the appropriate molecular weight ranges to probe for specific proteins and subsequently blocked using OneBlock Western-CL blocking buffer (Genesee Scientific). Blots were probed with the following primary antibodies, also diluted in OneBlock (note: all antibodies were rabbit anti-human): anti-PARP (Cell Signaling catalog no. 9532); anti-lamin A/C (Cell Signaling catalog no. 2032); anti-DJ-1 (Cell Signaling catalog no. 5560); anti-β-actin (Cell Signaling catalog no. 4970); and anti-DVL3 (Cell Signaling catalog no. 3218). Incubation with the primary antibody proceeded overnight at 4 °C with constant rotation. Blots were washed with 1× TBST three times for 5 min each and then probed with secondary goat anti-rabbit antibody (Jackson ImmunoResearch catalog no. 111-035-144) at 1:20,000 dilution for 1 h 15 min at room temperature. Blots were incubated with ProSignal Femto ECL Reagent (Genesee Scientific) for 2–3 min at room temperature and imaged using the ChemiDoc MP Imaging System (Bio-Rad). Blots were quantified using the Image Lab software (Bio-Rad) and normalized to the β-actin loading control.
H/Dx-mass spectrometry
Our H/Dx analyses were performed as reported previously with minor modifications (30, 55). H/Dx experiments were performed using a protein stock at the initial concentration of 30 μm of each of the caspase-6 variants or 30 μm WT caspase-6 with 30 μm lamin C in 20 mm Tris, pH 8.5, 200 mm NaCl, 5% glycerol, and 2 mm DTT in H2O. The protein samples were injected into the nanoACQUITY system equipped with H/Dx technology for UPLC separation (Waters Corp. (85)) to generate mapping experiments used to assess sequence coverage. Generated maps were used for all subsequent exchange experiments. H/Dx was performed by diluting the initial 30 μm protein stock 15-fold with D2O (Cambridge Isotopes) containing buffer (10 mm phosphate, pD 7.5, 200 mm NaCl) and incubated at 10 °C for various time points (0.17, 1, 10, 60, and 120 min). At the designated time point, an aliquot from the exchanging experiment was sampled and diluted into D2O quenching buffer containing (100 mm phosphate, pH 2.5, 200 mm NaCl) at 1 °C. The process was repeated at all time points, including for nondeuterated samples in H2O-containing buffers. Quenched samples were injected into a 5-μm BEH 2.1 × 30-mm Enzymate-immobilized pepsin column (Waters Corp.) at 100 μl/min in 0.1% formic acid at 10 °C and then incubated for 4.5 min for on-column digestion. Peptides were collected at 0 °C on a C18 VanGuard trap column (1.7 μm × 30 mm) (Waters Corp.) for desalting with 0.1% formic acid in H2O and then subsequently separated with an in-line 1.8 μm Hss T3 C18 2.1 × 30-mm nanoACQUITY UPLC column (Waters Corp.) for a 10-min gradient ranging from 0.1% formic acid to acetonitrile (7 min, 5–35%; 1 min, 35–85%; 2 min hold 85% acetonitrile) at 40 μl/min at 0 °C. Fragments were mass-analyzed using the Synapt G2Si ESL-Q-ToF mass spectrometer (Waters Corp.). Between injections, a pepsin-wash step was performed to minimize the previously determined caspase-6 peptide carryover. Mass and collision-induced dissociation in data-independent acquisition mode (MSE) and ProteinLynx Global Server (PLGS) version 3.0 software (Waters Corp.) were used to identify the peptides in the undeuterated mapping experiments and analyzed in the same fashion as H/Dx experiments. Mapping experiments generated from PLGS were imported into the DynamX version 3.0 (Waters Corp.) with quality thresholds of MS1 signal intensity of ≥5000, maximum sequence length of 25 amino acids, minimum products per amino acid of ≥0.1. Automated results were inspected manually to ensure the corresponding m/z and isotopic distributions at various charge states were assigned to the appropriate peptide. DynamX was utilized to generate the relative deuterium incorporation plots and H/Dx heat map for each peptide. The relative deuterium uptake of common peptides was determined by subtracting the weighted-average mass of the centroid of the undeuterated control samples from the deuterated samples at each time point. All experiments were made under the same experimental conditions negating the need for back-exchange calculations but therefore are reported as relative (56). All H/Dx experiments were performed twice, on 2 separate days, and a 98 and 95% confidence limit of uncertainty was applied to calculate the mean relative deuterium uptake of ± 0.42 or 0.73 for the R42–44A or R44K datasets, respectively. Mean relative deuterium uptake thresholds were calculated as described previously (86). Differences in deuterium uptake that exceeded the error of the datasets were considered significant.
Thermal stability analysis by differential scanning fluorimetry
The thermal stability of each caspase and respective variants was measured in the presence and absence of 3× molar excess of inhibitor N-acetyl-Val-Glu-Ile-Asp-aldehyde for 1 h at room temperature. Caspases were subsequently incubated at 8–10 μm in caspase-6 activity assay buffer (100 mm HEPES, pH 7.5, 100 mm NaCl, 0.1% CHAPS, 10% sucrose, 5 mm DTT) in the presence of 5× SYPRO® Orange dye (Sigma). Stability was measured using a CFX Connect Real-Time PCR detection system (Bio-Rad). Reactions were 50 μl in a 96-well plate. Fluorescence intensity was recorded at increasing temperatures at 0.5 °C intervals from 25 to 95 °C. Thermal melting points (Tm) values were calculated by Boltzmann sigmoidal curve-fitting analysis using Prism 5 (GraphPad) software, where V50 is equal to the Tm.
Fluorescence polarization
Fluorescence polarization protocol was adapted from Ref. 39. Caspase-6 C163S FL active-site knockout and variants R42–44A/C163S (R42A/R43A/R44A/C163S) and R44K/C163S were exchanged via filtration into a buffer containing 100 mm carbonate buffer, pH 9.0, 200 mm NaCl using Nap-5 (GE Healthcare) desalting columns, and then incubated with 20× molar ratio of FITC (Sigma Millipore) for 2 h at room temperature with constant stirring. After labeling, the enzymes were exchanged into a minimal buffer containing 50 mm Tris, pH 8.0, 200 mm NaCl by Nap-25 (GE Healthcare). Exchanged proteins were filtered through a 10,000 MWCO Amicon spin filter (EMD Millipore) to remove residual free fluorophore. 20 μl of the FITC-labeled caspase variants at 250 nm were added to 80 μl of a 2-fold serial titration containing the substrates of interest to achieve a 30 to 0 μm final concentration. Proteins were incubated at 37 °C and imaged every 15 min in a Spectramax M5 spectrometer (Molecular Devices) at excitation 495/emission 519 nm until equilibrium was reached. Data were analyzed using Prism 5 (GraphPad Prism) and fit to a Boltzmann sigmoidal equation to identify the KDapparent value of the interaction.
Caspase-6 expression and purification
Plasmids encoding human caspase-6 and all variants were transformed into BL21(DE3) E. coli cells. Single colony was picked and placed into a flask containing 50 ml of Lennox Broth (LB) media with ampicillin (100 μg/ml, ThermoFisher Scientific) to make a seed culture. Culture was grown overnight at 37 °C until dense. Large cultures were inoculated with 2–3 ml of dense seed culture and grown in 1000 ml of LB media with ampicillin (100 μg/ml, ThermoFisher Scientific) at 37 °C until they reached an OD600 of 0.6. The temperature was reduced to 20 °C, and cells were induced with 1 mm IPTG (Gold Biotechnology) for 3 h to express soluble proteins. Cell pellets were stored at −80 °C, freeze-thawed, and lysed in a microfluidizer (Microfluidics, Inc.) in a buffer containing 50 mm Tris, pH 8.5, 300 mm NaCl, 5% glycerol, and 2 mm imidazole. Lysed cells were centrifuged at 27,000 rcf to remove cellular debris. The supernatant was loaded onto a 5-ml HiTrap nickel-affinity column (GE Healthcare). The column was washed with a buffer of 50 mm Tris, pH 8.5, 300 mm NaCl, 5% glycerol, and 50 mm imidazole. Caspase-6 was eluted with a step gradient to 300 mm imidazole. The eluted fraction was diluted 6-fold into a buffer containing 20 mm Tris, pH 8.5, and 2 mm DTT to reduce the salt concentration. This protein was loaded onto a 5-ml HiTrap Q column (GE Healthcare). The column was developed with a linear NaCl gradient. Protein eluted in 120 mm NaCl and was assessed for purity by SDS-PAGE and stored at −80 °C in elution buffer.
Lamin C expression and purification
Plasmids encoding human lamin C were transformed into BL21(DE3) E. coli cells. Cultures were grown in LB media with ampicillin (100 μg/ml, ThermoFisher Scientific) at 37 °C until they reached an OD600 of 0.6. The temperature was reduced to 18 °C, and cells were induced with 0.1 mm IPTG (Gold Biotechnology) for 15 h to express soluble proteins. Cell pellets were stored at −80 °C, freeze-thawed, and lysed in a microfluidizer (Microfluidics, Inc.) in a buffer containing 100 mm phosphate, pH 8.0, 100 mm NaCl, and 2 mm imidazole. Lysed cells were centrifuged at 27,000 rcf to remove cellular debris. The supernatant was loaded onto a 5-ml HiTrap nickel-affinity column (GE Healthcare). The column was washed with a lysis buffer. Lamin C was eluted with a shallow gradient over 60 min to 300 mm imidazole. The eluted fractions were pooled and diluted 6-fold into a buffer containing 20 mm Tris, pH 8.0, 1 mm EDTA, and 2 mm DTT to reduce the salt concentration. This protein was loaded onto a 5-ml HiTrap Q column (GE Healthcare). The column was developed with a linear NaCl gradient. Protein eluted in 320 mm NaCl and was assessed for purity by SDS-PAGE. Fractions containing lamin C were pooled and concentrated to 1 ml using the Amicon Ultra-15 10,000 MWCO spin filter (Millipore). Proteins were loaded onto a 26/600 Superdex 200 pg column (GE Healthcare Inc,) and eluted at 2 ml/min in sizing buffer containing 20 mm Tris, pH 8.0, 100 mm NaCl, 2 mm EDTA, 5% glycerol, and 2 mm DTT. Fractions containing lamin C were eluted at ∼90 min/180 ml and were assessed for purity by SDS-PAGE. Protein was aliquoted and stored at −80 °C in sizing buffer.
Author contributions
D. J. M. and J. A. H. conceptualization; D. J. M. and J. A. H. data curation; D. J. M., C. L. M., M. J. O., and J. A. H. formal analysis; D. J. M., C. L. M., and M. J. O. investigation; D. J. M. and J. A. H. methodology; D. J. M. writing-original draft; D. J. M., C. L. M., M. J. O., and J. A. H. writing-review and editing; M. J. O. resources; M. J. O. software; J. A. H. supervision; J. A. H. funding acquisition; J. A. H. project administration.
Supplementary Material
Acknowledgments
We thank A. C. Clark for access to the CaspBase prior to publication. We thank Dr. Stephen J. Eyles for his continued and excellent support during all hydrogen–deuterium exchange experiments performed at the Mass Spectrometry Core Facility within the Institute for Applied Life Sciences at University of Massachusetts, Amherst.
This work was supported by National Institutes of Health Grant GM80532 (to J. A. H.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S8 and Table S1.
- NTD
- N-terminal domain
- amc
- 7-amino-4-methylcoumarin
- casp
- caspase
- FL
- full-length
- cho
- aldehyde
- H/Dx
- hydrogen/deuterium exchange
- MS
- mass spectrometry
- POOL
- Partial Order Optimum Likelihood
- THEMATICS
- THEoretical Microscopic Titration Curve Shapes
- CatK
- cathepsin K
- PDB
- Protein Data Bank
- PARP
- poly(ADP-ribose) polymerase
- IPTG
- isopropyl β-d-1-thiogalactopyranoside
- rcf
- relative centrifugal force
- CT
- constitutive two-chain
- CTD
- C-terminal domain.
References
- 1. Elmore S. (2007) Apoptosis: a review of programmed cell death. Toxicol. Pathol. 35, 495–516 10.1080/01926230701320337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pistritto G., Trisciuoglio D., Ceci C., Garufi A., and D'Orazi G. (2016) Apoptosis as anticancer mechanism: function and dysfunction of its modulators and targeted therapeutic strategies. Aging 8, 603–619 10.18632/aging.100934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Greenblatt M. S., Bennett W. P., Hollstein M., and Harris C. C. (1994) Mutations in the p53 tumor suppressor gene: clues to cancer etiology and molecular pathogenesis. Cancer Res. 54, 4855–4878 [PubMed] [Google Scholar]
- 4. Vaux D. L., Cory S., and Adams J. M. (1988) Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature 335, 440–442 10.1038/335440a0 [DOI] [PubMed] [Google Scholar]
- 5. Plati J., Bucur O., and Khosravi-Far R. (2008) Dysregulation of apoptotic signaling in cancer: molecular mechanisms and therapeutic opportunities. J. Cell. Biochem. 104, 1124–1149 10.1002/jcb.21707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Margolis R. L., Chuang D.-M., and Post R. M. (1994) Programmed cell death: implications for neuropsychiatric disorders. Biol. Psychiatry 35, 946–956 10.1016/0006-3223(94)91241-6 [DOI] [PubMed] [Google Scholar]
- 7. Ghavami S., Shojaei S., Yeganeh B., Ande S. R., Jangamreddy J. R., Mehrpour M., Christoffersson J., Chaabane W., Moghadam A. R., Kashani H. H., Hashemi M., Owji A. A., and Łos M. J. (2014) Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog. Neurobiol. 112, 24–49 10.1016/j.pneurobio.2013.10.004 [DOI] [PubMed] [Google Scholar]
- 8. Walensky L. D. (2006) BCL-2 in the crosshairs: tipping the balance of life and death. Cell Death Differ. 13, 1339–1350 10.1038/sj.cdd.4401992 [DOI] [PubMed] [Google Scholar]
- 9. Friedlander R. M. (2003) Apoptosis and caspases in neurodegenerative diseases. N. Engl. J. Med. 348, 1365–1375 10.1056/NEJMra022366 [DOI] [PubMed] [Google Scholar]
- 10. McIlwain D. R., Berger T., and Mak T. W. (2015) Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 7, a026716 10.1101/cshperspect.a026716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mahrus S., Trinidad J. C., Barkan D. T., Sali A., Burlingame A. L., and Wells J. A. (2008) Global sequencing of proteolytic cleavage sites in apoptosis by specific labeling of protein N termini. Cell 134, 866–876 10.1016/j.cell.2008.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Timmer J. C., and Salvesen G. S. (2011) N-Terminomics: a high-content screen for protease substrates and their cleavage sites. Methods Mol. Biol. 753, 243–255 10.1007/978-1-61779-148-2_16 [DOI] [PubMed] [Google Scholar]
- 13. Dix M. M., Simon G. M., Wang C., Okerberg E., Patricelli M. P., and Cravatt B. F. (2012) Functional interplay between caspase cleavage and phosphorylation sculpts the apoptotic proteome. Cell 150, 426–440 10.1016/j.cell.2012.05.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schilling O., and Overall C. M. (2008) Proteome-derived, database-searchable peptide libraries for identifying protease cleavage sites. Nat. Biotechnol. 26, 685–694 10.1038/nbt1408 [DOI] [PubMed] [Google Scholar]
- 15. Julien O., Zhuang M., Wiita A. P., O'Donoghue A. J., Knudsen G. M., Craik C. S., and Wells J. A. (2016) Quantitative MS-based enzymology of caspases reveals distinct protein substrate specificities, hierarchies, and cellular roles. Proc. Natl. Acad. Sci. U.S.A. 113, E2001–E2010 10.1073/pnas.1524900113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Julien O., and Wells J. A. (2017) Caspases and their substrates. Cell Death Differ. 24, 1380–1389 10.1038/cdd.2017.44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Slee E. A., Adrain C., and Martin S. J. (2001) Executioner caspase-3, -6, and -7 perform distinct, non-redundant roles during the demolition phase of apoptosis. J. Biol. Chem. 276, 7320–7326 10.1074/jbc.M008363200 [DOI] [PubMed] [Google Scholar]
- 18. McStay G. P., Salvesen G. S., and Green D. R. (2008) Overlapping cleavage motif selectivity of caspases: implications for analysis of apoptotic pathways. Cell Death Differ. 15, 322–331 10.1038/sj.cdd.4402260 [DOI] [PubMed] [Google Scholar]
- 19. Thomsen N. D., Koerber J. T., and Wells J. A. (2013) Structural snapshots reveal distinct mechanisms of procaspase-3 and -7 activation. Proc. Natl. Acad. Sci. U.S.A. 110, 8477–8482 10.1073/pnas.1306759110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jabaiah A. M., Getz J. A., Witkowski W. A., Hardy J. A., and Daugherty P. S. (2012) Identification of protease exosite-interacting peptides that enhance substrate cleavage kinetics. Biol. Chem. 393, 933–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Martini C., Bédard M., Lavigne P., and Denault J. B. (2017) Characterization of Hsp90 co-chaperone p23 cleavage by caspase-7 uncovers a peptidase-substrate interaction involving intrinsically disordered regions. Biochemistry 56, 5099–5111 10.1021/acs.biochem.7b00298 [DOI] [PubMed] [Google Scholar]
- 22. Boucher D., Blais V., and Denault J.-B. (2012) Caspase-7 uses an exosite to promote poly(ADP ribose) polymerase 1 proteolysis. Proc. Natl. Acad. Sci. U.S.A. 109, 5669–5674 10.1073/pnas.1200934109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stennicke H. R., Renatus M., Meldal M., and Salvesen G. S. (2000) Internally quenched fluorescent peptide substrates disclose the subsite preferences of human caspases 1, 3, 6, 7 and 8. Biochem. J. 350, 563–568 10.1042/0264-6021:3500563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Talanian R. V., Quinlan C., Trautz S., Hackett M. C., Mankovich J. A., Banach D., Ghayur T., Brady K. D., and Wong W. W. (1997) Substrate specificities of caspase family proteases. J. Biol. Chem. 272, 9677–9682 10.1074/jbc.272.15.9677 [DOI] [PubMed] [Google Scholar]
- 25. Stoch S. A., and Wagner J. A. (2008) Cathepsin K inhibitors: a novel target for osteoporosis therapy. Clin. Pharmacol. Ther. 83, 172–176 10.1038/sj.clpt.6100450 [DOI] [PubMed] [Google Scholar]
- 26. Gauthier J. Y., Chauret N., Cromlish W., Desmarais S., Duong L. T., Falgueyret J. P., Kimmel D. B., Lamontagne S., Léger S., LeRiche T., Li C. S., Massé F., McKay D. J., Nicoll-Griffith D. A., Oballa R. M., et al. (2008) The discovery of odanacatib (MK-0822), a selective inhibitor of cathepsin K. Bioorg. Med. Chem. Lett. 18, 923–928 10.1016/j.bmcl.2007.12.047 [DOI] [PubMed] [Google Scholar]
- 27. Leung P., Pickarski M., Zhuo Y., Masarachia P. J., and Duong L. T. (2011) The effects of the cathepsin K inhibitor odanacatib on osteoclastic bone resorption and vesicular trafficking. Bone 49, 623–635 10.1016/j.bone.2011.06.014 [DOI] [PubMed] [Google Scholar]
- 28. Merck Newsroom Home (2015) Merck Announces Data from Pivotal Phase 3 Fracture Outcomes Study for Odanacatib, an Investigational Oral, Once-Weekly Treatment for Osteoporosis, Whitehouse Station, NJ [Google Scholar]
- 29. Panwar P., Søe K., Guido R. V., Bueno R. V., Delaisse J. M., and Brömme D. (2016) A novel approach to inhibit bone resorption: exosite inhibitors against cathepsin K. Br. J. Pharmacol. 173, 396–410 10.1111/bph.13383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dagbay K. B., Bolik-Coulon N., Savinov S. N., and Hardy J. A. (2017) Caspase-6 undergoes a distinct helix-strand interconversion upon substrate binding. J. Biol. Chem. 292, 4885–4897 10.1074/jbc.M116.773499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nikolaev A., McLaughlin T., O'Leary D. D., and Tessier-Lavigne M. (2009) APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature 457, 981–989 10.1038/nature07767 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32. Guo H., Albrecht S., Bourdeau M., Petzke T., Bergeron C., and LeBlanc A. C. (2004) Active caspase-6 and caspase-6-cleaved Tau in neuropil threads, neuritic plaques, and neurofibrillary tangles of Alzheimer's disease. Am. J. Pathol. 165, 523–531 10.1016/S0002-9440(10)63317-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Graham R. K., Deng Y., Carroll J., Vaid K., Cowan C., Pouladi M. A., Metzler M., Bissada N., Wang L., Faull R. L., Gray M., Yang X. W., Raymond L. A., and Hayden M. R. (2010) Cleavage at the 586 amino acid caspase-6 site in mutant Huntingtin influences caspase-6 activation in vivo. J. Neurosci. 30, 15019–15029 10.1523/JNEUROSCI.2071-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Giaime E., Sunyach C., Druon C., Scarzello S., Robert G., Grosso S., Auberger P., Goldberg M. S., Shen J., Heutink P., Pouysségur J., Pagès G., Checler F., and Alves da Costa C. (2010) Loss of function of DJ-1 triggered by Parkinson's disease-associated mutation is due to proteolytic resistance to caspase-6. Cell Death Differ. 17, 158–169 10.1038/cdd.2009.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Robert G., Puissant A., Dufies M., Marchetti S., Jacquel A., Cluzeau T., Colosetti P., Belhacene N., Kahle P., Da Costa C. A., Luciano F., Checler F., and Auberger P. (2012) The caspase-6 derived N-terminal fragment of DJ-1 promotes apoptosis via increased ROS production. Cell Death Differ. 19, 1769–1778 10.1038/cdd.2012.55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pakavathkumar P., Noël A., Lecrux C., Tubeleviciute-Aydin A., Hamel E., Ahlfors J. E., and LeBlanc A. C. (2017) Caspase vinyl sulfone small molecule inhibitors prevent axonal degeneration in human neurons and reverse cognitive impairment in caspase-6-overexpressing mice. Mol. Neurodegener. 12, 22 10.1186/s13024-017-0166-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lee J. W., Kim M. R., Soung Y. H., Nam S. W., Kim S. H., Lee J. Y., Yoo N. J., and Lee S. H. (2006) Mutational analysis of the CASP6 gene in colorectal and gastric carcinomas. APMIS 114, 646–650 10.1111/j.1600-0463.2006.apm_417.x [DOI] [PubMed] [Google Scholar]
- 38. Hill M. E., MacPherson D. J., Wu P., Julien O., Wells J. A., and Hardy J. A. (2016) Reprogramming caspase-7 specificity by regio-specific mutations and selection provides alternate solutions for substrate recognition. ACS Chem. Biol. 11, 1603–1612 10.1021/acschembio.5b00971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Eron S. J., Raghupathi K., and Hardy J. A. (2017) Dual site phosphorylation of caspase-7 by PAK2 blocks apoptotic activity by two distinct mechanisms. Structure 25, 27–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Grinshpon R. D., Williford A., Titus-McQuillan J., and Clay Clark A. (2018) The CaspBase: a curated database for evolutionary biochemical studies of caspase functional divergence and ancestral sequence inference. Protein Sci. 27, 1857–1870 10.1002/pro.3494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sievers F., Wilm A., Dineen D., Gibson T. J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., Thompson J. D., and Higgins D. G. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Celniker G., Nimrod G., Ashkenazy H., Glaser F., Martz E., Mayrose I., Pupko T., and Ben-Tal N. (2013) ConSurf: using evolutionary data to raise testable hypotheses about protein function. Isr. J. Chem. 53, 199–206 10.1002/ijch.201200096 [DOI] [Google Scholar]
- 43. Ashkenazy H., Erez E., Martz E., Pupko T., and Ben-Tal N. (2010) ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 38, W529–W533 10.1093/nar/gkq399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Landau M., Mayrose I., Rosenberg Y., Glaser F., Martz E., Pupko T., and Ben-Tal N. (2005) ConSurf 2005: the projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res. 33, W299–W302 10.1093/nar/gki370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ashkenazy H., Abadi S., Martz E., Chay O., Mayrose I., Pupko T., and Ben-Tal N. (2016) ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 44, W344–W350 10.1093/nar/gkw408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Glaser F., Pupko T., Paz I., and Bell R. E. (2003) ConSurf: identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics 19, 163–164 10.1093/bioinformatics/19.1.163 [DOI] [PubMed] [Google Scholar]
- 47. Tong W., Wei Y., Murga L. F., Ondrechen M. J., and Williams R. J. (2009) Partial order optimum likelihood (POOL): maximum likelihood prediction of protein active site residues using 3D structure and sequence properties. PLoS Comput. Biol. 5, e1000266 10.1371/journal.pcbi.1000266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Somarowthu S., Yang H., Hildebrand D. G., and Ondrechen M. J. (2011) High-performance prediction of functional residues in proteins with machine learning and computed input features. Biopolymers 95, 390–400 10.1002/bip.21589 [DOI] [PubMed] [Google Scholar]
- 49. Han G. W., Ko J., Farr C. L., Deller M. C., Xu Q., Chiu H. J., Miller M. D., Sefcikova J., Somarowthu S., Beuning P. J., Elsliger M. A., Deacon A. M., Godzik A., Lesley S. A., Wilson I. A., and Ondrechen M. J. (2011) Crystal structure of a metal-dependent phosphoesterase (YP_910028.1) from Bifidobacterium adolescentis: computational prediction and experimental validation of phosphoesterase activity. Proteins 79, 2146–2160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Parasuram R., Coulther T. A., Hollander J. M., Keston-Smith E., Ondrechen M. J., and Beuning P. J. (2018) Prediction of active site and distal residues in E. coli DNA polymerase IIIα polymerase activity. Biochemistry 57, 1063–1072 10.1021/acs.biochem.7b01004 [DOI] [PubMed] [Google Scholar]
- 51. Ringe D., Wei Y., Boino K. R., and Ondrechen M. J. (2004) Protein structure to function: insights from computation. Cell. Mol. Life Sci. 61, 387–392 10.1007/s00018-003-3291-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Vaidya S., Velázquez-Delgado E. M., Abbruzzese G., and Hardy J. A. (2011) Substrate-induced conformational changes occur in all cleaved forms of caspase-6. J. Mol. Biol. 406, 75–91 10.1016/j.jmb.2010.11.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Takahashi A., Alnemri E. S., Lazebnik Y. A., Fernandes-Alnemri T., Litwack G., Moir R. D., Goldman R. D., Poirier G. G., Kaufmann S. H., and Earnshaw W. C. (1996) Cleavage of lamin A by Mch2α but not CPP32: multiple interleukin 1 β-converting enzyme-related proteases with distinct substrate recognition properties are active in apoptosis. Proc. Natl. Acad. Sci. U.S.A. 93, 8395–8400 10.1073/pnas.93.16.8395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ehrnhoefer D. E., Skotte N. H., Savill J., Nguyen Y. T., Ladha S., Cao L. P., Dullaghan E., and Hayden M. R. (2011) A quantitative method for the specific assessment of caspase-6 activity in cell culture. PLoS ONE 6, e27680 10.1371/journal.pone.0027680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dagbay K. B., and Hardy J. A. (2017) Multiple proteolytic events in caspase-6 self-activation impact conformations of discrete structural regions. Proc. Natl. Acad. Sci. U.S.A. 114, E7977–E7986 10.1073/pnas.1704640114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wales T. E., and Engen J. R. (2006) Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom. Rev. 25, 158–170 10.1002/mas.20064 [DOI] [PubMed] [Google Scholar]
- 57. Vaidya S., and Hardy J. A. (2011) Caspase-6 latent state stability relies on helical propensity. Biochemistry 50, 3282–3287 10.1021/bi2001664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Krissinel E., and Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 10.1016/j.jmb.2007.05.022 [DOI] [PubMed] [Google Scholar]
- 59. Feldman T., Kabaleeswaran V., Jang S. B., Antczak C., Djaballah H., Wu H., and Jiang X. (2012) A class of allosteric caspase inhibitors identified by high-throughput screening. Mol. Cell 47, 585–595 10.1016/j.molcel.2012.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Huntington J. A. (2005) Molecular recognition mechanisms of thrombin. J. Thromb. Haemost. 3, 1861–1872 10.1111/j.1538-7836.2005.01363.x [DOI] [PubMed] [Google Scholar]
- 61. Bode W. (2006) The structure of thrombin: A Janus-headed proteinase. Semin. Thromb. Hemost. 32, 16–31 10.1055/s-2006-939551 [DOI] [PubMed] [Google Scholar]
- 62. Sharma V., Panwar P., O'Donoghue A. J., Cui H., Guido R. V., Craik C. S., and Brömme D. (2015) Structural requirements for the collagenase and elastase activity of cathepsin K and its selective inhibition by an exosite inhibitor. Biochem. J. 465, 163–173 10.1042/BJ20140809 [DOI] [PubMed] [Google Scholar]
- 63. Du X., Chen N. L., Wong A., Craik C. S., and Brömme D. (2013) Elastin degradation by cathepsin V requires two exosites. J. Biol. Chem. 288, 34871–34881 10.1074/jbc.M113.510008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kornacker M. G., Lai Z., Witmer M., Ma J., Hendrick J., Lee V. G., Riexinger D. J., Mapelli C., Metzler W., and Copeland R. A. (2005) An inhibitor binding pocket distinct from the catalytic active site on human β-APP cleaving enzyme. Biochemistry 44, 11567–11573 10.1021/bi050932l [DOI] [PubMed] [Google Scholar]
- 65. Horiuchi K. Y., Scherle P. A., Trzaskos J. M., and Copeland R. A. (1998) Competitive inhibition of MAP kinase activation by a peptide representing the α(c) helix of ERK. Biochemistry 37, 8879–8885 10.1021/bi972731q [DOI] [PubMed] [Google Scholar]
- 66. Wu Q. Y., Sheehan J. P., Tsiang M., Lentz S. R., Birktoft J. J., and Sadler J. E. (1991) Single amino acid substitutions dissociate fibrinogen-clotting and thrombomodulin-binding activities of human thrombin. Proc. Natl. Acad. Sci. U.S.A. 88, 6775–6779 10.1073/pnas.88.15.6775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Coughlin S. R. (1999) How the protease thrombin talks to cells. Proc. Natl. Acad. Sci. U.S.A. 96, 11023–11027 10.1073/pnas.96.20.11023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Di Cera E. (2006) Thrombin: a paradigm for enzymes allosterically activated by monovalent cations. Rend. Lincei. 17, 97–113 10.1007/BF02904503 [DOI] [PubMed] [Google Scholar]
- 69. Wells C. M., and Di Cera E. (1992) Thrombin is a Na+-activated enzyme. Biochemistry 31, 11721–11730 10.1021/bi00162a008 [DOI] [PubMed] [Google Scholar]
- 70. Carter W. J., Cama E., and Huntington J. A. (2005) Crystal structure of thrombin bound to heparin. J. Biol. Chem. 280, 2745–2749 10.1074/jbc.M411606200 [DOI] [PubMed] [Google Scholar]
- 71. Dyson H. J. (2016) Making sense of intrinsically disordered proteins. Biophys. J. 110, 1013–1016 10.1016/j.bpj.2016.01.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Babu M. M., van der Lee R., de Groot N. S., and Gsponer J. (2011) Intrinsically disordered proteins: regulation and disease. Curr. Opin. Struct. Biol. 21, 432–440 10.1016/j.sbi.2011.03.011 [DOI] [PubMed] [Google Scholar]
- 73. Mollica L., Bessa L. M., Hanoulle X., Jensen M. R., Blackledge M., and Schneider R. (2016) Binding mechanisms of intrinsically disordered proteins: theory, simulation, and experiment. Front. Mol. Biosci. 3, 52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Shehadi I. A., Yang H., and Ondrechen M. J. (2002) Future directions in protein function prediction. Mol. Biol. Rep. 29, 329–335 10.1023/A:1021220208562 [DOI] [PubMed] [Google Scholar]
- 75. Brodkin H. R., DeLateur N. A., Somarowthu S., Mills C. L., Novak W. R., Beuning P. J., Ringe D., and Ondrechen M. J. (2015) Prediction of distal residue participation in enzyme catalysis. Protein Sci. 24, 762–778 10.1002/pro.2648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Velázquez-Delgado E. M., and Hardy J. A. (2012) Zinc-mediated allosteric inhibition of caspase-6. J. Biol. Chem. 287, 36000–36011 10.1074/jbc.M112.397752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Lee A. W., Champagne N., Wang X., Su X. D., Goodyer C., and Leblanc A. C. (2010) Alternatively spliced caspase-6B isoform inhibits the activation of caspase-6A. J. Biol. Chem. 285, 31974–31984 10.1074/jbc.M110.152744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Baumgartner R., Meder G., Briand C., Decock A., D'arcy A., Hassiepen U., Morse R., and Renatus M. (2009) The crystal structure of caspase-6, a selective effector of axonal degeneration. Biochem. J. 423, 429–439 10.1042/BJ20090540 [DOI] [PubMed] [Google Scholar]
- 79. Müller I., Lamers M. B., Ritchie A. J., Dominguez C., Munoz-Sanjuan I., and Kiselyov A. (2011) Structure of human caspase-6 in complex with Z-VAD-FMK: new peptide binding mode observed for the non-canonical caspase conformation. Bioorg. Med. Chem. Lett. 21, 5244–5247 10.1016/j.bmcl.2011.07.041 [DOI] [PubMed] [Google Scholar]
- 80. Wang X.-J., Cao Q., Liu X., Wang K.-T., Mi W., Zhang Y., Li L.-F., LeBlanc A. C., and Su X.-D. (2010) Crystal structures of human caspase-6 reveal a new mechanism for intramolecular cleavage self-activation. EMBO Rep. 11, 841–847 10.1038/embor.2010.141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Cao Q., Wang X. J., Li L. F., and Su X. D. (2014) The regulatory mechanism of the caspase-6 pro-domain revealed by crystal structure and biochemical assays. Acta Crystallogr. D Biol. Crystallogr. 70, 58–67 10.1107/S1399004713024218 [DOI] [PubMed] [Google Scholar]
- 82. Ondrechen M. J., Clifton J. G., and Ringe D. (2001) THEMATICS: a simple computational predictor of enzyme function from structure. Proc. Natl. Acad. Sci. U.S.A. 98, 12473–12478 10.1073/pnas.211436698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Capra J. A., Laskowski R. A., Thornton J. M., Singh M., and Funkhouser T. A. (2009) Predicting protein ligand binding sites by combining evolutionary sequence conservation and 3D structure. PLoS Comput. Biol. 5, e1000585 10.1371/journal.pcbi.1000585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Ko J., Murga L. F., André P., Yang H., Ondrechen M. J., Williams R. J., Agunwamba A., and Budil D. E. (2005) Statistical criteria for the identification of protein active sites using theoretical microscopic titration curves. Proteins Struct. Funct. Genet. 59, 183–195 10.1002/prot.20418 [DOI] [PubMed] [Google Scholar]
- 85. Wales T. E., Fadgen K. E., Gerhardt G. C., and Engen J. R. (2008) High-speed and high-resolution UPLC separation at zero degrees celsius. Anal. Chem. 80, 6815–6820 10.1021/ac8008862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Houde D., Berkowitz S. A., and Engen J. R. (2011) The utility of hydrogen/deuterium exchange mass spectrometry in biopharmaceutical comparability studies. J. Pharm. Sci. 100, 2071–2086 10.1002/jps.22432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Wei Y., Fox T., Chambers S. P., Sintchak J., Coll J. T., Golec J. M., Swenson L., Wilson K. P., and Charifson P. S. (2000) The structures of caspases-1,-3,-7 and-8 reveal the basis for substrate and inhibitor selectivity. Chem. Biol. 7, 423–432 10.1016/S1074-5521(00)00123-X [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.