

Abstract
Matriptase is a member of the type-II transmembrane serine protease (TTSP) family and plays a crucial role in the development and maintenance of epithelial tissues. As all chymotrypsin-like serine proteases, matriptase is synthesized as a zymogen (proform), requiring a cleavage event for full activity. Recent studies suggest that the zymogen of matriptase possesses enough catalytic activity to not only facilitate autoactivation, but also carry out its in vivo functions, which include activating several proteolytic and signaling cascades. Inhibition of zymogen matriptase may therefore be a highly effective approach for limiting matriptase activity. To this end, here we sought to characterize the catalytic activity of human zymogen matriptase and to develop mAb inhibitors against this enzyme form. Using a mutated variant of matriptase in which the serine protease domain is locked in the zymogen conformation, we confirmed that the zymogen form of human matriptase has catalytic activity. Moreover, the crystal structure of the catalytic domain of zymogen matriptase was solved to 2.5 Å resolution to characterize specific antibody-based matriptase inhibitors and to further structure-based studies. Finally, we describe the first antibody-based competitive inhibitors that target both the zymogen and activated forms of matriptase. We propose that these antibodies provide a more efficient way to regulate matriptase activity by targeting the protease both before and after its activation and may be of value for both research and preclinical applications.
Keywords: epitope mapping, cancer therapy, enzyme kinetics, enzyme inhibitor, surface plasmon resonance (SPR), serine protease, matriptase, multi-domain protein, TTSP, zymogen
Introduction
Matriptase (also known as MT-SP1) is an 855-amino acid member of the type-II transmembrane serine protease (TTSP)3 family, characterized by a single-span transmembrane region followed by a multidomain architecture and a C-terminal catalytic domain with trypsin-like features (Fig. 1A) (1). An essential physiological role of matriptase was established when it was observed that matriptase knockout mice die within 48 h of birth due to a detrimental lack of epidermal barrier function (2). Matriptase is almost exclusively expressed by epithelial cells and has been shown to play an important role in physiological processes such as development of epidermis and hair follicles (2, 3). In fact, a mutant of matriptase with reduced activity has been shown to cause skin disease in humans (4). Matriptase is proposed to activate several growth factors, receptors, ion channels, and proteases responsible for the maintenance of homeostasis and epithelial tightness. Targets include, among others, pro-hepatocyte growth factor (pro-HGF), involved in cellular proliferation, morphogenesis, and angiogenesis, and protease-activated receptor-2 (PAR-2), involved in inflammatory responses (5–9). In summary, based on the variety of interacting proteins, matriptase is likely a central activator of multiple signaling and proteolytic cascades (3).
Figure 1.

Matriptase protein and constructs. A, schematic drawing of full-length matriptase with the N-terminal transmembrane region (black), the SEA domain (green), two CUB domains (orange), four LDLRa domains (blue), and the C-terminal serine protease domain (purple). The cleaved activation loop rearranges to create the catalytically active protease. B, schematic drawing of the zymogen-locked form of matriptase characterized in this paper, consisting of only the serine protease domain with position R614A mutated to avoid activation (zSPD). C, a Coomassie-stained SDS-PAGE (10%) shows that zSPD (consisting of only the serine protease domain, as shown in B) is pure and the expected size (25–30 kDa). D, the sequence of the crystallized form of zSPD (zSPD-S805A) with an additional mutation in the active site, shown with matriptase numbering (sides) and chymotrypsin numbering (top). The mutations are color-coded: R614A (blue), N772Q (green), and S805A (blue).
Matriptase activity is regulated by the Kunitz-type standard mechanism inhibitors hepatocyte growth factor activator inhibitor (HAI)-1 and -2 (10–12). Unregulated matriptase activity has been shown to play a role in several pathological conditions, including cancer. In several cancers, an excess of matriptase compared with its inhibitors HAI-1 and HAI-2 is observed, which shifts the balance in favor of protease activity, causing detrimental pro-carcinogenic effects (13, 14). Moreover, elevated matriptase protein levels in tumors have been shown to correlate with a poor prognosis (15).
Classically, chymotrypsin-like serine proteases are synthesized as inactive precursors, termed zymogens. The zymogen is activated by a specific cleavage in a conserved activation loop, resulting in a well-described structural rearrangement leading to the formation of a functional protease (16–18). Although zymogens are considered to be inactive, exceptions exist. The zymogen form of tissue-type plasminogen activator has significant catalytic activity due to a unique structural feature in which a lysine side chain near the activation hole in the catalytic domain is able to substitute for the hydrogen bond that is normally not formed until activation cleavage results in the insertion of a newly released N terminus (19). Interestingly, the zymogen form of rat matriptase has been shown to harbor significant catalytic activity, although no obvious structural or sequence-based reason has yet been proposed (20). Several studies have also provided strong evidence for human zymogen matriptase being able to autoactivate, suggesting a basal activity for the matriptase zymogen (21, 22). Furthermore, we have previously shown that the zymogen form of matriptase reacts with a peptide inhibitor biotin-Arg-Gln-Arg-Arg chloromethyl ketone (CMK) (23). Finally, it was recently shown that a nonactivatable (zymogen-locked) version of mouse matriptase was able to support the natural function of matriptase in a transgenic mouse model system where natural matriptase had been ablated, in strong support of a physiological role of zymogen activity of matriptase (24).
As matriptase is an attractive molecular target in basic research and pharmacological intervention, there has been much interest in developing inhibitors. Currently, several active-site inhibitors against matriptase have been engineered, with varying success due to lack of affinity, specificity, or both. Active-site inhibitors include modified naturally occurring cyclopeptides and several monoclonal antibodies (25–27). To date, only one set of compounds has been reported to target the zymogen form of matriptase, the small organochemicals termed compounds 1–4, which all showed an improved inhibitory effect toward matriptase-mediated activation of prostasin as compared with an active-site inhibitor of activated matriptase. However, the reported IC50 values were in the low micromolar range, the best being 2.6 μm, and the information regarding specificity and mechanism remains to be addressed (28).
The present study was initiated to investigate the catalytic activity of the zymogen form of human matriptase. We show that a zymogen-locked mutant of human matriptase has significant catalytic activity. Furthermore, two monoclonal antibodies (aZ-mAb-6 and aZ-mAb-7) were developed as competitive inhibitors of both zymogen and activated matriptase. These antibodies bind with high affinity and specificity and represent new tools for pharmacological intervention by targeting both zymogen and activated matriptase to ensure efficient shutdown of all matriptase activity. In addition, the X-ray crystal structure of the catalytic domain of matriptase in its zymogen form was solved. This new structure facilitated the antibody characterization in the present study and will provide a structural basis for future studies of the zymogen form of matriptase.
Results
The zymogen form of matriptase displays catalytic activity
To characterize the catalytic activity of matriptase in its zymogen form, we prepared a zymogen variant of the serine protease domain (SPD) by recombinant expression in yeast (Pichia pastoris). The sequence Cys604–Val855 of human matriptase was fused to an N-terminal His tag, and the mutation N164Q was introduced to avoid N-glycosylation. An additional mutation in the activation cleavage site (R614A) created a variant of matriptase locked in the zymogen form (Fig. 1B). The resulting 29-kDa recombinant protein was used as a representative of the zymogen form of matriptase termed zSPD. Zymogen purity is >95% as demonstrated by a Coomassie-stained SDS-polyacrylamide gel (Fig. 1C). The WT SPD of matriptase was expressed in Escherichia coli and refolded in vitro. As this recombinant matriptase spontaneously autoactivated during purification, as was observed by SDS-PAGE band shift and verified by MS and chromogenic activity assays (data not shown), this is henceforth used as a representative of the activated form of matriptase termed acSPD. Hydrolysis of the chromogenic peptide substrate, isoleucil-prolyl-arginine-p-nitroaniline (S-2288, Chromogenix) by either zSPD or acSPD was measured, and the corresponding enzymatic parameters Km and kcat were calculated (Table 1 and Fig. 2A). The catalytic efficiencies (kcat/Km) were determined to be 3.0 × 107 ± 7 × 107 m−1 s−1 and 1.01 × 109 ± 217 × 109 m−1 s−1 for zSPD and acSPD, respectively. By calculating the ratio of the catalytic efficiencies, a zymogenicity factor of 33 ± 11 was obtained, which corresponds to zSPD having an apparent catalytic activity of ∼3% of that of acSPD.
Table 1.
The zymogenicity factor of matriptase
All values represent an average of at least three independent measurements.
| Km | Vmax | kcat | kcat/Km | Zymogenicity factor | |
|---|---|---|---|---|---|
| μm | μm s−1 | 104 s−1 | μm−1 s−1 | ||
| zSPD | 256 ± 54 | 295 ± 93 | 0.77 ± 0.09 | 30 ± 7 | 33 ± 11 |
| acSPD | 387 ± 74 | 391 ± 38 | 39 ± 4 | 1011 ± 217 |
Figure 2.

The activity of acSPD and zSPD and the corresponding inhibitory effects of monoclonal antibodies on activity. A, activity of acSPD and zSPD shown by a dose-dependent enzyme activity assay (top left and bottom left, respectively) and the corresponding Michaelis–Menten fit (right panels) for 1 nm acSPD (top right) and 10 nm zSPD (bottom right) using 0–0.4 or 2 mm S-2288 substrate. Each graph is one representative experiment from at least three independent experiments. The zSPD has a lower Km and kcat than acSPD as a consequence of the higher concentration and lower activity. B, initial screening of inhibitory effects of the nine monoclonal antibodies toward both acSPD (left) and zSPD (right). An increasing concentration of antibody was incubated with either acSPD (50 pm) or zSPD (20 nm), and the residual rate of substrate conversion was measured by chromogenic assays (n = 3 for each data point of the shown curves). Antibodies aZ-mAb-6 and -7 are the most efficient inhibitors. C, Ki determinations of aZ-mAb-6 and aZ-mAb-7. acSPD (50 pm) was preincubated with a concentration series of the antibodies for 1 h, and the conversion of the chromogenic substrate S-2288 was monitored as the change in absorbance at 405 nm at 37 °C for 1 h (left). The Ki values for aZ-mAb-6 and -7 were 21 ± 2 and 9 ± 2 nm, respectively. The values for apparent Ki were determined by fitting the calculated reaction rates to Equation 2 (right) for competitive inhibition and then converting into the Ki value using Equation 3.
Characterization of mAb-based inhibitors of zymogen and activated matriptase
Monoclonal antibodies were generated with zSPD as antigen. A panel of nine monoclonal antibodies (aZ-mAb-1 to -7, aZ-mAb-9, and aZ-mAb-10) was chosen for further examination. Surface plasmon resonance (SPR) was used to analyze the binding of the antibodies to zymogen and activated matriptase. All nine monoclonal antibodies bound both zSPD and acSPD with affinities (KD) in the low nanomolar range (Table S1, Fig. 3, and Figs. S1 and S2).
Figure 3.

Determination of the equilibrium constant by surface plasmon resonance experiments. KD measurements where the red lines represent the binding curves and the black lines represent the corresponding 1:1 fit were performed by the Biacore evaluation software. The individual monoclonal antibodies were captured on an anti-mouse IgG surface before acSPD (top) or zSPD (bottom) was injected. The concentration range of acSPD was 1.56–50 nm for aZ-mAb-6 and -7 and 0.78–25 nm for aZ-mAb-4. For zSPD, a concentration range of 0.94–30 nm was used for all three antibodies. Each sensorgram represents one representative of at least three independent experiments.
The nine monoclonal antibodies underwent further testing to establish any modulatory effect on the catalytic activity of zymogen or activated matriptase. The catalytic activity of zSPD or acSPD toward a peptide substrate was measured in the presence of each antibody (Fig. 2B). Our initial results showed that two antibodies, aZ-mAb-6 and aZ-mAb-7, dose-dependently inhibited the activity of both zSPD and acSPD. The remaining antibodies (aZ-mAb-1, -2, -3, -4, -5, -9, and -10) caused either no inhibition or only partial inhibition.
To characterize the mode of inhibition of the antibodies, the Vmax and Km of acSPD in the presence or absence of antibodies were determined (Table 2 and Figs. S3 and S4). We observed an unchanged Vmax and an increased Km in the presence of aZ-mAb-6 and aZ-mAb-7, suggesting a competitive mechanism of inhibition (24). For the antibodies aZ-mAb-1, -2, -3, -5, -9, and -10, little change in Km and Vmax was detected (Table 2), suggesting that they are noncompetitive or mixed inhibitors. As no change was observed for Km and Vmax in the presence of antibody aZ-mAb-4, the lack of inhibitory activity of this antibody was confirmed, and it was used as a comparative control in subsequent experiments. Next, the equilibrium constant (Ki) of aZ-mAb-6 and aZ-mAb-7 against activated matriptase was determined to 21 ± 2 and 9 ± 2 nm, respectively (Fig. 2C). A similar analysis was performed on zSPD, including aZ-mAb-6 and aZ-mAb-7 (Table 2). Again, we observed an unchanged Vmax and an increased Km in support of a competitive mechanism of inhibition for aZ-mAb-6 and aZ-mAb-7. As expected, there was no significant effect of aZ-mAb-4.
Table 2.
Summary of Vmax, Km, kcat, and Ki
| Vmax | Km | kcat | Ki | n | |
|---|---|---|---|---|---|
| μm s−1 | μm | 104 s−1 | nm | ||
| acSPD | |||||
| No antibody | 391 ± 38 | 387 ± 74 | 39 ± 4 | NAa | 5 |
| aZ-mAb-1 | 184 ± 38b | 197 ± 64b | 18 ± 4 | NA | 3 |
| aZ-mAb-2 | 348 ± 22 | 489 ± 45 | 35 ± 2 | NA | 3 |
| aZ-mAb-3 | 152 ± 27b | 166 ± 36b | 15 ± 3 | NA | 3 |
| aZ-mAb-4 | 349 ± 61 | 347 ± 87 | 35 ± 6 | NA | 4 |
| aZ-mAb-5 | 339 ± 60 | 421 ± 102 | 34 ± 6 | NA | 3 |
| aZ-mAb-6 | 371 ± 35 | 857 ± 84b | 37 ± 4 | 21 ± 2 | 3 |
| aZ-mAb-7 | 365 ± 19 | 1172 ± 255b | 37 ± 2 | 9 ± 2 | 3 |
| aZ-mAb-9 | 281 ± 58b | 424 ± 109 | 28 ± 6 | NA | 3 |
| aZ-mAb-10 | 308 ± 42b | 451 ± 102 | 31 ± 4 | NA | 3 |
| zSPD | |||||
| No antibody | 295 ± 93 | 256 ± 54 | 0.77 ± 0.09 | NA | 10 |
| aZ-mAb-4 | 445 ± 14b | 193 ± 9 | 0.89 ± 0.03 | NA | 3 |
| aZ-mAB-6 | 260 ± 14 | 901 ± 39b | 0.52 ± 0.03 | NA | 3 |
| aZ-mAb-7 | 346 ± 8 | 609 ± 14b | 0.69 ± 0.02 | NA | 3 |
a NA, not applicable.
b Statistically significant difference from “no antibody” by unpaired t test calculated using GraphPad QuickCalcs online software.
Antibodies aZ-mAb-6 and aZ-mAb-7 are not substrates for matriptase
To ensure that the difference in affinity of the antibodies toward acSPD and zSPD is not due to antibody cleavage catalyzed by matriptase, the stability of the antibodies was analyzed following prolonged incubation with matriptase. Antibodies aZ-mAb-6 and aZ-mAb-7 were incubated with acSPD at a 1:1 molar ratio before SDS-PAGE analysis. The gel showed no sign of matriptase-catalyzed antibody degradation (Fig. 4).
Figure 4.
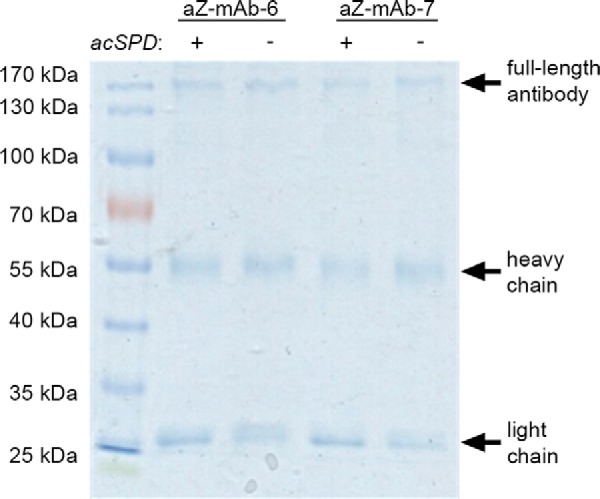
The monoclonal antibodies aZ-mAb-6 and aZ-mAb-7 are not substrates for matriptase. Antibodies aZ-mAb-6 and aZ-mAb-7 were incubated with or without acSPD at a 1:1 molar ratio for 1 h at 37 °C. No cleavage of the antibodies is seen after incubation with matriptase.
Antibodies aZ-mAb-6 and aZ-mAb-7 target human matriptase with high specificity
The active sites of serine proteases are generally highly conserved, and therefore inhibitory compounds often inhibit several related proteases. To gain insight about the inhibitory specificity of antibodies aZ-mAb-6 and aZ-mAb-7, three human serine proteases closely related to matriptase were tested: urokinase plasminogen activator (uPA), hepatocyte growth factor activator, and hepsin. Even at an antibody concentration of 400 nm, a 20-fold higher concentration than the inhibitory constant (Ki) toward matriptase, no inhibition was observed (Fig. 5). Furthermore, no inhibition was observed toward the mouse ortholog of matriptase (Fig. 5), whose catalytic domain shares 87% identity to the human protein. Together, these observations strongly suggest that antibodies aZ-mAb-6 and aZ-mAb-7 have high specificity toward human matriptase.
Figure 5.

The monoclonal antibodies aZ-mAb-6 and -7 specifically inhibit human matriptase. The ability of aZ-mAb-4, -6, and -7 to inhibit human matriptase and other closely related serine proteases was tested in an activity assay. For all assays, an excess of antibody (400 nm) was preincubated with the proteases for 1 h at 37 °C before the addition of substrate. Human matriptase, acSPD (100 pm), was almost completely inhibited by aZ-mAb-6 (green line) and aZ-mAb-7 (black line), but not by aZ-mAb-4 (red line). The activity of mouse matriptase SPD (100 pm), human uPA (1 nm), hepsin (1 nm), and HGFA (5 nm) were not significantly inhibited by any of the monoclonal antibodies. The graphs shown are representative of three replicates.
Antibodies aZ-mAb-6 and aZ-mAb-7 can identify and block the activity of WT full-length matriptase in complex biological samples
To examine the potential use of antibodies aZ-mAb-6 and aZ-mAb-7 for the study of matriptase activity in complex biological samples, an assay was established using cultured cells. HEK293 cells were transiently transfected with combinations of WT full-length membrane-anchored matriptase and two variants of its cognate inhibitor HAI-2. Matriptase needs co-expression with the inhibitors HAI-1 or HAI-2 to obtain significant levels of plasma membrane–bound matriptase. Either WT HAI-2 or a HAI-2 variant with compromised inhibitory activity (HAI-2 C47F) due to a mutation in the predicted P2 position of the inhibitory loop of Kunitz domain 1 was used (29). Transiently transfected cells were lysed, and the peptidolytic activity of the cell lysates was assayed with or without the antibodies aZ-mAb-6 and aZ-mAb-7 or a control antibody (anti-uPA) (Fig. 6A). Both antibodies aZ-mAb-6 and aZ-mAb-7 but not the control antibody inhibited the proteolytic turnover of the broad-spectrum serine protease substrate; this activity was interpreted as derived from unopposed matriptase in either the active zymogen form or the activated form. Little or no background activity was observed in mock-transfected cells or cells transfected with matriptase and WT HAI-2, suggesting that little to no unopposed matriptase is present under these conditions at our detection limit (<10 pm active sites as estimated from in vitro enzymatic assays). When matriptase was co-expressed with HAI-2 C47F, we observed a clear proteolytic activity, which could be silenced by aZ-mAb-6 and aZ-mAb-7 but not by the control antibody (anti-uPA), strongly suggesting the presence of unopposed matriptase activity under these conditions.
Figure 6.
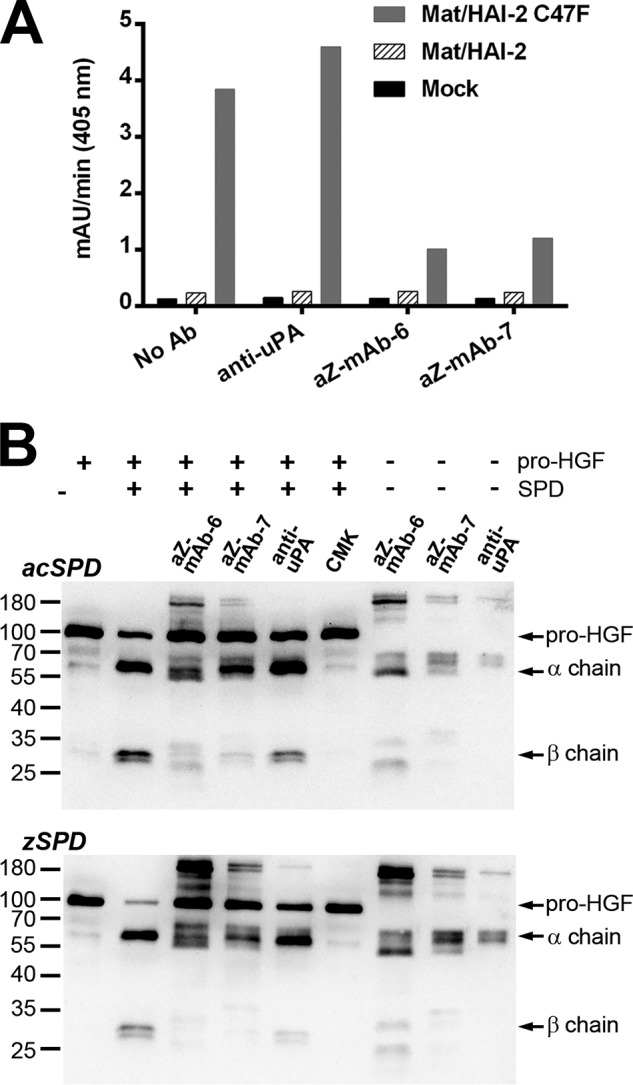
A, the monoclonal antibodies aZ-mAb-6 and -7 inhibit matriptase activity in the protein extracts of transiently transfected cells. HEK293 cells were transiently transfected with empty expression vector (mock) or expression vectors encoding WT full-length matriptase together with WT HAI-2 or mutant HAI-2 C47F, respectively. Protein extracts were preincubated for 1 h at room temperature with 500 ng of antibody (anti-uPA, aZ-mAb-6, aZ-mAb-7, or no antibody), and a peptidolytic activity assay was subsequently carried out with 300 μm chromogenic substrate S-2288. The absorbance of the reaction mixture was measured at 405 nm continuously every 5 min for 5 h, and the mean velocity of the substrate reaction (in milli-absorbance units/min) was calculated. The figure shown is representative of three replicates. B, aZ-mAb-6 and -7 inhibit matriptase-mediated pro-HGF cleavage. Pro-HGF (60 nm final concentration) was incubated for 4.5 h at 37 °C with either 200 nm zSPD or 1 nm acSPD that had been preincubated with aZ-mAb-6, aZ-mAb-7, anti-uPA, or biotin-RQRR-CMK, as indicated above the gels. The processing of pro-HGF was visualized on Western blotting using an antibody that recognizes pro-HGF as well as its α and β chains. Antibody-only samples were prepared in parallel to show the cross-reactivity of the antibodies themselves with the secondary antibody.
Antibodies aZ-mAb-6 and aZ-mAb-7 can inhibit matriptase catalyzed cleavage of a natural protein substrate
To examine the ability of antibodies aZ-mAb-6 and aZ-mAb-7 to inhibit the cleavage of a natural matriptase substrate, a pro-HGF cleavage assay was established. Pro-HGF was incubated with or without zSPD or acZPD in the presence or absence of inhibitors. Both antibodies aZ-mAb-6 and aZ-mAb-7, but not the control antibody (anti-uPA), inhibited the cleavage of pro-HGF by both acSPD and zSPD into its α and β chains (Fig. 6B, top and bottom, respectively). A peptide inhibitor, biotin-RQRR-CMK (23), was included as a positive control. These results strongly suggest that antibodies aZ-mAb-6 and aZ-mAb-7 inhibit matriptase-catalyzed cleavage of a physiologically relevant protein substrate.
Antibodies aZ-mAb-6 and aZ-mAb-7 bind near the active site of matriptase
As competitive inhibitors, we expect both antibodies to bind in or near the active site to directly compete with substrates. We presume that the epitopes of antibodies aZ-mAb-6 and aZ-mAb-7 are conserved between zymogen and activated matriptase, which is why some of the following experiments were only carried out only on activated protein. First, it was investigated whether mutation of the nucleophilic serine 805 (Ser195) in the active site of matriptase affected binding of the antibodies to zymogen or activated matriptase. Using SPR, no differences were seen in the KD for binding of antibodies aZ-mAb-6 and aZ-mAb-7 to zSPD, zSPD-S805A, or their activated counterparts (data not shown). This suggests that antibodies aZ-mAb-6 and aZ-mAb-7 do not interact directly with the nucleophile serine.
Next, possible interaction between the antibodies and the specificity pockets S2–S4 of matriptase, as S1 is not present in the zymogen, was probed by competition with either the peptide inhibitor biotin-RQRR-CMK (23) or the isolated inhibitor domain of HAI-1 termed IK1 (30). The peptide CMK inhibitor is a small molecule inhibitor of matriptase that binds covalently to the reactive histidine residue in the active site of matriptase via its CMK, whereas its peptide moiety is expected to occupy the S1–S4 specificity pockets adjacent to the active site. Neither aZ-mAb-6 nor aZ-mAb-7 bound to acSPD in the presence of the CMK inhibitor (Fig. 7A), whereas binding of the control noninhibitory antibody aZ-mAb-4 was not affected. The IK1 domain of HAI-1 binds with high affinity to the active site and covers an extended area in and around the active site and specificity pockets of matriptase (30, 31). SPR analysis showed that IK1 blocked the binding of not only aZ-mAb-6 and aZ-mAb-7 but also aZ-mAb-4 to acSPD (Fig. 7B). This suggests that all three epitopes must be near the active site of matriptase, as the larger footprint of IK1 prevents binding of all three antibodies. Finally, the three antibodies were shown to have overlapping epitopes by SPR, as each of the three antibodies block mutual binding of the other antibodies to zSPD (Fig. 7C).
Figure 7.

Epitope mapping of the monoclonal antibodies by SPR. A and B, antibodies (labeled above each panel) were captured by anti-mouse IgG directly immobilized on a CM5 chip before injection of the active site–binding peptide biotin-RQRR-CMK (A) or the larger active site–binding protein IK1 (B) in complex with acSPD. Both biotin-RQRR-CMK and IK1 block the binding of acSPD to aZ-mAb-6 and aZ-mAb-7. IK1 also blocks binding of acSPD to aZ-mAb-4. C, the antibodies aZ-mAb-4, -6, and -7 (labeled above) were individually immobilized on a CM5 chip surface, and antibody alone or in complex with zSPD was injected over the surface-bound antibodies. Antibodies injected alone as a control did not show any observable binding (not shown). Antibody was incubated with zSPD for 1 h at room temperature. All zSPD–antibody complexes have reduced binding, indicating that aZ-mAb-4, -6, and -7 have overlapping epitopes. The shown sensorgrams are representative of at least three replicates.
The structure of zymogen matriptase reveals a classical chymotrypsin-like zymogen fold
With the availability of zymogen-locked matriptase protein, the X-ray crystal structure was pursued with the intent to provide structural insight into the unusual activity of the zymogen as well as aid in the characterization of our antibodies. To prevent unwanted proteolytic degradation during crystallization, an additional mutation of the nucleophile Ser805 (Ser195) to alanine was introduced (zSPD-S805A, Fig. 1D). The atomic coordinates for the crystal structure are available in the Protein Data Bank under accession number 5LYO. The resulting 2.5 Å structure (Table 3) was that of a classical zymogen form of a chymotrypsin-like serine protease (Fig. 8A), in that important structural features for activity such as the S1 specificity pocket and oxyanion hole were absent. Interestingly, Asp804 (Asp194) is engaged in hydrogen bonding with His639 (His40), which again hydrogen-bonds to Ser631 (Ser32), forming what has previously been described as the zymogen triad (19), a feature that is observed in several catalytically inactive zymogens of this family and effectively adds an additional stabilizing interaction to the zymogen inactive conformation (Fig. 8C). Generally, most structural elements accessible for protein–protein interactions on the surface of the zSPD-S805A structure are identical to those observed in the structure of activated matriptase, especially within the N-terminal β-barrel; however, the C-terminal domain of zSPD-S805A contains several loop regions that deviate from the structure of activated matriptase (Fig. 8D). The electron density, although not well-defined, supports an intact and uncleaved activation loop as expected for the zymogen form (Fig. 8B).
Table 3.
Data collection and refinement statistics
Values in parentheses are for the highest-resolution shell.
| Data collection | |
| PDB accession code | 5LYO |
| Wavelength (Å) | 0.97625 |
| Space group | P3121 |
| a, b, c (Å) | 108.50, 108.50, 123.70 |
| α, β, γ (degrees) | 90, 90, 120 |
| Resolution (Å) | 50–2.50 (2.65–2.50) |
| Rmeas | 0.194 (1.116) |
| I/σI | 6.08 (1.27) |
| Completeness (%) | 97.6 (89.5) |
| Redundancy | 2.88 (2.58) |
| No. of unique reflections | 56,209 (8340) |
| CC(1/2) | 0.998 (0.529) |
| Refinement | |
| R factor | 0.2144 |
| Rfree | 0.2629 |
| Root mean square deviation | |
| Bond angles (degrees) | 0.020 |
| Bond lengths (Å) | 1.998 |
| Ramachandran plot (%) | |
| Favored | 95.89 |
| Allowed | 5.31 |
| Outliers | 0.80 |
Figure 8.

The structure of zymogen matriptase, zSPD-S805A. A, the solved structure of zSPD-S805A (wheat) with the 60 loop, the 70 loop, the 170 loop, and the activation loop specified together with the residues in the catalytic triad and the S1 pocket (PDB entry 5LYO). B, the intact activation loop for zSPD-S805A (wheat) and the cleaved activation loop for activated matriptase (red). C, the zymogen triad for zSPD-S805A and activated matriptase, showing rearrangement of Asp804 (Asp194). D, alignment of zSPD-S805A (wheat) and activated matriptase (gray), where the loops undergoing conformational changes are marked in red for activated matriptase. The S1 pocket is not formed in the zymogen.
Discussion
In this study, we used a zymogen-locked form of matriptase produced by mutating arginine 614 of the activation motif in the activation loop to an alanine (R15A), termed zSPD. This prevents activation by proteolytic cleavage by serine proteases with trypsin-like Arg/Lys specificity. We observed a lower but clearly detectable activity for zSPD versus the efficient catalytic activity of the activated matriptase (Fig. 2A, left). The zymogenicity factor was calculated to be 33 ± 11, equivalent to the zymogen preparation having ∼3% of the activity of an activated matriptase preparation. This result is consistent with the reported zymogenicity factor of 27 for rat matriptase (20). Our findings that zymogen matriptase possesses basal activity are in support of recent studies showing that the R614A (R15A) full-length WT matriptase variant is capable of performing its physiological catalytic functions required for homeostasis (24, 32). Besides being proposed to be at a pinnacle of cascades, matriptase may be transactivating other matriptase zymogens due to the match between the activation motif and the substrate specificity of matriptase itself, a unique feature among serine proteases (21, 33). This is also in line with the matriptase SPD being fully activated during the in vitro refolding procedure, as observed in our previous study (30).
According to the paradigm, a zymogen is thought to be in constant conformational transition equilibrium between two structural states with a presumed short-lived “active-like” conformation (34). Chymotrypsin-like serine proteases show strong preference for the inactive state and are thus virtually inactive (e.g. trypsinogen). For a rare subset of these proteases, the equilibrium appears to be shifted toward a more active-like zymogen population.
It is clear that many cells have zymogen matriptase on the plasma membrane and that this may have enough catalytic activity to control the activation of growth factors, signaling pathways, etc. to sustain survival (24, 32). Our working hypothesis is that zymogen matriptase, which is found in low concentrations on the cell surface, is a pinnacle protease in multiple cascades with enough catalytic activity to perform its physiological role. If the concentration of zymogen matriptase increases on the cell surface, they will begin to encounter one another, resulting in possible transactivation events. The activated matriptase molecules will then be immediately inhibited by its co-expressed (co-localized) inhibitors HAI-1/HAI-2 and targeted for endocytosis and degradation. Solid evidence suggests that if the equilibrium between matriptase and HAI-1/HAI-2 is out of balance in such a way that the total matriptase activity (sum of activity of zymogen and activated matriptase) becomes dominating, this accumulation of unopposed matriptase activity results in carcinogenesis (3, 35). Studies in mice have shown that both HAI-1 and HAI-2 can negate the oncogenic potential of matriptase when overexpressed together (13, 14).
In this study, we have generated two competitive antibodies (aZ-mAb-6 and aZ-mAb-7) that target the activity of both zymogen and activated matriptase. These inhibitory molecules represent, to the best of our knowledge, the first macromolecular inhibitors of any chymotrypsin-like serine protease in the zymogen form. The antibodies are specific for human matriptase, bind with affinities in the low nanomolar range, and are capable of inhibiting both glycosylated (full-length matriptase from a HEK293 cell culture system; Fig. 6A) and nonglycosylated matriptase. Although our antibody-based inhibitors may exhibit lower affinity relative to a naturally occurring Kunitz-type inhibitor such as HAI-1 or will eventually lose in competition with a covalent CMK-based inhibitor, the unmatched specificity of an antibody outweighs all when studying or targeting the activity of matriptase in complex mixtures such as in in vivo studies or in a clinical setting. It is likely that our antibodies can be used to study the in vivo importance of the activation process, as we expect these antibodies to block transactivation on the cell surface. Furthermore, aZ-mAb-6 and aZ-mAb-7 could possibly be used in research and diagnostics to evaluate the level of unopposed matriptase activity (matriptase activity not inhibited by inhibitors) in complex biological samples, as shown in Fig. 6, which has not been possible previously due to the lack of a specific substrate and/or inhibitors. The inhibitory antibodies described in the present study will also prove a tool to elucidate whether inhibition of plasma membrane–bound zymogen matriptase or activated matriptase is sufficient to prevent/inhibit cancer development.
The solved crystal structure of zymogen matriptase has an overall conformation that resembles the classical chymotrypsin-like zymogen form with loops in and around the active site deviating in conformation from that of the active structure, a zymogen triad and no oxyanion hole or S1 specificity pocket. In all, the structure of zymogen matriptase suggest that the crystallized form represents the inactive form of the unactivated zymogen and that future studies must be done to describe the apparent conformational liability of the zymogen form of matriptase that allow for its unusually high zymogen activity.
The structural information from available structures of the catalytic domain of active and now zymogen matriptase was combined with the observations from the biochemical and biophysical analyses to propose the possible epitopes of the antibodies. Comparable binding affinities of the antibodies against both forms of matriptase suggest that the antibodies mainly bind in a region of the structure with a conformation that is invariant before and after activation. Comparing the zymogen and active structure (Fig. 8D), the N-terminal subdomain appears to present the most promising invariant area near the active site. The antibodies do not appear to bind directly in the active site because binding is unaffected by mutation of the active-site serine. However, reduced binding to acSPD in the presence of the biotin-RQRR-CMK inhibitor showed that aZ-mAb-6 and aZ-mAb-7 most likely bind in close proximity to the active site. The fact that the antibodies bind zSPD to the same response on the surface plasmon resonance sensor chip (total associated mass) as acSPD and that both zymogen and activated matriptase are inhibited suggests that the antibodies must bind both the “active-like” zymogen conformation and the prevailing inactive zymogen conformation. This means that inactive zymogen can interact with the inhibitory antibodies even before transition to the “active” zymogen form has taken place, which makes them very efficient inhibitors. This also suggests that binding of the antibodies does not require the presence of the S1 pocket, which is not formed in the true zymogen form, thus excluding the area around the S1 pocket from the epitopes. This is a rather unusual feature for a competitive inhibitor of a serine protease, which often relies on direct interaction with the S1 pocket for specificity and affinity (36). Furthermore, based on the fact that the epitope of aZ-mAb-4 overlaps the epitopes of both aZ-mAb-6 and aZ-mAb-7 and all three antibodies compete with binding of the IK1 fragment of HAI-1, we propose that aZ-mAb-6 and aZ-mAb-7 are binding mainly to the N-terminal conformationally invariant subdomain near, but not in, the S1 pocket, effectively covering most of the noncatalytic parts of the active site of matriptase. This is also compatible with our observation that both antibodies inhibit matriptase glycosylated on the α-helix adjacent to the 170 loop (Fig. 8D, top right). During this study, an extensive attempt to map the exact epitopes was carried out; however, mutating solvent-exposed side chains in the area surrounding the active site of matriptase resulted in loss of either protein expression or folding efficiency (data not shown). Future co-crystallization studies may be able to reveal more structure-based insight into epitopes and the inhibitory mechanism of the antibodies.
Experimental procedures
Recombinant proteins
Production of activated matriptase catalytic domain (acSPD) in E. coli
A pT7 vector containing the human matriptase sequence Gly596–Val855 (ST14 gene, GenBankTM accession number NG_012132.1) and a C602S mutation to remove the cysteine between the activation loop and SPD was transformed into competent BL21 (DE3) E. coli cells (30). Cells were cultured under agitation at 37 °C in 2xYT media supplemented with 100 μg/ml ampicillin and 34 μg/ml chloramphenicol. A 20-ml overnight culture was transferred into 1 liter of 2xYT medium and grown until the absorbance at 600 nm was >0.6 before induction with 1 mm isopropyl β-d-1-thiogalactopyranoside for 4 h. The cells were pelleted by centrifugation at 5000 × g for 20 min and frozen at −80 °C until purification.
Purification and refolding of the catalytic domain of matriptase (acSPD) from E. coli
Cell pellets were resuspended in sonication buffer (50 mm Tris-HCl, pH 8.0, 0.5 m NaCl, 10% glycerol, 1 mm β-mercaptoethanol, 1 mm EDTA) and sonicated on ice. Lysates were pelleted at 10,000 × g for 30 min, and the insoluble fraction was washed in several steps using wash buffer 1 (50 mm Tris-HCl, pH 8.0, 0.5 m NaCl, 10% glycerol, 1 mm β-mercaptoethanol, 1 mm EDTA, 1% Triton X-100), wash buffer 2 (50 mm Tris-HCl, pH 8.0, 0.5 m NaCl, 10% glycerol, 1 mm β-mercaptoethanol, 1 mm EDTA, 0.25% Triton X-100), and wash buffer 3 (50 mm Tris-HCl, pH 8.0, 0.5 m NaCl, 10% glycerol, 1 mm β-mercaptoethanol, 1 mm EDTA). After each wash, the sample was centrifuged as described above, and the supernatant was discarded. The insoluble matter was resuspended in denaturing buffer (50 mm Tris-HCl, pH 8.0, 100 mm NaCl, 10 mm β-mercaptoethanol containing 6 m urea, 1 mm EDTA) by slowly stirring on ice for 30 min. The resuspended pellet was then centrifuged as described above, and the supernatant was incubated with nickel-nitrilotriacetic acid–agarose containing 20 mm imidazole by slow stirring on ice for 1 h. Elution was performed with denaturing buffer supplemented with 400 mm imidazole, and fractions containing protein were dialyzed against denaturing buffer. The protein concentration was measured at an absorbance of 280 nm with an extinction coefficient of the catalytic domain of 53,200 m−1 cm−1. The concentration was adjusted to 0.1–0.2 mg/ml using denaturing buffer. Protein-containing fractions were dialyzed overnight at 4 °C against refolding buffer (50 mm Tris-HCl, pH 8, 10% glycerol, 1 mm β-mercaptoethanol, and 3 m urea). Autoactivation of the protease occurs during the overnight dialysis against storage buffer (50 mm Tris, pH 8.0, 10% glycerol) at 4 °C, which was confirmed by increasing activity against a chromogenic substrate and by SDS-PAGE. Activated proteins were purified over a benzamidine-Sepharose (GE Healthcare) column that had been equilibrated with storage buffer. Bound protease was eluted with storage buffer supplemented with 1 m arginine. Proteins were further purified by size-exclusion chromatography (Superdex75 HR10/30 column, GE Healthcare) using storage buffer. Protein-containing fractions were pooled, quantitated, and stored at −80 °C.
Production of the catalytic domain of WT and mutant forms of zymogen matriptase in P. pastoris
The catalytic domain of WT and mutant forms of zymogen matriptase were expressed in P. pastoris. The DNA sequence encoding the matriptase catalytic domain (Cys604–Val855) was amplified from full-length human matriptase cDNA. The two mutations R614A (R15A) and N772Q (N164Q), with the corresponding standard chymotrypsin numbering given in parentheses, were introduced to lock the zymogen conformation and to remove a potential glycosylation site, respectively (zSPD). A construct with an additional mutation in S805A (S195A), which would render the resulting protein product inactive, was also constructed for crystallization (zSPD-S805A). The DNA constructs were subcloned into the XhoI-SalI sites of the pPICαA expression vector (Invitrogen) with an α-factor secretion signal sequence and an N-terminal His tag. Plasmid DNA was linearized by digestion with SacI prior to transformation into P. pastoris strain X-33 (Invitrogen) and cultured on YPD plates at 30 °C supplemented with 100 μg/ml zeocin for 3 days to screen for positive colonies. A positive colony was inoculated into a starter culture of 5 ml of YPD and grown for 24 h. The starter culture was transferred into 200 ml of BMGY for an additional 24 h before further transferred into 1 liter of BMMY induction medium supplemented daily with 1% methanol for 4 days.
Purification of the zymogen form of the catalytic domain of matriptase
After 4 days of methanol induction, the medium was harvested from yeast cells by centrifugation and filtrated using 0.22-μm membranes. The filtered medium was loaded onto a nickel-nitrilotriacetic acid column equilibrated with wash buffer 4 (20 mm Tris-HCl, pH 7.4, 150 mm NaCl), washed with wash buffer 4 supplemented with 20 mm imidazole, and bound protein was eluted by increasing the imidazole concentration to 300 mm. Eluted protein was dialyzed against 20 mm Tris-HCl, pH 7.4, 150 mm NaCl and concentrated before further purification by size-exclusion chromatography (Superdex75 HR10/30 column, GE Healthcare) in 20 mm Tris-HCl, pH 7.4, 150 mm NaCl. Fractions were pooled, quantitated, and stored at −80 °C. Purity (>95%) was verified by SDS-PAGE.
Generation of monoclonal antibodies
Mice (female Naval Medical Research Institute mice) were immunized three times with 25 μg of matriptase zymogen adsorbed onto Al(OH)3 in Gerbu adjuvant at 2-week intervals. An intravenous injection of 25 μg of antigen in saline was given to the mice 3 days prior to the fusion together with adrenaline. The fusion was performed as described (37) and later modified as described (38) with PEG as fusogen and the SP2/0-AG14 myeloma cell line as the fusion partner. Identification of matriptase-binding antibodies was carried out by ELISA screening using MaxiSorp polystyrene plates (NUNC, Roskilde, Denmark) coated with the matriptase zymogen. Ten of 40 clones positive for binding were taken out for cloning by the limiting dilution method. Selected single clones were grown in culture flasks in RPMI + 10% fetal calf serum.
Purification of monoclonal antibodies aZ-mAb-4, -6, and -7
Antibodies were purified by Protein G affinity chromatography on a column equilibrated with 20 mm Tris-HCl, pH 8, 500 mm NaCl. Bound antibody was eluted with 0.1 m glycine, pH 2.5, and fractions were pH-neutralized by adding 10% of 1 m Tris-HCl, pH 9. The fractions with highest A280 nm were pooled and dialyzed against 20 mm HEPES-NaOH, pH 7.4, 150 mm NaCl, 1 mm CaCl2 overnight at 4 °C. Purity of the antibodies was validated by SDS-PAGE, and the antibodies were stored at 4 °C in dialysis buffer supplemented with 300 μm NaN3.
SPR measurements
All SPR experiments were performed on a Biacore T200 (GE Healthcare) at 25 °C with a flow rate of 30 μl/min unless otherwise stated. A standard CM5 chip (GE Healthcare) was prepared by immobilizing covalently ∼6000 response units (RU) of anti-mouse IgG antibodies (GE Healthcare) on both the active and reference flow cell according to the manufacturer's protocol (GE Healthcare). The amine coupling protocol (Biacore, GE Healthcare) was used. The individual monoclonal antibodies were captured on the active flow cell anti-mouse IgG surface in running buffer (20 mm HEPES-NaOH, pH 7.4, 150 mm NaCl, 1 mm CaCl2, 0.1% BSA) to ∼250 RU. To correct for bulk effects, the signal from the reference surface was subtracted from the data of the active flow cell. acSPD, zSPD, and mutants thereof were injected in varying concentrations with an association phase of 120 s and allowed to dissociate for 180 s. The anti-mouse IgG surface was regenerated between binding cycles by a 120-s pulse of regeneration buffer (10 mm glycine-HCl, pH 1.7; GE Healthcare).
For the KD determinations by SPR, six different concentrations of SPD in a 2-fold dilution series were used. The following concentrations of acSPD were used: 1.56–50 nm for aZ-mAb-6 and -7 and 0.78–25 nm aZ-mAb-4. Concentrations of zSPD were 0.94–30 nm for aZ-mAb-6 and -7.
To determine binding in the presence of the biotin-RQRR-CMK inhibitor, acSPD at a concentration of either 50 nm (for aZ-mAb-6 and -7) or 6 nm (for aZ-mAb-4) was mixed with a high excess of biotin-RQRR-CMK (5 μm) immediately before injection, as the biotin-RQRR-CMK has a short half-life in aqueous solutions. The SPD–RQRR-CMK complex or SPD-DMSO was injected over an aZ-mAb-4, -6, or -7 surface. RQRR-CMK was stored in DMSO and was therefore added in the reference SPD solution. Buffer was applied as control.
To determine binding in the presence of the IK1 inhibitor fragment of HAI-1, acSPD (50 nm) was preincubated with IK1 (100 nm) at 37 °C for 2 h. Binding of the complex was measured on either an aZ-mAb-4, -6, or -7 surface. The binding was determined relative to acSPD (50 nm) and the two controls IK1 (100 nm) and HBS buffer supplemented with 1 mm CaCl2 and 0.1% BSA.
To test for overlapping epitopes of anti-zymogen monoclonal antibodies, antibodies aZ-mAb-4, -6, and -7 and the control anti-HAI1–31 (matriptase-independent antibody targeting HAI-1) were immobilized on a CM5 chip at pH 4.5 following the amine coupling protocol (GE Healthcare). The instrument was set to aim for a capture level of 1000 RU per flow cell of antibody (30 μg/ml). acSPD and zSPD (25 nm) were preincubated with the aZ-mAb-4, -6, and -7 (100 nm) for 1 h at room temperature. The matriptase–antibody complexes were added to the four antibody-immobilized flow cells and were compared against the binding of acSPD (25 nm) or zSPD (25 nm). The controls were single-antibody solutions (100 nm) and buffer. The dissociation time was increased to 600 s to ensure a clean surface.
Determination of mAb inhibition
Unless stated otherwise, all chromogenic assays were performed at 37 °C in HBS-PEG buffer supplemented with CaCl2 and PEG-8000 (20 mm HEPES-NaOH, pH 7.4, 150 mm NaCl, 1 mm CaCl2, 0.05% PEG 8000). Plates were read at 405 nm in a MultiskanTM GO microplate spectrophotometer (Thermo Scientific).
IC50 determinations were performed by preincubating acSPD (50 pm) or zSPD (20 nm) with aZ-mAb-4 (0–500 nm) and aZ-mAb-6 and -7 (0–1000 nm) for 1 h at 37 °C. The reaction was initiated by adding S-2288 (500 μm) and measured for 1 h (acSPD) or 5 h (zSPD).
Determination of the Km, Vmax, and Ki for aZ-mAb
The maximal velocity rate Vmax and the Michaelis constant Km are given by the equations,
| (Eq. 1) |
| (Eq. 2) |
| (Eq. 3) |
where [S] is the substrate concentration, [I] is the inhibitor concentration, Ki is the inhibitor's dissociation constant, Kiapp, is the apparent Ki, V0 is the initial reaction rate, Vmax is the maximum reaction rate, and Km is the concentration of substrate needed to reach half-maximal velocity (39). All Km and Vmax determinations were made by fitting to Equation 1 in GraphPad. aZ-mAb-4 (20 nm) or aZ-mAb-6 and -7 (60 nm) were preincubated with zSPD (10 nm) or acSPD (1 nm) for 1 h before the reactions were initiated by adding S-2288 (0–2 mm). The progress of substrate cleavage was measured for 1 or 5 h for acSPD and zSPD, respectively.
Determination of aZ-mAb inhibition constants
For a competitive inhibitor, Kiapp is given by Equation 2, and to correct for competition with the substrate, Equation 3 was applied to obtain the actual Ki value. To determine the Ki values, aZ-mAb-4, -6, and -7 (0–400 nm) were preincubated with matriptase (100 pm) for 1 h at 37 °C before the reactions were initiated with S-2288 (500 μm). The reaction progress was monitored for 1 h.
Determination of specificity of aZ-mAbs
Specificity assays were carried out in HBS-PEG buffer or HBS-BSA buffer (10 mm HEPES-NaOH, pH 7.4, 140 mm NaCl, 0.1% BSA). The proteases and their respective chromogenic substrates in HBS-PEG buffer were as follows: acSPD (100 pm) and mouse matriptase catalytic domain (100 pm; R&D Systems) with S-2288 (500 μm); full-length human uPA (1 nm; ProSpec) with S-2444 (500 μm); full-length human HGFA (5 nm; Sino Biological Inc.) with Spectrozyme FVIIa (500 μm), where HGFA was activated by thrombin (1:100 molar ratio) at 37 °C for 1 h until thrombin was blocked with 10 μg/ml Dextran Sulfate; and human hepsin (1 nm) with S-2366 (500 μm) and in HBS-BSA buffer. All proteins were incubated with aZ-mAb-4, -6, or -7 (400 nm) for 1 h at 37 °C. All samples were measured for 30 min except for HGFA, which was measured for 60 min.
Zymogen matriptase crystallization and structure model construction
The active site–mutated zymogen-locked form of matriptase was used for crystallization, containing three mutations: R614A, N772Q, and S805A (zSPD-S805A; Fig. 1D). Initial crystallization conditions were screened using commercial kits (Clear Strategy I, Clear Strategy II, Structure Screen I + II, and JCSG-plus). 0.2 μl of protein was mixed with 0.2 or 0.1 μl of mother liquid using a Mosquito crystallization robot by sitting-drop vapor diffusion at 20 °C. The diffracted crystal was obtained in 0.1 m MES, pH 6.5, 2.0 m ammonium sulfate. For X-ray data collection, the crystal was transferred to a cryoprotectant solution containing 20% glycerol, 0.1 m MES, pH 6.5, 2.0 m ammonium sulfate and flash-frozen in liquid nitrogen. The diffraction data set was collected at a Micro-focus beam line in the European Synchrotron Radiation Facility. The data were processed to a resolution of 2.5 Å using XDS. Data collection statistics and cell parameters are summarized in Table 3. The diffraction data were indexed to the space group P 3(1)21 with a corresponding Matthews coefficient indicating that the crystals contained three molecules in the asymmetric unit (87 kDa). The first two molecules were found by molecular replacement using BALBES and the active matriptase catalytic domain as search model (PDB entry 4ISN) with initial Rfree of 0.42. Based on the phase from the first two molecules, the missing third molecule was found by Molrep using fast rotation function and phased translation function. The model was built using manual building in COOT and refined in Phenix by applying NCS and TLS throughout the refinement cycles. The final structure was refined to the R and Rfree values of 0.214 and 0.263 and a tight stereochemistry with root mean square deviations of bond lengths and bond angles of 0.02 Å and 2.00°, respectively.
Matriptase activity in cell lysates
HEK293 cells were transiently transfected with empty expression vector (mock) or expression vectors encoding matriptase together with WT HAI-2 or mutant HAI-2 C47F, respectively, using Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer's protocol. The constructs encoding full-length human WT matriptase and HAI-2 were inserted into pcDNA3.1 plasmid vectors (Thermo Fisher Scientific) as described previously by Nonboe et al. (29). HAI-2 was co-expressed to ensure stable matriptase expression, and the HAI-2 C47F mutant was included to allow measurable matriptase activity. Cells were grown for ∼48 h before they were lysed on ice in 150 μl of lysis buffer (PBS including 1% Triton X-100 and 0.5% sodium deoxycholate). Protein extracts were prepared by centrifuging the lysates at 20,000 × g for 20 min at 4 °C to remove cell debris. 5 μg of antibody (anti-uPA, aZ-mAB-6, aZ-mAB-7, or no antibody) was added to 60 μl of the protein extracts and incubated at room temperature for 1 h. Samples were subsequently added to substrate S-2288 in HBS including 0.1% BSA to a final concentration of 300 μm in a total reaction volume of 200 μl. Absorbance was measured in a plate reader at 405 nm continuously every 5 min for 5 h, and the mean velocity of the substrate reaction (in milli-absorbance units/min) was calculated.
Matriptase-mediated pro-HGF cleavage
zSPD (200 nm) or acSPD (1 nm) was preincubated with either 20 times the Ki of aZ-mAb-6 or -7 (420 and 180 nm, respectively), 420 nm anti-uPA, or 5 μm biotin-RQRR-CMK for 1 h at 37 °C in PBS, pH 7.4, including 0.1% Triton X-100. Recombinant human pro-HGF (75 ng, R&D Systems) was added, and the samples were incubated for 4.5 h at 37 °C before the addition of reducing loading dye (including 200 mm DTT). Samples containing antibodies alone were prepared in parallel. The Western blotting was performed as described by Nonboe et al. (29) using anti-HGF antibody (R&D Systems, AF-294-SP) and anti-goat horseradish peroxidase–conjugated antibody (R&D Systems, HAF017).
Author contributions
T. T., Z. H., D. D. S., S. S., D. M. D., L. V., and K. S. data curation; T. T., Z. H., S. S., D. M. D., L. K. V., and J. K. J. formal analysis; T. T., Z. H., S. S., and C. R. S. visualization; T. T., C. R. S., L. K. V., and J. K. J. writing-original draft; Z. H., D. M. D., L. V., K. S., L. K. V., and J. K. J. methodology; D. M. D., L. V., and L. K. V. resources; C. R. S., L. K. V., and J. K. J. writing-review and editing; L. K. V. and J. K. J. supervision; L. K. V. and J. K. J. funding acquisition; J. K. J. conceptualization.
Supplementary Material
This work was supported by the Danish Cancer Society and the Danish Cancer Research Foundation. The authors declare that they have no conflicts of interest with the contents of this article.
This work is dedicated to our dear colleague and friend, Prof. Peter A. Andreasen, who passed away during this project.
This article contains Table S1 and Figs. S1–S4.
The atomic coordinates and structure factors (code 5LYO) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- TTSP
- type II transmembrane serine protease
- HAI-1
- hepatocyte growth factor activator inhibitor-1
- HAI-2
- hepatocyte growth factor activator inhibitor-2
- pro-HGF
- pro-hepatocyte growth factor
- HGFA
- hepatocyte growth factor activator
- IK1
- HAI-1 protein fragment consisting of internal (N235Q) domain and Kunitz-1
- SPD
- serine protease domain
- acSPD
- serine protease domain of activated human matriptase
- zSPD
- serine protease domain of zymogen human matriptase
- SPR
- surface plasmon resonance
- RU
- resonance units
- CMK
- chloromethylketone
- PDB
- Protein Data Bank
- TLS
- translation/libration/screw
- NCS
- noncrystallographic symmetry
- uPA
- urokinase plasminogen activator
- HBS
- Hepes-buffered saline.
References
- 1. Bugge T. H., Antalis T. M., and Wu Q. (2009) Type II transmembrane serine proteases. J. Biol. Chem. 284, 23177–23181 10.1074/jbc.R109.021006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. List K., Haudenschild C. C., Szabo R., Chen W., Wahl S. M., Swaim W., Engelholm L. H., Behrendt N., and Bugge T. H. (2002) Matriptase/MT-SP1 is required for postnatal survival, epidermal barrier function, hair follicle development, and thymic homeostasis. Oncogene 21, 3765–3779 10.1038/sj.onc.1205502 [DOI] [PubMed] [Google Scholar]
- 3. List K. (2009) Matriptase: a culprit in cancer? Future Oncol. 5, 97–104 10.2217/14796694.5.1.97 [DOI] [PubMed] [Google Scholar]
- 4. Basel-Vanagaite L., Attia R., Ishida-Yamamoto A., Rainshtein L., Ben Amitai D., Lurie R., Pasmanik-Chor M., Indelman M., Zvulunov A., Saban S., Magal N., Sprecher E., and Shohat M. (2007) Autosomal recessive ichthyosis with hypotrichosis caused by a mutation in ST14, encoding type II transmembrane serine protease matriptase. Am. J. Hum. Genet. 80, 467–477 10.1086/512487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lee S. L., Dickson R. B., and Lin C. Y. (2000) Activation of hepatocyte growth factor and urokinase/plasminogen activator by matriptase, an epithelial membrane serine protease. J. Biol. Chem. 275, 36720–36725 10.1074/jbc.M007802200 [DOI] [PubMed] [Google Scholar]
- 6. Ustach C. V., Huang W., Conley-LaComb M. K., Lin C. Y., Che M., Abrams J., and Kim H. R. (2010) A novel signaling axis of matriptase/PDGF-D/ss-PDGFR in human prostate cancer. Cancer Res. 70, 9631–9640 10.1158/0008-5472.CAN-10-0511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sales K. U., Friis S., Konkel J. E., Godiksen S., Hatakeyama M., Hansen K. K., Rogatto S. R., Szabo R., Vogel L. K., Chen W., Gutkind J. S., and Bugge T. H. (2015) Non-hematopoietic PAR-2 is essential for matriptase-driven pre-malignant progression and potentiation of ras-mediated squamous cell carcinogenesis. Oncogene 34, 346–356 10.1038/onc.2013.563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Miller G. S., and List K. (2013) The matriptase-prostasin proteolytic cascade in epithelial development and pathology. Cell Tissue Res. 351, 245–253 10.1007/s00441-012-1348-1 [DOI] [PubMed] [Google Scholar]
- 9. Wu C. J., Feng X., Lu M., Morimura S., and Udey M. C. (2017) Matriptase-mediated cleavage of EpCAM destabilizes claudins and dysregulates intestinal epithelial homeostasis. J. Clin. Invest. 127, 623–634 10.1172/JCI88428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Szabo R., Hobson J. P., List K., Molinolo A., Lin C. Y., and Bugge T. H. (2008) Potent inhibition and global co-localization implicate the transmembrane Kunitz-type serine protease inhibitor hepatocyte growth factor activator inhibitor-2 in the regulation of epithelial matriptase activity. J. Biol. Chem. 283, 29495–29504 10.1074/jbc.M801970200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Szabo R., Hobson J. P., Christoph K., Kosa P., List K., and Bugge T. H. (2009) Regulation of cell surface protease matriptase by HAI2 is essential for placental development, neural tube closure and embryonic survival in mice. Development 136, 2653–2663 10.1242/dev.038430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Szabo R., Molinolo A., List K., and Bugge T. H. (2007) Matriptase inhibition by hepatocyte growth factor activator inhibitor-1 is essential for placental development. Oncogene 26, 1546–1556 10.1038/sj.onc.1209966 [DOI] [PubMed] [Google Scholar]
- 13. Sales K. U., Friis S., Abusleme L., Moutsopoulos N. M., and Bugge T. H. (2015) Matriptase promotes inflammatory cell accumulation and progression of established epidermal tumors. Oncogene 34, 4664–4672 10.1038/onc.2014.391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. List K., Szabo R., Molinolo A., Sriuranpong V., Redeye V., Murdock T., Burke B., Nielsen B. S., Gutkind J. S., and Bugge T. H. (2005) Deregulated matriptase causes ras-independent multistage carcinogenesis and promotes ras-mediated malignant transformation. Genes Dev. 19, 1934–1950 10.1101/gad.1300705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tanabe L. M., and List K. (2017) The role of type II transmembrane serine protease mediated signaling in cancer. FEBS J. 284, 1421–1436 10.1111/febs.13971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huber R., and Bode W. (1978) Structural basis of the activation and action of trypsin. Acc. Chem. Res. 11, 114–122 10.1021/ar50123a006 [DOI] [Google Scholar]
- 17. Wang D., Bode W., and Huber R. (1985) Bovine chymotrypsinogen A. X-ray crystal structure analysis and refinement of a new crystal form at 1.8 Å resolution. J. Mol. Biol. 185, 595–624 10.1016/0022-2836(85)90074-9 [DOI] [PubMed] [Google Scholar]
- 18. Berg J. M., Tymoczko J. L., and Stryer L. (2012) Biochemistry, 7th Ed W.H. Freeman & Co., New York [Google Scholar]
- 19. Madison E. L., Kobe A., Gething M. J., Sambrook J. F., and Goldsmith E. J. (1993) Converting tissue plasminogen activator to a zymogen: a regulatory triad of Asp-His-Ser. Science 262, 419–421 10.1126/science.8211162 [DOI] [PubMed] [Google Scholar]
- 20. Inouye K., Yasumoto M., Tsuzuki S., Mochida S., and Fushiki T. (2010) The optimal activity of a pseudozymogen form of recombinant matriptase under the mildly acidic pH and low ionic strength conditions. J. Biochem. 147, 485–492 10.1093/jb/mvp190 [DOI] [PubMed] [Google Scholar]
- 21. Oberst M. D., Williams C. A., Dickson R. B., Johnson M. D., and Lin C.-Y. (2003) The activation of matriptase requires its noncatalytic domains, serine protease domain, and its cognate inhibitor. J. Biol. Chem. 278, 26773–26779 10.1074/jbc.M304282200 [DOI] [PubMed] [Google Scholar]
- 22. Tseng I. C., Xu H., Chou F. B., Li G., Vazzano A. P., Kao J. P. Y., Johnson M. D., and Lin C. Y. (2010) Matriptase activation, an early cellular response to acidosis. J. Biol. Chem. 285, 3261–3270 10.1074/jbc.M109.055640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Godiksen S., Soendergaard C., Friis S., Jensen J. K., Bornholdt J., Sales K. U., Huang M., Bugge T. H., and Vogel L. K. (2013) Detection of active matriptase using a biotinylated chloromethyl ketone peptide. PLoS One 8, e77146 10.1371/journal.pone.0077146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Friis S., Tadeo D., Le-Gall S. M., Jürgensen H. J., Sales K. U., Camerer E., and Bugge T. H. (2017) Matriptase zymogen supports epithelial development, homeostasis and regeneration. BMC Biol. 15, 46 10.1186/s12915-017-0384-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gray K., Elghadban S., Thongyoo P., Owen K. A., Szabo R., Bugge T. H., Tate E. W., Leatherbarrow R. J., and Ellis V. (2014) Potent and specific inhibition of the biological activity of the type-II transmembrane serine protease matriptase by the cyclic microprotein MCoTI-II. Thromb. Haemost. 112, 402–411 10.1160/TH13-11-0895 [DOI] [PubMed] [Google Scholar]
- 26. Colombo E., Désilets A., Duchêne D., Chagnon F., Najmanovich R., Leduc R., and Marsault E. (2012) Design and synthesis of potent, selective inhibitors of matriptase. ACS Med. Chem. Lett. 3, 530–534 10.1021/ml3000534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schneider E. L., Lee M. S., Baharuddin A., Goetz D. H., Farady C. J., Ward M., Wang C. I., and Craik C. S. (2012) A reverse binding motif that contributes to specific protease inhibition by antibodies. J. Mol. Biol. 415, 699–715 10.1016/j.jmb.2011.11.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xu Z., Chen Y. W., Battu A., Wilder P., Weber D., Yu W., Mackerell A. D., Chen L. M., Chai K. X., Johnson M. D., and Lin C. Y. (2011) Targeting zymogen activation to control the matriptase-prostasin proteolytic cascade. J. Med. Chem. 54, 7567–7578 10.1021/jm200920s [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nonboe A. W., Krigslund O., Soendergaard C., Skovbjerg S., Friis S., Andersen M. N., Ellis V., Kawaguchi M., Kataoka H., Bugge T. H., and Vogel L. K. (2017) HAI-2 stabilizes, inhibits and regulates SEA-cleavage-dependent secretory transport of matriptase. Traffic 18, 378–391 10.1111/tra.12482 [DOI] [PubMed] [Google Scholar]
- 30. Hong Z., De Meulemeester L., Jacobi A., Pedersen J. S., Morth J. P., Andreasen P. A., and Jensen J. K. (2016) Crystal structure of a two-domain fragment of hepatocyte growth factor activator inhibitor-1: functional interactions between the Kunitz-type inhibitor domain-1 and the neighboring polycystic kidney disease-like domain. J. Biol. Chem. 291, 14340–14355 10.1074/jbc.M115.707240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhao B., Yuan C., Li R., Qu D., Huang M., and Ngo J. C. K. (2013) Crystal structures of matriptase in complex with its inhibitor hepatocyte growth factor activator inhibitor-1. J. Biol. Chem. 288, 11155–11164 10.1074/jbc.M113.454611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Friis S., Uzzun Sales K., Godiksen S., Peters D. E., Lin C.-Y., Vogel L. K., and Bugge T. H. (2013) A matriptase-prostasin reciprocal zymogen activation complex with unique features: prostasin as a non-enzymatic co-factor for matriptase activation. J. Biol. Chem. 288, 19028–19039 10.1074/jbc.M113.469932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Takeuchi T., Harris J. L., Huang W., Yan K. W., Coughlin S. R., and Craik C. S. (2000) Cellular Localization of membrane-type serine protease 1 and identification of protease-activated receptor-2 and single-chain urokinase-type plasminogen activator as substrates. J. Biol. Chem. 275, 26333–26342 10.1074/jbc.M002941200 [DOI] [PubMed] [Google Scholar]
- 34. Pozzi N., Chen Z., Zapata F., Niu W., Barranco-Medina S., Pelc L. A., and Di Cera E. (2013) Autoactivation of thrombin precursors. J. Biol. Chem. 288, 11601–11610 10.1074/jbc.M113.451542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kataoka H., Kawaguchi M., Fukushima T., and Shimomura T. (2018) Hepatocyte growth factor activator inhibitors (HAI-1 and HAI-2): emerging key players in epithelial integrity and cancer. Pathol. Int. 68, 145–158 10.1111/pin.12647 [DOI] [PubMed] [Google Scholar]
- 36. Farady C. J., and Craik C. S. (2010) Mechanisms of macromolecular protease inhibitors. ChemBioChem 11, 2341–2346 10.1002/cbic.201000442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Köhler G., and Milstein C. (1975) Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 256, 495–497 10.1038/256495a0 [DOI] [PubMed] [Google Scholar]
- 38. Reading C. L. (1982) Theory and methods for immunization in culture and monoclonal antibody production. J. Immunol. Methods 53, 261–291 10.1016/0022-1759(82)90175-2 [DOI] [PubMed] [Google Scholar]
- 39. Copeland R. A. (2000) Enzymes: A Practical Introduction to Structure, Mechanism and Data Analysis, 2nd Ed., Wiley-VCH, New York [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.