

Abstract
Background:
Atherosclerotic occlusions decrease blood flow to the lower limbs causing ischemia and tissue loss in patients with peripheral artery disease (PAD). Currently, no effective medical therapies are available to induce angiogenesis and promote perfusion recovery in patients with severe PAD. Clinical trials aimed at inducing VEGF-A levels, a potent pro-angiogenic growth factor to induce angiogenesis and perfusion recovery were not successful. Alternate splicing in the exon-8 of VEGF-A results in the formation of VEGFxxxa (VEGF165a) and VEGFxxxb (VEGF165b) isoforms with existing literature focusing on VEGF165b’s role in inhibiting VEGFR2 dependent angiogenesis. However, we have recently shown that VEGF165b blocks VEGF-A induced endothelial VEGFR1 activation in ischemic muscle to impair perfusion recovery. Since macrophage secreted VEGF165b has been shown to decrease angiogenesis in peripheral artery disease and macrophages were well known to play important roles in regulating ischemic muscle vascular remodeling, we examined the role of VEGF165b in regulating macrophage function in PAD.
Methods:
Femoral artery ligation and resection was used as an in vivo preclinical PAD model and hypoxia serum starvation was used as an in vitro model for PAD. Experiments including laser-doppler perfusion imaging, adoptive cell transfer to ischemic muscle, immunoblot analysis, enzyme-linked immunosorbent assays, Immunostainings, flow cytometry, qPCR analysis and RNA-Seq analysis were performed to determine a role of VEGF165b in regulating macrophage phenotype and function in PAD.
Results:
First, we found increased VEGF165b-expression with increased M1-like-macrophages in PAD vs. non-PAD (controls) muscle-biopsies. Next, using in vitro hypoxia serum starvation (HSS), in vivo pre-clinical PAD models and adoptive-transfer of VEGF165b-expressing bone marrow-derived macrophages (BMDM) or VEGFR1+/− BMDM (M1-like-phenotype), we demonstrate that VEGF165b inhibits VEGFR1-activation to induce an M1-like-phenotype that impairs ischemic-muscle neovascularization. Subsequently, we found S100A8/S100A9 as VEGFR1 downstream regulators of macrophage-polarization by RNA-Seq analysis of HSS-VEGFR1+/+ vs. HSS-VEGFR1+/− BMDM.
Conclusion:
In our current study, we demonstrate that increased VEGF165b-expression in macrophages induces an anti-angiogenic M1-like-phenotype that directly impairs angiogenesis. VEGFR1 inhibition by VEGF165b results in S100A8/S100A9 mediated calcium influx to induce an M1-like-phenotype that impairs ischemic-muscle revascularization and perfusion recovery.
Keywords: Alternative splicing, Growth factors, Therapeutic Angiogenesis, Autocrine signaling, Paracrine effects, Cell communication
Introduction
In patients with peripheral arterial disease (PAD), atherosclerotic-occlusions in the blood vessels leads to tissue ischemia in the lower limbs with leg pain while walking, rest and tissue loss. Severe PAD (critical limb ischemia-CLI) often results in tissue necrosis and limb amputation1. Approximately 500–1000 patients per million PAD patients over 5–8 years develop CLI2 resulting in approximately 200,000 amputations in the US alone. In the frequent setting of complete blockades in the blood vessels that supply the leg, perfusion to the limb becomes dependent on the extent of vascular remodeling from angiogenesis and arteriogenesis in the ischemic-muscle3. Currently, no therapies can directly increase the blood flow to the ischemic-muscle in PAD patients and new therapies are being actively sought4.
VEGF-A, a potent angiogenic molecule is upregulated in ischemic-muscle and its upregulation can contribute to tissue revascularization5–8. The role of VEGF-A function and signaling in angiogenesis grew more complex by the discovery of alternative splicing in its 8th exon that results in the occurrence of pro-angiogenic VEGFxxxa (xxx for the number of amino acids) and anti-angiogenic VEGFxxxb (VEGF165b) isoform families9–11. We and others have demonstrated that VEGF165b can impair perfusion-recovery in experimental-PAD12,13. However, contrary to expectations, we showed that expression of VEGF165b impaired the extent of perfusion-recovery following experimental-PAD by blocking a novel VEGFR1-STAT3 signaling in ischemic endothelial cells but not through inhibiting VEGFR2-activation12.
Previous publications have demonstrated that VEGFR1 regulates the mobilization of bone marrow cells of macrophage lineage to stimulate solid tumor growth14; and VEGFR1-signaling in metastasis-associated macrophages induces inflammatory signaling that promotes breast cancer metastasis15. In human and experimental-PAD, macrophages were described to play important roles in vascular remodeling16,17. Macrophages that infiltrate the ischemic-muscle attain a pro-inflammatory, cytotoxic and anti-angiogenic M1-phenotype due to the ischemic-muscle environment and aggravate tissue damage by severely hindering reparative/regenerative process and/or adaptive processes18,19. We have recently demonstrated that increased M1-like-macrophages in the distal ischemic leg (where the bulk of angiogenesis occur) decrease angiogenic-remodeling20. So far, macrophage-specific function of VEGFR1-activation and the downstream signaling in PAD is not known. Based on our recent report demonstrating an enhanced VEGFR1-activation post VEGF165b-inhibition promotes perfusion-recovery in experimental-PAD8, we investigated the mechanistic role of VEGF165b-VEGFR1 signaling in regulating macrophage-polarization and ischemic-muscle revascularization in experimental-PAD.
Methods
Expanded methods can be found in the online-only Data supplement. The data, analytic methods, and study materials will be made available to other researchers for purposes of reproducing/replicating the procedure/results. Requests can be made to the corresponding author who manages the information.
Animal Model of Hind-Limb Ischemia (HLI):
Unilateral femoral artery ligation and excision was performed on age- and sex-matched 12–16-week-old animals12,20,21. All animal experiments were approved and performed according to University of Virginia Institutional Animal Care Committee and conformed to the Guide for the Care and Use of Laboratory Animals published by the NIH.
Human Samples:
The Institutional Review boards at Duke University, University of Colorado, and the University of Virginia approved the research protocols with human samples. All subjects gave informed consent.
Cell Transfections:
Plasmid transfections were performed by Lipofectamine-3000 according to the manufacturers’ instructions.
Statistics:
GraphPad Prism 7 was used to analyze the statistical significance of the data. Statistical test accompanied each figure legend. P<0.05 considered significant for all experiments. Data are presented as Mean ± Standard Error.
Results
1. Macrophage-specific VEGF165b-expression inversely correlates with VEGFR1-activation and directly correlates with M1-like-phenotype in ischemic-muscle.
A recent study by Kikuchi et al13 suggested that monocyte/macrophage secreted VEGF165b increased circulating VEGF165b levels in PAD patients’ serum and inhibited endothelial angiogenic potential by inhibiting VEGFR2-activation. In order to asses a quantitative correlation between VEGF165b levels and PAD severity, we performed VEGF165b ELISA (lower detection limit 62.5pg/ml) in plasma samples from PAD patients, and age and gender-matched controls. We found that VEGF165b levels were detectable only in ~25% (5 out of total 22 samples) of control human plasma samples (range-13.2 to 25.2pg/ml), ~10% (2 in 20 samples) in intermittent claudication (IC, 18.8pg/ml) and none in (0 out of 19 samples) CLI (Fig-1A).
Figure-1:

Increased VEGF165b-expression inversely correlates with decreased VEGFR1-activation and directly with M1-like macrophage phenotype. A) VEGF165b ELISA in human normal, intermittent claudication (IC) and critical limb ischemia (CLI) patients’ plasma samples. Control n=24, IC n=20, CLI n=19. Non-parametric one-way ANOVA Kruskal-Wallis test with Dunn’s multiple comparisons test and Geisser-Greenhouse correction was used to determine statistical significance. B, C) VEGF165b levels in conditioned-medium (medium) and lysates from control and hypoxia serum starved (HSS) (B) human peripheral blood monocyte derived macrophages (HPBMDM) and (C) mouse bone marrow derived macrophages (BMDM). VEGF165b levels in conditioned-medium was normalized to the corresponding cell lysate total protein levels. n=4. A-C) One way ANOVA with Dunnett’s multiple test correction was used to determine statistical significance. D) VEGF165b levels in conditioned-medium and lysates from bone marrow monocytes from non-ischemic leg (NIL-Mo) and monocytes from ischemic leg (IL-Mo). VEGF165b levels in conditioned-medium was normalized to the corresponding cell lysate total protein levels. n=4. E-F) Flowcytometric analysis of E) VEGF165b and F) pVEGFR1Y1333 expression in macrophages from non-ischemic gastrocnemius (NGA) and ischemic gastrocnemius (IGA) muscle. n=4. G-H) Flowcytometric analysis of pVEGFR1 expression in G) VEGF165b+ve (VEGF165b positive) and H) VEGF165b−ve (VEGF165b negative) macrophages from NGA and IGA. n=4. I-J) Flowcytometric analysis of I) CD80high M1-like macrophages and J) Arg-1+ M2-like macrophages from NGA and IGA. n=4. D-J) Unpaired T-test was used to determine statistical significance. Increased VEGF165b-expression correlate with higher CD68+CD80+ M1-like macrophages in human PAD. Immunostaining VEGF165b, CD68, CD206 and CD80 in normal (NL) and peripheral arterial disease (PAD) muscle-biopsies. Quantification of K) VEGF165b expressing (VEGF165b+ve) cells, L) total macrophages (CD68+), M) macrophages expressing CD80 (CD68+CD80+) and N) macrophages expressing CD206 (CD68+CD206+). Normal n=5, PAD n=7. K-N) Unpaired T-test was used to determine statistical significance. *P<0.05 considered significant.
We sought to determine to what extent, or whether, monocytes/macrophages contribute to circulating VEGF165b levels. We isolated human peripheral blood monocyte-derived macrophages (HPBMDMs) as previously described20 and cultured them under normal or hypoxia serum-starvation (HSS). HPBMDM cell-lysates and conditioned media were examined for VEGF165b levels. While we did not observe any detectable levels of VEGF165b in conditioned media from control or HSS-challenged HPBMDMs, a significantly higher VEGF165b levels was observed in HSS-challenged HPBMDMs cell-lysates vs. controls (control 32.8±2.5pg/mg vs. 6hr-HSS 78.8±1.8pg/mg (P<0.05 vs. control), 12hr-HSS 58.7±7.3pg/mg and 24hr-HSS 23.8±2.2pg/mg, Fig-1B).
We also isolated bone marrow-derived macrophages (BMDMs) from mice using methods previously described20. Similar to the data from HPBMDMs, VEGF165b was not detectable from conditioned-medium from BMDMs from Balb/c mice under HSS or control conditions (Fig-1C). However, in cell-lysates a significant increase in VEGF165b levels was observed in BMDM under HSS vs. control (control 39.6±1.2pg/mg vs. 6hr-HSS 40.9±5.1pg/mg, 12hr-HSS 52.7±4.4pg/mg vs. 24hr-HSS 67.7±2.9pg/mg (P<0.05 vs. Control), Fig-1C). To confirm whether there is a difference in monocyte vs. macrophage-specific VEGF165b secretion pattern, we isolated bone marrow (BM) monocytes from non-ischemic and ischemic Balb/c legs at day-3 post-HLI and examined for VEGF165b secretion into culture-medium. Consistent with the data from HPBMDMs and Balb/c-BMDM, VEGF165b was not detected in culture-medium from non-ischemic or ischemic-leg BM (Fig-1D). However, in cell-lysates, a significant increase in VEGF165b levels was observed in ischemic vs. non-ischemic leg BM monocytes (Non-ischemic leg BM 8.8±1.7pg/mg vs. Ischemic-leg BM 18.5±3.2pg/mg, P=0.03, Fig-1D).
Knowing that ischemic monocytes/macrophages express but do not secrete VEGF165b protein and higher VEGF165b levels are linked to a poorer response to experimental-PAD13, we hypothesized that ‘VEGF165b induction serves an autocrine role in macrophages that have the ability to impair ischemic-muscle tissue perfusion in PAD’. To confirm our hypothesis, we next examined macrophage-specific VEGF165b-expression in Balb/c ischemic-muscle. Flow cytometry analysis (gating strategy presented in Supplemental-Figure-112,20) showed significantly higher macrophage-specific VEGF165b-expression in ischemic (IGA) vs. non-ischemic (NGA) gastrocnemius muscle (NGA:0.8±0.7% vs. IGA:12.7±1.7%, P=0.0007, Fig-1E). We have recently shown that ischemia-induced VEGF165b-expression inversely correlated with VEGFR1-activation in human and experimental-PAD muscle12,22. Hence, we next examined whether increased macrophage-specific VEGF165b-expression in ischemic-muscle correlates with changes in the extent of VEGFR1-activation. Flow cytometry analysis showed that VEGF165b-induction inversely correlated with significantly lower VEGFR1-activation in ischemic-muscle macrophages vs. non-ischemic groups (P<0.03, Fig-1F). Furthermore, VEGF165b-expression was 12.7% in 15.5% of total VEGF-A expression in ischemic-muscle macrophages (VEGF165a=~2.8%, Supplemental-Figure-2) indicating that a significant increase in VEGF165b-expression over VEGF165a contributes to lower VEGFR1-activation in ischemic-muscle macrophages vs. non-ischemic
We next gated macrophages from non-ischemic and ischemic-muscle as VEGF165b-expressing (CD11b+F4/80+VEGF165b+ve) and VEGF165b non-expressing (CD11b+F4/80+VEGF165b−ve) populations and examined the extent of VEGFR1-activation within these populations12. Flow cytometry analysis showed that while VEGF165b+ve macrophages in ischemic-muscle have significantly lower VEGFR1-activation (~30X) vs. non-ischemic (P=0.001, Fig-1G), VEGF165b−ve macrophages showed no significant differences in VEGFR1-activation vs. non-ischemic (Fig-1H). Since VEGFR1-activity has been associated with changes in macrophage-phenotype23, we hypothesized that VEGF165b inhibits VEGFR1-activation to induce a pro-inflammatory phenotype. Consistent with our hypothesis, increased VEGF165b-expression in macrophages correlated with a significant increase (~6X) in M1-like-phenotype24 (CD45+CD11b+F4/80+CD80high, P=0.001, Fig-1I) but not with M2-like-phenotype24 (CD45+CD11b+F4/80+Arg1+, Fig-1J) in IGA vs. NGA. These data showed that VEGF165b regulates macrophage-phenotype by modulating the extent of VEGFR1-activation in distal ischemic-muscle.
2. VEGF165b-expression in human-PAD correlates with increased M1-like-macrophages in PAD muscle-biopsies
To examine the translational relevance of our murine findings, we next examined age- and gender-matched, human, normal and PAD muscle-biopsies12 that were used in our previous publications12,20,22,25 and correlated VEGF165b-expression with the number of CD80+ and CD206+ cells. Immunohistochemistry of VEGF165b in normal (n=5) vs. PAD (n=7) muscle-biopsies showed a significant increase (~2.5X, NL-7.6±0.8 vs. PAD-19.7±1.1) in VEGF165b+ve cells in PAD muscle-biopsies vs. normal (P<0.0001, Fig-1K). We next performed immunofluorescence analysis to determine differences in CD206+ (M2-like) and CD80+ (M1-like) macrophages in normal and PAD muscle-biopsies. A significant increase in total macrophages (CD68+, a pan-macrophage marker) was observed (~2X, NL-16±2.1 vs. PAD-33.8±1.6) in PAD muscle-biopsies vs. normal (Fig-1L). Subsequent analysis of CD80+ and CD206+ macrophages in total CD68+-macrophages showed a significant increase (~3X) in CD80+-macrophages in PAD-muscle vs. normal (NL-9±3.1 vs. PAD-27.5±2.4, P=0.0004, Fig-1M); but no significant difference in CD206+-macrophages in total CD68+-macrophages (NL-7.7±2.9, PAD-12.7±1.0, Fig-1N). Immunostaining with CD163+ (M2-marker) also showed no significant difference in CD163+-macrophages between PAD-muscle vs. normal (Supplemental-Figure-3).
We next quantified CD133+ cells to determine whether immature progenitor cells contributed to changes in macrophage subsets in PAD muscle biopsies. While total CD133+ cells are significantly lower in PAD-muscle vs. normal (indicating that increased total or M1-like-macrophages are not due to CD133+ progenitors in PAD-muscle, Supplemental-Figure-4), no significant difference between VEGF165b+CD133+, CD68+CD133+ or VEGF165b+CD133+CD68+ cells was observed between PAD-muscle vs. normal (data not shown).
We have previously shown that higher VEGF165b protein in PAD muscle-biopsies correlated with decreased VEGFR1-activation12 and our current data show that VEGF165b-induction correlates with increased M1-like-phenotype in PAD muscle-biopsies. Hence, we conclude that VEGF165b-induction in ischemic-muscle decreases macrophage-specific VEGFR1-activation to induce an M1-like-phenotype in PAD patients.
3. VEGF165b skews macrophage phenotype to M1-like in preclinical-PAD
To determine the causal role of VEGF165b in modulating macrophage-phenotype, we performed loss of function experiments by intramuscular delivery of VEGF165b-Ab (or isotype-matched IgG (control)) in ischemic-muscle; and gain of function experiments by intramuscular delivery of VEGF165b-expressing plasmid (or negative-plasmid (control), Supplemental-Figure-5) in non-ischemic-muscle and examined differences in macrophage-phenotype based on CD80 or Arg-1-expression24
Loss of VEGF165b function:
VEGF165b-expression in ischemic-muscle was inhibited by intramuscular delivery of VEGF165b-Ab into ischemic-muscle immediately after HLI as previously described12. Flow cytometry analysis showed no significant difference in the total macrophage numbers between VEGF165b-Ab vs. IgG treated ischemic-muscle (IGA) at day-3 post-HLI (Fig-2A). VEGF165b-inhibition in IGA significantly increased Arg-1+-macrophages (~2X, IgG-10.2±1.8 vs. VEGF165b-Ab-20±3.3%, P<0.05, Fig-2B) with a concomitant decrease in CD80high-macrophages (IgG-36.6±7.7 vs. VEGF165b-Ab-11.4±3.7%, P<0.03, Fig-2C) vs. IgG.
Figure-2.

VEGF165b regulates macrophage phenotype. A-C) Flowcytometric analysis of A) total (F4/80+), B) Arg-1+ M2-like (F4/80+Arg-1+) and C) CD80high M1-like (F4/80+CD80high) macrophages from ischemic gastrocnemius muscle (IGA) treated with IgG or VEGF165b-antibody (VEGF165b-Ab). n=4. Unpaired T-test was used to determine statistical significance. D-F) Flowcytometric analysis of D) total (F4/80+), E) Arg-1+ M2-like (F4/80+Arg-1+) and F) CD80high M1-like (F4/80+CD80high) macrophages from non-ischemic gastrocnemius muscle (NGA) treated with control plasmid (Neg Pld) or VEGF165b-expressing plasmid (V165b pld). n=4. Unpaired T-test was used to determine statistical significance. G) Flowcytometric analysis of Arg-1 expression in bone marrow derived macrophages (BMDM) treated with IgG (10ug/ml), V165b (VEGF165b, 50ng/ml) or VEGF165b-Ab (10ug/ml). n=3. One Way ANOVA with Dunnett’s multiple test correction was used to determine statistical significance. *P<0.05 considered significant.
Gain of VEGF165b function:
VEGF165b overexpression in non-ischemic leg did not induce differences in total macrophage numbers in NGA (Fig-2D). However, a significant decrease in Arg-1+-macrophages (control-plasmid-9.9±0.4 vs. VEGF165b-plasmid-5.9±1.1%, P=0.02, Fig-2E) and a significant increase in CD80high-macrophages (control-plasmid-17.1±2.5 vs. VEGF165b-plasmid-24.6±1.5%, P=0.04, Fig-2F) was observed in NGA treated with VEGF165b-expressing vs. control-plasmid.
We next confirmed our in vivo results in vitro using Balb/c-BMDMs. In vitro, VEGF165b significantly decreased Arg-1-expression (~2X) and VEGF165b-inhibition significantly induced Arg-1-expression (~1.5X) vs. IgG in BMDMs (P<0.05, Fig-2G). Thrombospondin-1 is a known Anti-angiogenic molecule that has been shown to induce M1-like-macrophages26. We next determined whether increased M2-like-macrophages post VEGF165b-inhibition is due to decreased thrombospondin-1 levels. Immunoblot analysis showed no significant difference in thrombospondin-1-expression either in ischemic muscle or HSS BMDMs treated with IgG or VEGF165b-Ab (Supplemental-Figure-6).
4. VEGF165b-expressing M1-like-macrophages impair perfusion in preclinical-PAD in a paracrine manner.
Since C57Bl6 mice exhibit a striking ability to favorably recover from HLI (i.e. a better recovery mouse strain than the Balb/c mouse strain in preclinical-PAD)27,28, we examined whether VEGF165b-expressing macrophages can impair angiogenesis and perfusion-recovery in C57Bl/6 ischemic-muscle. First, we collected conditioned-medium from 1) BMDMs transfected with negative-plasmid or VEGF165b-expressing plasmid (transfection efficiency presented in Supplemental-Figure-2) and 2) HSS-challenged BMDMs treated with IgG or VEGF165b-Ab in vitro. While conditioned-medium from C57Bl/6-BMDM transfected with VEGF165b-expressing plasmid significantly decreased capillary-like tube formation on growth factor reduced matrigel (GFRM) vs. C57Bl/6-BMDM transfected with negative-plasmid (P<0.05, Fig-3A), conditioned-medium from HSS-challenged C57Bl/6-BMDM treated with VEGF165b-Ab increased capillary-like tube formation on GFRM vs. IgG (~3X, P<0.05, Fig-3B).
Figure-3.

VEGF165b expressing macrophages inhibit angiogenesis and perfusion recovery. A, B) In vitro angiogenesis assay of endothelial cells treated with A) conditioned-medium (CM) from C57Bl/6-bone marrow derived macrophages transfected with negative plasmid (Neg Pld) or VEGF165b-expressing plasmid (VEGF165b-Pld). B) Conditioned-medium from HSS-challenged BMDM treated with IgG or VEGF165b-Ab. n=6. Unpaired T-test to determine statistical significance. C) Laser Doppler perfusion imaging of C57Bl/6 ischemic-muscle treated with BMDM-transfected with negative plasmid or VEGF165b-expressing plasmid. n≥5. Repeated measures ANOVA with Dunnett’s multiple test correction was used to determine statistical significance. *P<0.05 considered significant from day 0, #P<0.05 considered significant between groups at that specific day (Unpaired T-test was used to determine statistical significance). D) CD31 immunostaining in ischemic-muscle treated with BMDM transfected with negative plasmid or VEGF165b-expressing plasmid. n≥5. Unpaired T-test was used to determine statistical significance. *P<0.05 considered significant.
We next adoptively transferred C57Bl6-BMDM transfected with control or VEGF165b-expressing plasmid into C57Bl/6 ischemic-muscle immediately after HLI and examined angiogenesis and perfusion-recovery. Laser Doppler imaging showed a significant decrease in the perfusion-recovery in the ischemic-muscle treated with VEGF165b-expressing macrophages vs. control (d14: Negative-plasmid BMDM 86.5±5.8 vs. VEGF165b-plasmid BMDM 66.9±6.1, Fig-3C). Immunohistochemistry showed a significant decrease in CD31+ (endothelial) cells in the ischemic-muscle treated with VEGF165b-expressing BMDM vs. control (Fig-3D). These results indicated that increased VEGF165b-expression in macrophages induces an M1-like-phenotype to impair angiogenesis and perfusion-recovery in preclinical-PAD.
5. VEGF165b induces an M1-like-macrophage phenotype by inhibiting VEGFR1-activation in preclinical-PAD.
We next examined the role of VEGF165b-VEGFR1 interactions in regulating macrophage-polarization. Flow cytometry analysis showed that VEGF165b-inhibition in ischemic-muscle significantly induced pVEGFR1Y1333-activation vs. IgG (P=0.004, Fig-4A, left panel) and immunofluorescence analysis of F4/80 and pVEGFR1Y1333 visually confirmed greater VEGFR1Y1333-activation in VEGF165b-Ab treated ischemic-muscle macrophages (Supplemental-Figure-7). Subsequently, flow cytometry analysis showed that VEGF165b-overexpression in non-ischemic-muscle significantly decreased pVEGFR1Y1333-activation (P=0.01, Fig-4A, right panel) in total macrophages vs. negative-plasmid controls. Correlating with increased Arg-1+-macrophages in VEGF165b-Ab treated ischemic-muscle (Fig-2B), VEGF165b-inhibition was associated with significantly greater pVEGFR1Y1333-activation in Arg-1+-macrophages (P=0.005, Fig-4B, left-panel); which correlated with decreased Arg-1+-macrophages in VEGF165b-plasmid treated non-ischemic-muscle (Fig-2E), VEGF165b-overexpression significantly decreased pVEGFR1Y1333-activation in Arg-1+-macrophages (P=0.03, Fig-4B, right-panel) vs. respective controls. VEGF165b-inhibition or VEGF165b-overexpression did not change pVEGFR1Y1333-activation in CD80high-macrophages vs. respective controls (Fig-4C). Placental growth factor (PLGF), a VEGFR1 specific ligand has shown to induce M2-like-macrophage polarization29, 30. Hence, we determined whether increased Arg-1+-macrophages post VEGF165b-inhibition is due to increased PGLF levels. Immunoblot analysis showed no significant difference in PLGF levels either in ischemic-muscle or HSS BMDMs treated with IgG or VEGF165b-Ab (Supplemental-Figure-8).
Figure-4.

VEGF165b inhibits macrophage pVEGFR1Y1333 activation. A-C) Flowcytometric analysis of pVEGFR1Y1333 activation in A) total, B) Arg-1+ M2-like and C) CD80high M1-like macrophages in ischemic gastrocnemius muscle (IGA) treated with IgG or VEGF165b-antibody (VEGF165b-Ab); or non-ischemic-muscle treated with control plasmid (Neg Pld) or VEGF165b-expressing plasmid (V165b pld). n=4. Unpaired T-test was used to determine statistical significance. D-F) Flowcytometric analysis of D) total, E) Arg-1+ M2-like and F) CD80high M1-like macrophages in the IGA of wild type littermates (WT, VEGFR1+/+), VEGFR1+/− + IgG and VEGFR1+/− + VEGF165b-Ab. n=4, One way ANOVA with Dunnett’s multiple test correction was used to determine statistical significance. *P<0.05 considered significant.
To confirm the role of VEGFR1 in regulating macrophage-polarization, we used VEGFR1+/− mice (VEGFR1−/− are embryonic lethal) backcrossed to Balb/c background that cannot up-regulate VEGFR1 in ischemic-muscle to the same extent as VEGFR1+/+ mice (~50% reduction in VEGFR1 induction in VEGFR1+/− ischemic-muscle vs. VEGFR1+/+ ischemic-muscle12). Flow cytometry analysis showed a significant decrease (~3X) in total macrophage numbers in VEGFR1+/− ischemic-muscle (IGA:VEGFR1+/+ 19.9±1.0 vs. VEGFR1+/− 5.4±0.9%, P=0.0002, Fig-4D) vs. VEGFR1+/+. Furthermore, a significant decrease (~2X) in Arg-1+-macrophages (IGA: VEGFR1+/+ 15.8±1.8 vs. VEGFR1+/− 8.2±1.2%, P<0.03, Fig-4E) with a concomitant increase in CD80high-macrophages (IGA: VEGFR1+/+ 19.4±5.9 vs. VEGFR1+/− 48.0±7.8%, P<0.04, Fig-4F) was observed in VEGFR1+/− ischemic-muscle vs. VEGFR1+/+. VEGF165b-Ab did not significantly change the numbers of total, M1 or M2-like-macrophages in VEGFR1+/− ischemic-muscle vs. IgG treated VEGFR1+/− ischemic-muscle (Figure-4). These results confirmed that VEGFR1 plays an important role in VEGF165b induced M1-like-polarization in preclinical-PAD.
We next examined VEGF165b-VEGFR1 interactions in macrophages in vitro. While VEGF165b treatment significantly decreased pVEGFR1Y1333-activation, VEGF165b-inhibition significantly induced pVEGFR1Y1333 activation vs. IgG-treated controls (P<0.05, Supplemental-Figure-9A), correlating with their M1- or M2-like-phenotypes indicating that VEGF165b induced M1-like-polarization involves pVEGFR1Y1333-inhibition.
VEGF165b has been predicted to be a competitive inhibitor of VEGF165a for VEGFR2 in endothelial cells11. However, in general, macrophages primarily express VEGFR1. Hence, to understand the molecular interactions that result in VEGFR1-activation post VEGF165b-inhibition, we pulled down VEGFR1 from normal or HSS-BMDM that were treated with IgG of VEGF165b-Ab and examined the bound fractions of VEGF-A and VEGF165b. immunoblot analysis of VEGFR1-pull-down complexes showed a significant increase in the bound fraction of VEGF165a to VEGFR1 post VEGF165b-inhibition under normal (P=0.005, Supplemental-Figure-9B) and HSS conditions (P=0.0004, Supplemental-Figure-9C) indicating that increased VEGF165b binding, over VEGF165a, to VEGFR1 decreases VEGFR1-activation.
To confirm our results, we treated BMDM with VEGF165a, VEGF165b or a combination of VEGF165a and VEGF165b at equimolar concentrations and examined the changes in VEGFR1Y1333-activation. Flow cytometry analysis showed that while VEGF165a significantly induced VEGFR1-activation in BMDM, VEGF165b significantly decreased VEGF165a induced VEGFR1-activation under HSS conditions (Supplemental-Figure-9D, E), indicating that VEGF165b is a competitive inhibitor of VEGFR1 at Y1333 site.
6. VEGFR1+/− macrophages impair perfusion vs. VEGFR1+/+ macrophages in preclinical-PAD
Since VEGF165b induced VEGFR1 dependent M1-like-polarization, we next examined the role of macrophage-specific VEGFR1 in regulating perfusion-recovery in experimental-PAD. qPCR analysis showed that while HSS significantly induced VEGFR1-expression in VEGFR1+/+ BMDM vs. control (~5.5X), no significant difference in VEGFR1-expression was observed in VEGFR1+/− under HSS vs. control (Supplemental-Figure-10). We next examined BMDMs from VEGFR1+/+ and VEGFR1+/− for their relative M1 and M2-like-states. At baseline, no significant difference in either Arg-1 or iNos-expression was observed between VEGFR1+/+ and VEGFR1+/− BMDM. However, under HSS, qPCR analysis showed that VEGFR1+/− BMDM express significantly lower Arg-1 (6h-HSS ~16.5X, P<0.05) and significantly higher iNos (6h-HSS~3X, 12h-HSS~3X, 24h-HSS~2.4X, P<0.05) levels vs. VEGFR1+/+ (Fig-5A) indicating that VEGFR1 plays an important role in regulating macrophage-polarization. Functional analysis of VEGFR1+/− BMDM showed that conditioned-medium from normal VEGFR1+/− BMDMs did not change endothelial capillary-like tube formation on GFRM (Fig-5B), however, HSS-VEGFR1+/− BMDMs significantly decreased endothelial capillary-like tube formation on GFRM vs. conditioned-medium from HSS-VEGFR1+/+ BMDMs (P=0.02, Fig-5C). After confirming that HSS induces M1-like-phenotype in VEGFR1+/− BMDM that can inhibit endothelial angiogenic potential, in a paracrine manner, in vitro, we next examined their functional role in regulating perfusion in vivo. BMDM from VEGFR1+/− and VEGFR1+/+ were adoptively transferred into ischemic-muscle of WT mice immediately after HLI. Laser Doppler imaging showed that ischemic-muscle that received VEGFR1+/− BMDM have significantly lower perfusion vs. VEGFR1+/+-BMDM (d14: VR1+/+ BMDM+VR1+/+ ischemic-muscle 63.4+/−1.9 vs. VR1+/− BMDM+VR1+/+ ischemic-muscle 48.8+/−3.5 Fig-5D). Immunohistochemistry showed a significant decrease in vascular density (CD31+) in ischemic-muscle that received VEGFR1+/− BMDM vs. VEGFR1+/+ BMDM (Fig-5E). This data indicated that VEGF165b-inhibition of VEGFR1 plays a role in regulating macrophage paracrine effects on ischemic vasculature.
Figure-5.
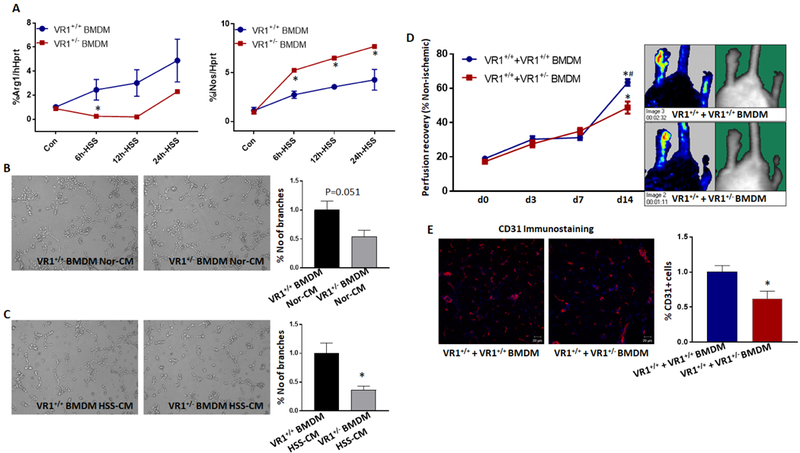
VEGFR1+/− macrophages inhibit angiogenesis and perfusion recovery. A) qPCR analysis of temporal changes in Arg-1 and iNos expression in VEGFR1+/+ and VEGFR1+/− BMDM under normal and hypoxia serum starvation (HSS) conditions. n=6. One way ANOVA with Dunnett’s multiple test correction was used to determine statistical significance. B-C) In vitro angiogenesis assay of endothelial cells treated with conditioned-medium (CM) from B) normal or C) HSS-challenged VEGFR1+/+ (VR1+/+) or VEGFR1+/− (VR1+/−) bone marrow derived macrophages. n=6. Unpaired T-test was used to determine statistical significance. D) Laser Doppler perfusion imaging of WT (VR1+/+) ischemic-muscle treated with VEGFR1+/+ BMDM or VEGFR1+/− BMDM. n≥5. Repeated measures ANOVA with Dunnett’s multiple test correction was used to determine statistical significance. *P<0.05 considered significant from day 0, #P<0.05 considered significant between groups at that specific day (Unpaired T-test was used to determine statistical significance). E) CD31 immunostaining in WT ischemic-muscle treated with VEGFR1+/+ or VEGFR1+/− BMDM. n≥5. Unpaired T-test was used to determine statistical significance. *P<0.05 considered significant.
7. VEGFR1-STAT3 interactions regulate macrophage-phenotype in preclinical-PAD
We next wanted to determine the signaling processes/intermediates downstream of VEGFR1Y1333-activation that plays a role in macrophage-polarization. We have previously shown that VEGFR1 can regulate STAT3-activation12. Hence, we examined whether STAT3 plays a role in VEGFR1 downstream signaling in regulating macrophage-polarization.
Flow cytometry analysis showed that VEGF165b-inhibition induced a significant increase in pSTAT3-activation in total (IgG 8.3±1.7 vs. VEGF165b-Ab 14.4±1.2, P=0.02, Fig-6A, left panel) and Arg-1+-macrophages (IgG 10.9±1.2 vs. VEGF165b-Ab 16.4±1.3, P=0.02, Fig-6B, left panel) in Balb/c-IGA vs. IgG. No significant difference in pSTAT3-activation was observed in CD80high-macrophages from Balb/c-IGA treated with VEGF165b-Ab vs. IgG
Figure-6.

VEGF165b inhibits VEGFR1-STAT3 signaling. A) Flowcytometric analysis of pSTAT3 activation in A) total, B) Arg-1+ M2-like and C) CD80high M1-like macrophages in ischemic gastrocnemius muscle (IGA) treated with IgG or VEGF165b-Ab; or non-ischemic gastrocnemius muscle (NGA) treated with control plasmid (Neg Pld) or VEGF165b-expressing plasmid (VEGF165b Pld). n=4. Unpaired T-test was used to determine statistical significance. D-F) Flowcytometric analysis of pSTAT3 activation in D) total, E) Arg-1+ M2-like and F) CD80high M1-like macrophages in IGA of wild type littermates (WT, VEGFR1+/+), VEGFR1+/−+ IgG and VEGFR1+/−+ VEGF165b-Ab. n=4, One way ANOVA with Dunnett’s multiple test correction was used to determine statistical significance. *P<0.05 considered significant.
VEGF165b-overexpression (by VEGF165b-plasmid) significantly decreased pSTAT3-activation in total (control plasmid 1.0±0.01 vs. VEGF165b-plasmid 0.6±0.08, P=0.03, Fig-6A, right panel) and Arg-1+-macrophages (control plasmid 0.6±0.07 vs. VEGF165b-plasmid 0.4±0.01, P=0.01, Fig-6B, right panel) in Balb/c-NGA vs. control plasmid. No significant differences in pSTAT3-activation was observed in CD80high-macrophages from Balb/c-NGA treated with VEGF165b-expressing plasmid vs. control-plasmid (Fig-6C).
We next examined the causal role of VEGFR1 in regulating STAT3-activation to modulate macrophage-polarization using VEGFR1+/− mice. Flow cytometry analysis showed a significant decrease in STAT3-activation in total macrophages from VEGFR1+/− ischemic-muscle (IGA~2X, P<0.03, Fig-6D) vs. VEGFR1+/+. A significant decrease in Arg-1+-macrophages in VEGFR1+/− ischemic-muscle was associated with decreased STAT3-activation (IGA~10X, P=0.001, Fig-6E). A significant increase in CD80high-macrophages in VEGFR1+/− ischemic-muscle was associated with decreased STAT3-activation (IGA~3X, P=0.01, Fig-6F) when vs. VEGFR1+/+.
Based on our recent data that show VEGFR1 when activated by VEGF165a interacts with STAT3 to induce STAT3-activation in endothelial cells12, we next examined the role of VEGF165b in regulating VEGFR1-STAT3 interactions in macrophages. In vitro, immunoblotting of VEGFR1-immunoprecipitated complexes from VEGF165b-Ab treated HSS-challenged BMDMs showed a significant increase in STAT3 binding vs. IgG (P=0.03, Supplemental-Figure-11A). This data indicated that VEGF165b competes with VEGF165a to bind and inhibit VEGFR1-STAT3 signaling activation in ischemic macrophages.
Flow cytometry analysis of BMDM treated with VEGF165b had significantly decreased pVEGFR1+pSTAT3+ vs. controls correlating with CD80high-macrophages vs. control; and BMDM treated with VEGF165b-Ab treatment significantly induced pVEGFR1+pSTAT3+ correlating with Arg-1+macrophages vs. IgG in vitro (Supplemental-Figure-11B). We next examined whether pVEGFR1+pSTAT3+ distinguishes M1 vs. M2-like-macrophages post VEGF165b-inhibition in vivo. Flow cytometry analysis showed a significant increase in pVEGFR1+pSTAT3+ expression in total macrophages (~3X, IgG 1.3±0.4 vs. VEGF165b-Ab 3.7±0.7%, P<0.04, Supplemental-Figure-11C), in ischemic-muscle treated with VEGF165b-Ab vs. IgG. This increase correlated with Arg-1+-macrophages (~2X, IgG 23.2±2.5 vs. VEGF165b-Ab 54.4±5.5%, P=0.0009, Supplemental-Figure-11D) but not with CD80high-macrophages (Supplemental-Figure-11E). These results indicated that VEGF165b-inhibition induced pVEGFR1+pSTAT3+ double positive Arg-1+-macrophages in ischemic-muscle vs. IgG.
8. S100A8/S100A9 downstream of VEGFR1 regulate macrophage-polarization.
Unlike VEGFR2, information on genes that regulate VEGFR1 downstream processes is limited31,32. Hence, to identify the VEGFR1 downstream signaling genes that regulate macrophage-polarization we performed RNA-Seq analysis of HSS-challenged VEGFR1+/− and VEGFR1+/+ BMDMs (Supplemental-table-1, 2). Volcano plots, principal component analysis (PCA) analysis33 and hallmark pathway analysis34 (GSEA) of the RNA-Seq data is presented in Supplemental-Figure-12. RNA-Seq analysis showed that S100A8 and S100A9, members of S100 family that regulate intracellular calcium levels35,36 are significantly upregulated in HSS-challenged VEGFR1+/− BMDM vs. VEGFR1+/+ (within top 5 genes, q<0.05, Supplemental-Figure-12, Supplemental-Table-1). qPCR analysis of S100A8 and S100A9 in normal and HSS-challenged VEGFR1+/+ and VEGFR1+/− BMDM showed no significant difference in S100A8 or S100A9-expression between normal VEGFR1+/− and VEGFR1+/+ BMDM, but a significant increase in HSS-VEGFR1+/− BMDM vs. HSS- VEGFR1+/+ BMDM confirming the RNA-Seq data (Fig-7A, B). Furthermore, a significant increase in S100A8 and S100A9-expression was also observed in VEGFR1+/− ischemic-muscle (Fig-7C, D) and HSS VEGFR1+/− skeletal muscle endothelial cells (Supplemental-Figure-13) vs. respective controls. No significant difference in S100A8 and S100A9-expression was observed between VEGFR1+/+ vs. VEGFR1+/− non-ischemic-muscle (Fig-7C, D) or normal VEGFR1+/+ vs. VEGFR1+/− skeletal muscle endothelial cells (Supplemental-Figure-13).
Figure-7.

VEGFR1 regulates S100A8/S100A9 expression in macrophages. A-B) qPCR analysis of A) S100A8 and B) S100A9 expression in VEGFR1+/+ (VR1+/+), VEGFR1+/− (VR1+/−) bone marrow derived macrophages (BMDM) under normal or HSS conditions. n=6. One way ANOVA with Bonferroni select pair comparison was used to determine statistical significance. C-D) qPCR analysis of C) S100A8 and D) S100A9 expression in VEGFR1+/+ (VR1+/+), VEGFR1+/− (VR1+/−) non-ischemic (NGA) and ischemic gastrocnemius muscle (NGA). n=5. One way ANOVA with Bonferroni select pair comparison was used to determine statistical significance. E-H) qPCR analysis of arginase-1 (Arg-1) and inducible nitric oxide synthase (iNos) expression in macrophages (Raw264.7) transfected with (E-F) S100A8 or (G-H) S100A9 under normal or HSS conditions. n=6. Unpaired T-test was used to determine statistical significance. I-J) qPCR analysis Arg-1:iNos expression in I-K) S100A8 or J-L) S100A9 transfected macrophages treated with BAPTA-AM under normal or HSS conditions. n=6. Unpaired T-test was used to determine statistical significance. M-N) In vitro angiogenesis assay of endothelial cells treated with conditioned-medium (CM) from M) normal or N) HSS-challenged macrophages transfected with control plasmid (Neg Pld), S100A8 plasmid (S100A8 Pld) or S100A9 plasmid (S100A9 Pld). n=4. One way ANOVA with Dunnett’s multiple test correction was used to determine statistical significance. *P<0.05 considered significant.
We next examined S100A8 and S100A9 function in regulating macrophage-polarization and angiogenesis in vitro. qPCR analysis of Arg-1 and iNos in macrophages overexpressing S100A8 or S100A9 showed a significant decrease in Arg-1-expression (S100A8: Nor ~9X (P=0.051), HSS ~1X (P=0.041); S100A9: Nor ~7X (P=0.0087), HSS ~5.5X (P<0.0001)) with a concomitant increase in iNos-expression (S100A8: Nor ~3.5X (P=0.003), HSS ~2X (P=0.002); S100A9: Nor ~4X (P=0.001) HSS (P=0.02)) under normal and in HSS conditions (Fig-7E-H). Transfection efficacy of S100A8 presented in Supplemental-Figure-14. Ct values of S100A9 in negative-plasmid transfected macrophages (Raw264.7) was higher than 50 cycles and S100A9 plasmid transfection decreased the Ct values to a mean of 22±0.17.
Since S100A8 and S100A9 were shown to induce calcium influx in neutrophils37, we next examined whether calcium influx due to increased S100A8 and S100A9-expression drive M1-like-phenotype. To confirm, we overexpressed S100A8 or S100A9 in macrophages and treated them with or without BAPTA-AM (1,2-Bis (2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester), a cell-permeable calcium chelator38 under normal or HSS conditions. Interestingly, BAPTA-AM induced greater Arg1:iNos expression in macrophages under normal conditions but no significant difference in Arg1:iNos expression was observed under HSS conditions (Supplemental-Figure-15). However, BAPTA-AM not only blocked S100A8 and S100A9 induced M1-like-phenotype but also promoted an M2-like-phenotype both under normal (Arg1:iNos: S100A8 ~1X (P=0.03), S100A9 ~3X (P=0.0003), Fig-7I, J) and HSS (Arg1:iNos: S100A8 (P=0.03), S100A9 (P=0.01), Fig-7K, L) conditions. These data suggested that while HSS induces an M1-like-phenotype, this phenotype may not be due to increased calcium levels; but S100A8 and S100A9 induced M1-like-phenotype is via increased calcium influx.
We next examined the role of S100A8 and S100A9 in endothelial cells. Overexpressing either S100A8 or S100A9 showed no significant difference in capillary-like tube formation on GFRM in vitro (Supplemental-Figure-16). However, conditioned-medium from macrophages transfected with S100A8 or S100A9 under normal and HSS conditions significantly decreased endothelial tube-like formation on GFRM (Fig-7M, N). These data suggested that increased S100A8 and S100A9-expression in macrophages inhibit endothelial angiogenic potential in a paracrine manner.
Since earlier reports have indicated that TLR-signaling regulates S100A8-expression via STAT3 pathway in macrophages39, we next examined whether STAT3 regulates S100A8 in macrophages under HSS conditions. qPCR analysis of S100A8 in macrophages treated with or without STAT3 inhibitor (100uM) showed no significant difference in S100A8-expression (Supplemental-Figure-17) indicating that in macrophages STAT3 is not a regulator of S100A8. Furthermore, our data also indicate that VEGFR1-activation induces distinct signaling independent of STAT3-activation to inhibit S100A8 and S100A9-expression in macrophages.
9. VEGF165b regulates S100A8/S100A9 expression in preclinical-PAD
We next wanted to obtain a direct evidence of VEGF165b in regulating macrophage-specific S100A8 and S100A9-expression. Since S100A8 and S100A9-expression is significantly higher in ischemic vs. non-ischemic-muscle (Supplemental-Figure-18, correlating with increased VEGF165b levels), we performed flow cytometry analysis of ischemic-muscle treated with IgG or VEGF165b-Ab. Flow cytometry analysis showed that VEGF165b-inhibition significantly decreased ischemia-induced total tissue (Fig-8A), endothelial (Supplemental-Figure-19) and macrophage (Fig-8B) specific S100A8/S100A9-expression vs. IgG. Furthermore, gating on S100A8/S100A9+ macrophages showed that VEGF165b-inhibition significantly decreased S100A8/S100A9+ CD80+-macrophages (Fig-8C) in ischemic-muscle vs. IgG. No significant difference in S100A8/S100A9+ Arg-1+-macrophages was observed.
Figure-8.

VEGF165b regulates S100A8/S100A9 expression in ischemic-muscle. A-B) Flowcytometric analysis S100A8/S100A9 expression in A) non-ischemic (NGA), ischemic-muscle treated with IgG or ischemic-muscle treated with VEGF165b-Ab, B) macrophages from NGA, IGA+IgG or IGA+VEGF165b-Ab. C) CD80high M1-like macrophages in NGA, IGA+IgG or IGA+VEGF165b-Ab. A-C) n=5, One way ANOVA with Dunnett’s multiple test correction was used to determine statistical significance. D-E) qPCR analysis of D) Arg-1 and E) iNos expression in macrophages (Raw264.7) transfected with S100A8 or S100A9 plasmid and treated with IgG or VEGF165b-Ab under normal or HSS conditions. n=6. Unpaired T-test was used to determine statistical significance. *P<0.05 considered significant. F) Schematic of VEGF165b induced macrophage M1 like polarization that inhibits perfusion recovery in PAD.
To confirm the in vivo data, we next overexpressed S100A8 or S100A9 in macrophages and treated them with IgG or VEGF165b-Ab under normal or HSS conditions. qPCR analysis showed that VEGF165b-inhibition significantly induced Arg-1-expression (S100A8: Nor ~6X, HSS ~9X; S100A9: Nor ~6.5X, HSS~14.5X, Fig-8D) in macrophages both under normal and HSS conditions. No significant difference in iNos-expression was observed between IgG or VEGF165b-Ab treated S100A8 or S100A9 transfected macrophages under normal or HSS conditions (Fig-8E). This data indicated that VEGFR1-activation post VEGF165b-inhibition decreases S100A8/S100A9-expression to induce M2-like-polarization in pre-clinical PAD. Schematic of the VEGF165b signaling that regulates macrophage-polarization in ischemic-muscle is presented in Fig-8F.
Discussion
The results of our study demonstrate for the first time that VEGF165b, a hereunto unrecognized splice variant within a family of critical growth factors, is capable and sufficient to alter macrophage-polarization in whole or in part through S100A8/S100A9. The effects of VEGF165b on macrophages have not been previously reported. We showed that VEGF165b binds to VEGFR1 on macrophages and this binding has two functions. First, this polarizes macrophages toward an M1 (inflammatory)-like-phenotype and this binding prevents the binding of VEGFR1-activating ligands, which can promote an M2 (reparative)-like-phenotype. Our findings in the area of ischemic-muscle revascularization including human-PAD-biopsies; mouse preclinical-PAD models that utilized VEGF165b-depletion and over-expression strategies, adoptive cell transfer experiments; complimentary in vitro experiments and RNA-Seq experiments showed that increased anti-angiogenic VEGF165b isoform expression in macrophages directly and competitively inhibits VEGFR1-S100A8/S100A9 signaling to induce an M1-like-phenotype that inhibits ischemic-muscle angiogenesis and perfusion-recovery in PAD.
Our understanding of the function of the alternatively spliced VEGF165b isoform is expanding but studies to date have largely focused on its role in blocking VEGFR2-activation in endothelial cells. Our current data show that in ischemic macrophages VEGF165b induces an autocrine effect that inhibits VEGFR1Y1333-activation that results in an M1-like-phenotype which in-turn exerts a paracrine effect on hypoxia-dependent angiogenesis. We have recently shown that VEGF165b exhibits differential effects on VEGFR1 and VEGFR2-activation i.e. it functions as an inhibitory isoform for VEGFR1Y1333, and functions as an activator of VEGFR2Y1175 in endothelial cells12 (contrary to the existing paradigm that VEGF165b largely functions as a competitive blocker of VEGF165a induced VEGFR2-activation9,11,31,40–43). While VEGFR2-expression is largely confined to endothelial cells, VEGFR1 is expressed on non-endothelial cell types44,45, including progenitor cells, macrophages, and inflammatory cells suggesting that the function of VEGF165b may well extend beyond endothelial cells. However, unlike extensive literature that exists on the VEGFR2-signaling networks, far less is known of VEGFR1-signaling and function in cardiovascular diseases44,46. Our finding of an increased M1-like-phenotype with VEGFR1-inhibition suggested that VEGF165b may function as more than an ‘inert blocker’.
We have previously shown that VEGF165b modulates VEGFR1-STAT3 activation in endothelial cells to increase angiogenesis. However, in macrophages, VEGF165b not only regulates VEGFR1-STAT3 signaling but also a VEGFR1-S100A8/S100A9 signaling to regulate an M1-like-phenotype. We identified S100A8/S100A9 as downstream signaling intermediates of VEGFR1 by RNA-Seq analysis of HSS VEGFR1+/+ BMDM vs. VEGFR1+/− BMDM. We used HSS VEGFR1+/+ and HSS VEGFR1+/− BMDM because no difference in VEGFR1-expression was observed between VEGFR1+/+ and VEGFR1+/− BMDM under normal conditions. Even though we identified S100A8/S100A9 downstream of VEGFR1 in macrophages, we observed significantly higher S100A8/S100A9 levels in HSS VEGFR1+/− ECs vs. HSS VEGFR1+/+ ECs, indicating that VEGFR1 regulates S100A8/S100A9-expression in endothelial cells also. Based on the limited amount of literature that exists on VEGFR1-signaling intermediates this data should advance the study of VEGFR1-biology.
Recent literature has shown that STAT3 can bind to the promoter of S100A8 in macrophages to induce S100A8-expression39; and increased S100A8-expression can induce STAT3 levels in lung injury models by LPS or cPG-DNA47 indicating a potentially positive feedback loop between S100A8 and STAT3 to regulate macrophage-phenotype. However, in culture, STAT3-inhibition did not change S100A8 levels, indicating that STAT3 regulation of S100A8 is cell and context dependent. These data expanded our recent findings and showed that a novel VEGFR1 dependent but STAT3 independent signaling regulates S100A8/S100A9-expression in macrophages that converge on regulating M2-like-phenotype. These data demonstrate that removal of VEGF165b inhibitory effect on VEGFR1 regulates M2-like-phenotype in (at least) two distinct ways. One involves VEGFR1-activation of STAT3, another signaling involves VEGFR1-inhibition of S100A8/S100A9, which converges on inducing an M2-like-phenotype that promotes angiogenesis and tissue recovery in distal ischemic-muscle. Additional work will be required to delineate the molecular signaling that connects VEGFR1-S100A8/S100A9 signaling.
Interestingly, while S100A8 or S100A9 overexpression did not directly modulate endothelial angiogenic potential under normal or HSS conditions; conditioned-medium from S100A8 or S100A9 transfected macrophages significantly decreased endothelial angiogenic potential. Even though S100A8 or S100A9 did not directly modulate endothelial angiogenic potential in vitro, it is possible that S100A8/S100A9 can modulate other endothelial properties such as vascular integrity/permeability, adhesion molecule expression and/or immune cells migration48,49. Furthermore, while conditioned-medium from VEGF165b transfected and HSS-VEGFR1+/− BMDM significantly decreased endothelial angiogenic capacity, conditioned-medium from BMDM treated with VEGF165b-Ab significantly induced endothelial angiogenic capacity indicating that increased VEGF165b-expression in ischemic macrophages inhibits angiogenesis in a paracrine manner. In addition, adoptive transfer of VEGF165b transfected BMDM and VEGFR1+/− BMDM significantly decreased angiogenesis and perfusion in the ischemic-muscle in vivo indicating that macrophage-specific VEGFR1-signaling plays a major role in regulating PAD muscle perfusion-recovery. Consistent with our murine findings, our recently published human data from PAD and control (age and gender matched) biopsies showed that greater VEGF165b binding to VEGFR1 correlated with decreased VEGFR1-activation12; and in our current work we show that VEGF165b induction correlated with increased CD80+-macrophages in human-PAD. This data showed that VEGF165b inhibits VEGFR1 to induce M1-like-phenotype in human-PAD. However, the markers used to distinguish M1 vs. M2-macrophages24 are not binary. While flow cytometry analysis will provide a better delineation of these cell populations with a more restricted marker expression, muscle availability from human muscle-biopsies limited us for studies such as double immunofluorescence analysis of CD68 and CD80 or CD206.
Kikuchi et al13 have previously shown that VEGF165b levels are significantly higher in the serum of PAD vs normal patients by western blot analysis. Serum preparation involves platelet activation to form a blood clot and it is possible that VEGF165b is released by circulating monocytes or perhaps activated platelets in this process in PAD patients. Furthermore, HSS macrophages expressed higher VEGF165b levels but this remains cell-associated and is not secreted, suggesting an autocrine effect. Unlike the freely circulating VEGF121a isoform, VEGF165a is predominantly membrane-bound and indeed Guzman-Hernandez et al50 showed that VEGF165a is prominently present on the outer surface of the cells. The membrane binding properties of VEGF165a is due to the heparin-binding sites in exon-7, which is same in VEGF165b isoforms. As such, we predict that VEGF165b similar to VEGF165a is inherently membrane bound. Whether and to what extent the 6-aminoacid switch in VEGF165b isoforms confer increased membrane binding properties resulting in decreased secretion is not clear. Such studies will advance our understanding of the mechanism of VEGF165b action.
The novelty of these findings on VEGF165b and its role on macrophages are relevant not only for PAD but also across other human cardiovascular diseases.
Supplementary Material
Clinical Perspective.
What is new?
While known functions of VEGF165b, an anti-angiogenic VEGF-A isoform has been confined to endothelial cells, we show that VEGF165b induces an anti-angiogenic, cytotoxic M1-like phenotype in macrophages that inhibits ischemic muscle angiogenesis in Peripheral artery disease (PAD)
In ischemic macrophages, VEGF165b silences VEGFR1 by an autocrine mechanism to induce an M1-like phenotype that inhibits angiogenesis by a paracrine mechanism in ischemic muscle
We identified S100A8/A9 as a novel VEGF165b-VEGFR1 downstream signaling intermediate that induces M1-like phenotype in ischemic macrophages in PAD
What are the clinical implications?
Therapies that can induce and sustain an M2-phenotype in ischemic-muscle were not yet successful due to the reversible nature of macrophage-polarization states.
Our current work showed that targeting anti-angiogenic VEGF165b isoforms have the translational potential to induce and sustain M2-like-macrophage phenotype in ischemic-muscle to promote therapeutic angiogenesis and enhance tissue recovery
Acknowledgments
Funding Sources: This study was supported by an American Heart Association Scientist Development Grant 16SDG30340002 to VCG and National Institutes of Health grants 1R01 HL116455, 1R01 HL121635 and 2R01 HL101200 from to BHA
Footnotes
Disclosures: Authors declare no conflict of interest.
Part of this study was presented as an abstract (poster) in IVBM-2016, NAVBO Scientific Sessions, Boston, Massachusetts, 30 October to 3 November 2016.
References
- 1.Annex BH. Therapeutic angiogenesis for critical limb ischaemia. Nat Rev Cardiol 2013;10:387–396. [DOI] [PubMed] [Google Scholar]
- 2.O’Riordain DS and O’Donnell JA. Realistic expectations for the patient with intermittent claudication. Br J Surg 1991;78:861–863. [DOI] [PubMed] [Google Scholar]
- 3.Simons M and Ware JA. Therapeutic angiogenesis in cardiovascular disease. Nat Rev Drug Discov 2003;2:863–871. [DOI] [PubMed] [Google Scholar]
- 4.Kullo IJ and Leeper NJ. The genetic basis of peripheral arterial disease: current knowledge, challenges, and future directions. Circ Res 2015;116:1551–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mohler ER III, Rajagopalan S, Olin JW, Trachtenberg JD, Rasmussen H, Pak R and Crystal RG. Adenoviral-mediated gene transfer of vascular endothelial growth factor in critical limb ischemia: safety results from a phase I trial. Vasc Med 2003;8:9–13. [DOI] [PubMed] [Google Scholar]
- 6.Rajagopalan S, Mohler E III, Lederman RJ, Saucedo J, Mendelsohn FO, Olin J, Blebea J, Goldman C, Trachtenberg JD, Pressler M, Rasmussen H, Annex BH and Hirsch AT. Regional Angiogenesis with Vascular Endothelial Growth Factor (VEGF) in peripheral arterial disease: Design of the RAVE trial. Am Heart J 2003;145:1114–1118. [DOI] [PubMed] [Google Scholar]
- 7.Rajagopalan S, Mohler ER III, Lederman RJ, Mendelsohn FO, Saucedo JF, Goldman CK, Blebea J, Macko J, Kessler PD, Rasmussen HS and Annex BH. Regional angiogenesis with vascular endothelial growth factor in peripheral arterial disease: a phase II randomized, double-blind, controlled study of adenoviral delivery of vascular endothelial growth factor 121 in patients with disabling intermittent claudication. Circulation 2003;108:1933–1938. [DOI] [PubMed] [Google Scholar]
- 8.Rasmussen HS, Rasmussen CS and Macko J. VEGF gene therapy for coronary artery disease and peripheral vascular disease. Cardiovasc Radiat Med 2002;3:114–117. [DOI] [PubMed] [Google Scholar]
- 9.Peiris-Pages M The role of VEGF 165b in pathophysiology. Cell Adh Migr 2012;6:561–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qiu Y, Hoareau-Aveilla C, Oltean S, Harper SJ and Bates DO. The anti-angiogenic isoforms of VEGF in health and disease. Biochem Soc Trans 2009;37:1207–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Woolard J, Wang WY, Bevan HS, Qiu Y, Morbidelli L, Pritchard-Jones RO, Cui TG, Sugiono M, Waine E, Perrin R, Foster R, Digby-Bell J, Shields JD, Whittles CE, Mushens RE, Gillatt DA, Ziche M, Harper SJ and Bates DO. VEGF165b, an inhibitory vascular endothelial growth factor splice variant: mechanism of action, in vivo effect on angiogenesis and endogenous protein expression. Cancer Res 2004;64:7822–7835. [DOI] [PubMed] [Google Scholar]
- 12.Ganta VC, Choi M, Kutateladze A and Annex BH. VEGF165b Modulates Endothelial VEGFR1-STAT3 Signaling Pathway and Angiogenesis in Human and Experimental Peripheral Arterial Disease. Circ Res 2017;120:282–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kikuchi R, Nakamura K, MacLauchlan S, Ngo DT, Shimizu I, Fuster JJ, Katanasaka Y, Yoshida S, Qiu Y, Yamaguchi TP, Matsushita T, Murohara T, Gokce N, Bates DO, Hamburg NM and Walsh K. An antiangiogenic isoform of VEGF-A contributes to impaired vascularization in peripheral artery disease. Nat Med 2014;20:1464–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Muramatsu M, Yamamoto S, Osawa T and Shibuya M. Vascular endothelial growth factor receptor-1 signaling promotes mobilization of macrophage lineage cells from bone marrow and stimulates solid tumor growth. Cancer Res 2010;70:8211–8221. [DOI] [PubMed] [Google Scholar]
- 15.Qian BZ, Zhang H, Li J, He T, Yeo EJ, Soong DY, Carragher NO, Munro A, Chang A, Bresnick AR, Lang RA and Pollard JW. FLT1 signaling in metastasis-associated macrophages activates an inflammatory signature that promotes breast cancer metastasis. J Exp Med 2015;212:1433–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heilmann C, Beyersdorf F and Lutter G. Collateral growth: cells arrive at the construction site. Cardiovasc Surg 2002;10:570–578. [DOI] [PubMed] [Google Scholar]
- 17.Seaman SA, Cao Y, Campbell CA and Peirce SM. Macrophage Recruitment and Polarization During Collateral Vessel Remodeling in Murine Adipose Tissue. Microcirculation 2016;23:75–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kharraz Y, Guerra J, Mann CJ, Serrano AL and Munoz-Canoves P. Macrophage plasticity and the role of inflammation in skeletal muscle repair. Mediators Inflamm 2013;2013:491497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rigamonti E, Zordan P, Sciorati C, Rovere-Querini P and Brunelli S. Macrophage plasticity in skeletal muscle repair. Biomed Res Int 2014;2014:560629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ganta VC, Choi MH, Kutateladze A, Fox TE, Farber CR and Annex BH. A MicroRNA93-IRF9-IRG1-Itaconic Acid Pathway Modulates M2-like-Macrophage Polarization to Revascularize Ischemic Muscle. Circulation 2017;135:2403–2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Y, Hazarika S, Xie D, Pippen AM, Kontos CD and Annex BH. In mice with type 2 diabetes, a vascular endothelial growth factor (VEGF)-activating transcription factor modulates VEGF signaling and induces therapeutic angiogenesis after hindlimb ischemia. Diabetes 2007;56:656–665. [DOI] [PubMed] [Google Scholar]
- 22.Jones WS, Duscha BD, Robbins JL, Duggan NN, Regensteiner JG, Kraus WE, Hiatt WR, Dokun AO and Annex BH. Alteration in angiogenic and anti-angiogenic forms of vascular endothelial growth factor-A in skeletal muscle of patients with intermittent claudication following exercise training. Vasc Med 2012;17:94–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Incio J, Tam J, Rahbari NN, Suboj P, McManus DT, Chin SM, Vardam TD, Batista A, Babykutty S, Jung K, Khachatryan A, Hato T, Ligibel JA, Krop IE, Puchner SB, Schlett CL, Hoffmman U, Ancukiewicz M, Shibuya M, Carmeliet P, Soares R, Duda DG, Jain RK and Fukumura D. PlGF/VEGFR-1 Signaling Promotes Macrophage Polarization and Accelerated Tumor Progression in Obesity. Clin Cancer Res 2016;22:2993–3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ambarus CA, Krausz S, van EM, Hamann J, Radstake TR, Reedquist KA, Tak PP and Baeten DL. Systematic validation of specific phenotypic markers for in vitro polarized human macrophages. J Immunol Methods 2012;375:196–206. [DOI] [PubMed] [Google Scholar]
- 25.Duscha BD, Robbins JL, Jones WS, Kraus WE, Lye RJ, Sanders JM, Allen JD, Regensteiner JG, Hiatt WR and Annex BH. Angiogenesis in skeletal muscle precede improvements in peak oxygen uptake in peripheral artery disease patients. Arterioscler Thromb Vasc Biol 2011;31:2742–2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Y, Qi X, Tong X and Wang S. Thrombospondin 1 activates the macrophage Toll-like receptor 4 pathway. Cell Mol Immunol 2013;10:506–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chalothorn D and Faber JE. Strain-dependent variation in collateral circulatory function in mouse hindlimb. Physiol Genomics 2010;42:469–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dokun AO, Keum S, Hazarika S, Li Y, Lamonte GM, Wheeler F, Marchuk DA and Annex BH. A quantitative trait locus (LSq-1) on mouse chromosome 7 is linked to the absence of tissue loss after surgical hindlimb ischemia. Circulation 2008;117:1207–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Incio J, Tam J, Rahbari NN, Suboj P, McManus DT, Chin SM, Vardam TD, Batista A, Babykutty S, Jung K, Khachatryan A, Hato T, Ligibel JA, Krop IE, Puchner SB, Schlett CL, Hoffmman U, Ancukiewicz M, Shibuya M, Carmeliet P, Soares R, Duda DG, Jain RK and Fukumura D. PlGF/VEGFR-1 Signaling Promotes Macrophage Polarization and Accelerated Tumor Progression in Obesity. Clin Cancer Res 2016;22:2993–3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li X, Jin Q, Yao Q, Zhou Y, Zou Y, Li Z, Zhang S and Tu C. Placental Growth Factor Contributes to Liver Inflammation, Angiogenesis, Fibrosis in Mice by Promoting Hepatic Macrophage Recruitment and Activation. Front Immunol 2017;8:801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harper SJ and Bates DO. VEGF-A splicing: the key to anti-angiogenic therapeutics? Nat Rev Cancer 2008;8:880–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shibuya M Vascular Endothelial Growth Factor (VEGF) and Its Receptor (VEGFR) Signaling in Angiogenesis: A Crucial Target for Anti- and Pro-Angiogenic Therapies. Genes Cancer 2011;2:1097–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Calabrese G, Mesner LD, Foley PL, Rosen CJ and Farber CR. Network Analysis Implicates Alpha-Synuclein (Snca) in the Regulation of Ovariectomy-Induced Bone Loss. Sci Rep 2016;6:29475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sangwung P, Zhou G, Nayak L, Chan ER, Kumar S, Kang DW, Zhang R, Liao X, Lu Y, Sugi K, Fujioka H, Shi H, Lapping SD, Ghosh CC, Higgins SJ, Parikh SM, Jo H and Jain MK. KLF2 and KLF4 control endothelial identity and vascular integrity. JCI Insight 2017;2:e91700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Averill MM, Kerkhoff C and Bornfeldt KE. S100A8 and S100A9 in cardiovascular biology and disease. Arterioscler Thromb Vasc Biol 2012;32:223–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Katashima T, Naruko T, Terasaki F, Fujita M, Otsuka K, Murakami S, Sato A, Hiroe M, Ikura Y, Ueda M, Ikemoto M and Kitaura Y. Enhanced expression of the S100A8/A9 complex in acute myocardial infarction patients. Circ J 2010;74:741–748. [DOI] [PubMed] [Google Scholar]
- 37.Ryckman C, Vandal K, Rouleau P, Talbot M and Tessier PA. Proinflammatory activities of S100: proteins S100A8, S100A9, and S100A8/A9 induce neutrophil chemotaxis and adhesion. J Immunol 2003;170:3233–42. [DOI] [PubMed] [Google Scholar]
- 38.Urbano FJ and Buno W. BAPTA-AM blocks both voltage-gated and Ca2+-activated K+ currents in cultured bovine chromaffin cells. Neuroreport 1998;9:3403–3407. [DOI] [PubMed] [Google Scholar]
- 39.Hsu K, Chung YM, Endoh Y and Geczy CL. TLR9 ligands induce S100A8 in macrophages via a STAT3-dependent pathway which requires IL-10 and PGE2. PLoS One 2014;9:e103629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kawamura H, Li X, Harper SJ, Bates DO and Claesson-Welsh L. Vascular endothelial growth factor (VEGF)-A165b is a weak in vitro agonist for VEGF receptor-2 due to lack of coreceptor binding and deficient regulation of kinase activity. Cancer Res 2008;68:4683–4692. [DOI] [PubMed] [Google Scholar]
- 41.Konopatskaya O, Churchill AJ, Harper SJ, Bates DO and Gardiner TA. VEGF165b, an endogenous C-terminal splice variant of VEGF, inhibits retinal neovascularization in mice. Mol Vis 2006;12:626–632. [PubMed] [Google Scholar]
- 42.Ngo DT, Farb MG, Kikuchi R, Karki S, Tiwari S, Bigornia SJ, Bates DO, LaValley MP, Hamburg NM, Vita JA, Hess DT, Walsh K and Gokce N. Antiangiogenic actions of vascular endothelial growth factor-A165b, an inhibitory isoform of vascular endothelial growth factor-A, in human obesity. Circulation 2014;130:1072–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Varey AH, Rennel ES, Qiu Y, Bevan HS, Perrin RM, Raffy S, Dixon AR, Paraskeva C, Zaccheo O, Hassan AB, Harper SJ and Bates DO. VEGF 165 b, an antiangiogenic VEGF-A isoform, binds and inhibits bevacizumab treatment in experimental colorectal carcinoma: balance of pro- and antiangiogenic VEGF-A isoforms has implications for therapy. Br J Cancer 2008;98:1366–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koch S and Claesson-Welsh L. Signal transduction by vascular endothelial growth factor receptors. Cold Spring Harb Perspect Med 2012;2:a006502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shibuya M Vascular endothelial growth factor receptor-1 (VEGFR-1/Flt-1): a dual regulator for angiogenesis. Angiogenesis 2006;9:225–230. [DOI] [PubMed] [Google Scholar]
- 46.Shibuya M Vascular endothelial growth factor and its receptor system: physiological functions in angiogenesis and pathological roles in various diseases. J Biochem 2013;153:13–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hiroshima Y, Hsu K, Tedla N, Wong SW, Chow S, Kawaguchi N and Geczy CL. S100A8/A9 and S100A9 reduce acute lung injury. Immunol Cell Biol 2017;95:461–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ehlermann P, Eggers K, Bierhaus A, Most P, Weichenhan D, Greten J, Nawroth PP, Katus HA and Remppis A. Increased proinflammatory endothelial response to S100A8/A9 after preactivation through advanced glycation end products. Cardiovasc Diabetol 2006;5:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang L, Luo H, Chen X, Jiang Y and Huang Q. Functional characterization of S100A8 and S100A9 in altering monolayer permeability of human umbilical endothelial cells. PLoS One 2014;9:e90472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guzman-Hernandez ML, Potter G, Egervari K, Kiss JZ and Balla T. Secretion of VEGF-165 has unique characteristics, including shedding from the plasma membrane. Mol Biol Cell 2014;25:1061–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.