

Abstract
Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is well known as a transmembrane cytokine and has been proposed as one of the most effective anti-cancer therapeutic agents, owing to its efficiency to selectively induce cell death in a variety of tumor cells. Suppression of autophagy flux has been increasingly acknowledged as an effective and novel therapeutic intervention for cancer. The present study demonstrated that the anti-cancer and anti-inflammatory drug celastrol, through its anti-metastatic properties, may initiate TRAIL-mediated apoptotic cell death in lung cancer cells. This sensitization was negatively affected by N-acetyl-l-cysteine, which restored the mitochondrial membrane potential (ΔΨm) and inhibited reactive oxygen species (ROS) generation. Notably, treatment with celastrol caused an increase in microtubule-associated proteins 1A/1B light chain 3B-II and p62 levels, whereas co-treatment of celastrol and TRAIL increased active caspase 3 and 8 levels compared with the control, confirming inhibited autophagy flux. The combined use of TRAIL with celastrol may serve as a safe and adequate therapeutic technique for the treatment of TRAIL-resistant lung cancer, suggesting that celastrol-mediated autophagy flux inhibition sensitized TRAIL-initiated apoptosis via regulation of ROS and ΔΨm.
Keywords: celastrol, autophagy flux, tumor necrosis factor-related apoptosis-inducing ligand, cancer, apoptosis
Introduction
Advanced tumors are occasionally non-responsive to mono-therapies. By contrast, combination chemotherapy may stimulate the regression of specific types of tumors even at progressive stages, and this particularly has served an important role in cancer treatment for a number of years (1,2). Furthermore, it has been suggested that effective and favorable drug combinations should be explored to improve combination chemotherapeutics, which may markedly improve the current outcomes of cancer treatment.
Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is a well-known transmembrane cytokine, and a member of the tumor necrosis factor family that mediates apoptosis in a variety of tumor cell types (3,4). TRAIL has been proposed as one of the most effective anti-cancer therapeutic agents, owing to its efficiency to selectively induce cell death in a variety of tumor cells whilst exhibiting minimal toxicity in the majority of normal cells (5,6). The binding of TRAIL to death receptors may also engage the extrinsic apoptotic pathway and trigger apoptotic signaling (7). TRAIL induces apoptosis by binding to death receptors and activating the Fas-associated death domain protein, ultimately leading to the activation of the effector caspase cascade, involving caspase-8, −9, −10, and −3 (8,9). A previous study has demonstrated that a number of cancer cells, together with lung A549 cells, acquire resistance to the apoptotic effects of TRAIL (10).
Celastrol is a triterpenoid, initially isolated from the Chinese herb Tripterygium wilfordii, or ‘Thunder God Vine’, that has been extensively applied in the treatment of autoimmune and neurodegenerative diseases (11–14). Previous studies have revealed that celastrol possesses multiple biological and pharmacological functions, in vitro and in vivo, including anti-microbial, anti-inflammatory and anti-cancer activities against prostate, breast and pancreatic cancer cells (15,16). It was also identified to have multiple mechanisms or signaling pathways that contribute to its anti-cancer activity, based on its efficiency to selectively initiate apoptosis in numerous tumor cell types (15,17).
Autophagy is an intracellular catabolic mechanism, whose primary functions are to sustain cellular homeostasis by recycling long-lived unrequired proteins, remove exhausted, redundant and defective cellular elements, and promote cell survival during brief periods of nutrient-starvation and other stressors (18,19). During autophagy, double membrane vesicles develop to form autophagic vacuoles named autophagosomes. These fuse with lysosomes to create auto-lysosomes, wherein sequestered segments are degraded by lysosomal enzymes (20,21). The autophagosome formation is mediated by the ubiquitin-like protein ATG12-autophagy protein (Atg)5-Atg16 complex and microtubule-associated proteins 1A/1B light chain 3B (LC3)-II, an LC3-I-phospholipid conjugate widely applied as an autophagy marker (22,23). Sequestosome 1 (p62) is a ubiquitin-binding protein and a well-known autophagy marker. It organizes into autophagosomes by directly interacting with LC3, and is completely degraded by autophagy. Inhibition of autophagy results in low levels of p62 accumulation (19). It was previously revealed that the inhibition of autophagy flux sensitizes cancer cells to conventional radio and chemotherapy treatment (24–26), confirming that autophagy flux inhibition may be an appropriate and effective technique for cancer treatment. The antimalarial drug chloroquine (CQ), which functions as an autophagy inhibitor, prevents lysosome acidification and lysosomal fusion with autophagosomes, and also inhibits the degradation of metabolic stress, thereby inducing apoptosis (27–30).
The mitochondria serve a pivotal role in the intrinsic pathway of cell death by transmitting apoptotic signals to the cytosol, leading to the activation of the caspase cascade. Changes in mitochondria appear following the activation of cell death signals, including the formation of reactive oxygen species (ROS), loss of mitochondrial membrane potential (ΔΨm), opening of the permeability alteration pore, and release of apoptotic bodies into the cytosol (31–36). ROS-inducing agents have been applied to kill tumor cells in cancer therapy (37,38).
The present study aimed to explore the use of celastrol as a sensitizing agent to TRAIL-initiated apoptosis in A549 cells. It was revealed that a combined regimen of celastrol and TRAIL resulted in an improved outcome compared with treatment with either celastrol or TRAIL alone.
Materials and methods
Cell culture
Human lung A549, HCC-15 and Calu-3 cell lines (American Type Culture Collection, Manassas, VA, USA) were maintained in RPMI-1640 medium (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) containing with 10% fetal bovine serum (FBS; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany). During experimentation, cells were switched to RPMI containing 1% FBS.
Reagents
Recombinant celastrol and chloroquine (20 µM) were obtained from Sigma-Aldrich; Merck KGaA (Darmstadt, Germany). TRAIL (200 ng/ml) was purchased from Abfrontier; Young in Frontier, Co., Ltd. (Seoul, South Korea).
Cell viability assay
A549, HCC-15 and Calu-3 cells were plated in 12-well plates and pre-exposed to celastrol (1, 2 and 4 µM) for 12 h in 37°C incubator, and were then additionally treated with TRAIL protein (200 ng/ml) for an additional 2 h. Cell morphology was assessed under a light microscope (Nikon Corporation, Tokyo, Japan, magnification, ×100), and cell viability was evaluated by a crystal violet staining method. Cells were stained with a staining solution (0.5% crystal violet in 30% ethanol and 3% formaldehyde) for 10 min at room temperature, washed four times with PBS, and were dried. Cells were then lysed with 1% SDS solution. The absorbance value was then measured at wavelength of 550 mm using a plate reader. Cell viability was expressed as relative dye intensity compared with that of the control.
Trypan blue exclusion assay
Cell viability was evaluated by trypan blue exclusion assay (Sigma-Aldrich; Merck KGaA), using a hemocytometer. Following each treatment, the cells at 1.0×104 cells/well in 12-well plates, were trypsinized and re-suspended in PBS. Trypan blue dye solution (0.4%) was added to the cell suspension for 5 min at room temperature. Unstained cells were viable and stained cells were dead. The total cell number and trypan blue-positive cells were counted using a light microscope in a blinded manner. The percentage of surviving cells was calculated using the formula: Number of stained cells/number of total cells ×100. Each treatment was performed in triplicate.
ROS determination
A549 cells (1.0×104 cells/well) in 12-well plates, were pre-incubated with N-acetyl cysteine (NAC) for 1 h and then incubated with TRAIL alone or combined with CQ (10 mM). The formation of ROS was ascertained through application of the cell permeable fluorescent marker dihydroethidium (DHE). Briefly, cells were treated with 5 µM DHE for 30 min at 37°C in the dark. The fluorescence was then measured using a fluorescence plate reader at excitation and emission wavelengths of 518 and 605 nm, respectively.
ΔΨm analysis
The changes in ΔΨm were assessed using a cationic fluorescent marker. A549 cells were maintained on cover slips in a 24-well plate, incubated with 10 ml JC-1 at 37°C for 30 min and then washed with PBS. The cells were then mounted with DakoCytomation fluorescent mounting medium (Dako; Agilent Technologies, Inc., Santa Clara, CA, USA) and visualized under a fluorescence microscope (magnification, ×400).
Immunofluorescent staining
A549 cells (5.0×103 cells/well) were cultured on poly-L-lysine coated coverslips. Following differentiation and specific treatment, the cells were fixed with 4% paraformaldehyde at room temperature 15 min and permeabilized with 0.1% Triton X-100. The cells were then incubated for 60 min at room temperature with blocking solution (5% FBS in Tris-buffered saline) followed by overnight incubation at 4°C with anti-p62 (1:250; cat. no. PA5-20839; Invitrogen; Thermo Fisher Scientific, Inc.) and cleaved caspase-3 [(1:250; cat. no. 9665; Cell Signaling Technology, Inc., Danvers, MA, USA) antibodies. Subsequent to washing with PBS, the cells were incubated with secondary antibody (Alexa Fluor® 488-conjugated donkey polyclonal anti-rabbit; 1:500; cat. no. A-21206; Thermo Fisher Scientific, Inc.; and Texas Red-X-conjugated goat polyclonal anti-mouse; 1:500; cat. no. T-6390; Thermo Fisher Scientific, Inc.) for 2 h in the dark. Finally, immunostaining was visualized under a fluorescence microscope (magnification, ×400).
Western blot analysis
Western blot analysis was performed as described previously (39). Briefly, immunoprecipitation assay buffer (Qiagen, Inc, Valencia, CA, USA) was used to extract total protein in A549 cells. The supernatant was collected by centrifugation (13,282 × g; 4°C; 10 min). The protein concentration was determined using the Pierce Bicinchoninic Protein Assay kit (Thermo Fisher Scientific, Inc.). Proteins (30 µg) were separated on 10% SDS-PAGE gels and blotted onto polyvinylidene fluoride membranes. Membranes were blocked with 5% non-fat dried milk at 25°C for 1 h, followed by incubation with primary antibodies overnight at 4°C. Antibodies against β-actin were purchased from Sigma-Aldrich; Merck KGaA, and those against cleaved caspase-8 were obtained from BD Pharmingen (BD Biosciences, Franklin Lakes, NJ, USA). Cleaved caspase-3 and LC3 antibodies were purchased from Cell Signaling Technology, Inc., and p62 antibodies were purchased from EMD Millipore (Billerica, MA, USA). Membranes were incubated with horseradish peroxidase conjugated secondary antibody (cat. no. 4410; 1:2,000; Cell Signaling Technology, Inc.) at 25°C for 1 h. The immune-reactive protein bands were visualized using an enhanced chemiluminescence detection system (GE Healthcare Life Sciences, Chalfont, UK). Densitometry of the signal bands was conducted using the Bio-1D densitometer (Vilber Lourmat, Eberhardzell, Germany). Images were examined using a Fusion-FX7 imaging system (Vilber Lourmat).
Statistical analysis
Statistical analysis was performed using GraphPad Prim (version 5.03; GraphPad Software, Inc., La Jolla, CA, USA). All experiments were performed in triplicate, and the data are expressed as the mean ± standard error. Significant differences between control and treated samples were analyzed using one-way analysis of variance followed by Duncan's post hoc test.
Results
Celastrol sensitizes TRAIL-initiated apoptosis in A549 cells
Alterations in cell morphologies were examined under a light microscope. Treatment with either celastrol or TRAIL alone did not or only marginally induced cell death (Fig. 1), and no morphological changes were observed. However, a combined regimen of TRAIL and different concentrations of celastrol markedly increased the number of apoptotic cell deaths compared with celastrol or TRAIL alone (Fig. 1A-D). These data indicate that celastrol promoted TRAIL-initiated apoptosis in A549 cells.
Figure 1.

Cela sensitizes TRAIL-initiated apoptosis in A549 cells. A549 cells were treated with different dose of cela (1, 2 and 4 µM) for 12 h and then exposed to TRAIL (200 ng/ml) for 2 h. (A) The morphology of the cells was evaluated by interference light microscopy (magnification, ×100). (B) Cellular viability was assessed with crystal violet staining. (C) Quantification data for the crystal violet staining cell viability. (D) Cellular viability was evaluated with trypan blue dye exclusion assays. *P<0.05 and **P<0.01 vs. TRAIL untreated group and only TRAIL treatment group. Cela, celastrol; TRAIL, tumor necrosis factor-related apoptosis-inducing ligand.
Autophagy flux is inhibited by celastrol in A549 cells
LC3-II and p62 expression increased following celastrol treatment (Fig. 2A-C). Immunofluorescence staining also demonstrated that varying doses of celastrol increased p62 protein levels (Fig. 2D). The combined regimen of TRAIL and celastrol increased active (Ac)-caspase 3 and Ac-caspase 8 levels (Fig. 2E-G). Immunofluorescent staining results also suggested that Ac-caspase 3 levels of combined celastrol and TRAIL treatment were increased than only TRAIL treatment (Fig. 2H), indicating that autophagy flux was inhibited by celastrol.
Figure 2.

Autophagy flux is induced by cela in A549 cells. A549 cells were treated with cela (4 µM) for 12 h and then exposed to TRAIL for an additional 1 h. (A) LC3-II and p62 protein levels were assessed by western blot analysis. Bar charts indicate the (B) LC3-II/LC3-I and (C) p62/β-actin ratios. (D) Cells were immunostained with p62 (red) and evaluated using fluorescence microscopy. Scale bar=50 µm. (E) Ac-cas3 and Ac-cas8 protein levels were assessed by western blot analysis. Bar charts indicate the (F) Ac-cas8/β-actin and (G) Ac-cas3/β-actin ratios. (H) Cells were immunostained with Ac-cas3 (green) antibody and evaluated using fluorescence microscopy. Scale bar=50 µm. β-actin was used as a control. **P<0.01 vs. TRAIL untreated group and only TRAIL treatment group. Cela, celastrol; TRAIL, tumor necrosis factor-related apoptosis-inducing ligand; LC3, microtubule-associated proteins 1A/1B light chain 3B; p62, Sequestosome 1; Ac-cas, active caspase.
Celastrol increases TRAIL-mediated cell death by attenuating autophagy flux
The cell morphology results indicated that increased apoptosis was achieved by co-treatment of TRAIL and celastrol or CQ (Fig. 3A). The combined regimen of CQ and TRAIL markedly increased cell death and attenuated cell viability (Fig. 3B and C). These results suggested that celastrol sensitized TRAIL-initiated cell death by attenuating autophagy flux.
Figure 3.
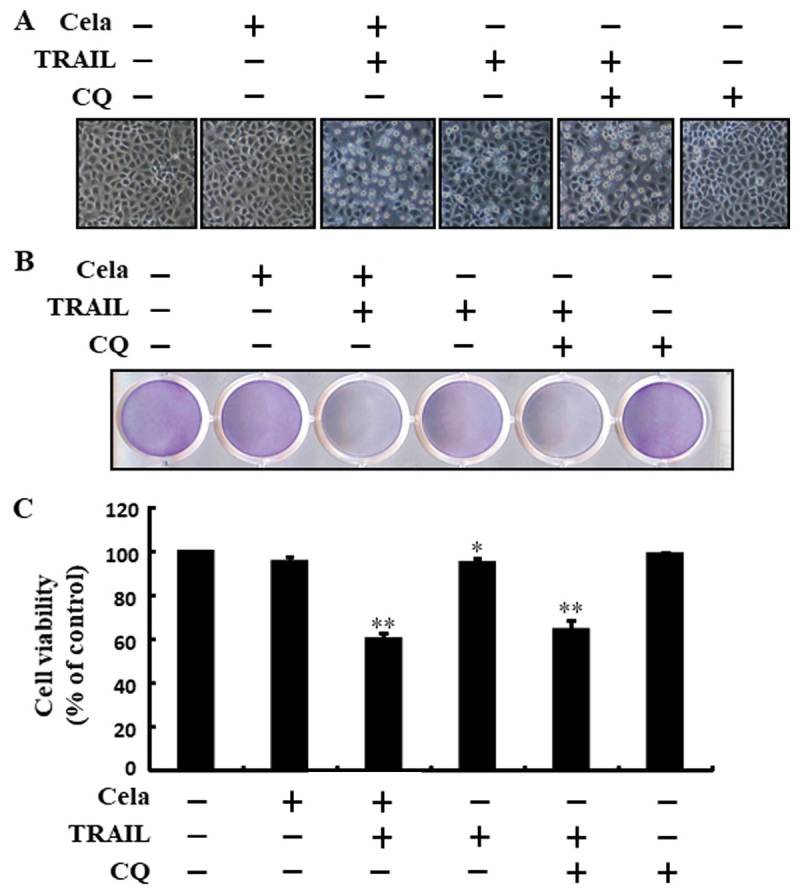
Cela improves TRAIL-induced cell death by attenuating autophagy flux. A549 cells were pretreated with CQ prior to exposure to cela (4 µM) for 12 h and then exposed to TRAIL for an additional 2 h. (A) The morphology of the cells was evaluated by interference light microscopy (magnification, ×100) (B) Cellular viability was assessed with crystal violet staining. (C) Bar diagram demonstrating the average density of crystal violet staining. *P<0.05 and **P<0.01 vs. TRAIL untreated group and only TRAIL treatment group. Cela, celastrol; TRAIL, tumor necrosis factor-related apoptosis-inducing ligand; CQ, chloroquine.
Celastrol-mediated autophagy flux inhibition promotes TRAIL-initiated apoptosis
LC3-II and p62 expression were markedly increased in A549 cells treated with celastrol or CQ alone, confirming that celastrol inhibits autophagy flux (Fig. 4A-C). Immunofluorescence staining results also demonstrated that p62 protein levels were increased (Fig. 4D). The combined regimen of TRAIL and CQ increased Ac-caspase 3 and Ac-caspase 8 protein levels (Fig. 4E-G). The immunofluorescent staining results also demonstrated that Ac-caspase 3 expression was increased (Fig. 4H). These results also indicated that the promotion of the TRAIL-initiated apoptotic mechanism by celastrol was due to the attenuation of autophagy flux.
Figure 4.

Cela induces a promotion of the TRAIL-initiated apoptotic mechanism by attenuating autophagy flux. A549 cells were pretreated with CQ prior to exposure to cela (4 µM) for 12 h and then exposure to TRAIL for an additional 1 h. (A) LC3-II and p62 protein levels were assessed by western blot analysis. Bar charts indicate the (B) LC3-II/LC3-I and (C) p62/β-actin ratios. (D) Cells were immunostained with p62 (red) and evaluated using fluorescence microscopy. Scale bar=50 µm. (E) Ac-cas3 and Ac-cas8 protein levels were assessed by western blot analysis. Bar charts indicate the (F) Ac-cas8/β-actin and (G) Ac-cas3/β-actin ratios. (H) Cells were immunostained with Ac-cas3 (green) antibody and evaluated using fluorescence microscopy. Scale bar=50 µm. β-actin was used as a control. **P<0.01 vs. TRAIL untreated group and only TRAIL treatment group. Cela, celastrol; TRAIL, tumor necrosis factor-related apoptosis-inducing ligand; CQ, chloroquine; LC3, microtubule-associated proteins 1A/1B light chain 3B; p62, Sequestosome 1; Ac-cas, active caspase.
Attenuation of autophagy stimulates ROS generation and changes in ΔΨm
As demonstrated in Fig. 5A, ROS levels were increased in cells treated with celastrol combined with TRAIL or CQ. These increases were effectively attenuated when the cells were pre-incubated with N-acetyl cysteine (NAC) for 1 h. Green fluorescence observed in the cells treated with celastrol combined with TRAIL or CQ suggested decreased ΔΨm values compared with that from the TRAIL alone treatment, but treatment with NAC restored these ΔΨm values (Fig. 5B). These data suggest that celastrol-mediated autophagy flux attenuation increased TRAIL-initiated apoptosis via increases in ROS generation and decreasing ΔΨm.
Figure 5.

Attenuation of autophagy stimulates ROS generation and changes of mitochondrial membrane potential. A549 cells were pretreated with NAC (10 mM) and CQ prior to exposure to cela (4 µM) for 12 h and then treatment with TRAIL for an additional 2 h. (A) The ROS level was assessed using a spectrofluorometer. (B) JC-1 aggregate forms (red) and mono forms (green) were assessed by confocal microscopy (magnification, ×400). *P<0.05 and **P<0.01 vs. TRAIL untreated group and only TRAIL treatment group. Cela, celastrol; TRAIL, tumor necrosis factor-related apoptosis-inducing ligand; CQ, chloroquine; NAC, N-acetyl cysteine; ROS, reactive oxygen species.
Celastrol sensitizes TRAIL-initiated apoptosis in different lung cells
Celastrol or TRAIL treatment alone did not, or only marginally affected, the cell viability of Calu-3 and HCC-15 cells (Fig. 6). Nevertheless, the combined TRAIL and celastrol regimen significantly decreased the cell viability of Calu-3 and HCC-15 cells (Fig. 6A-F). These results suggested that celastrol significantly increased TRAIL-initiated apoptosis in Calu-3 and HCC-15 cells.
Figure 6.

Cela sensitizes TRAIL-initiated apoptosis in different lung cell lines. (A) Calu-3 morphology was evaluated by interference light microscopy (magnification, ×100). (B) Calu-3 viability as assessed with crystal violet staining and (C) percentage of crystal violet positive cells. (D) HCC-15 morphology was evaluated by interference light microscopy (magnification, ×100). (E) HCC-15 viability as assessed with crystal violet staining and (F) percentage of crystal violet positive cells. *P<0.05 and **P<0.01 vs. TRAIL untreated group and only TRAIL treatment group. Cela, celastrol; TRAIL, tumor necrosis factor-related apoptosis-inducing ligand.
Discussion
The aim of the present study was to determine the function of celastrol with or without TRAIL, in lung cancer cells. The results revealed that celastrol-mediated autophagy flux inhibition sensitized TRAIL-mediated apoptosis via regulation of ROS and ΔΨm.
TRAIL is considered one of the most promising anti-cancer agents identified in the previous 25 years, owing to the specificity of its action, initiating apoptosis in specific cell types (5,40–44) and prompting the destruction of cancer cells without causing toxicity to normal cells. Previous studies have confirmed that the repeated administration of TRAIL proteins adequately attenuated tumor growth without causing serious side effects (6,45). In this manner, considerable attention has been paid to TRAIL as a anti-cancer therapy for human cancer. The antitumor activities of celastrol are not only limited to in vitro systems, as this triterpenoid was also identified to inhibit the uncontrolled growth and metastasis of melanoma in xenograft mouse models (46). Previous studies have demonstrated that multiple mechanisms or signaling pathways lead to celastrol-induced cancer cell death (15,17,47). Autophagy flux is a complete system of autophagy, starting with the amalgamation of the autophagosome with the lysosome, resulting in the degradation and recycling of their cargo (18,48). ROS serves an important role in response to cancer therapeutics, considering that ROS formation is associated with cancer initiation and progression.
A previous study have attempted to examine TRAIL sensitizers that are capable of overcoming TRAIL resistance in cancer cells (49). The present study revealed that a combined regimen of TRAIL and different doses of celastrol significantly increased the level of apoptotic cell death compared with treatment with celastrol or TRAIL alone. A previous study has suggested that celastrol treatment attenuated cell proliferation and the induction of apoptosis and autophagy in gastric cancer cells (50). However, the results from the present study indicated that LC3-II and p62 expression increased following celastrol treatment, where co-treatment of celastrol and TRAIL increased Ac-cas3 and Ac-cas8 levels. It has bene demonstrated previously that treatment of celastrol in MCF-7 cells induces apoptosis through mitochondrial caspase-dependent and independent signaling pathways (51). Furthermore, Kannaiyan et al (52) identified that celastrol treatment inhibits proliferation and stimulates apoptosis in RPMI-8226 cells, alongside the activation of c-Jun N-terminal kinase and attenuation of the phosphoinositide 3-kinase/protein kinase B signaling pathways. The present study also demonstrated that the combined regimen of TRAIL and celastrol or chloroquine significantly attenuated cell viability and increased cell death. Previous studies have suggested that the inhibition of autophagy stimulates dysfunctional mitochondria and ROS generation in prostate cancer cells (53,54). The data from the present study suggest that celastrol-mediated autophagy flux inhibition increased TRAIL-induced apoptosis via ROS generation and decreases in ΔΨm. In conclusion, the present study demonstrated that celastrol improved TRAIL-initiated apoptosis via inhibiting autophagy flux. In addition, the use of TRAIL in combination with celastrol may provide an adequate therapeutic strategy for safe treatment of TRAIL-resistant lung cancer, suggesting that celastrol-mediated autophagy flux inhibition sensitized TRAIL-initiated apoptosis via the regulation of ROS and the ΔΨm.
Acknowledgements
Not applicable.
Funding
The present study was supported by a grant from the National Research Foundation of Korea, funded by the Korean Government (Grant no. 2016R1A2B2009293).
Availability of data and materials
The analyzed data sets generated during the study are available from the corresponding author on reasonable request.
Authors' contributions
UMN and SYP designed the study. UMN and HHY performed the experiments. UMN and SYP analyzed the data and wrote the manuscript. UMN, HHY and SYP revised and approved the final version of the manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent of publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Garcia G, Odaimi M. Systemic combination chemotherapy in elderly pancreatic cancer: A review. J Gastrointest Cancer. 2017;48:121–128. doi: 10.1007/s12029-017-9930-0. [DOI] [PubMed] [Google Scholar]
- 2.Sun W, Sanderson PE, Zheng W. Drug combination therapy increases successful drug repositioning. Drug Discov Today. 2016;21:1189–1195. doi: 10.1016/j.drudis.2016.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aggarwal BB. Signalling pathways of the TNF superfamily: A double-edged sword. Nat Rev Immunol. 2003;3:745–756. doi: 10.1038/nri1184. [DOI] [PubMed] [Google Scholar]
- 4.Pitti RM, Marsters SA, Ruppert S, Donahue CJ, Moore A, Ashkenazi A. Induction of apoptosis by Apo-2 ligand, a new member of the tumor necrosis factor cytokine family. J Biol Chem. 1996;271:12687–12690. doi: 10.1074/jbc.271.22.12687. [DOI] [PubMed] [Google Scholar]
- 5.Ashkenazi A, Pai RC, Fong S, Leung S, Lawrence DA, Marsters SA, Blackie C, Chang L, McMurtrey AE, Hebert A, et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. J Clin Invest. 1999;104:155–162. doi: 10.1172/JCI6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bellail AC, Qi L, Mulligan P, Chhabra V, Hao C. TRAIL agonists on clinical trials for cancer therapy: The promises and the challenges. Rev Recent Clin Trials. 2009;4:34–41. doi: 10.2174/157488709787047530. [DOI] [PubMed] [Google Scholar]
- 7.Wang S, El-Deiry WS. TRAIL and apoptosis induction by TNF-family death receptors. Oncogene. 2003;22:8628–8633. doi: 10.1038/sj.onc.1207232. [DOI] [PubMed] [Google Scholar]
- 8.Gonzalvez F, Ashkenazi A. New insights into apoptosis signaling by Apo2L/TRAIL. Oncogene. 2010;29:4752–4765. doi: 10.1038/onc.2010.221. [DOI] [PubMed] [Google Scholar]
- 9.Johnstone RW, Frew AJ, Smyth MJ. The TRAIL apoptotic pathway in cancer onset, progression and therapy. Nat Rev Cancer. 2008;8:782–798. doi: 10.1038/nrc2465. [DOI] [PubMed] [Google Scholar]
- 10.Jin CY, Park C, Hwang HJ, Kim GY, Choi BT, Kim WJ, Choi YH. Naringenin up-regulates the expression of death receptor 5 and enhances TRAIL-induced apoptosis in human lung cancer A549 cells. Mol Nutr Food Res. 2011;55:300–309. doi: 10.1002/mnfr.201000024. [DOI] [PubMed] [Google Scholar]
- 11.Li H, Zhang YY, Huang XY, Sun YN, Jia YF, Li D. Beneficial effect of tripterine on systemic lupus erythematosus induced by active chromatin in BALB/c mice. Eur J Pharmacol. 2005;512:231–237. doi: 10.1016/j.ejphar.2005.02.030. [DOI] [PubMed] [Google Scholar]
- 12.Xu X, Wu Z, Xu C, Ren Y, Ge Y. Observation on serum anti-double stranded DNA antibodies of tripterine in systemic lupus erythematosus of (NZBxW)F1 mice. Ann Rheum Dis. 2003;62:377–378. doi: 10.1136/ard.62.4.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pinna GF, Fiorucci M, Reimund JM, Taquet N, Arondel Y, Muller CD. Celastrol inhibits pro-inflammatory cytokine secretion in Crohn's disease biopsies. Biochem Biophys Res Commun. 2004;322:778–786. doi: 10.1016/j.bbrc.2004.07.186. [DOI] [PubMed] [Google Scholar]
- 14.Cleren C, Calingasan NY, Chen J, Beal MF. Celastrol protects against MPTP- and 3-nitropropionic acid-induced neurotoxicity. J Neurochem. 2005;94:995–1004. doi: 10.1111/j.1471-4159.2005.03253.x. [DOI] [PubMed] [Google Scholar]
- 15.Yang H, Chen D, Cui QC, Yuan X, Dou QP. Celastrol, a triterpene extracted from the Chinese ‘Thunder of God Vine,’ is a potent proteasome inhibitor and suppresses human prostate cancer growth in nude mice. Cancer Res. 2006;66:4758–4765. doi: 10.1158/0008-5472.CAN-05-4529. [DOI] [PubMed] [Google Scholar]
- 16.Zhang T, Hamza A, Cao X, Wang B, Yu S, Zhan CG, Sun D. A novel Hsp90 inhibitor to disrupt Hsp90/Cdc37 complex against pancreatic cancer cells. Mol Cancer Ther. 2008;7:162–170. doi: 10.1158/1535-7163.MCT-07-0484. [DOI] [PubMed] [Google Scholar]
- 17.Nagase M, Oto J, Sugiyama S, Yube K, Takaishi Y, Sakato N. Apoptosis induction in HL-60 cells and inhibition of topoisomerase II by triterpene celastrol. Biosci Biotechnol Biochem. 2003;67:1883–1887. doi: 10.1271/bbb.67.1883. [DOI] [PubMed] [Google Scholar]
- 18.Mizushima N. Autophagy: Process and function. Genes Dev. 2007;21:2861–2873. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- 19.Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Todde V, Veenhuis M, van der Klei IJ. Autophagy: Principles and significance in health and disease. Biochim Biophys Acta. 2009;1792:3–13. doi: 10.1016/j.bbadis.2008.10.016. [DOI] [PubMed] [Google Scholar]
- 21.Mizushima N, Komatsu M. Autophagy: Renovation of cells and tissues. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 22.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tanida I, Minematsu-Ikeguchi N, Ueno T, Kominami E. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy. 2005;1:84–91. doi: 10.4161/auto.1.2.1697. [DOI] [PubMed] [Google Scholar]
- 24.Apel A, Herr I, Schwarz H, Rodemann HP, Mayer A. Blocked autophagy sensitizes resistant carcinoma cells to radiation therapy. Cancer Res. 2008;68:1485–1494. doi: 10.1158/0008-5472.CAN-07-0562. [DOI] [PubMed] [Google Scholar]
- 25.Carew JS, Espitia CM, Esquivel JA II, Mahalingam D, Kelly KR, Reddy G, Giles FJ, Nawrocki ST. Lucanthone is a novel inhibitor of autophagy that induces cathepsin D-mediated apoptosis. J Biol Chem. 2011;286:6602–6613. doi: 10.1074/jbc.M110.151324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boya P, Gonzalez-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N, Métivier D, Meley D, Souquere S, Yoshimori T, et al. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol. 2005;25:1025–1040. doi: 10.1128/MCB.25.3.1025-1040.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Poole B, Ohkuma S. Effect of weak bases on the intralysosomal pH in mouse peritoneal macrophages. J Cell Biol. 1981;90:665–669. doi: 10.1083/jcb.90.3.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fan C, Wang W, Zhao B, Zhang S, Miao J. Chloroquine inhibits cell growth and induces cell death in A549 lung cancer cells. Bioorg Med Chem. 2006;14:3218–3222. doi: 10.1016/j.bmc.2005.12.035. [DOI] [PubMed] [Google Scholar]
- 29.Jiang PD, Zhao YL, Deng XQ, Mao YQ, Shi W, Tang QQ, Li ZG, Zheng YZ, Yang SY, Wei YQ. Antitumor and antimetastatic activities of chloroquine diphosphate in a murine model of breast cancer. Biomed Pharmacother. 2010;64:609–614. doi: 10.1016/j.biopha.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 30.Yoon YH, Cho KS, Hwang JJ, Lee SJ, Choi JA, Koh JY. Induction of lysosomal dilatation, arrested autophagy, and cell death by chloroquine in cultured ARPE-19 cells. Invest Ophthalmol Vis Sci. 2010;51:6030–6037. doi: 10.1167/iovs.10-5278. [DOI] [PubMed] [Google Scholar]
- 31.Kroemer G, Reed JC. Mitochondrial control of cell death. Nat Med. 2000;6:513–519. doi: 10.1038/74994. [DOI] [PubMed] [Google Scholar]
- 32.Lim ML, Minamikawa T, Nagley P. The protonophore CCCP induces mitochondrial permeability transition without cytochrome c release in human osteosarcoma cells. FEBS Lett. 2001;503:69–74. doi: 10.1016/S0014-5793(01)02693-X. [DOI] [PubMed] [Google Scholar]
- 33.Linsinger G, Wilhelm S, Wagner H, Häcker G. Uncouplers of oxidative phosphorylation can enhance a Fas death signal. Mol Cell Biol. 1999;19:3299–3311. doi: 10.1128/MCB.19.5.3299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mlejnek P. Caspase-3 activity and carbonyl cyanide m-chlorophenylhydrazone-induced apoptosis in HL-60. Altern Lab Anim. 2001;29:243–249. doi: 10.1177/026119290102900313. [DOI] [PubMed] [Google Scholar]
- 35.Nagata S. Apoptosis by death factor. Cell. 1997;88:355–365. doi: 10.1016/S0092-8674(00)81874-7. [DOI] [PubMed] [Google Scholar]
- 36.Newmeyer DD, Ferguson-Miller S. Mitochondria: Releasing power for life and unleashing the machineries of death. Cell. 2003;112:481–490. doi: 10.1016/S0092-8674(03)00116-8. [DOI] [PubMed] [Google Scholar]
- 37.Zou ZZ, Nie PP, Li YW, Hou BX, Rui-Li, Shi XP, Ma ZK, Han BW, Luo XY. Synergistic induction of apoptosis by salinomycin and gefitinib through lysosomal and mitochondrial dependent pathway overcomes gefitinib resistance in colorectal cancer. Oncotarget. 2017;8:22414–22432. doi: 10.18632/oncotarget.5628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mi YJ, Geng GJ, Zou ZZ, Gao J, Luo XY, Liu Y, Li N, Li CL, Chen YQ, Yu XY, Jiang J. Dihydroartemisinin inhibits glucose uptake and cooperates with glycolysis inhibitor to induce apoptosis in non-small cell lung carcinoma cells. PLoS One. 2015;10:e0120426. doi: 10.1371/journal.pone.0120426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nazim UM, Moon JH, Lee YJ, Seol JW, Park SY. PPARγ activation by troglitazone enhances human lung cancer cells to TRAIL-induced apoptosis via autophagy flux. Oncotarget. 2017;8:26819–26831. doi: 10.18632/oncotarget.21754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kischkel FC, Lawrence DA, Chuntharapai A, Schow P, Kim KJ, Ashkenazi A. Apo2L/TRAIL-dependent recruitment of endogenous FADD and caspase-8 to death receptors 4 and 5. Immunity. 2000;12:611–620. doi: 10.1016/S1074-7613(00)80212-5. [DOI] [PubMed] [Google Scholar]
- 41.Van Geelen CM, de Vries EG, de Jong S. Lessons from TRAIL-resistance mechanisms in colorectal cancer cells: Paving the road to patient-tailored therapy. Drug Resist Updat. 2004;7:345–358. doi: 10.1016/j.drup.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 42.Srivastava RK. TRAIL/Apo-2L: Mechanisms and clinical applications in cancer. Neoplasia. 2001;3:535–546. doi: 10.1038/sj.neo.7900203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shankar S, Srivastava RK. Enhancement of therapeutic potential of TRAIL by cancer chemotherapy and irradiation: Mechanisms and clinical implications. Drug Resist Updat. 2004;7:139–156. doi: 10.1016/j.drup.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 44.LeBlanc H, Lawrence D, Varfolomeev E, Totpal K, Morlan J, Schow P, Fong S, Schwall R, Sinicropi D, Ashkenazi A. Tumor-cell resistance to death receptor-induced apoptosis through mutational inactivation of the proapoptotic Bcl-2 homolog Bax. Nat Med. 2002;8:274–281. doi: 10.1038/nm0302-274. [DOI] [PubMed] [Google Scholar]
- 45.Walczak H, Miller RE, Ariail K, Gliniak B, Griffith TS, Kubin M, Chin W, Jones J, Woodward A, Le T, et al. Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat Med. 1999;5:157–163. doi: 10.1038/5517. [DOI] [PubMed] [Google Scholar]
- 46.Abbas S, Bhoumik A, Dahl R, Vasile S, Krajewski S, Cosford ND, Ronai ZA. Preclinical studies of celastrol and acetyl isogambogic acid in melanoma. Clin Cancer Res. 2007;13:6769–6778. doi: 10.1158/1078-0432.CCR-07-1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sethi G, Ahn KS, Pandey MK, Aggarwal BB. Celastrol, a novel triterpene, potentiates TNF-induced apoptosis and suppresses invasion of tumor cells by inhibiting NF-kappaB-regulated gene products and TAK1-mediated NF-kappaB activation. Blood. 2007;109:2727–2735. doi: 10.1182/blood-2006-10-050807. [DOI] [PubMed] [Google Scholar]
- 48.Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–175. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dimberg LY, Anderson CK, Camidge R, Behbakht K, Thorburn A, Ford HL. On the TRAIL to successful cancer therapy? Predicting and counteracting resistance against TRAIL-based therapeutics. Oncogene. 2013;32:1341–1350. doi: 10.1038/onc.2012.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee HW, Jang KS, Choi HJ, Jo A, Cheong JH, Chun KH. Celastrol inhibits gastric cancer growth by induction of apoptosis and autophagy. BMB Rep. 2014;47:697–702. doi: 10.5483/BMBRep.2014.47.12.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang HS, Kim JY, Lee JH, Lee BW, Park KH, Shim KH, Lee MK, Seo KI. Celastrol isolated from Tripterygium regelii induces apoptosis through both caspase-dependent and -independent pathways in human breast cancer cells. Food Chem Toxicol. 2011;49:527–532. doi: 10.1016/j.fct.2010.11.044. [DOI] [PubMed] [Google Scholar]
- 52.Kannaiyan R, Manu KA, Chen L, Li F, Rajendran P, Subramaniam A, Lam P, Kumar AP, Sethi G. Celastrol inhibits tumor cell proliferation and promotes apoptosis through the activation of c-Jun N-terminal kinase and suppression of PI3 K/Akt signaling pathways. Apoptosis. 2011;16:1028–1041. doi: 10.1007/s10495-011-0629-6. [DOI] [PubMed] [Google Scholar]
- 53.Saleem A, Dvorzhinski D, Santanam U, Mathew R, Bray K, Stein M, White E, DiPaola RS. Effect of dual inhibition of apoptosis and autophagy in prostate cancer. Prostate. 2012;72:1374–1381. doi: 10.1002/pros.22487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang J, Tan X, Yang Q, Zeng X, Zhou Y, Luo W, Lin X, Song L, Cai J, Wang T, Wu X. Inhibition of autophagy promotes apoptosis and enhances anticancer efficacy of adriamycin via augmented ROS generation in prostate cancer cells. Int J Bioch Cell Biol. 2016;77:80–90. doi: 10.1016/j.biocel.2016.05.020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The analyzed data sets generated during the study are available from the corresponding author on reasonable request.