

Abstract
Fusion transcripts are chimeric RNAs generated as a result of fusion either at DNA or RNA level. These novel transcripts have been extensively studied in the case of human cancers but still remain underexamined in plants. In this study, we introduce the first plant-specific database of fusion transcripts named AtFusionDB (http://www.nipgr.res.in/AtFusionDB). This is a comprehensive database that contains the detailed information about fusion transcripts identified in model plant Arabidopsis thaliana. A total of 82 969 fusion transcript entries generated from 17 181 different genes of A. thaliana are available in this database. Apart from the basic information consisting of the Ensembl gene names, official gene name, tissue type, EricScore, fusion type, AtFusionDB ID and sample ID (e.g. Sequence Read Archive ID), additional information like UniProt, gene coordinates (together with the function of parental genes), junction sequence, expression level of both parent genes and fusion transcript may be of high utility to the user. Two different types of search modules viz. ‘Simple Search’ and ‘Advanced Search’ in addition to the ‘Browse’ option with data download facility are provided in this database. Three different modules for mapping and alignment of the query sequences viz. BLASTN, SW Align and Mapping are incorporated in AtFusionDB. This database is a head start for exploring the complex and unexplored domain of gene/transcript fusion in plants.
Introduction
The origin and evolution of new genes are the constant sources of evolutionary renovation and adaptation. Gene duplication, the de novo origination of gene, transposition, fission and fusion are the major processes leading to the genesis of new genes (1, 2). Fusion transcripts illustrate an event in which a hybrid RNA is composed of transcripts from two separate genes (3). This can be accomplished by translocation of the original genes at the DNA level or post-transcriptionally during splicing events, and it has been documented in diverse life forms (4). The formation of fusion transcripts can occur either by gene or chromosomal rearrangements (gene fusion at DNA level by translocation, deletion and inversion) or by intergenic RNA cis-splicing and trans-splicing events, i.e. transcript fusion at RNA level (5–7). The fusion transcripts formed post-transcriptionally are more common (8). The splicing event is said to be of ‘cis-type’ when the two exons derived from two neighboring genes transcribe simultaneously or ‘trans-type’ when the two exons originated from two separate premature mRNAs. Nonetheless, both of these aforementioned types of splicing events are mediated by spliceosome complex (9). Read-through fusion transcripts are generated by fusion of the two adjoining individually spliced genes in the same orientation and from the same strand, resembling alternative splicing (10, 11). Likewise, the fusion transcripts that are originated as a result of the hybridization of transcripts from two nearby genes located in opposite strands give rise to cis-acting chimeric transcripts (12). Intra-chromosomal fusion transcripts are generated by fusion of genes or transcripts coming from the same chromosome while inter-chromosomal chimeric transcripts are formed as a result of gene or transcript fusion from different chromosomes (13). The different fusion transcript types viz. read-through, cis-acting, intra-chromosomal and inter-chromosomal transcripts have been illustrated in Figure 1.
Figure 1.
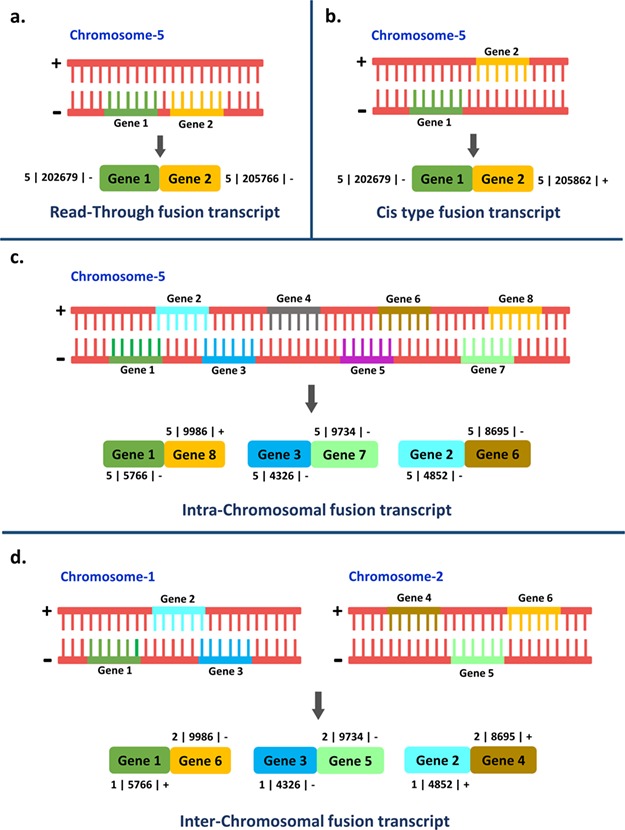
Representation of the four different types of fusion transcripts.
The existence and impact of fusion transcripts at molecular and physiological levels have been studied in eukaryotes including Drosophila (14), zebrafish (15), plants (16–18) and humans (19, 20). The role of gene fusion in promoting hematological and solid cancers has been well established in humans (21, 22), and this has paved way to further inquire about their biological relevance in other organisms as well. The very famous and extensively studied BCR-ABL1 fusion transcript is involved in promoting malignancy in the case of chronic myelogenous leukemia (23). These fusion transcripts have been exploited as biomarkers in cancer prediction and targets of molecular therapeutics (24). Chimeric transcripts may either act as long non-coding RNAs or can encode novel chimeric proteins (25), thus can alter cellular signaling and overall functioning in diverse organisms. The emergence of high-throughput technologies has led to the accumulation of enormous sequencing data, which has eased the understanding of the molecular mechanism behind this complex event, and its implications are being attempted to be elucidated in eukaryotic organisms including plants (21). The currently available fusion transcript databases such as ChiTaRS (26), FusionCancer (24), ChimerDB (27), Mitelman Database (28) and FusionHub (29) harbor information related to fusion transcripts reported in human cancers, mouse and flies. Till date, only scarce knowledge about fusion transcripts is available for plants (30–32). A freely available fusion transcript database of plants is currently unavailable to our best of knowledge. In this study, we have developed a database named AtFusionDB which is the plant-exclusive knowledge base for fusion transcripts predicted in the model plant Arabidopsis thaliana (the thale cress or mouse-ear cress). The overall structure and major elements of the AtFusionDB are highlighted in Figure 2. Gene fusion is believed to be a major factor for controlling morphology, physiology and phenotypic character in an organism as well as a major contributor for adaptive evolution. Thus, this attempt will unravel new directions for exploring the impact and consequences of gene-fusion events in the plant kingdom and elucidating the significance of shuffling and fusion of transcripts on the physiology of plants.
Figure 2.

Overall representation of AtFusionDB.
Materials and methods
Data retrieval
We have downloaded all the paired-end RNA-Seq data of A. thaliana available at the Sequence Read Archive (SRA) of NCBI (https://www.ncbi.nlm.nih.gov/sra). SRA data were further converted into the FASTQ format using ‘fastq-dump’ utility of SRA Toolkit version 2.8.2-1 (https://www.ncbi.nlm.nih.gov/sra/docs/toolkitsoft/).
Identification of fusion transcripts
The FASTQ files of paired-end RNA-Seq run obtained from the previous step were given as an input to ‘EricScript-Plants’ (https://github.com/asherkhb/EricScript-Plants) for the identification of fusion transcripts in A. thaliana. EricScript-Plants is a modified version of EricScript (33) to work for all the plant species available at Ensembl Plants (34). This version of EricScript was downloaded using the command ‘git clone’ (https://github.com/asherkhb/EricScript-Plants.git). EricScript is a freely available software package (https://sites.google.com/site/bioericscript) for the identification of fusion transcripts from paired-end RNA-Seq data sets. It is developed in Practical Extraction and Reporting Language (PERL) and requires several other dependencies, i.e. R (http://cran.r-project.org/), ada package (http://cran.r-project.org/web/packages/ada/index.html), BWA (35), SAMtools version >0.1.17 (36), bedtools version >2.15 (37), BLAT (38) and seqtk (https://github.com/lh3/seqtk). The major limitation of this script is the use of transcriptome instead of a reference genome for the mapping of sequencing reads. The output of the EricScript-Plants reports the candidate fusions in two tab-delimited files; the first file (e.g. samplename.results.total.tsv) contains all the identified fusions, whereas the other file (e.g. samplename.results.filtered.tsv) reports the fusions with ‘EricScore’ >0.5. In a quality-filtering approach, EricScript exploits three different scores viz. genuine junction score, edge score and uniformity score. Using an AdaBoost qualifier (39), these three scores are unified into a single score, called ‘EricScore’, which assigns each candidate fusion a probability score of ‘well’ pattern, and thus classifying all the fusions for discriminating between real transcripts and false-positive events (33). The value of ‘EricScore’ determines the probability of fusion transcripts to be real, with the score ranging from 0.01 to 0.99. The transcripts with the highest EricScore represents the highest possibility to be fusion transcripts. In order to eliminate the false positives, we have only considered the fusion transcripts whose ‘EricScore’ values were >0.5 (e.g. results in the file samplename.results.filtered.tsv). The detailed explanation of the EricScript output files is given at the web page (https://sites.google.com/site/bioericscript/).
The Bioconductor (https://www.bioconductor.org/) packages, SRAdb (40) and GEOmetadb (41) were utilized for the retrieval of tissue-type information of RNA-Seq samples analyzed in this study and combined with each entry of AtFusionDB.
AtFusionDB web interface development
After the compilation of all the information, AtFusionDB web interface was developed using Hypertext Mark-up Language, Cascading Style Sheets, Structured Query Language, Java scripting language, PERL and Hypertext Preprocessor on Apache Hypertext Transfer Protocol server. The gene coordinates of each individual gene were prepared using the chromosome no., breakpoint and strand sense information separated by ‘|’ like chromosome no. | breakpoint | strand (e.g. 5 | 25563 | +).
Database organization
The entire data stored in AtFusionDB are organized at different levels. At the most basic or primary level, the user can search by using simple keywords such as ‘gene name’, ‘chromosome’, ‘tissue’, ‘fusion-name’, ‘SRA-ID’, ‘AtFusionDB-ID’ etc. as per the requirement. The information will be displayed in tabular form according to the number of display fields selected by the user. The secondary data can be accessed to gain further information on sequencing experiments and detailed information on fusion transcripts by clicking on the hyperlinks of ‘SRA-ID’ and ‘fusion’ on search result pages, respectively. At the tertiary level, additional information about the contributing genes giving rise to chimeric transcripts can be accessed by clicking on the hyperlinks of the UniProt ID(s) and EnsemblPlants ID(s) on the fusion information page. We have made efforts to make the database easy and convenient to access and fetch the information supplemented with downloadable links.
AtFusionDB web interface features
AtFusionDB provides the two user-friendly ‘Search’ options viz. ‘simple’ and ‘advanced’ for searching fusion transcript information by using different types of keywords. The ‘Simple Search’ option facilitates the user to fetch fusion transcript information by providing different search terms like gene name, chromosome number, tissue etc. To provide flexibility, two options i.e. ‘containing’ and ‘exact’ have been incorporated for search terms. This option also facilitates the user to select the fields to be displayed. The ‘Advanced Search’ option provides the facility to make the user-built query using up to 11 different combinations of keywords. The keywords (e.g. fusion, tissue, chromosomes, genes, ‘EricScore’ etc.) can be defined to be included together or searched alternatively or excluded using ‘add’ and ‘remove facility’. The conditional operators viz. ‘=’, ‘Like’ and ‘!=’ and two logical operators ‘OR’ and ‘AND’ can be used as per the need of the users. For convenient browsing, ‘Browse’ section is also available for the user. This section enables the user to browse the database by the following categories: fusion type, tissue type, ‘EricScore’ range, chromosome and frequency. The frequency of occurrence of fusion transcripts can be further browsed with respect to the tissue type, condition and fusion type separately.
The ‘Tools’ section of AtFusionDB facilitates the user to extract useful information regarding fusion transcript by providing input query to the following tools: ‘SW Align’, ‘Mapping’ and ‘BLAST’ (42). ‘SW Align’ allows the user to align their query sequence with the fusion transcript junction sequences available in AtFusionDB database. This option helps the user to identify and characterize their sequence of interest. Here, we have incorporated ‘WATER’ utility of EMBOSS-6.6.0 package, following the Smith–Waterman Algorithm (43). ‘Mapping’ option facilitates the user to map all the fusion junction sequences from AtFusionDB database to the gene sequences as query provided by the user and only those sequences from AtFusionDB matching 100% with the query sequences are displayed. This module is useful for the detection of fusion transcripts in newly assembled genome drafts or novel gene sequences. In this module, we have incorporated BLASTN (44) option of the BLAST software package. ‘BLAST’ module is helpful to find the regions of similarity between the user input FASTA sequences and AtFusionDB database sequences using BLASTN with the option to change Expect value (E value). The respective ID(s) of sequences from AtFusionDB producing significant alignments with the query sequences are further hyperlinked to display their detailed information.
The ‘Method’ section explains the pipeline opted for the identification of fusion transcripts. The ‘Statistics’ page graphically represents the total and unique fusion transcripts incorporated in AtFusionDB on the basis of fusion and tissue types. The user can also visualize a pie chart depicting the distribution of unique and common fusion transcripts found in SRA samples. ‘Help/Guide’ section is useful for the user to understand AtFusionDB database and use it effectively.
Results and discussion
We have downloaded and analyzed a total of 4697 paired-end RNA-Seq data sets of A. thaliana. We could not found the fusion transcripts in 1036 samples because of different logistic reasons (e.g. the quality of data, the quantity of data, the absence of fusion transcripts etc.). Out of remaining 3661 samples, 141 samples have only the fusion transcripts with low EricScore (e.g. <0.5), not considered for further analyses. Finally, the most probable fusion transcripts (with EricScore >0.5) from 3520 samples were incorporated in AtFusionDB database. These 82 969 fusion transcripts with ‘EricScore’ >0.5 were considered for further data processing and refinement in AtFusionDB. Altogether, 17 181 genes were involved in fusion transcript generation. Rank-wise distribution of total predicted fusion transcripts in accordance with ‘EricScore’ range has been listed in the table available at the ‘Statistics’ section of the database.
A total of 82 969 fusion transcript entries of AtFusionDB are represented by 71 920 unique fusion transcripts. A total of 41 838 fusion transcripts were nonrecurrent and found only in one RNA-Seq sample. However, numerous transcripts were observed to be common in two or more than two samples that is graphically represented in the ‘Statistics’ section of AtFusionDB. The fusion transcripts (total and unique) were also categorized and distributed on the basis of their aforementioned fusion types. It was noticed that inter-chromosomal transcripts were the most abundant and cis-acting transcripts were least in number. The graphical representation showing their distribution has been provided in the Statistics section of AtFusionDB. The three most frequently occurring (e.g. recurrent) fusion transcripts in all analyzed samples were ATPB_ATPE, RPL22_RPS3 and PSBE_PSBF. All of these transcripts were of read-through type and originating from chloroplast. Multifold expression of these specific fusion transcripts in contrast to the significantly low expression of their individual contributing genes indicates that they might have a distinct role in governing cellular dynamics.
The tissue-wise study of the total fusion transcript entries, as well as unique chimeric transcripts, was also carried out on different tissues and developmental stages in the life cycle of A. thaliana. All the 3520 samples predicted with reliable fusion transcripts were distributed on the basis of their developmental stages and tissue origin. It was observed that most chimeric transcripts were derived from seedling, seed, root and whole plant RNA samples. It was noted that few genes commonly contributing in chimeric transcripts generation in the majority of tissue types were elongation factor 1-alpha 3/4 (A1_A1), chloroplastic ATP synthase subunit beta and ATP synthase epsilon chain (ATPB_ATPE) and Chlorophyll a-b binding protein 3 (LHCB1.1_LHCB1.3) and Tubulin alpha chain and Tubulin alpha-2 chain (TUBA6_TUBA4). The most frequently occurring fusion transcripts on the basis of fusion type and tissue origin along with their respective frequencies are shown in Table 1. The tissue-wise distribution of SRA samples is graphically represented in Figure 3. The samples were also categorized on the basis of different experimental conditions of abiotic and biotic stresses together with their respective frequency as demonstrated in Table 2.
Table 1.
A list of the most frequently occurring fusion transcripts on the basis of fusion type and tissue type
| Fusion type | |||
|---|---|---|---|
| Cis (5168) | Read-through (9313) | Intra-chromosomal (16 853) | Inter-chromosomal (51 635) |
| AT1G66900_AT1G66910 (67) AT1G29920_AT1G29930 (65) AT5G19770_AT5G19780 (57) AT1G29930_AT1G29920 (56) |
ATCG00480_ATCG00470 (268) ATCG00810_ATCG00800 (184) AT3G59330_AT3G59320 (131) ATCG00580_ATCG00570 (115) |
AT2G07725_AT2G07715 (95) ATCG00065_ATCG01230 (82) AT1G07940_AT1G07920 (80) AT2G36070_AT2G20510 (57) |
AT1G07590_AT5G10100 (102) AT4G14960_AT1G50010 (95) AT5G38410_AT1G67090 (88) AT2G45330_AT5G23600 (78) |
| Tissue type | |||
| Seed (8608) | Seedling (36 530) | Leaf (7126) | Stem (831) |
| AT4G11845_AT4G11840 (15) AT4G14960_AT1G50010 (11) AT1G28660_AT1G28670 (9) AT2G15890_AT2G15880 (8) |
ATCG00480_ATCG00470 (134) AT5G38410_AT1G67090 (76) ATCG00810_ATCG00800 (74) ATCG00580_ATCG00570 (68) |
ATCG00480_ATCG00470 (26) AT1G66900_AT1G66910 (22) ATCG00810_ATCG00800 (18) AT3G59330_AT3G59320 (16) |
ATCG00480_ATCG00470 (9) AT1G29920_AT1G29930 (7) AT1G29910_AT1G29920 (7) AT5G38410_AT1G67090 (6) |
| Root (8402) | Flower and floral parts (4176) | Whole plant (11 514) | Other (2910) |
| AT2G07725_AT2G07715 (33) ATCG00480_ATCG00470 (21) AT4G27090_AT5G10100 (19) AT4G18730_AT5G10100 (16) |
AT4G35450_AT4G35460 (7) AT4G25290_AT4G25280 (7) ATCG00810_ATCG00800 (6) ATCG00190_ATCG00180 (6) |
AT1G07590_AT5G10100 (88) ATCG00480_ATCG00470 (59) ATCG00810_ATCG00800 (52) AT3G02080_AT5G10100 (46) |
AT3G59330_AT3G59320 (12) AT4G14960_AT1G50010 (11) AT3G61780_AT5G28400 (10) ATCG00580_ATCG00570 (9) |
Figure 3.
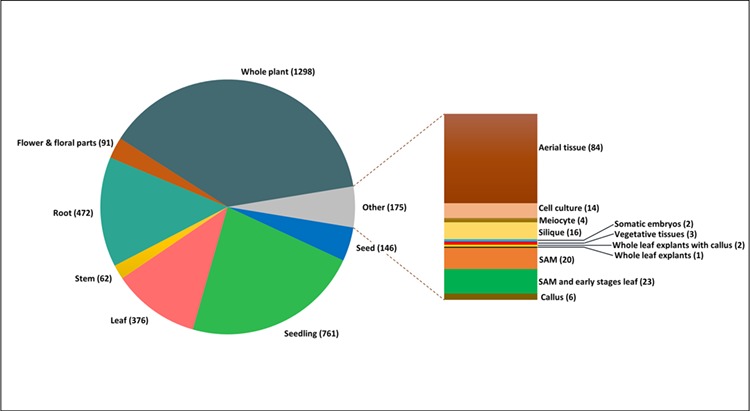
Tissue-wise distribution of total 3520 experimental samples analysed for incorporation into AtFusionDB.
Table 2.
Condition-wise study of frequently occurring fusion transcripts
| Condition | Number of samples | Samples having fusion | Ensembl gene1_gene2 | Fusion type | Tissue type |
|---|---|---|---|---|---|
| Wild | 138 | 6 | AT1G04820_AT4G14960 | Inter-chromosomal | 14-day-old seedling, root, rosette leaf, seedling and silique |
| 6 | AT2G36070_AT2G20510 | Intra-chromosomal | 14-day-old seedling, anther, hypocotyl, seed and seedling | ||
| 6 | AT4G11845_AT4G11840 | Read-through | 14-day-old seedling, root, shoot, cotyledon, whole leaf explants with callus, whole plant and whole seedling | ||
| 6 | ATCG00820_ATCG00810 | Read-through | Callus, cell culture, root, rosette leaf and seedling | ||
| 7 | AT4G30220_AT2G14285 | Inter-chromosomal | Anther and rosette leaf | ||
| 7 | ATCG00480_ATCG00470 | Read-through | 14-day-old seedling, cell culture, hypocotyl, Rosette leaf, seedling, shoot and cotyledon | ||
| Dark-adapted | 5 | 2 | ATCG00350_ATCG00340 | Read-through | Cell culture and seedling |
| 2 | ATCG00570_ATCG00560 | Read-through | Cell culture | ||
| 2 | ATCG00790_ATCG00780 | Read-through | Cell culture | ||
| 2 | ATCG00810_ATCG00800 | Read-through | Cell culture and seedling | ||
| 2 | ATCG00820_ATCG00810 | Read-through | Cell culture | ||
| 2 | ATCG01120_ATCG01110 | Read-through | Cell culture and seedling | ||
| 3 | ATCG00130_ATCG00120 | Read-through | Cell culture and seedling | ||
| Drought stress | 74 | 4 | AT2G05070_AT2G05100 | Read-through | Leaf and rosette leaf |
| 4 | AT3G02065_AT3G02070 | Read-through | Rosette leaf | ||
| 4 | AT3G59330_AT3G59320 | Read-through | Rosette leaf | ||
| 4 | AT3G62310_AT2G47250 | Inter-chromosomal | Rosette leaf | ||
| 4 | AT4G08480_AT4G08470 | Read-through | Rosette leaf | ||
| 5 | AT1G60560_AT1G60590 | Intra-chromosomal | Rosette leaf | ||
| Heat stress | 141 | 12 | ATCG00270_ATCG00280 | Read-through | Aerial seedling, flower bud and seedling |
| 15 | AT1G73320_AT1G73310 | Read-through | Aerial seedling, flower bud and root | ||
| 16 | AT3G09162_AT3G09160 | Read-through | Aerial seedling, rosette leaf and seedling | ||
| 17 | ATCG00810_ATCG00800 | Read-through | Aerial seedling and flower bud | ||
| 18 | AT3G59330_AT3G59320 | Read-through | Aerial seedling, flower bud, Shoot Apical Meristem (SAM) and early stages leaf and seedling | ||
| 28 | ATCG00480_ATCG00470 | Read-through | Aerial seedling, seedling and whole plant | ||
| Oxidative stress | 3 | 2 | AT1G53670_AT1G53680 | Read-through | Root |
| 2 | AT3G25597_AT3G25600 | Read-through | Root | ||
| 2 | AT4G24413_AT4G24410 | Read-through | Root | ||
| 2 | AT5G56720_AT5G56730 | Cis | Root | ||
| 2 | ATCG00340_ATCG00330 | Read-through | Root | ||
| 2 | ATMG00410_AT2G07707 | Inter-chromosomal | Root | ||
| 2 | ATMG00600_AT2G07648 | Inter-chromosomal | Root | ||
| 3 | AT5G38344_AT5G38350 | Read-through | Root | ||
| Nematode Heterodera schachtii infection | 4 | 2 | AT1G23870_AT1G23880 | Read-through | Root |
| 2 | AT1G29470_AT3G61260 | Inter-chromosomal | Root | ||
| 2 | AT5G24520_AT5G24510 | Read-through | Root | ||
| Pseudomonas aeruginosa infection | 16 | 3 | AT1G76690_AT1G76680 | Read-through | Root and shoot |
| 3 | AT2G04100_AT2G04090 | Read-through | Root and shoot | ||
| 3 | AT2G05100_AT2G05070 | Read-through | Shoot | ||
| 3 | AT2G47110_AT3G52590 | Inter-chromosomal | Root and shoot | ||
| Turnip crinkle virus infection | 6 | 2 | AT1G72610_AT1G29920 | Intra-chromosomal | Leaf |
| 2 | AT3G12120_AT2G34420 | Inter-chromosomal | Leaf | ||
| 2 | AT3G26395_AT5G27730 | Inter-chromosomal | Leaf | ||
| 2 | AT3G59330_AT3G59320 | Read-through | Leaf | ||
| 2 | AT4G01590_AT4G35685 | Intra-chromosomal | Leaf | ||
| 2 | AT4G15000_AT1G29910 | Inter-chromosomal | Leaf | ||
| 2 | AT5G19770_AT5G19780 | Cis | Leaf | ||
| 2 | ATCG00350_ATCG00340 | Read-through | Leaf | ||
| 2 | ATCG00480_ATCG00470 | Read-through | Leaf | ||
| 2 | ATCG00810_ATCG00800 | Read-through | Leaf | ||
| Spaceflight grown | 139 | 8 | ATCG00480_ATCG00470 | Read-through | Root |
| 9 | AT2G25210_AT4G31985 | Inter-chromosomal | Root | ||
| 9 | AT4G27090_AT5G10100 | Inter-chromosomal | Root | ||
| 13 | AT2G07725_AT2G07715 | Intra-chromosomal | Root | ||
| Nutrient deficient | 22 | 4 | AT1G26250_AT1G26255 | Cis | Root |
| 4 | AT4G24413_AT4G24410 | Read-through | Root | ||
| 6 | AT2G27010_AT2G27000 | Read-through | Root | ||
| Abscisic acid treated | 12 | 3 | AT2G41200_AT2G41210 | Read-through | Seedling |
| 3 | AT5G22794_AT1G49940 | Inter-chromosomal | Seedling | ||
| 3 | ATCG00810_ATCG00800 | Read-through | Leaf and seedling |
By comparing fusion transcripts from the study done on rice by Zhang and his team in 2010 (16), we found 31 fusion-contributing genes from AtFusionDB homologous to fusion-contributing genes in rice (Supplementary Data Table 1.1). Further, we also found two fusion genes viz. AK101547_AK121590 and AK121590_AK101547 from rice that was homologous to 18 fusion genes in our database (Supplementary Data Table 1.2). Similar comparative studies were also performed in Nicotiana tabacum (tobacco) and it was observed that 35 different genes from tobacco were homologous to 10 fusion contributing genes from AtFusionDB. It was noticed that the genes expressing ribosomal proteins, tubulin and glyceraldehyde-3 phosphate dehydrogenase were common in all three plants considered for the study, thereby indicating their vital roles as fusion transcripts in governing the physiology of plants. The BLAST data results and gene list supporting homology studies along with their respective functions have been provided in the supplementary files (Supplementary Data Tables 1.1, 1.3, 1.4 and 1.5). Thus, our study has confirmed the previous reports of the existence of chimeric transcripts in rice and also indicates the significance of these novel fusion transcripts in other plants as well.
Despite the continuous efforts from researchers for understanding the origin of life, the evolution of genes, genomes and organisms, innumerable questions related to the birth and evolution of genes, which are the structural and functional unit of life, still remains unanswered. However, the rise of Big Data Era has provided burgeons of sequencing data that can be exploited for a better understanding of diverse mechanisms of origin of new genes and the impact of fusion of genes on each and every aspect of growth, development, physiology and adaptive evolution of the parental organisms as well as their progenies. Although a plethora of gene fusion transcripts has been predicted, an in-depth study, validation and functional characterization of the transcripts together with their encoded products are yet to be accomplished. AtFusionDB is the first attempt to gather and store information related to fusion transcripts in plants. This database will make it easy to explore the significance of gene/transcript fusion in plants. It will prove to be beneficial for the biologists in gaining knowledge of this rarely explored domain in the plant kingdom.
Authors contributions
D.D. and A.S. developed the web interface of the database. D.D., A.S., S.Z. and S.K. collected and compiled the data and performed the analysis. S.Z. and S.K. wrote the manuscript. S.K. conceived the idea and coordinated the project.
Supplementary Material
Acknowledgements
The authors are thankful to the Department of Biotechnology (DBT)-eLibrary Consortium, India, for providing access to e-resources. S.Z. and A.S. are thankful to the Council of Scientific and Industrial Research, India, for research fellowships. Authors are thankful to the Distributed Information Sub-Centers of DBT at National Institute of Plant Genome Research.
Database URL: http://www.nipgr.res.in/AtFusionDB
Funding
Core research grant of National Institute of Plant Genome Research, India.
Conflict of interest. None declared.
References
- 1. Epstein C.J. (1971) Evolution by gene duplication. Am. J. Hum. Genet. 23, 541. [Google Scholar]
- 2. Williford A. and Betrán E. (2013) Gene Fusion. In: eLS. John Wiley & Sons, Ltd, Chichester, UK. [Google Scholar]
- 3. Kumar S., Razzaq S.K., Duy Vo A.D. et al. (2016) Identifying fusion transcripts using next generation sequencing advanced review Wiley Interdiscip. Rev. RNA. 7, 811–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kaessmann H. (2010) Origins, evolution, and phenotypic impact of new genes. Genome Res., 20, 1313–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Frenkel-Morgenstern M., Lacroix V., Ezkurdia I. et al. (2012) Chimeras taking shape: potential functions of proteins encoded by chimeric RNA transcripts. Genome Res., 22, 1231–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Qin F., Song Z., Babiceanu M. et al. (2015) Discovery of CTCF-sensitive cis-spliced fusion RNAs between adjacent genes in human prostate cells. PLoS Genet., 11, e1005001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li H., Wang J., Ma X. et al. (2009) Gene fusions and RNA trans-splicing in normal and neoplastic human cells. Cell Cycle, 8, 218–222. [DOI] [PubMed] [Google Scholar]
- 8. Latysheva N.S. and Babu M.M. (2016) Discovering and understanding oncogenic gene fusions through data intensive computational approaches. Nucleic Acids Res., 44, 4487–4503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Di Segni G., Gastaldi S. and Tocchini-Valentini G.P. (2008) Cis- and trans-splicing of mRNAs mediated by tRNA sequences in eukaryotic cells. Proc. Natl. Acad. Sci. USA, 105, 6864–6869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nacu S., Yuan W., Kan Z. et al. (2011) Deep RNA sequencing analysis of readthrough gene fusions in human prostate adenocarcinoma and reference samples. BMC Med. Genomics, 4, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Varley K.E., Gertz J., Roberts B.S. et al. (2014) Recurrent read-through fusion transcripts in breast cancer. Breast Cancer Res. Treat., 146, 287–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang Y., Gong M., Yuan H. et al. (2012) Chimeric transcript generated by cis-splicing of adjacent genes regulates prostate cancer cell proliferation. Cancer Discov., 2, 598–607. [DOI] [PubMed] [Google Scholar]
- 13. Maher C.A., Kumar-Sinha C., Cao X. et al. (2009) Transcriptome sequencing to detect gene fusions in cancer. Nature, 458, 97–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rogers R.L., Bedford T., Lyons A.M. et al. (2010) Adaptive impact of the chimeric gene Quetzalcoatl in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA, 107, 10943–10948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fu B., Chen M., Zou M. et al. (2010) The rapid generation of chimerical genes expanding protein diversity in zebrafish. BMC Genomics, 11, 657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang G., Guo G., Hu X. et al. (2010) Deep RNA sequencing at single base-pair resolution reveals high complexity of the rice transcriptome. Genome Res., 20, 646–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Koller B., Fromm H., Galun E. et al. (1987) Evidence for in vivo trans splicing of pre-mRNAs in tobacco chloroplasts. Cell, 48, 111–119. [DOI] [PubMed] [Google Scholar]
- 18. Kawasaki T., Okumura S., Kishimoto N. et al. (1999) RNA maturation of the rice SPK gene may involve trans-splicing. Plant J., 18, 625–632. [DOI] [PubMed] [Google Scholar]
- 19. Edwards P.A. (2010) Fusion genes and chromosome translocations in the common epithelial cancers. J. Pathol., 220, 244–254. [DOI] [PubMed] [Google Scholar]
- 20. Mertens F., Johansson B., Fioretos T. et al. (2015) The emerging complexity of gene fusions in cancer. Nat. Rev. Cancer, 15, 371–381. [DOI] [PubMed] [Google Scholar]
- 21. Annala M.J., Parker B.C., Zhang W. et al. (2013) Fusion genes and their discovery using high throughput sequencing. Cancer Lett., 340, 192–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nothwang H.G., Kim H.G., Aoki J. et al. (2001) Functional hemizygosity of PAFAH1B3 due to a PAFAH1B3-CLK2 fusion gene in a female with mental retardation, ataxia and atrophy of the brain. Hum. Mol. Genet., 10, 797–806. [DOI] [PubMed] [Google Scholar]
- 23. Shtivelman E., Lifshitz B., Gale R.P. et al. . Fused transcript of abl and bcr genes in chronic myelogenous leukaemia. Nature, 315, 550–554. [DOI] [PubMed] [Google Scholar]
- 24. Wang Y., Wu N., Liu J. et al. (2015) FusionCancer: a database of cancer fusion genes derived from RNA-seq data. Diagn. Pathol., 10, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jia Y., Xie Z. and Li H. (2016) Intergenically spliced chimeric RNAs in cancer. Trends Cancer, 2, 475–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gorohovski A., Tagore S., Palande V. et al. (2017) ChiTaRS-3.1—the enhanced chimeric transcripts and RNA-seq database matched with protein–protein interactions. Nucleic Acids Res., 45, D790–D795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kim N., Kim P., Nam S. et al. (2006) ChimerDB—a knowledgebase for fusion sequences. Nucleic Acids Res., 34, D21–D24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mitelman F., Johansson B. and Mertens F. (2007) The impact of translocations and gene fusions on cancer causation. Nat. Rev. Cancer, 7, 233–245. [DOI] [PubMed] [Google Scholar]
- 29. Panigrahi P., Jere A. and Anamika K. (2018) FusionHub: a unified web platform for annotation and visualization of gene fusion events in human cancer. PLoS One, 13, e0196588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen J.J. (1997) A gene fusion at a homeobox locus: alterations in leaf shape and implications for morphological evolution. Plant Cell, 9, 1289–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang W., Zheng H., Fan C. et al. (2006) High rate of chimeric gene origination by retroposition in plant genomes. Plant Cell, 18, 1791–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen C.J., Liu Q., Zhang Y.C. et al. (2011) Genome-wide discovery and analysis of microRNAs and other small RNAs from rice embryogenic callus. RNA Biol., 8, 538–547. [DOI] [PubMed] [Google Scholar]
- 33. Benelli M., Pescucci C., Marseglia G. et al. (2012) Discovering chimeric transcripts in paired-end RNA-seq data by using EricScript. Bioinformatics, 28, 3232–3239. [DOI] [PubMed] [Google Scholar]
- 34. Bolser D., Staines D.M., Pritchard E. et al. (2016) Ensembl Plants: Integrating Tools for Visualizing, Mining, and Analyzing Plant Genomics Data., Methods in Molecular Biology (Clifton, N.J.), 1374, 115–140. [DOI] [PubMed] [Google Scholar]
- 35. Li H. and Durbin R. (2009) Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li H., Handsaker B., Wysoker A. et al. (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics, 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Quinlan A.R. and Hall I.M. (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics, 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kent W.J. (2002) BLAT—the BLAST-like alignment tool. Genome Res., 12, 656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. McPherson A., Hormozdiari F., Zayed A. et al. (2011) deFuse: an algorithm for gene fusion discovery in tumor RNA-Seq data. PLoS Comput. Biol., 7, e1001138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhu Y., Stephens R.M., Meltzer P.S. et al. (2013) SRAdb: query and use public next-generation sequencing data from within R. BMC Bioinformatics, 14, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhu Y., Davis S., Stephens R. et al. (2008) GEOmetadb: powerful alternative search engine for the Gene Expression Omnibus. Bioinformatics, 24, 2798–2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Altschul S.F., Gish W., Miller W. et al. (1990) Basic local alignment search tool. J. Mol. Biol., 215, 403–410. [DOI] [PubMed] [Google Scholar]
- 43. Smith T.F. and Waterman M.S. (1981) Identification of common molecular subsequences. J. Mol. Biol., 147, 195–197. [DOI] [PubMed] [Google Scholar]
- 44. Chen Y., Ye W., Zhang Y. et al. (2015) High speed BLASTN: an accelerated MegaBLAST search tool. Nucleic Acids Res., 43, 7762–7768. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.