

Abstract
Mitogen-activated protein kinase kinase (MEK) small molecule inhibitors have been investigated in preclinical or clinical trials for the treatment of cancer. In the present study the genetic test results of 120 patients with colorectal cancer (CRC) were screened and the mutation rate of MEK1 was identified to be 1.67%. MEK inhibition by U0126 significantly decreased the growth of SW48 cells that harbored the MEK1 Q56P mutation, although it did not evidently affect the growth of NCI-H508 cells with MEK1 wild-type. In addition, U0126 increased the sensitivity of SW48 cells to 5-fluorouracil (5-FU) and oxaliplatin by producing more γH2AX foci and decreasing the expression of excision repair cross-complementation group 1 and thymidylate synthase. The results suggested that MEK inhibitors in combination with oxaliplatin/5-FU may offer an improved therapeutic effect in patients with MEK-mutant CRC.
Keywords: colorectal cancer, mitogen-activated protein kinase kinase inhibitor, oxaliplatin, 5-fluorouracil
Introduction
Colorectal cancer (CRC) is one of the leading causes of cancer-associated mortality worldwide. Despite advances in treatment methods, patients with CRC have a poor 5-year survival rate (1). Currently, 5-fluorouracil (5-FU) and oxaliplatin serve pivotal roles in treatment regimens for CRC (2). 5-FU increases DNA damage via inhibition of thymidylate synthase (TYMS). Oxaliplatin is a third-generation platinum-containing compound that may induce DNA cross-links, leading to DNA double-strand breaks (DSBs) (3). DSBs are one of the most important factors that threaten the integrity of the genome.
Mitogen-activated protein kinase kinase 1 (MEK1), also known as MAP2K1, is a protein kinase that is a known downstream target of Raf-1 proto-oncogene serine/threonine kinase and is upstream of extracellular signal-regulated kinase (ERK). A variety of small molecule inhibitors of MEK are currently investigated in preclinical or clinical trials for the treatment of malignancies (4). The small-molecule compound U0126 has been identified as a MEK1/2 inhibitor that directly suppresses MEK1/2 activation with well characterized off-target effects (5). Although the pharmacological characteristics of U0126 indicate render it unsuitable for clinical use, it has been demonstrated to be an efficient inhibitor in vitro and in vivo to study the functions of MEK1/2 (6).
Combination therapy is a common approach in cancer chemotherapy. However, the effect of an MEK inhibitor combined with oxaliplatin or 5-FU in MEK-mutant colorectal cells and the underlying mechanism remain unclear. Therefore, the present study aimed to evaluate the combined effect of the MEK inhibitor U0126 with oxaliplatin and 5-FU in CRC cells, and to further explore the underlying mechanisms involved.
Materials and methods
Reagents and cell culture
The human SW48 (cat. no. CCL-231) and NCI-H508 (cat. no. CCL-253) cell lines were obtained from the ATCC (Manassas, VA, USA). SW48 cells were cultured in Leibovitz's L-15 medium (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin and 50 µmol/l β-mercaptoethanol, and were maintained in a tissue-culture incubator (Thermo Fisher Scientific, Inc.) without CO2. NCI-H508 cells were cultured in an environment of 5% CO2 at 37°C in Gibco RPMI-1640 medium (Thermo Fisher Scientific, Inc.) supplemented with 10% FBS.
U0126 was purchased from Selleck Chemicals (cat. no. S1102; Houston, TX, USA). 5-FU and oxaliplatin were obtained from Tongtai Medicine Co., Ltd. (Shandong, China) and Chenxin Medicine Co., Ltd. (Shandong, China), respectively. Antibodies against Akt (cat. no. 4691S; dilution, 1:2,000), phospho-Akt (cat. no. 4060S; dilution, 1:2,000), p44/42 MAPK (cat. no. 4695; dilution, 1:2,000), phospho-p44/42 MAPK (cat. no. 4370; dilution, 1:2,000), H2AX (cat. no. 9718; dilution, 1:1,000) and phospho-H2AX (cat. no. 7631; dilution, 1:1,000) were obtained from Cell Signaling Technology, Inc. (Danvers, MA, USA). β-actin polyclonal antibody (cat. no. AP0060; dilution, 1:2,000) was purchased from Bioworld Technology, Inc. (St. Louis Park, MN, USA), and goat anti-rabbit secondary antibody was obtained from Signalway Antibody LLC (cat. no. L3012; College Park, MD, USA). Excision repair cross-complementation group 1 (ERCC1) rabbit polyclonal antibody (cat. no. 14586-1-AP) and TYMS rabbit polyclonal antibody (cat. no. 15047-1-AP) were purchased from ProteinTech Group, Inc. (Wuhan, China).
Patient samples
A total of 120 CRC patients, including 72 males and 48 females with a mean age of 62.3, were enrolled into the present study. Tumor tissues were obtained in Jiangsu Institute of Cancer Research (Nanjing, Jiangsu, China) between 2016 and 2017. The histological diagnosis of all samples was confirmed by pathologists. TNM classification of malignant tumors was used to determine tumor stage (7). Inclusion criteria were: i) Histologically confirmed diagnosis of CRC, ii) age ≥18 years, iii) availability of tumor tissue for next-generation analyses and iv) no prior therapy for CRC except surgery or radiotherapy, or any adjuvant chemotherapy had ceased for >12 months. Patients with incomplete records, no available tumor tissue or any other malignancy during the last 5 years were also excluded. All detailed information was recorded and summarized in Table I. All patients who participated in the study provided signed informed consent. The research using human tissue received approval from the Ethics Committee of Jiangsu Institute of Cancer Research.
Table I.
Patient characteristics (n=120).
| Variable | N |
|---|---|
| Sex | |
| Male | 72 |
| Female | 48 |
| Age, years | |
| <60 | 45 |
| ≥60 | 75 |
| Histopathological grading | |
| High/moderate | 39 |
| Low | 81 |
| TNM staging | |
| I–II | 46 |
| III–IV | 74 |
| Distant metastasis | |
| Yes | 33 |
| No | 87 |
Mutation detection of cells and tissues by next-generation sequencing
Gene mutations in formalin-fixed and paraffin-embedded tissues of 120 CRC patients were detected using Ion AmpliSeq™ Colon and Lung Cancer Panel (Ion AmpliSeq™ Community 4571815; Thermo Fisher Scientific, Inc.), including mutations in KRAS, EGFR, BRAF, PIK3CA, ALK, NRAS, ERBB2, MET, MEK1, PTEN, SMAD4, STK11, FBXW7, ERBB4, DDR2, CTNNB1, AKT1, NOTCH1, FGFR1, FGFR2 and FGFR3. Next, DNA was extracted with E.Z.N.A.® Tissue DNA kit (Omega Bio-Tek Inc., Norcross, GA, USA), and DNA concentration was determined by Qubit® 2.0 fluorometer dsDNA HS assay kit (Thermo Fisher Scientific, Inc.). A total of 15 ng DNA was then amplified, fragmented, ligated to adapters, barcoded and clonally amplified onto beads to create DNA libraries using Ion PGM™ ampliSeq kit 2.0 and IonXpress barcode adapters kit (Thermo Fisher Scientific, Inc.), according to the manufacturer's protocol. Subsequently, library mixtures were enriched on an Ion OneTouch system with Ion PGM™ Hi-Q OT2 kit (Thermo Fisher Scientific, Inc.). Finally, the library pool was sequenced with Ion PGM™ Hi-Q sequencing kit (Thermo Fisher Scientific, Inc.) using Ion Torrent PGM system. The Ion Torrent variant caller plugin (version 4.0) was used to align reads to the reference genome hg19. The sequencing coverage of the tested genomic regions was >1,000 and the uniformity was >90%.
Cell viability assay
The cells were plated onto 96-well plates at a density of ~5,000 cells per well. After 24 h, different concentrations of oxaliplatin (0.5, 1,5, 10, 20 and 50 µg/ml), 5-FU (0.5, 1,5, 10, 20 and 50 µg/ml) or U0126 (0.1, 0.5, 1,5, 10 and 20 µM) were added to the cells for 72 h. Cell Counting Kit-8 (CCK-8) reagent (Dojindo Molecular Technologies, Inc., Kumamoto, Japan) was then added to each well, and the optical density (OD) value was measured at a wavelength of 450 nm with an absorbance reader (BioTek ELx800; BioTek Instruments, Inc., Winooski, VT, USA). The viability of untreated cells was set to 100%, and the data of treated cells are expressed as a percentage of the control.
The calculation of combination index (CI)
The interactions between drugs were presented in terms of the combination index (CI), which was calculated by dividing the expected growth inhibition rate by the observed growth inhibition rate. A value of CI<1.0 was considered to indicate a synergistic interaction, while CI>1.0 indicated antagonistic drug effects. The CI analysis was performed using CalcuSyn software (version 1.0, Biosoft, Cambridge, UK).
Flow cytometry analysis
SW48 cells were seeded in 6-well plates at a concentration of 1×106 cells per well. Next, the cells were incubated with oxaliplatin, 5-FU or U0126 alone, or with a combination of the drugs. Finally, the cells were stained with a FITC-Annexin V apoptosis detection kit (BD Biosciences, San Jose, CA, USA), and the apoptosis rate was detected with a BD Accuri C6 flow cytometer (BD Biosciences).
Western blot analysis
Subsequent to harvesting, cells were lysed in radioimmunoprecipitation assay buffer (Beyotime Institute of Biotechnology, Jiangsu, China), and then lysates were centrifuged at 12,000 × g for 20 min at 4°C. Protein content was determined by DC Protein Assay kit (Bio-Rad Laboratories, Inc., Hercules, CA, USA) and protein extracts (50 µg) were subjected to electrophoresis on a NuPAGE 10% Bis-Tris gel (Thermo Fisher Scientific, Inc.). Following protein transfer onto polyvinylidene difluoride membranes (EMD Millipore, Billerica, MA, USA), membranes were incubated in 5% bovine serum albumin for 1 h and then incubated overnight at 4°C with the primary antibodies mentioned in the reagents section. Subsequently, the membranes were incubated for 1 h at room temperature with a horseradish peroxidase-conjugated secondary antibody and visualized with an enhanced chemiluminescence solution (EMD Millipore) and a BioRad ChemiDoc™ XRS+ system (Bio-Rad Laboratories, Inc.).
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR) assay
Total RNA was extracted from cells with TRIzol® reagent (Thermo Fisher Scientific, Inc.) and RNA concentration was measured by OD-1000+ (Wuyi Technology Co., Ltd., Nanjing, China). Following RT with Takara PrimeScript™ RT Master Mix kit (cat. no. RR036Q; Takara Bio Inc., Otsu, Japan), the PowerUp™ SYBR® Green Master Mix (cat. no. A25742; Thermo Fisher Scientific, Inc.) and an Applied Biosystems 7300 Real-Time PCR system were applied for qPCR analysis. The cycling conditions comprised 2 min at 50°C, 10 min at 95°C and 40 cycles at 95°C for 15 sec and 60°C for 60 sec. Experiments were conducted in triplicate, and β-actin was used as an internal control. The primer sequences used in this assay were as follows: ERCC1, 5′-GGCGACGTAATTCCCGACTAT-3′ (forward) and 5′-GGATGTAGTCTGGGTGCAGGTT-3′ (reverse); TYMS, 5′-TTTGGAGGAGTTGCTGTGGTT-3′ (forward) and 5′-GATCCATTGGCATCCCAGAT-3′ (reverse); and β-actin, 5′-TTCTACAATGAGCTGCGTGTG-3′ (forward) and 5′-CAGCCTGGATAGCAACGTACA-3′ (reverse). The relative expression of RNA was calculated using the comparative Cq method (8). The experiments were replicated three times.
Immunofluorescence staining
Cells were seeded on coverslips, which were kept in a 24-well plate at a concentration of 1×104 cells for 24 h before treatment, and treated with different drugs for 72 h. Next, the cells were washed and fixed in 4% paraformaldehyde in PBS for 1 h at 37°C. The coverslips were then washed three times with PBS and blocked in immunofluorescence staining blocking buffer (cat. no. P0102; Beyotime Institute of Biotechnology) for 1 h. Subsequently, cells were incubated with a primary antibody against rabbit anti-phospho-H2AX (1:200) at 4°C overnight and washed three times with 0.3% Triton X-100 in PBS. FITC-labeled Goat Anti-Rabbit IgG (cat. no. A0562; JingAn Biological, Jiangsu, China) was used for visualization of phospho-H2AX staining, while the nuclei were stained with 4′,6-diamidino-2-phenylindole. The samples were immediately examined using a fluorescence microscope (Carl Zeiss AG, Oberkochen, Germany).
Statistical analysis
Statistical analysis was performed with IBM SPSS software (version 20.0; IBM Corp., Armonk, NY, USA). Comparisons between pairs were performed using a Student's t-test, while multiple comparisons between the groups were analyzed using one-way analysis of variance followed by a Student-Newman-Keuls test. All the results are presented as the mean ± standard deviation of at least three independent experiments. P<0.05 was considered to indicate a statistically significant difference.
Results
MEK1 gene mutations in CRC cell lines and patients
The colon and lung panel was used to screen gene mutations in SW48 and NCI-H508 cell lines. All gene alterations are listed in Table II. The MEK1 Q56P mutation was identified in SW48 cells. In addition, the presence of this mutation of MEK in patients with CRC was examined in the current study by retrospectively summarizing the genetic test results of 120 patients with CRC. Genomic profiling of these 120 samples revealed two MEK1 mutations in the included CRC patients, including p.D67N and p.Q56P. The total mutation rate of MEK1 was 1.67%.
Table II.
Gene mutations detected by next-generation sequencing.
| Cell line | Gene | Mutation site | Protein position |
|---|---|---|---|
| SW48 | EGFR | c.2155G>A | p.G719S |
| CTNNB1 | c.98C>A | p.S33Y | |
| MEK1 | c.167A>C | p.Q56P | |
| NCI-H508 | BRAF | c.1786G>C | p.G596R |
| PIK3CA | c.1633G>A | p.E545K |
U0126 effectively inhibits the growth of SW48 cells
To verify the role of MEK1 mutation, SW48 and NCI-H508 cells were stimulated with a concentration gradient of U0126 (1, 5, 10 and 20 µM) for 72 h, and the cell viability was measured by a CCK-8 assay. Cell growth profiles demonstrated that inhibition of MEK by U0126 treatment significantly decreased the growth of SW48 cells, whereas U0126 exerted little effect on the growth of NCI-H508 cells (Fig. 1A). Approximately 82.8% of NCI-H508 cells survived with stimulation of 20 µM U0126. Therefore, the SW48 cell line was selected for use in subsequent investigations. Western blot analysis revealed that U0126 exposure decreased the phosphorylation of ERK in a dose-dependent manner, whereas Akt phosphorylation was not evidently affected (Fig. 1B). Furthermore, treatment with various concentrations of oxaliplatin or 5-FU, the most frequently used chemotherapeutic agents in CRC, was found to induce dose-dependent growth inhibition in SW48 cells (Fig. 1C and D).
Figure 1.

Effects of U0126, oxaliplatin and 5-FU on SW48 cells. Cell viability was measured using CCK-8 assay and is represented as the percentages of the untreated group value. (A) CCK-8 was performed following the treatment of SW48 and NCI-H508 cells with increasing concentrations of U0126 for 72 h. There was a statistical difference between the two groups (*P<0.05). (B) After 72 h of U0126 exposure, the cells were lysed and subjected to western blot analysis with relevant antibodies. Cell viability of cells treated with a concentration gradient of (C) oxaliplatin (0.5, 1, 5, 10, 20 and 50 µg/ml) and (D) 5-FU (0.5, 1, 5, 10, 20 and 50 µg/ml) for 3 days was assessed by CCK-8 assay. Values are expressed as the mean ± standard deviation of three individual measurements. *P<0.05 and **P<0.01, vs. the untreated control group. 5-FU, 5-fluorouracil; CCK-8, Cell Counting Kit-8; ERK, extracellular signal-regulated kinase; t-, total; p-, phosphorylated.
Combined effect of MEK1 inhibitor with oxaliplatin and 5-FU
Compared with the control group (100%), the cell viability after stimulation with 2 and 5 µg/ml oxaliplatin decreased to 81.43±0.95 and 70.03±2.61%, respectively. However, the combination of U0126 (2 µM) with 2 or 5 µg/ml oxaliplatin significantly reduced cellular proliferation to 62.07±0.65 and 59.17±1.16%, respectively (Fig. 2A). Similarly, the cytotoxic effect in cells co-treated with U0126 and 5-FU (0.5 and 1 µg/ml) was increased compared with that in cells treated with either U0126 or 5-FU alone (Fig. 2B). Furthermore, the CI values, shown in Table III, were both <1.0 for combined treatment with U0126 and oxaliplatin, and combined treatment with U0126 and 5-FU, indicating synergism between the MEK inhibitor and two drugs.
Figure 2.

U0126 increased oxaliplatin and 5-FU toxicity in SW48 cells. (A) Cytotoxic effect in SW48 cells treated with or without 2 µM U0126 in combination with different concentrations of (A) oxaliplatin or (B) 5-FU for 72 h, as determined by CCK-8 assay. (C) Flow cytometry results, representative of three separate experiments. (D) Apoptotic rates of SW48 cells following exposure to U0126 and/or oxaliplatin/5-FU are presented. The abbreviation U in the figure stands for U0126. Data are expressed as the mean ± standard deviation of three independent experiments, performed in triplicate. *P<0.05 and **P<0.01, vs. control group; ##P<0.01 vs. treatment with oxaliplatin/5-FU alone. 5-FU, 5-fluorouracil; CCK-8, Cell Counting Kit-8.
Table III.
Combination index (CI) values for combination of U0126 with oxaliplatin or 5-FU.a
| Drug 1 | Drug 2 | CI |
|---|---|---|
| U0126 | Oxaliplatin 2 µg/ml | 0.72±0.03 |
| Oxaliplatin 2 µg/ml | 0.89±0.07 | |
| U0126 | 5-FU 0.5 µg/ml | 0.77±0.06 |
| 5-FU 1 µg/ml | 0.86±0.02 |
Data was presented as the mean ± standard deviation.
The results of the apoptosis assay (Fig. 2C and D) were similar to those of the CCK-8 experiment. Compared with the control group (8.70±1.56%), single treatment with U0126, 2 or 5 µg/ml oxaliplatin, and 0.5 or 1 µg/ml 5-FU induced significant cell apoptosis (16.30±0.42, 16.90±1.27, 19.65±0.78, 12.95±0.78 and 20.40±1.41%, respectively). However, the combination of U0126 (2 µM) and oxaliplatin (2 or 5 µg/ml) significantly enhanced cell apoptosis (26.30±0.57 and 32.75±0.92%, respectively). Similarly, treatment with 0.5 or 1 µg/ml 5-FU combined with U0126 markedly increased the cell apoptosis rate (25.43±0.90 and 38.63±0.75%, respectively; Fig. 2C and D).
Combination of U0126 and oxaliplatin or 5-FU triggers the formation of γH2AX foci
To further reveal potential mechanisms underlying the effect of U0126 on oxaliplatin/5-FU therapeutic efficacy in SW48 cells, the present study detected the effect of combination therapy on DNA repair. According to the results of CCK-8 and flow cytometry analysis, the doses of 2 µM U0126 combined with 5 µg/ml oxaliplatin or 1 µg/ml 5-FU were selected for subsequent investigations. Histone H2AX is a variant of the H2A histone family that is involved in chromosomal stability (9). H2AX is phosphorylated at serine 139 when cells are induced by irradiation or cytotoxic drugs, and is a key protein for DNA repair and genomic stability (10). In the current study, γH2AX foci were analyzed by immunofluorescence staining following single or combined treatment with U0126, oxaliplatin or 5-FU. There were significant differences between the cells treated with U0126/oxaliplatin/5-FU alone and control cells, suggesting that all three drugs were able to induce DSBs. Additionally, cells exposed to oxaliplatin or 5-FU combined with U0126 exhibited significantly more γH2AX foci compared with cells treated with monotherapy (Fig. 3A). The number of foci in 100 cells of each sample was calculated, and the mean number of foci per cell is shown in Fig. 3B. Treatment with U0126, oxaliplatin and 5-FU induced comparable amounts of γH2AX foci per cell (11.13±4.65 for U0126, 19.07±6.09 for oxaliplatin and 25.36±8.12 for 5-FU). Furthermore, the induction of γH2AX was accelerated in SW48 cells treated with U0126 and oxaliplatin/5-FU together. The average number of foci following stimulation with U0126 and oxaliplatin was 33.96±6.53, whereas the number of foci was 38.58±10.53 following U0126 and 5-FU treatment (Fig. 3B).
Figure 3.
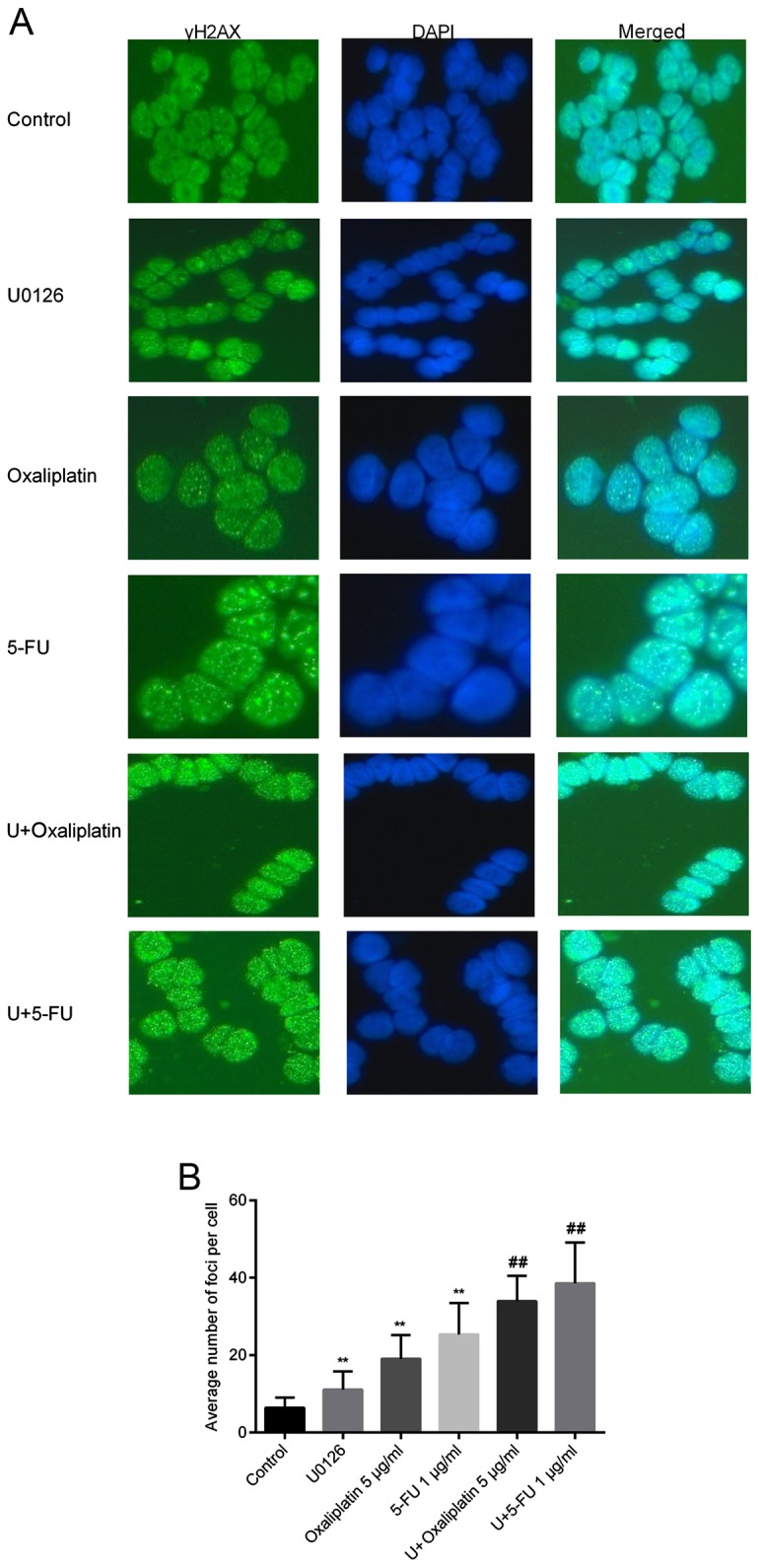
Immunofluorescence staining for γH2AX. (A) γH2AX foci images at a magnification of ×400. Cells were treated with or without 2 µM U0126, combined with oxaliplatin or 5-FU for 72 h. γH2AX expression was detected by immunofluorescence staining and a fluorescence microscope. Cell nuclei were stained with DAPI. (B) Distribution of foci in SW48 cells following exposure to U0126 and/or oxaliplatin/5-FU. **P<0.01 vs. control group; ##P<0.01 vs. oxaliplatin/5-FU-treated group. 5-FU, 5-fluorouracil.
U0126 decreases ERCC1 expression induced by oxaliplatin and enhances the inhibition of TYMS expression when combined with 5-FU
Removal of adducts from genomic DNA is mediated by the enzyme ERCC1, which serves an important role in the rate-limiting step or regulation of nucleotide excision repair (11). Increased expression of ERCC1 caused by platinum in several cancer types has been associated with improvement of DNA repair capacity and resistance to platinum-based drugs (12,13). In the current study, it was observed that oxaliplatin exposure increased ERCC1 expression by 1.62±0.02-fold. By contrast, the combination of U0126 and oxaliplatin significantly reduced ERCC1 mRNA levels to 0.86±0.06-fold (Fig. 4A). U0126 and 5-FU treatment alone inhibited TYMS expression to 0.36±0.07-fold and 0.45±0.02-fold, respectively. Furthermore, the combination of U0126 and 5-FU decreased TYMS expression by 0.21±0.03-fold (Fig. 4B). The results of protein expression were consistent with the mRNA expression (Fig. 4C and D).
Figure 4.

Gene expression induced by U0126 combined with or without oxaliplatin/5-FU. (A) ERCC1 mRNA levels in SW48 cells were examined by RT-qPCR following treatment with U0126 and/or oxaliplatin. (B) TYMS mRNA levels in SW48 cells were examined by RT-qPCR following treatment with U0126 and/or 5-FU. The graph depicts the fold change in ERCC1/TYMS levels normalized to β-actin levels. Data are presented as means ± standard deviation. Western blot analysis results of (C) ERCC1 and (D) TYMS protein expression levels in cells treated with U0126 in combination with oxaliplatin or 5-FU, respectively. **P<0.01 vs. control group; ##P<0.01 vs. oxaliplatin/5-FU-treated group. 5-FU, 5-fluorouracil; ERCC1, excision repair cross-complementation group 1; TYMS, thymidylate synthase; RT-qPCR, reverse transcription-quantitative polymerase chain reaction.
Discussion
Individualized therapy facilitates the selection of the most suitable drug therapy for each patient according to differences in the gene composition or alterations in expression levels. Molecular targeted drugs selectively destroy tumor cells with specific mutated genes, leading to their death, without damaging the cells of the surrounding normal tissue (14). Selection of the appropriate molecular targeted drug subsequent to relevant gene testing can result in accurate and timely individualized treatment for patients.
In 2014, the United States Food and Drug Administration (https://www.fda.gov/) approved the combination of the MEK inhibitor trametinib with the BRAF inhibitor dabrafenib to treat patients with inoperable or metastatic melanoma with BRAF V600E or V600K mutation. MEK1 mutations are present in 1.5% of CRCs and the majority of mutations cause constitutive activation of this kinase (15). A comprehensive study of MEK1 somatic mutations in lung adenocarcinoma revealed that overexpression of MEK1 proteins with mutations in F53L, Q56P and K57N causes phosphorylation of ERK and increased colony formation, which may be inhibited by a MEK1/2 inhibitor (16). Transfection with MEK1 expression vectors illustrated that mutations in this gene, including Q56P and S72G, induced the phosphorylation of ERK1/2 and had a transforming potential, enhancing the tumorigenicity. It was also observed that use of a MEK inhibitor decreased the phosphorylation of ERK1/2 and induced apoptosis of cell lines with MEK1 mutations (17). In the present study, the mutation rate of MEK1 was observed to be 1.67%. MEK inhibition by U0126 significantly decreased the growth of SW48 cells that harbored a MEK1 Q56P mutation, although the effect on the growth of NCI-H508 cells with MEK1 wild-type was not marked. The results, to a certain extent, suggested that CRC patients with such oncogenic MEK1 mutations may be suitable for targeted therapy with MEK inhibitors.
Studies have reported that activation of MEK/ERK signaling is associated with increased resistance to fluoropyrimidines in breast and hepatocellular carcinoma (18,19). In recent years, chemotherapy regimens consisting of 5-FU in combination with oxaliplatin or irinotecan have served as the first-line options for treatment of metastatic CRC. However, 5-FU and oxaliplatin therapy exhibits problems with chemosensitivity (20). A MEK1/2 inhibitor, GSK1120212, exhibited an additive effect in combination with 5-FU or oxaliplatin, and a synergistic effect in combination with SN-38 in HT-29 cells (21). GSK1120212 and other retinoblastoma gene-reactivating agents have been reported to reduce thymidylate synthase expression and enhance the efficacy of 5-FU in BRAF-mutated CRC (22). Furthermore, MEK/ERK pathway inhibition enhanced the sensitivity to 5-FU in KRAS-mutated and murine colorectal tumor xenograft models (23–25). Consistent with the findings of previous studies, the current study demonstrated that the MEK1 inhibitor U0126 increased sensitivity to 5-FU and oxaliplatin treatment in MEK1-mutated CRC. The purpose of the present study was to provide experimental evidence for the combination of a MEK1 inhibitor and chemotherapeutic agents in patients with CRC harboring mutations in MEK1.
γH2AX foci were also detected by immunofluorescence staining to further investigate the mechanism of action of the investigated drugs. γH2AX is a topic of primary research interest in the study of DNA damage stress responses. In 1998, Rogakou et al (26) reported that irradiation and other treatments may cause rapid phosphorylation of Ser residues in H2XA, termed γH2AX. γH2AX foci may be used to estimate the kinetics of DSBs rejoining and has become the gold standard for the detection of DSBs. It has further been revealed that there is no cell specificity for the formation of γH2AX foci, and that induced DSBs are accompanied by H2AX phosphorylation and clustering (27). In the present study, SW48 cells treated with U0126, oxaliplatin or 5-FU alone exhibited an increased number of γH2AX foci compared with the control cells. Combination treatment of U0126 with produced more γH2AX foci as compared with the single drug treatments, which may be associated with the induction of DSBs. Phosphorylation of H2AX indicated the presence of a large number of DSBs and slow repair in SW48 cells exposed to combination therapy.
The present study demonstrated that combination treatment with the MEK inhibitor and oxaliplatin/5-FU increased DNA damage. Therefore, the expression levels of ERCC1 and TYMS, two critical enzymes involved in the nucleotide excision repair pathway, were detected. High expression of ERCC1 is associated with resistance to platinum-based chemotherapy (28). In addition, TYMS is considered an important predictor of 5-FU sensitivity (29) and it is important for maintaining the dTMP pool for DNA synthesis and repair. TYMS inhibitors, including fluorinated pyrimidine derivatives, are capable of inhibiting the activity of TYMS; thus, TYMS expression is associated with in vivo chemosensitivity to such inhibitors. Improved efficacy of 5-FU may be achieved by increasing and prolonging thymidylate synthase inhibition (30). The present study results confirmed that oxaliplatin increased ERCC1 expression, while 5-FU inhibited TYMS expression. Furthermore, U0126 decreased ERCC1 expression induced by oxaliplatin and enhanced the inhibition of TYMS expression when combined with 5-FU, indicating that the inhibition of ERCC1 and TYMS expression levels by U0126 may contribute to increased sensitivity to oxaliplatin/5-FU therapy and severe DNA damage.
However, the present study has certain limitations. Only one cell line was used to study the combination effect of MEK1 inhibitor and chemotherapeutic agents in vitro. Further investigations are required to examine the detailed underlying mechanisms.
In conclusion, a MEK1 inhibitor may be an effective candidate for use with oxaliplatin and 5-FU, particularly for MEK1-mutated cases of CRC. However, the future clinical utility of MEK1 inhibitors in combination with chemotherapeutic agents is limited by a lack of in vivo experimental results. In the future, further investigations should be conducted to explore the synergistic effect of MEK inhibitors with oxaliplatin/5-FU in vivo.
Acknowledgements
Not applicable.
Funding
This study was supported by the Program of the Department of Health in Jiangsu Province (grant no. Z201602) and the Science Foundation of Jiangsu Province (grant no. BE2016795).
Availability of data and material
The materials described in the manuscript, including all relevant raw data, will be freely available to any researcher wishing to use them for non-commercial purposes, without breaching participant confidentiality. The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
CJ and HL wrote the manuscript and designed the study. YD and HC performed the PCR and western blotting experiments. SL, RM and ZW performed immunofluorescence staining and flow cytometry FCM. JW and JF contributed to the design of the study. All authors read and approved the final version of the manuscript.
Ethics approval and consent
All patients who participated in the study provided signed informed consent. The research using human tissue received approval from Jiangsu Institute of Cancer Research Ethics Committee.
Patient consent for publication
All patients participated in the study signed informed consent.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clini. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 2.Meyerhardt JA, Mayer RJ. Systemic therapy for colorectal cancer. N Engl J Med. 2005;352:476–487. doi: 10.1056/NEJMra040958. [DOI] [PubMed] [Google Scholar]
- 3.Chiu SJ, Lee YJ, Hsu TS, Chen WS. Oxaliplatin-induced gamma-H2AX activation via both p53-dependent and -independent pathways but is not associated with cell cycle arrest in human colorectal cancer cells. Chem Biol Interact. 2009;182:173–182. doi: 10.1016/j.cbi.2009.08.019. [DOI] [PubMed] [Google Scholar]
- 4.Fremin C, Meloche S. From basic research to clinical development of MEK1/2 inhibitors for cancer therapy. J Hematol Oncol. 2010;3:8. doi: 10.1186/1756-8722-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, Van Dyk DE, Pitts WJ, Earl RA, Hobbs F, et al. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem. 1998;273:18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- 6.Stepanenko A, Andreieva S, Korets K, Mykytenko D, Huleyuk N, Vassetzky Y, Kavsan V. Step-wise and punctuated genome evolution drive phenotype changes of tumor cells. Mutat Res. 2015;771:56–69. doi: 10.1016/j.mrfmmm.2014.12.006. [DOI] [PubMed] [Google Scholar]
- 7.Rusch VW, Rice TW, Crowley J, Blackstone EH, Rami-Porta R, Goldstraw P. The seventh edition of the American joint committee on cancer/international union against cancer staging manuals: The new era of data-driven revisions. J Thorac Cardiovasc Surg. 2010;139:819–821. doi: 10.1016/j.jtcvs.2010.02.013. [DOI] [PubMed] [Google Scholar]
- 8.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 9.Motoyama N, Naka K. DNA damage tumor suppressor genes and genomic instability. Curr Opin Genet Dev. 2004;14:11–16. doi: 10.1016/j.gde.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 10.Rogakou EP, Nieves-Neira W, Boon C, Pommier Y, Bonner WM. Initiation of DNA fragmentation during apoptosis induces phosphorylation of H2AX histone at serine 139. J BiolChem. 2000;275:9390–9395. doi: 10.1074/jbc.275.13.9390. [DOI] [PubMed] [Google Scholar]
- 11.Niedernhofer LJ, Odijk H, Budzowska M, van Drunen E, Maas A, Theil AF, de Wit J, Jaspers NG, Beverloo HB, Hoeijmakers JH, Kanaar R. The structure-specific endonuclease Ercc1-Xpf is required to resolve DNA interstrand cross-link-induced double-strand breaks. Mol Cell Biol. 2004;24:5776–5787. doi: 10.1128/MCB.24.13.5776-5787.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ronchi CL, Sbiera S, Kraus L, Wortmann S, Johanssen S, Adam P, Willenberg HS, Hahner S, Allolio B, Fassnacht M. Expression of excision repair cross complementing group 1 and prognosis in adrenocortical carcinoma patients treated with platinum-based chemotherapy. Endocr-Relat Cancer. 2009;16:907–918. doi: 10.1677/ERC-08-0224. [DOI] [PubMed] [Google Scholar]
- 13.Lu M, Gao J, Wang XC, Shen L. Expressions of thymidylate synthase, thymidine phosphorylase, class III beta-tubulin, and excision repair cross-complementing group 1predict response in advanced gastric cancer patients receiving capecitabine plus paclitaxel or cisplatin. Chin J Cancer Res. 2011;23:288–294. doi: 10.1007/s11670-011-0288-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grady WM, Pritchard CC. Molecular alterations and biomarkers in colorectal cancer. Toxicol Pathol. 2014;42:124–139. doi: 10.1177/0192623313505155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cancer Genome Atlas Network: Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arcila ME, Drilon A, Sylvester BE, Lovly CM, Borsu L, Reva B, Kris MG, Solit DB, Ladanyi M. MAP2K1 (MEK1) mutations define a distinct subset of lung adenocarcinoma associated with smoking. Clin Cancer Res. 2015;21:1935–1943. doi: 10.1158/1078-0432.CCR-14-2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sogabe S, Togashi Y, Kato H, Kogita A, Mizukami T, Sakamoto Y, Banno E, Terashima M, Hayashi H, de Velasco MA, et al. MEK inhibitor for gastric cancer with MEK1 gene mutations. Mol Cancer Ther. 2014;13:3098–3106. doi: 10.1158/1535-7163.MCT-14-0429. [DOI] [PubMed] [Google Scholar]
- 18.Jin W, Wu L, Liang K, Liu B, Lu Y, Fan Z. Roles of the PI-3K and MEK pathways in Ras-mediated chemoresistance in breast cancer cells. Br J Cancer. 2003;89:185–191. doi: 10.1038/sj.bjc.6601048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yan Y, Li J, Han J, Hou N, Song Y, Dong L. Chlorogenic acid enhances the effects of 5-fluorouracil in human hepatocellular carcinoma cells through the inhibition of extracellular signal-regulated kinases. Anticancer Drugs. 2015;26:540–546. doi: 10.1097/CAD.0000000000000218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu X, Li Z, Yu J, Chan MT, Wu WK. MicroRNAs predict and modulate responses to chemotherapy in colorectal cancer. Cell Prolif. 2015;48:503–510. doi: 10.1111/cpr.12202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamaguchi T, Kakefuda R, Tajima N, Sowa Y, Sakai T. Antitumor activities of JTP-74057 (GSK1120212), a novel MEK1/2 inhibitor, on colorectal cancer cell lines in vitro and in vivo. Int J Oncol. 2011;39:23–31. doi: 10.3892/ijo.2011.1015. [DOI] [PubMed] [Google Scholar]
- 22.Watanabe M, Sowa Y, Yogosawa M, Sakai T. Novel MEK inhibitor trametinib and other retinoblastoma gene (RB)-reactivating agents enhance efficacy of 5-fluorouracil on human colon cancer cells. Cancer Sci. 2013;104:687–693. doi: 10.1111/cas.12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pereira DM, Simões AE, Gomes SE, Castro RE, Carvalho T, Rodrigues CM, Borralho PM. MEK5/ERK5 signaling inhibition increases colon cancer cell sensitivity to 5-fluorouracil through a p53-dependent mechanism. Oncotarget. 2016;7:34322–34340. doi: 10.18632/oncotarget.9107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Urick ME, Chung EJ, Shield WP III, Gerber N, White A, Sowers A, Thetford A, Camphausen K, Mitchell J, Citrin DE. Enhancement of 5-fluorouracil-induced in vitro and in vivo radiosensitization with MEK inhibition. Clin Cancer Res. 2011;17:5038–5047. doi: 10.1158/1078-0432.CCR-11-0358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gong J, Chen Y, Yang L, Pillai R, Shirasawa S, Fakih M. MEK162 enhances antitumor activity of 5-fluorouracil and trifluridine in KRAS-mutated human colorectal cancer cell lines. Anticancer Res. 2017;37:2831–2838. doi: 10.21873/anticanres.11634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 27.MacPhail SH, Banath JP, Yu TY, Chu EH, Lambur H, Olive PL. Expression of phosphorylated histone H2AX in cultured cell lines following exposure to X-rays. Int J Radiat Biol. 2003;79:351–358. doi: 10.1080/0955300032000093128. [DOI] [PubMed] [Google Scholar]
- 28.Li J, Li ZN, Yu LC, Bao QL, Wu JR, Shi SB, Li XQ. Association of expression of MRP1, BCRP, LRP and ERCC1 with outcome of patients with locally advanced non-small cell lung cancer who received neoadjuvant chemotherapy. Lung Cancer. 2010;69:116–122. doi: 10.1016/j.lungcan.2009.09.013. [DOI] [PubMed] [Google Scholar]
- 29.Wakasa K, Kawabata R, Nakao S, Hattori H, Taguchi K, Uchida J, Yamanaka T, Maehara Y, Fukushima M, Oda S. Dynamic modulation of thymidylate synthase gene expression and fluorouracil sensitivity in human colorectal cancer cells. PLoS One. 2015;10:e0123076. doi: 10.1371/journal.pone.0123076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peters GJ, Backus HH, Freemantle S, van Triest B, Codacci-Pisanelli G, van der Wilt CL, Smid K, Lunec J, Calvert AH, Marsh S, et al. Induction of thymidylate synthase as a 5-fluorouracil resistance mechanism. Biochimica Biophysica Acta. 2002;1587:194–205. doi: 10.1016/S0925-4439(02)00082-0. [DOI] [PubMed] [Google Scholar]