

Abstract
MHC class I-specific reagents such as fluorescently-labeled multimers (e.g., tetramers) have greatly advanced the understanding of CD8+ T cells under normal and diseased states. However, recombinant MHC class I components (comprising MHC class I heavy chain and β2 microglobulin) are usually produced in bacteria following a lengthy purification protocol that requires additional non-covalent folding steps with exogenous peptide for complete molecular assembly. We have provided an alternative and rapid approach to generating soluble and fully-folded MHC class I molecules in eukaryotic cell lines (such as CHO cells) using a Sleeping Beauty transposon system. Importantly, this method culminates in generating stable cell lines that reliably secrete epitope-defined MHC class I molecules into the tissue media for convenient purification and eventual biotinylation/multimerization. Additionally, MHC class I components are covalently linked, providing the opportunity to produce a diverse set of CD8+ T cell-specific reagents bearing peptides with various affinities to MHC class I.
Keywords: Major histocompatibility complex (MHC) class I, single-chain trimer, tetramer, dextramer, Sleeping Beauty transposon system, stable cell line generation
INTRODUCTION
The major histocompatibility complex (MHC) class I and II molecules play an integral role in T cell development and peripheral effector responses (Alcover et al., 2018). MHC class I is retained on the plasma membrane of nucleated cells and consists of a multi-unit heavy chain whose tertiary structure is stabilized by β2 microglobulin through non-covalent forces (Wieczorek et al., 2017). To provide specific binding to antigen specific CD8+ T cells, MHC class I usually retains a short 8–10 amino acid peptide within the MHC peptide binding groove that is derived from degraded intracellular proteins (hereafter referred to as peptide/MHC).
Our understanding of basic T cell properties and dynamics under a variety of normal and diseased settings has been greatly advanced by the ability to produce and purify peptide/MHC for use in assays to specifically engage the T cell receptor (TCR). Arguably the most widespread approach incorporates fluorochrome-conjugated peptide/MHC multimers (e.g., tetramers) for analyzing or isolating antigen-specific CD8+ T cells from biological samples (Khairnar et al., 2018; Soen et al., 2003). Peptide/MHC generation has continued similarly to the process outlined in the landmark work by Altman and colleagues (Altman et al., 1996). Briefly, β2 microglobulin and MHC class I heavy chain (containing a BirA tail) are individually expressed in E. coli and later purified from inclusion bodies through a laborious lysis/solubilization process. A defined MHC class I peptide is then added alongside β2 microglobulin and heavy chain in a precise folding reaction mixture that requires several days to complete prior to affinity chromatography (AC) purification of properly folded peptide/MHC and later biotinylation steps. Although this standard production process works to eventually yield excellent reagents for immunologic assays, there exist a number of major disadvantages. Namely, the standard method is [i] time consuming, [ii] requires substantial levels of raw ingredients (particularly purified MHC class I peptide), and [iii] cannot guarantee large-scale production of properly folded peptide/MHC molecules based on predicted peptide binders. For example, it is extremely difficult to stably produce MHC molecules bearing peptides with low-to-moderate affinity to the MHC peptide binding groove.
To circumvent these perceived drawbacks (particularly in stabilizing peptide binding to the MHC peptide binding groove), previous efforts have revealed the ability to engineer and produce peptide/MHC molecules in bacteria by covalently joining the MHC class I peptide, β2 microglobulin, and heavy chain with discrete amino acid linkers (designated single-chain trimers [SCTs]) (Yu et al., 2002). For most SCTs reported, these engineered proteins fold correctly and specifically engage CD8+ T cells as tetramers (Mitaksov et al., 2007), irrespective of the artificial linker design (Hansen et al., 2009). However, this particular SCT method still utilizes a bacterial expression system and requires substantial purification and refolding efforts.
We, therefore, sought an alternative method to potentially improve the production of peptide/MHC based on the SCT approach. Our current work highlights the ability to rapidly generate eukaryotic cell lines that stably express and secrete peptide/MHC into the tissue media for purification and biotinylation. This modified protocol could potentially provide a much faster/convenient route to generating properly folded peptide/MHC with minimal user intervention, especially for MHC class I targets with high demand (such as the model OVA epitope SIINFEKL). Since we have adopted the SCT strategy, MHC molecules presenting a range of class I peptides (i.e., low-to-high binding affinity) can also be reliably generated. Additionally, it remains possible that these eukaryotic-derived peptide/MHC molecules more accurately recapitulate binding dynamics with TCRs in downstream assays (Schmidt and Lill 2018).
MATERIALS AND METHODS
Mice
Female 6–8-week-old C57BL/6J (stock #000664) and OT-1 (stock #003831) mice were purchased from The Jackson Laboratory (Bar Harbor, Maine, USA) and maintained in micro-isolator cages under sterile conditions. Animals were humanely euthanized and spleens/lymph nodes harvested and combined for Ficoll gradient centrifugation (GE HealthCare, Piscataway, NJ). The lymphocyte interphase was then subjected to ACK lysis and eventual CD8+ T cell purification using MACS bead positive selection as instructed by the manufacturer (Miltenyi Biotec, Cambridge, MA). Purified CD8+ T cells were aliquoted in 90% FBS/10% DMSO and stored in liquid nitrogen until use. All mouse procedures were followed in accordance with TTUHSC IACUC-approved protocols.
Cell lines and culture
FreeStyle™ Chinese Hamster Ovary (CHO-S) (Thermo Fisher Scientific, Waltham, MA) and 4T1 (ATCC, Manassas, VA) cells were utilized for in vitro studies. CHO-S cells were passaged in FreeStyle™ CHO Expression Medium (Thermo Fisher Scientific) according to the manufacturer’s recommendations. 4T1 cells are naturally deficient in H-2Kb expression and were grown in RPMI 1640 supplemented with 10% heat-inactivated FBS, 100 U/ml penicillin, 100 μg/ml streptomycin, and 10 mmol/l L-glutamine (all from Thermo Fisher Scientific). All cell lines were maintained in vented flasks at 37 °C with 5% CO2.
Cloning strategy and construction of transposon expression vectors
Full-length mouse β2 microglobulin (NCBI Reference Sequence: NM_009735.3) and MHC class I heavy chain (H-2Kb) (NCBI Reference Sequence: NM_001001892.2) cDNA were synthesized from C57BL/6J splenocytes following TRIzol lysis (Thermo Fisher Scientific) and RT-PCR using oligo(dT) primers (SuperScript IV First-Strand Synthesis Kit; Thermo Fisher Scientific). Secretory MHC class I heavy chain was designed to not include the transmembrane domain. The leader signal, SIINFEKL epitope, and Gly/Ser amino acid linkers were ultimately added by overhang PCR as previously reported (Hansen et al., 2009) using the Phusion High-Fidelity DNA Polymerase (Thermo Scientific Fisher). PCR fragments were gel excised/purified and ligated (LigaFast™ Rapid DNA Ligation System; Promega, Madison, WI) into puc19 (NEB, Ipswich, MA) via SacI/HindIII restriction enzyme sites and fragments pieced together using the unique NheI/BamHI sites of the synthetic peptide/MHC sequence (Supplementary Figure 2). The BirA AviTag™ amino acid sequence (GLNDIFEAQKIEWHE) was subsequently added to the construct’s terminus by overlap-extension PCR and cloned into a separate puc19 holding vector. Full-length peptide/MHC was then amplified and cloned into the pSBbi-pur transposon vector (kindly provided by Dr. Eric Kowarz [Addgene plasmid # 60523]) using the SfiI restriction enzyme sites (Kowarz et al., 2015). Plasmid transformations were carried out in chemically-competent NEB-5 alpha E. coli (NEB) using ampicillin selection. All vector constructs were confirmed by restriction enzyme digestion and DNA sequencing.
Sleeping Beauty (SB) transposon system
Parental cell lines were transiently transfected with transposon-related vectors using Lipofectamine reagent (Thermo Fisher Scientific) as directed by the manufacturer. A plasmid encoding the SB 100X transposase (pCMV[CAT]T7-SB100; designated Vector 1), a gift from Dr. Zsuzsanna Izsvak (Addgene plasmid # 34879), and a transposon plasmid containing the necessary inverted terminal repeats (pSBbi-pur; designated Vector 2) were used. Both vectors were provided concurrently to stably integrate peptide/MHC transgenes into cells. Briefly, 1×105 cells were plated in 24-well plates (Corning, Corning NY) and exposed to Vector 1 (12.5 ng)/Vector 2 (488 ng) in a total volume of 500 µl media as similarly described (Kowarz et al., 2015). Culture media was replenished with 2 ml fresh media after 24 hrs and 48 hrs, whereupon cells were exposed to lethal doses of puromycin (CHO-S - 10 μg/ml; 4T1 – 5 μg/ml) (Invivogen, San Diego, CA). Fresh media containing puromycin was provided every 2–3 days as needed. Generally, actively dividing cells were ready for expansion by 2 weeks, with virtually all cell clones expressing peptide/MHC molecules.
SDS-PAGE and western blot
To remove extraneous tissue media components such as surfactants, CHO cell supernatants were passed onto PD10 columns (GE Healthcare) and concentrated/washed in PBS using a 30 MWCO Amicon centrifugal unit (MilliporeSigma, Burlington, MA). Protein concentration was determined using a BCA protein assay kit (Thermo Scientific Fisher) and sample aliquots stored at −80 °C until use. Protein purity was assessed with SDS-PAGE and coomassie blue staining while protein identity was confirmed through western blotting. Briefly, proteins were resolved on a 4%/12% polyacrylamide gel, transferred to a PVDF membrane (Amersham Hybond, 0.2μm; GE Healthcare), and blocked with 5% milk in PBST (0.1% Tween 20 in PBS) for 1 hr at RT. Membranes were then incubated with various primary antibodies (1 μg/ml) in block solution with rocking at 4 °C overnight. Specific primary reagents included anti-mouse β2 microglobulin (clone 893803; R&D Systems, Minneapolis, MN) or anti-BirA tail (clone Abc; Avidity, Aurora, CO) antibodies. Blots were then washed extensively with PBST and incubated in block with secondary HRP-conjugated goat antibodies specific to rat IgG (H+L) or mouse IgG (H+L) (Thermo Scientific Fisher) for 1 hr at RT. Washed blots were finally developed with a SignalFire ECL reagent (Cell Signaling, Danvers, MA) and exposed/imaged on a ChemiDoc™ Touch Imaging System (Bio-Rad, Hercules, CA). In separate experiments, blots containing biotinylated protein were probed with a Streptavidin-HRP reagent (Thermo Scientific Fisher) for 1 hr at RT to confirm streptavidin binding potential.
Immunoprecipitation
A Pierce Classic IP kit (Thermo Scientific Fisher) was used to investigate ligand binding of soluble peptide/MHC protein. Peptide/MHC protein was incubated overnight at 4 °C with 2 μg of the anti-SIINFEKL (clone eBio25-D1.16; Thermo Scientific Fisher) or mouse IgG isotype (clone MOPC-21; MP Biomedicals, Santa Ana, CA) antibodies. The anti-SIINFEKL antibody functions as a TCR-like antibody (Lowe et al., 2017), and binds SIINFEKL/H-2Kb much like SIINFEKL-reactive T cells such as OT-1 CD8+ T cells. Following purification of IgG containing complexes, samples were boiled and analyzed by western blot as described above.
Affinity chromatography (AC)
The MHC class I-reactive M1/42 antibody (Bio X Cell, West Lebanon, NH) was covalently bound to an NHS-activated agarose bead column (HiTrap™ NHS-activated HP; GE Healthcare) manually as directed by the manufacturer. The column was attached to an ÄKTA™ start chromatography system (GE Healthcare) for automatic operation and equilibrated with 20 mM sodium phosphate, pH 7.0 (binding buffer). Cell-free supernatants were first desalted in PBS using PD-10 columns and diluted 1:2 in binding buffer prior to AC. Samples were then applied to the M1/42 column, unbound material washed away with binding buffer, and peptide/MHC molecules eluted using 0.1 M glycine, pH 2.7. Eluates were dispensed in tubes containing 1 M Tris, pH 9 and relevant fractions combined and concentrated/washed in PBS as explained above.
Biotinylation and tetramerization
Peptide/MHC was biotinylated using 2.5 μg BirA ligase (Avidity) overnight at 4 °C according to the manufacturer’s guidelines. Reaction components were removed by successive washes in PBS using a 30 MWCO centrifugal filter (MilliporeSigma). Alternatively, larger reaction mixtures could be exclusively polished through size exclusion chromatography (Altman and Davis 2016) using a HiPrep™ 16/60 Sephacryl™ S-200 HR column (GE Healthcare) as indicated in Figure 1B. In order to produce small-scale tetramer batches, biotinylated peptide/MHC was incubated with PE-conjugated streptavidin (BD Biosciences, San Jose, CA) at a 4:1 molar ratio in the dark for 30 min at RT. Tetramers were then stored at 4 °C shielded from light.
Figure 1.
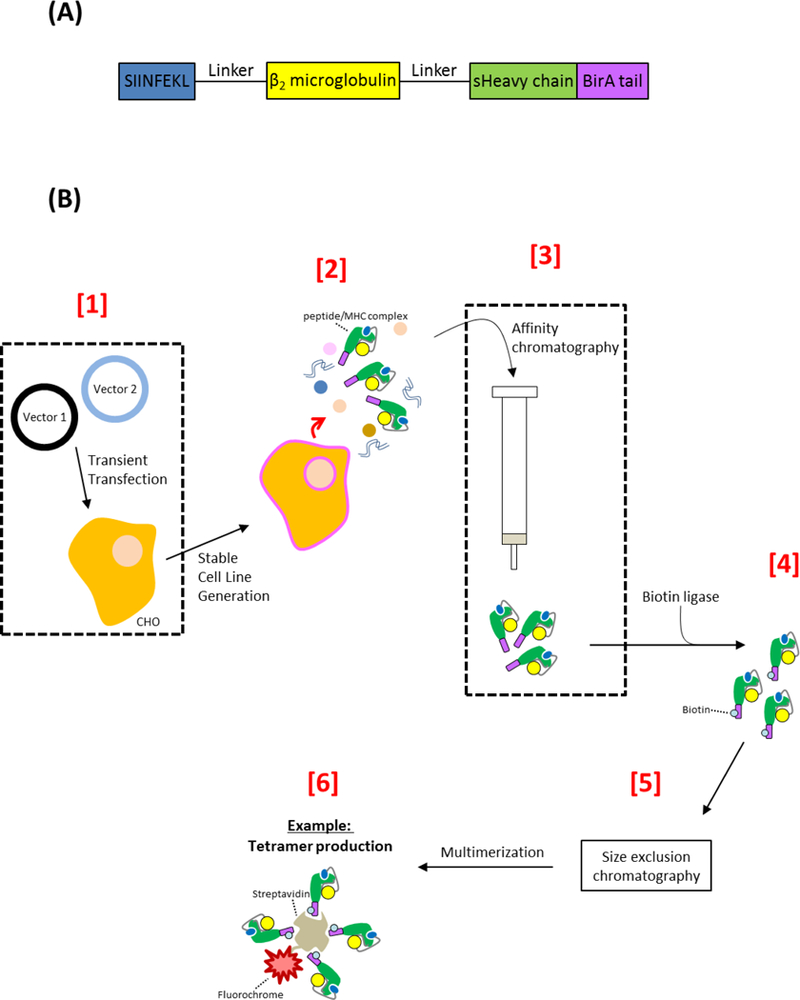
(A) Representative DNA schematic of the synthetic peptide/MHC complex. The designed peptide/MHC molecule contains distinct peptide, beta-2-microglobulin, and MHC heavy chain regions that are linked via explicit glycine/serine amino acids. The MHC heavy chain lacks a transmembrane domain (designated sHeavy chain), ensuring that properly folded peptide/MHC protein is secreted from cells into the culture medium. The peptide/MHC construct also contains a terminal BirA tail for enzymatic conjugation of biotin. (B) Standard workflow for expressing, purifying, and biotinylating eukaryotic-derived peptide/MHC molecules. [1] A suitable eukaryotic host cell line (such as CHO cells) is transiently transfected with two vectors that allow for transposon-directed integration of genes of interest. Vector 1 encodes the SB transposase (SB100X) while vector 2 is a transposon-compatible plasmid that encodes the synthetic peptide/MHC complex and puromycin resistance transgenes. [2] A stable cell line secreting peptide/MHC complexes is generated in as little as 2 weeks following antibiotic selection and expansion. [3] Spent culture media is processed through AC (against the sHeavy chain) to selectively purify peptide/MHC complexes. [4] A biotin ligase may then be employed to enzymatically conjugate biotin to the BirA tail of the synthetic peptide/MHC protein. [5] Following an additional polishing step such as size exclusion chromatography and [6] multimerization steps, the peptide/MHC reagent can be incorporated into immunologically-relevant assays that, for example, detect antigen-specific T cells. Abbreviation used: secretory MHC class I heavy chain (sHeavy chain), major histocompatibility complex (MHC)
ELISA
To investigate the success of peptide/MHC biotinylation, a 96-well high protein binding plate (Corning) was first coated overnight at 4 °C with 4 μg/ml streptavidin (Promega) in 0.1 M sodium carbonate and then blocked (3% BSA/PBS) for 1 hr at RT. Biotinylated protein samples were diluted in block and added to wells at various dilutions and incubated at RT for 1 hr. Wells were washed extensively with PBST and then exposed to 4 μg/ml of either a MHC class I-reactive (clone M1/42; Biolegend) or rat isotype control IgG (Biolegend) antibody in block. In separate wells, a positive control biotinylated irrelevant rat antibody (Biolegend) was incorporated to validate the extent of streptavidin binding. The plate was again washed with PBST and relevant wells provided a goat anti-rat HRP (Thermo Scientific Fisher) or goat anti-mouse FcγR-specific HRP (Jackson ImmunoResearch, West Grove, PA) antibody in block for 1 hr at RT. Wells were washed with PBST and developed for 5 min after the addition of 200 μl 1-Step Ultra TMB (Thermo Scientific Fisher). Reactions were terminated with 100 μl TMB stop solution (KPL), and the absorbance immediately read at 450 nm using a Cytation 5 Imaging Reader (Biotek, Winooski, VT).
Flow cytometry
To confirm gene expression, the 4T1 cell line (1×105 cells) was stained with relevant primary antibodies (anti-SIINFEKL/H-2Kb or mouse IgG isotype antibodies at 2 μg/ml) in FACS buffer (0.5% BSA/0.1% NaN3 in PBS) for 20 min at 4 °C, washed, incubated with a PE-conjugated anti-mouse secondary antibody (Jackson ImmunoResearch), washed again, and resuspended in FACS buffer for analysis (Supplementary Figure 1). To ensure proper orientation, biotinylated peptide/MHC was incubated with 5 μm PMMA beads conjugated to streptavidin (PolyAn, Berlin, Germany), washed, and incubated at 4 °C with correct pairs of primary and PE-conjugated secondary antibodies (as indicated in Figure 4) prior to analysis in FACS buffer. Frozen CD8+ T cells were thawed, washed, and resuspended in FACS buffer so that each stain consisted of 2×105 viable CD8+ T cells. Cells were exposed to 50 nM dasatinib (Selleck Chemicals, Houston, TX) for 30 min at 37 °C and promptly incubated with peptide/MHC tetramers for 20 min at RT in the dark (Dolton et al., 2015). Cells were washed with FACS buffer and labeled with an anti-CD8-FITC antibody (clone 53–6.7; BioLegend, San Diego, CA) at 4 °C for 20 min shielded from light. After additional wash steps, cells were resuspended in fix solution (1% FBS, 2.5% formaldehyde in PBS) and assessed by flow cytometry using a BD LSRFortessa. Single and double color stain analysis was carried out using FlowJo software. In the case of tetramer analysis, PE/FITC compensated events were first gated based on FSC/SSC profiles and subsequently evaluated by PE and FITC fluorescence.
Figure 4.
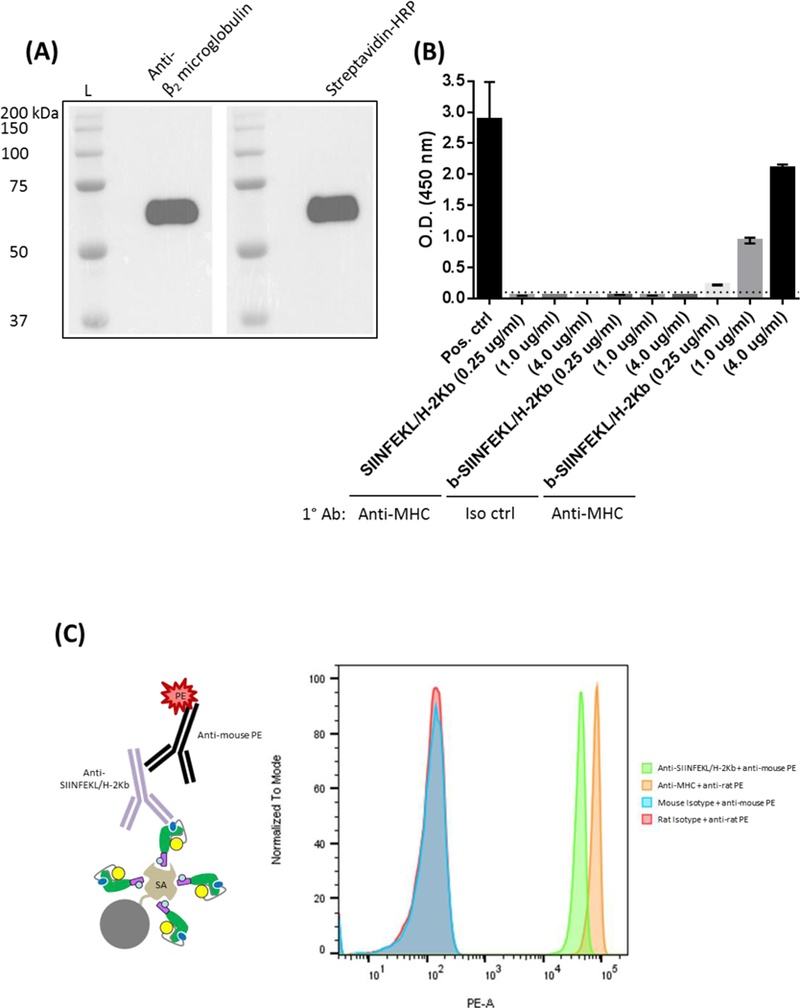
Peptide/MHC was biotinylated as detailed in the Materials and Methods and specific streptavidin binding initially confirmed through (A) western blot and (B) ELISA using wells coated with streptavidin. (C) Biotinylated peptide/MHC was also incubated with streptavidin-conjugated 5 μm beads and washed extensively. Beads were then exposed to isotype, anti-SIINFEKL/H-2Kb, or anti-MHC monoclonal antibodies followed by washing steps and incubation with relevant secondary PE-conjugated antibodies. Specific ligand reactivity was subsequently determined by flow cytometry. Abbreviations used: protein ladder (L), SIINFEKL epitope + MHC class I (SIINFEKL/H-2Kb), biotinylated SIINFEKL/H-2Kb (b-SIINFEKL/H-2Kb), positive (Pos), isotype (Iso), control (ctrl), major histocompatibility complex (MHC), horseradish peroxidase (HRP), streptavidin (SA), primary antibody (1° Ab)
RESULTS
Cloning, expression, and purification strategy for soluble eukaryotic-derived peptide/MHC
The design of linked peptide/MHC class I molecules closely followed the previously reported generation of SCTs in bacteria (Hansen et al., 2009). Essentially, as presented in Figure 1A, flexible glycine/serine linkers join a particular peptide epitope, β2 microglobulin, and MHC class I heavy chain. A BirA tail was also genetically encoded to the 3ʹ end of the molecule for enzyme-directed biotinylation of secreted protein. Yet, a notable difference of our design involved omitting the transmembrane region of MHC class I (designated sHeavychain) to allow for functional and fully-folded secreted protein. Our conceptual process for eukaryotic expression centered on the use of a SB transposon system to stably integrate transgene content into relevant cell lines (Figure 1B). Therefore, CHO cells would be transiently transfected with transposon-relevant plasmids (encoding separately a SB transposase and synthetic peptide/MHC molecule) and stable cells generated through antibiotic selection. After expanding relevant CHO cell clones, secreted peptide/MHC could be purified by AC, biotinylated, and multimerized to produce, for example, tetramers capable of binding antigen-specific T cells.
Purification of secreted and fully-folded peptide/MHC protein
We first determined the suitability of the SB transposon system to stably integrate the peptide/MHC transgene since we were unaware of previously published work using this arrangement of techniques. However, this particular peptide/MHC construct differed from the aforementioned soluble design in Figure 1A by encoding full-length MHC class I heavy chain (i.e., the murine MHC haplotype H-2Kb), thereby, ensuring cell surface expression for relative ease of detection. Briefly, 4T1 (H-2Kb null) cells were exposed to transposon vectors that expressed either a null or SIINFEKL/H-2Kb construct and puromycin resistant clones selected and expanded in culture. Cells were then stained with isotype or anti-SIINFEKL/H-2Kb antibodies and analyzed by flow cytometry using a PE-conjugated secondary antibody. In comparison to 4T1-null cells, 4T1-SIINFEKL/H-2Kb cells expressed clear and robust levels of synthetic peptide/MHC (Supplementary Figure 1). These confirmatory results were expected, given the widespread use of the SB approach to induce expression of various protein classes in established cell lines and primary cells (Kebriaei et al., 2017).
CHO cells were next transfected with SB-related vectors as described in Figure 1 to induce stable expression of soluble peptide/MHC. Parental CHO cells exhibited enhanced resistance to puromycin, requiring sustained culturing in high concentrations of puromycin at 10 μg/ml. However, we were able to expand puromycin-resistant clones by 2 weeks post plasmid transfection. Cells were then grown to saturating conditions in culture for at least 4 days to generate suitable whole protein concentrations from a total volume of 20 ml media. Yet, our choice of serum-free media contained a number of proprietary agents such as surfactants that could potentially interfere with protein purification and validation assays. We, therefore, took the precautionary step of desalting and concentrating cell-free supernatants in PBS prior to downstream analysis. Our initial assessment of CHO-derived extracellular proteins by SDS-PAGE (under reducing conditions) and coomassie blue staining revealed distinct protein bands around a predicted 51 kDa molecular weight for synthetic peptide/MHC (Figure 2A) that was not evident from CHO cells expressing a null construct (data not shown). This specific protein band was subsequently confirmed following AC as exhibited in Figure 2B. That is, cell-free supernatants were passed onto an agarose bead column containing the MHC class I-reactive M1/42 antibody and bound material eluted and ultimately assessed again by SDS-PAGE. We typically harvested at least 100 μg/ml AC-purified peptide/MHC from small-scale culturing efforts. However, the described protocol can be scaled-up (or down) depending on the desired peptide/MHC total yield. Although beyond the scope of this report, alternative culturing parameters and/or leader signals (Haryadi et al., 2015) may potentially enhance overall CHO secretion of peptide/MHC. Our AC procedure appeared to provide substantial protein purity based on obtaining [i] one distinct chromatogram elution peak and [ii] a protein band comprising > 95% of detectable protein by coomassie blue staining.
Figure 2.
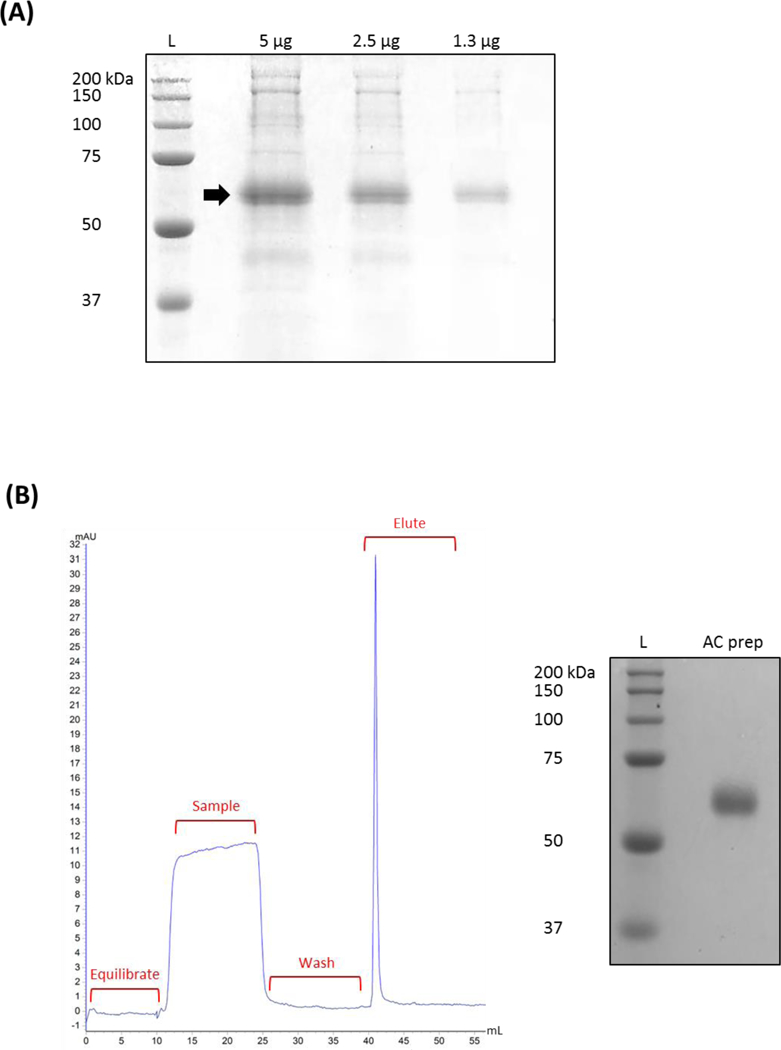
Stable CHO cells lines were established by a SB transposon system to secrete peptide/MHC molecules into culture media. (A) Evidence of extracellular peptide/MHC protein was first determined from cell-free supernatants by SDS-PAGE and coomassie blue staining. (B) Representative chromatogram of small scale AC purification of peptide/MHC-containing media. Cell-free supernatant was passed through an equilibrated agarose bead column containing the MHC class I-reactive antibody M1/42. After washing away unbound material, peptide/MHC protein was eluted, buffer-exchanged, and concentrated. Protein purity was then assessed using SDS-PAGE and coomassie blue staining. Arrow inset indicating peptide/MHC around the predicted molecular weight. Abbreviation used: protein ladder (L), affinity chromatography (AC), preparation (prep)
Determination of peptide/MHC identity and upstream binding potential
We next confirmed the identity of AC purified protein by western blot separate from MHC class I reactivity. Based on the linked design of polychain peptide/MHC molecules (Figure 1A), we incorporated monoclonal antibodies specific to mouse β2 microglobulin and the BirA tail peptide GLNDIFEAQKIEWHE. Developed blots clearly verified the integrity of synthetic peptide/MHC molecule expression, indicating no observable issues in CHO cells secreting these artificial proteins (Figure 3A). An immunoprecipitation reaction was subsequently attempted in order to demonstrate ligand binding potential of linked SIINFEKL to the MHC class I binding groove. SIINFEKL/H-2Kb was incubated with 2 μg of anti-SIINFEKL/H-2Kb or isotype control antibodies overnight. As detailed in the Materials and Methods, IgG containing material was purified using protein A/G-complexed agarose and resolved by SDS-PAGE under reducing conditions. The presence of SIINFEKL/H-2Kb was further established through western blot using an anti-mouse β2 microglobulin antibody (Figure 3B). Overall, these results help further validate the identity of soluble peptide/MHC from CHO cells and establish that these secreted proteins retain ligand binding specificity at the peptide/MHC class I interface.
Figure 3.
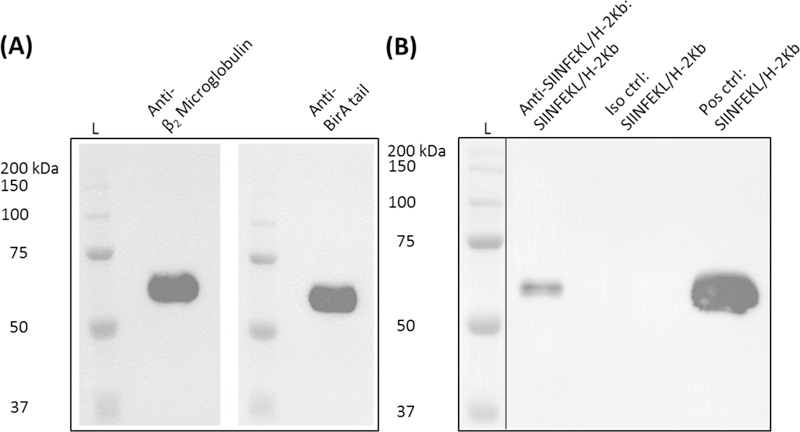
(A) Purified peptide/MHC (i.e., SIINFEKL/H-2Kb) identify was confirmed by western blot using monoclonal antibodies reactive to the β2 microglobulin and BirA tail regions of the design molecule as depicted in Figure 1A. (B). Peptide/MHC ligand binding was determined through immunoprecipitation and western blot following incubation of SIINFEKL/H-2Kb and a TCR-like antibody specific to this particular peptide/MHC class I complex. AC-purified peptide/MHC was also incorporated as a positive control. Abbreviations used: protein ladder (L), SIINFEKL epitope + MHC class I (SIINFEKL/H-2Kb), isotype (Iso), positive (Pos), control (ctrl)
Multimerization and functional assessment of eukaryotic-derived peptide/MHC
Biotinylated peptide/MHC arguably provides the greatest convenience to users for downstream assays. We, therefore, designed a BirA tail to the terminal end of the peptide/MHC transgene by using a particular BirA tail peptide, GLNDIFEAQKIEWHE, which offers a highly targeted site for enzymatic conjugation of biotin with minimal footprint (Beckett et al., 1999). AC purified peptide/MHC was subjected to BirA ligase activity in the presence of free biotin overnight. Excess biotin and other reaction components were removed by excessive washing in PBS using a 30 MWCO centrifugal unit, and the extent of biotinylation first assessed by western blot and ELISA. In the case of western blot analysis, SDS-PAGE-resolved biotinylated protein was incubated with HRP-conjugated streptavidin to generate a specific peptide/MHC signal (Figure 4A). Likewise, ELISA determination of biotinylated peptide/MHC clearly confirmed the functionality of the BirA tail region (Figure 4B). Briefly, wells were coated overnight with streptavidin followed by various concentrations of peptide/MHC and biotinylated peptide/MHC. Detection was finally determined indirectly by MHC class I reactivity using the M1/42 antibody. Biotinylated SIINFEKL/H-2Kb (as low as 0.25 μg/ml protein) was clearly evident in appropriate wells, with negligible reactivity occurring for conditions containing either unbiotinylated SIINFEKL/H-2Kb or biotinylated SIINFEKL/H-2Kb incubated with an isotype control. We then reconfirmed the ability of immobilized/properly oriented biontinylated peptide/MHC to specifically engage ligands through the MHC class I peptide binding site. Streptavidin was covalently attached to PMMA 5 μM beads and incubated with saturating conditions of biotinylated SIINFEKL/H-2Kb. Beads were washed, incubated with appropriate primary reagents (i.e., anti-SIINFEKL/H-2Kb and anti-MHC class I agents), and the extent of MHC ligand binding determined by flow cytometry using secondary PE-conjugated antibodies. Ultimately, as initially validated by our immunoprecipitation efforts in Figure 3B, biotinylated peptide/MHC bound streptavidin and specifically interacted with a TCR-like antibody that recapitulates CD8+ T cell interactions (Figure 4C), indicating no observable functional issues with peptide/MHC post biotinylation and multimerization.
We next assessed the ability of fluorescently-labeled multimers to specifically detect antigen-specific CD8+ T cells. Splenocytes and lymph nodes were harvested from wild-type and OT-1 transgenic (i.e., SIINFEKL-reactive) mice and CD8+ T cells subsequently purified through magnetic bead selection. Biotinylated SIINFEKL/H-2Kb was then incubated with PE-conjugated streptavidin following a 4:1 molar ratio, thereby, generating tetramers. In order to stabilize membrane dynamics of TCRs, CD8+ T cells were exposed to dasatinib (as previously described [Dolton et al., 2015]), followed by incubation with tetramers. After suitable wash steps, cells were specifically labeled with a FITC-conjugated anti-mouse CD8 antibody that displays minimal interference with TCR:MHC binding (Clement et al., 2011). Cells were fixed and double-positive events (CD8+/tetramer+) assessed by flow cytometry. As detailed in Figure 5, CD8+ OT-1 cells clearly bound PE-labeled tetramers, with most cells displaying CD8 and tetramer positivity (in comparison to wild-type mice). Altogether, these data help support our overarching strategy (as outlined in Figure 1) of stably producing eukaryotic-derived peptide/MHC that can be multimerized and used for immunologically-relevant assays such as CD8+ T cell identification.
Figure 5.
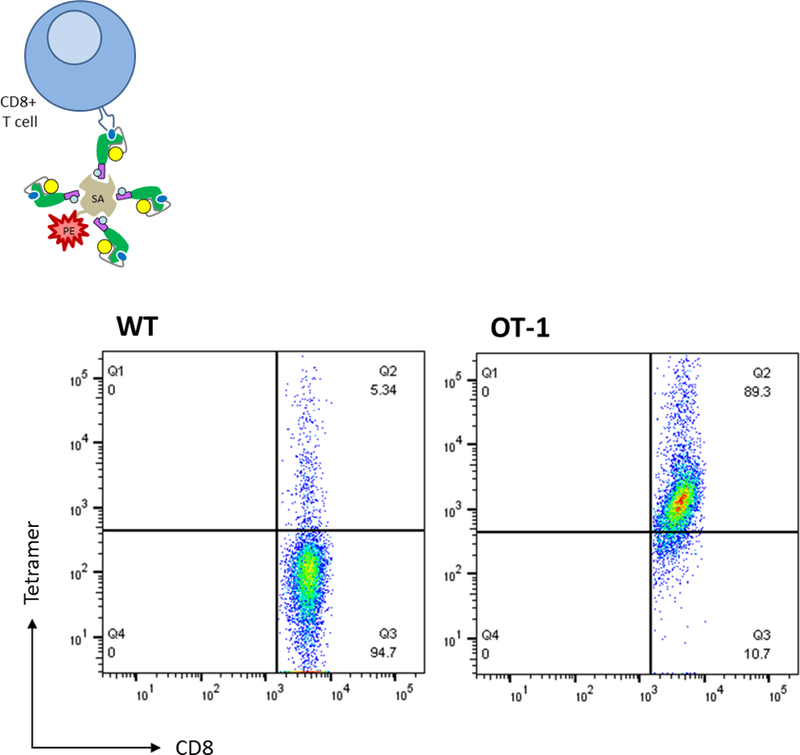
Biotinylated peptide/MHC was incubated with PE-conjugated streptavidin to produce “small-scale” batches of SIINFEKL-reactive tetramers. Purified CD8+ T cells harvested from either wild-type or OT-1 mice were incubated with tetramers, washed, and stained with an anti-CD8 FITC antibody. Cells were again washed, fixed, and analyzed by flow cytometry for ligand binding. Abbreviations used: wild-type (WT), streptavidin (SA)
DISCUSSION
Although MHC class I peptide candidates can be easily identified through in silico prediction methods (Andreatta and Nielsen 2016), free peptide occupancy of MHC class I molecules tends to be a rate limiting step in successfully generating stable peptide/MHC molecules from bacteria (Altman and Davis 2016). Considering the vital role MHC plays in human health (Cho and Sprent 2018), an inability to produce certain peptide/MHC reagents may adversely impact efforts on a number of fronts including diagnostics, therapies, and generalized scientific endeavors. For example, the burgeoning field of neoepitope identification for personalized cancer therapy could be stalled by those unique patient epitopes that exhibit high dissociation rates from MHC class I (Hu et al., 2018). One workaround to the issue of peptide occupancy has been in designing and expressing polychain SCTs that ensure full assembly of peptide, β2 microglobulin, and MHC class I heavy chain through flexible linkers (Hansen et al., 2009). SCTs retained on the plasma membrane of eukaryotic cells maintain their native conformation and are highly resistant to exogenous peptide binding (Yu et al., 2002). Additionally, membrane-bound SCTs serve as effective targets for CD8+ T cell priming (Hung et al., 2007) and destruction (Yu et al., 2002). In the case of diagnostic determination of CD8+ T cell frequencies by tetramers, SCTs can be modified for expression/secretion in bacteria and biotinylation by way of an explicit amino acid sequence that directs BirA enzyme function (Mitaksov et al., 2007).
We have provided a unique protocol to establish eukaryotic cell lines (such as CHO suspension cells) in as little as two weeks that stably secrete peptide/MHC molecules through transposon-directed delivery. Soluble peptide/MHC may then be biotinylated and utilized as multimers (via streptavidin), particularly for CD8+ T cell relevant assays. Currently, CHO cells are an industry standard in producing FDA approved therapeutic recombinant proteins (Kuo et al., 2018). We sought the advantages of the CHO cell line in order to [i] instigate post-translational modifications and [ii] be easily grown at high density under serum-free conditions in suspension cultures. However, other common cell line “protein workhorses” (e.g., HEK-293 cells) should be amenable to the expression and characterization techniques outlined. We are only aware of two reports that partly resemble our described method. White and colleagues designed a soluble HIV-reactive MHC class I molecule (consisting of free heavy chain + linked peptide-β2 microglobulin) for expression in baculovirus, which was capable of biotinylation/multimerization and identifying a particular T cell hybridoma by flow cytometry (White et al., 1999). An additional approach by Greten and colleagues performed standard plasmid DNA transfection to produce a peptide-β2 microglobulin-heavy chain linked protein in J588L cells that was only capable of dimerization due to mouse IgG fusion (Greten et al., 2002). Yet, the work outlined in this current report provides a previously unrecognized approach of combining the technical ease of CHO cell line cultivation and long-term expression of soluble covalently-linked peptide/MHC molecules for multimerization based on the end user’s needs.
The rapid development of stable CHO cells secreting peptide/MHC would not be possible without the SB transposon system. Our studies incorporated the SB transposase SB100X, which has a high gene insertion efficiency at close-to-random chromosomal sites (Mátés et al., 2009). The SB approach provides the advantageous properties of viral transduction to insert transgenes without the disadvantages of either maintaining genomic material episomally (e.g., adeno-associated viruses) or near/in proto-oncogenes (e.g., retroviral vectors) (Kebriaei et al., 2017). Additionally, manufacturing high-quality viral particles can be a cumbersome task fraught with regulatory obstacles. We generally experienced little difficulty in developing and expanding stable lines from parental cells after transiently transfecting plasmids that propagated the SB transposon system. The protocol is also amenable to freezing material at convenient stopping points. There was no apparent adverse effects to generating multimerized reagents when either cell-free CHO-derived supernatants or biotinylated peptide/MHC was frozen long-term at −80 °C. Although we did not formally test freezing conditions, these materials did not appear to require specialized conditions such as stabilizing agents. Importantly, our described method is compatible with downstream tetramer production with the added benefits of convenient peptide/MHC expression that can incorporate a range of peptide affinities to the MHC peptide binding groove. Traditionally, to circumvent the low intrinsic affinity of the TCR with peptide/MHC, tetravalent multimers have been utilized to increase T cell avidity. However, these soluble peptide/MHC molecules could be utilized for higher order reagents to better discriminate low frequency T cells (or bind “difficult” TCRs) such as fluorochrome-conjugated dextramers since the production process involves incubating biotinylated peptide/MHC with a dextran backbone containing streptavidin (Dolton et al., 2014). Overall, this report should provide a starting point (with necessary validation steps) to reliably produce a range of soluble eukaryotic-derived peptide/MHC molecules for diagnostic, therapeutic, and investigative purposes.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by startup funds (Dodge Jones Foundation – Abilene) and NIH (R15 CA215874) and DOD (W81XWH-18-1-0293) grants to D.B.L.
Abbreviations:
- MHC
Major histocompatibility complex
- TCR
T cell receptor
- SCT
single-chain trimer
- CHO
Chinese hamster ovary
- SB
sleeping beauty
- sHeavy
secretory MHC class I heavy chain
- AC
affinity chromatography
- Ab
antibody
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors have no competing interests to declare.
REFERENCES
- Alcover A, Alarcón B, Di Bartolo V. Cell Biology of T Cell Receptor Expression and Regulation. Annu Rev Immunol 2018. April 26;36:103–125. [DOI] [PubMed] [Google Scholar]
- Altman JD, Davis MM. MHC-Peptide Tetramers to Visualize Antigen-Specific T Cells. Curr Protoc Immunol 2016. November 1;115:17.3.1–17.3.44. [DOI] [PubMed] [Google Scholar]
- Altman JD, Moss PA, Goulder PJ, Barouch DH, McHeyzer-Williams MG, Bell JI, McMichael AJ, Davis MM. Phenotypic analysis of antigen-specific T lymphocytes. Science 1996. October 4;274(5284):94–6. [DOI] [PubMed] [Google Scholar]
- Andreatta M, Nielsen M. Gapped sequence alignment using artificial neural networks: application to the MHC class I system. Bioinformatics 2016. February 15;32(4):511–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckett D, Kovaleva E, Schatz PJ. A minimal peptide substrate in biotin holoenzyme synthetase-catalyzed biotinylation. Protein Sci 1999. April;8(4):921–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho JH, Sprent J. TCR tuning of T cell subsets. Immunol Rev 2018. May;283(1):129–137. [DOI] [PubMed] [Google Scholar]
- Clement M, Ladell K, Ekeruche-Makinde J, Miles JJ, Edwards ES, Dolton G, Williams T, Schauenburg AJ, Cole DK, Lauder SN, Gallimore AM, Godkin AJ, Burrows SR, Price DA, Sewell AK, Wooldridge L. Anti-CD8 antibodies can trigger CD8+ T cell effector function in the absence of TCR engagement and improve peptide-MHCI tetramer staining. J Immunol 2011. July 15;187(2):654–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolton G, Lissina A, Skowera A, Ladell K, Tungatt K, Jones E, Kronenberg-Versteeg D, Akpovwa H, Pentier JM, Holland CJ, Godkin AJ, Cole DK, Neller MA, Miles JJ, Price DA, Peakman M, Sewell AK. Comparison of peptide-major histocompatibility complex tetramers and dextramers for the identification of antigen-specific T cells. Clin Exp Immunol 2014. July;177(1):47–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolton G, Tungatt K, Lloyd A, Bianchi V, Theaker SM, Trimby A, Holland CJ, Donia M, Godkin AJ, Cole DK, Straten PT, Peakman M, Svane IM, Sewell AK. More tricks with tetramers: a practical guide to staining T cells with peptide-MHC multimers. Immunology 2015. September;146(1):11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greten TF, Korangy F, Neumann G, Wedemeyer H, Schlote K, Heller A, Scheffer S, Pardoll DM, Garbe AI, Schneck JP, Manns MP. Peptide-beta2-microglobulin-MHC fusion molecules bind antigen-specific T cells and can be used for multivalent MHC-Ig complexes. J Immunol Methods 2002. December 20;271(1–2):125–35. [DOI] [PubMed] [Google Scholar]
- Hansen T, Yu YY, Fremont DH. Preparation of stable single-chain trimers engineered with peptide, beta2 microglobulin, and MHC heavy chain. Curr Protoc Immunol 2009. November;Chapter 17:Unit17.5. [DOI] [PubMed]
- Hu Z, Ott PA, Wu CJ. Towards personalized, tumour-specific, therapeutic vaccines for cancer. Nat Rev Immunol 2018. March;18(3):168–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung CF, Calizo R, Tsai YC, He L, Wu TC. A DNA vaccine encoding a single-chain trimer of HLA-A2 linked to human mesothelin peptide generates anti-tumor effects against human mesothelin-expressing tumors. Vaccine 2007. January 2;25(1):127–35. [DOI] [PubMed] [Google Scholar]
- Haryadi R, Ho S, Kok YJ, Pu HX, Zheng L, Pereira NA, Li B, Bi X, Goh LT, Yang Y, Song Z. Optimization of heavy chain and light chain signal peptides for high level expression of therapeutic antibodies in CHO cells. PLoS One 2015. February 23;10(2):e0116878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kebriaei P, Izsvák Z, Narayanavari SA, Singh H, Ivics Z. Gene Therapy with the Sleeping Beauty Transposon System. Trends Genet 2017. November;33(11):852–870. [DOI] [PubMed] [Google Scholar]
- Khairnar V, Duhan V, Patil AM, Zhou F, Bhat H, Thoens C, Sharma P, Adomati T, Friendrich SK, Bezgovsek J, Dreesen JD, Wennemuth G, Westendorf AM, Zelinskyy G, Dittmer U, Hardt C, Timm J, Göthert JR, Lang PA, Singer BB, Lang KS. CEACAM1 promotes CD8+ T cell responses and improves control of a chronic viral infection. Nat Commun 2018. July 2;9(1):2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowarz E, Löscher D, Marschalek R. Optimized Sleeping Beauty transposons rapidly generate stable transgenic cell lines. Biotechnol J 2015. April;10(4):647–53. [DOI] [PubMed] [Google Scholar]
- Kuo CC, Chiang AW, Shamie I, Samoudi M, Gutierrez JM, Lewis NE. The emerging role of systems biology for engineering protein production in CHO cells. Curr Opin Biotechnol 2018. June;51:64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe DB, Bivens CK, Mobley AS, Herrera CE, McCormick AL, Wichner T, Sabnani MK, Wood LM, Weidanz JA. TCR-like antibody drug conjugates mediate killing of tumor cells with low peptide/HLA targets. MAbs 2017. May/June;9(4):603–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mátés L, Chuah MK, Belay E, Jerchow B, Manoj N, Acosta-Sanchez A, Grzela DP, Schmitt A, Becker K, Matrai J, Ma L, Samara-Kuko E, Gysemans C, Pryputniewicz D, Miskey C, Fletcher B, VandenDriessche T, Ivics Z, Izsvák Z. Molecular evolution of a novel hyperactive Sleeping Beauty transposase enables robust stable gene transfer in vertebrates. Nat Genet 2009. June;41(6):753–61. [DOI] [PubMed] [Google Scholar]
- Mitaksov V, Truscott SM, Lybarger L, Connolly JM, Hansen TH, Fremont DH. Structural engineering of pMHC reagents for T cell vaccines and diagnostics. Chem Biol 2007. August;14(8):909–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M, Lill JR. MHC class I presented antigens from malignancies: A perspective on analytical characterization & immunogenicity. J Proteomics 2018. April 24 pii: S1874–3919(18)30181–7. [DOI] [PubMed]
- Soen Y, Chen DS, Kraft DL, Davis MM, Brown PO. Detection and characterization of cellular immune responses using peptide-MHC microarrays. PLoS Biol 2003. December;1(3):E65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White J, Crawford F, Fremont D, Marrack P, Kappler J. Soluble class I MHC with beta2-microglobulin covalently linked peptides: specific binding to a T cell hybridoma. J Immunol 1999. March 1;162(5):2671–6. [PubMed] [Google Scholar]
- Wieczorek M, Abualrous ET, Sticht J, Álvaro-Benito M, Stolzenberg S, Noé F, Freund C. Major Histocompatibility Complex (MHC) Class I and MHC Class II Proteins: Conformational Plasticity in Antigen Presentation. Front Immunol 2017. March 17;8:292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu YY, Netuschil N, Lybarger L, Connolly JM, Hansen TH. Cutting edge: single-chain trimers of MHC class I molecules form stable structures that potently stimulate antigen-specific T cells and B cells. J Immunol 2002. April 1;168(7):3145–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.