


Abstract
Whole-exome and whole-genome sequencing have revealed millions of somatic mutations associated with different human cancers, and the vast majority of them are located outside of coding sequences, making it challenging to directly interpret their functional effects. With the rapid advances in high-throughput sequencing technologies, genome-scale long-range chromatin interactions were detected, and distal target genes of regulatory elements were determined using three-dimensional (3D) chromatin looping. Herein, we present OncoBase (http://www.oncobase.biols.ac.cn/), an integrated database for annotating 81 385 242 somatic mutations in 68 cancer types from more than 120 cancer projects by exploring their roles in distal interactions between target genes and regulatory elements. OncoBase integrates local chromatin signatures, 3D chromatin interactions in different cell types and reconstruction of enhancer-target networks using state-of-the-art algorithms. It employs informative visualization tools to display the integrated local and 3D chromatin signatures and effects of somatic mutations on regulatory elements. Enhancer-promoter interactions estimated from chromatin interactions are integrated into a network diffusion system that quantitatively prioritizes somatic mutations and target genes from a large pool. Thus, OncoBase is a useful resource for the functional annotation of regulatory noncoding regions and systematically benchmarking the regulatory effects of embedded noncoding somatic mutations in human carcinogenesis.
INTRODUCTION
Noncoding variants are capable of causing common diseases and account for the vast majority of heritability (1). To date, a majority of studies have focused exclusively on the effects of missense variants in coding regions (2–4) that comprise <2% of the human genome (5). Mapping variants to the whole genome indicate that disease-associated single nucleotide polymorphisms (SNPs) are strongly enriched in regulatory elements, especially those activated in relevant cell types (6). Moreover, numerous studies have shown that associated variants for a particular trait/disease are significantly enriched in certain regulatory regions of relevant tissues/cell types (7). Importantly, the noncoding regions possess many functional elements based on one dimensional (1D) epigenomic features and three-dimensional (3D) spatial long-range interactions that could help to build accurate enhancer-promoter regulatory pairs; therefore, integrating noncoding variants with 1D coordinated epigenetic profiles and 3D long-range interactions in specific tissue/cell types will provide a promising direction to fine-map causal regulatory variants and understand underlying regulatory mechanisms in human diseases.
Recent discoveries, including the identification of recurrent somatic mutations in the TERT promoter in multiple cancer types (8–11), have supported the idea that somatic mutations in noncoding regions also play vital roles in tumor development (12,13). More than 98% of somatic mutations in most cancers are located in non-coding regions, and some have been identified as putative driver mutations (14). Several databases and computational tools have been developed for annotating noncoding SNPs based on their local genomic 1D features (15–19) and/or their 3D chromatin interactions (20–24), but few tools for annotating noncoding somatic mutations have been designed specifically for human cancers. Moreover, most regulatory elements are widely dispersed across the genome (21), and regulatory somatic mutations are highly outnumbered by neutral passenger mutations due to intratumoral heterogeneity (25,26). Therefore, it is challenging to interpret the effects of noncoding somatic mutations in regulating their target genes in human cancers.
Fortunately, ENCODE (27), Roadmap Epigenomics (28) projects and studies on individual groups (29–31) have revealed the landscape of 1D regulatory elements across the human genome. The rapid development of chromosome conformation capture (3C)-based technologies, such as ChIA-PET (32,33), 5C (34) and Hi-C (35–37), has provided increased datasets on the 3D architecture of the human genome. Studies based on these technologies have uncovered models on how regulatory elements regulate the expression of distal target genes (36,38,39). Regulatory elements, such as enhancers, insulators and protein-binding sites, are anchored to the promoter regions of genes via chromatin looping to orchestrate gene transcription. Chromatin loops identified by Hi-C frequently link enhancers to promoters and are conserved across human cell lines (36) and tissues (38). In addition, enhancer-like elements frequently contact transcriptionally active genes, while potential long-range silencers interact with transcriptionally inactive genes. Furthermore, the interacting loci are enriched for disease-associated variants, suggesting that distal somatic mutations may disrupt the regulation of relevant genes (39). Recent studies have made strong cases for using 3D genome information to interpret noncoding, disease-associated variants (39–43). A system-level understanding of how cancer mutations affect signaling networks is pivotal for interpreting the complex genotype-to-phenotype relationship in terms of tumor behavior and patient outcomes (44). This sophisticated, functional understanding of somatic mutations is key for distinguishing driver mutations from non-pathogenic passengers (26,45). Therefore, it is essential to link noncoding regulatory somatic mutations to target genes by integrating 3D chromatin interactions and 1D chromatin signatures.
A large number of tumor somatic mutations have been identified by TCGA (46), ICGC (47), COSMIC (48) and ClinVar (49,50) but the potential functions of most of these noncoding somatic mutations remain unknown. In this study, we built the platform OncoBase to decipher tumOr NonCoding sOmatic mutations by Base-pAir reSolution Estimation (Figure 1). OncoBase provides comprehensive annotations and predictions of regulatory somatic mutations by employing state-of-the-art methods for target predictions, gene or mutation prioritizations and functional predictions. OncoBase integrates genotype data, phenotype data, 3D chromatin interactions, and important genomic features, including chromatin states, histone modifications, gain/loss of TFBS motifs, and multiple concepts of QTLs (eQTL, dsQTLs, hQTLs and mQTLs), across a broad range of cell types. OncoBase provides a series of informative tables, publishable figures and a network diffusion scoring system to help researchers discover the regulatory roles of noncoding somatic mutations in human cancers based on their 1D and 3D genomic features.
Figure 1.

Workflow of OncoBase construction.
MATERIALS AND METHODS
Somatic mutations and tumor types
To curate as many somatic mutations as possible, we collected somatic mutations from four databases (Table 1), including 1 823 191 somatic mutations in 36 cancer types from TCGA (46), 77 460 941 somatic mutations in 57 cancer types from ICGC (47), 20 909 477 somatic mutations from COSMIC (48), and 345 756 clinical variants from ClinVar (49,50). In total, we collected 81 385 242 somatic mutations in more than 120 types of cancer projects. These somatic variants including noncoding somatic mutations and coding somatic mutations were annotated by ANNOVAR (51).
Table 1.
Summary of data sources of OncoBase
| Type | Source | Cell-types/tissues | Number of regions/sites | Record of mutation | #Unique mutation |
|---|---|---|---|---|---|
| Somatic mutation | ICGC | 57 | - | 77 460 941 | 81 385 242 |
| TCGA | 36 | - | 1 823 191 | ||
| COSMIC | - | - | 20 909 477 | ||
| Mutation | ClinVar | - | - | 345 756 | 345 756 |
| Regulatory elements | ENCODE | 16 | 3 824 829 | 21 640 293 | 57 344 038 |
| RoadMap | 111 | ||||
| Cistrome | 352 | 133 638 513 | 44 139 519 | ||
| GTRD | 1 355 | 38 291 345 | 28 171 788 | ||
| Mutation_Target_Pairs | EpiTensor | 127 | 394 060 | 7 371 607 | 49 110 107 |
| JEME | 127 | 288 882 | 6 793 562 | ||
| GeneHancer | - | 233 757 | 9 065 807 | ||
| 4DGenome | 53 | 1 151 004 | 45 199 484 | ||
| Enhancer/promoter | EnhancerAtlas | 71 | 577 992 | 22 452 362 | 30 145 302 |
| dbSUPER | 99 | 65 213 | 11 435 320 | ||
| SEA | 15 | 2 283 | 548 744 | ||
| UCNEbase | - | 4 315 | 35 389 | ||
| HoneyBadger (enhancer) | 127 | 1 598 323 | 7 023 417 | ||
| HoneyBadger (promoter) | 127 | 56 893 | 1 063 507 | ||
| QTL | GTEx_eQTL | 48 | 341 316 | 86 548 | 3 190 193 |
| PancanQTL | 33 | 1 412 029 | 647 927 | ||
| Other eQTL | 9 | 2 612 515 | 536 714 | ||
| mQTL | 5 | 14 217 993 | 3 135 075 | ||
| dsQTL | 1 | 214 522 | 59 782 | ||
| hQTLs(distal) | 1 | 9 972 | 244 | ||
| hQTLs(local) | 1 | 37 287 | 13 070 | ||
| Motif | motifbreak | - | - | 74 141 414 | 74 141 414 |
| Expression | TCGA | 36 | - | - | - |
| GTEx | 48 | - | - | - | |
| Conservation | UCSC | - | - | - | - |
One-dimensional chromatin features
A variety of local chromatin signatures were used to annotate the regulatory functions of somatic mutations, including predicted chromatin states, histone modifications, DNase I hypersensitivity sites (DHSs) and transcription factor binding sites (TFBSs). Chromatin states were predicted using the core 25-state ChromHMM model (52,53) trained on the imputed data for 12 marks, H3K4me1, H3K4me2, H3K4me3, H3K9ac, H3K27ac, H4K20me1, H3K79me2, H3K36me3, H3K9me3, H3K27me3, H2A.Z and DNase I hypersensitive sites (DHSs), across all 127 reference epigenomes. Regulatory somatic mutations were assigned to corresponding enhancers or promoters with chromatin states by intersecting intervals by coordinate. The binding sites of 153 transcription factors in 91 human cell lines and DHSs in 125 cell lines were obtained from ENCODE and intersected with somatic mutations for annotation. In addition, 1034 epigenetic profiles of 29 main human tissues or cells were implemented to allow for a well-organized visualization by using the JBrowse Genome Browser.
Prediction of the effects of somatic mutations on transcription factor binding
First, 2 817 position weight matrices (PWMs) of transcription factors from the HOCOMOCO (54), FactorBook (55), Homer (56) and ENCODE motif (57) were collected by motifbreakR (58) were used for further prediction of the effects of somatic mutations. In contrast to 3DSNP (21), we employed motifbreakR (58) to measure the effects of somatic mutations on TF binding motifs by using a highly efficient information content-based algorithm to discriminate between truly disruptive versus neutral variants. In contrast to the TFM-Scan software employed by 3DSNP (21), motifbreakR scores and reports the reference and alternate alleles of the sequence and the effect (strong, weak or neutral) according to P-values for PWM match.
Sequence conservation of regulatory somatic mutations
The conservation of somatic mutations was measured by the PhyloP scores obtained from the UCSC Genome Browser (59). The PhyloP scores were calculated from multiple alignments of 100 vertebrate genomes. The absolute values of the PhyloP scores represent -log(P-values) under a null hypothesis of neutral evolution, and sites predicted to be conserved are assigned positive scores, while sites predicted to be fast-evolving are assigned negative scores.
Reconstruction of high-resolution 3D chromatin interactions
Hi-C sequencing datasets were chosen as the main sources for deciphering 3D chromatin interactions in OncoBase because Hi-C measures all pair-wise interaction frequencies across the entire genome, and detection is not dependent on any specific transcription factor. As the linear increase in resolution requires a quadratic increase in the total number of sequencing reads as well as sequencing cost, most available Hi-C datasets have a relatively low resolution, such as 25 or 40 kb. These low-resolution Hi-C datasets can be used to define large-scale genomic patterns, such as A/B compartments or topologically associating domains (TADs), but cannot be used to identify more refined structures, such as enhancer–promoter interactions or sub-domains (60). Therefore, it is urgent to reconstruct chromatin interactions at the gene level with less than a 1 kb resolution. Here, we employed a novel algorithm named EpiTensor (61) to reconstruct chromatin interactions to investigate the regulatory roles of somatic mutations located in interacting loci. EpiTensor can capture spatial associations between distal loci at a 200 bp resolution by using tensor decomposition analysis of TADs and multi-dimensional epigenomes. To obtain higher resolution chromatin interactions of the human genome in different cell lines or tissues, we collected 80 TADs and 127 epigenomes of different cell lines or tissues from the 3DIV database (24) and RoadMap epigenomics project (28), respectively. The spatial and epigenomic datasets were then used to reconstruct chromatin interactions by EpiTensor. Finally, high-resolution interactions were constructed and classified into three types of TSS to enhancer, TSS to TSS, and enhancer to enhancer in 127 cell lines or tissues. Furthermore, high-resolution interactions were marked by 25 chromatin states predicted by ChromHMM to determine whether they are active, inactive or poised. In addition, we also collected 1 981 153 chromatin interacting pairs from the 4DGeneome (62) to expand the annotation of chromatin interactions from 53 tissues or cells by 3C, 4C, 5C, ChIA-PET and IM-PET.
Tissue/cell type-specific enhancers/promoters and super-enhancers
Thanks to the rapid development of high-throughput sequencing technology, genome annotation consortia—e.g. ENCODE (27) and NIH Epigenome Roadmap (28)—have generated massive amounts of different types of sequencing data, making it possible to identify enhancers on a genome-wide scale. The current release of OncoBase enables the availability of a total of 30 145 302 total putative enhancers/promoters related to somatic mutations collected from 5major databases of enhancers/promoters or super-enhancers: 577 992 enhancers in 71 tissues or cell types from EnhancerAtlas (63), 65 213 super-enhancers in 99 tissues or cell types from dbSUPER (64), 2283 super-enhancers in 15tissues or cell types from SEA (65), 4 315 ultra-conserved non-coding elements (UCNEs) that typically function as enhancers in various developmental contexts (66) from UCNEbase (67), and 1 598 323 enhancers and 56 893 promoters from HoneyBadger of Reg2Map project.
Targets of tissue/cell type-specific enhancers and super-enhancers
Although the databases mentioned here have been set up for enhancers in the human genome, they provide only limited, basic information about enhancers, such as their coordinates, cell or tissue types, and nearby genes; therefore, we employed EpiTensor to obtain 25 222 085 high-resolution (∼200 bp) chromatin interactions, including 2 847 794, 5 691 699, and 16 682 592 interactions for promoter to promoter, enhancer to promoter and enhancer to enhancer, respectively. Moreover, we curated predictions of target genes by other two algorithms: 9 879 737 enhancer-target networks in 935 samples by JEME (68) and 284 834 links of enhancers to genes by GeneHancer (69). In total, we deposited 35 386 656 enhancer-target pairs, including 19 472 521 enhancer-promoter pairs, from more than 1000 human samples.
Interactive circular visualization of various biological data
The high-resolution chromatin interactions, clusters of transcription factor binding, somatic mutations, enhancers or super-enhancers and their predicted targets were illustrated in a circular ideogram layout by BioCircos (70), which is a useful tool implemented to circular visualization of various biological data, such as genomic features, genetic variations, gene expression and biomolecular interactions.
Expression of quantitative trait loci
The effects of genetic variants on gene regulation could be interpreted by correlations between genotype and tissue-specific gene expression levels. Expression quantitative trait loci (eQTLs) are genomic loci that regulate gene expression levels and play a crucial role in deciphering gene regulation and spatio-temporal specificity (71). We collected a total of 341 316 significant SNP-gene pairs (FDR ≤ 0.05) in 48 human tissues from the GTEx project version 7 (72). Nominal eQTL P-values and the effect sizes were obtained for each SNP-gene pair to measure the significance of eQTLs. Nominal eQTL P-values were generated using a two-tailed t test to test the alternative hypothesis that the beta deviates from the null hypothesis of β = 0. The effect size of the eQTLs is defined as the slope (‘β’) of the linear regression and is computed as the effect of the alternative allele (ALT) relative to the reference allele (REF) in the human genome. Most importantly, we collected 1 412 029 significant cis-eQTLs- and trans-eQTLs-gene pairs in 33 cancer types from PancanQTL database (73). In addition, Oncobase also included eQTLs from experimentally supported eQTL databases (74,75) and the eQTL browser (http://eqtl.uchicagoedu/cgi-bin/gbrowse/eqtl/) (76) to provide association labels for somatic mutations. Tissue and developmental-stage information were labeled according to the cell type from which eQTL was identified. The statistical test to measure significance is similar to that used for GTEx eQTLs.
Methylation, DNase I sensitivity and histone markers quantitative trait loci
The effects of genetic variants on DNA methylation, DNase I sensitivity and histone modifications could also aid in deciphering the function of regulatory somatic mutations in epigenetic regulation and molecular processes. Genomic loci that affect DNA methylation, DNase I sensitivity and histone modifications are called mQTLs, dsQTLs and hQTLs, respectively. DNA methylation contains significant heritable components that are highly stable across the lifespan and may have a causal role in complex traits (77). We used 14 217 993 mQTL-CpG pairs of human blood at five different life stage from mQTLdb (FDR<0.05) (77) to annotate regulatory somatic mutations. As DNase I sensitivity QTLs are a major determinant of human expression variation, we collected 214 522 dsQTL-peak pairs (FDR<0.1) in lymphoblastoid cell lines (LCLs) to annotate regulatory somatic mutations (78). In addition, we annotated the regulatory somatic mutations by using 47 259 hQTL-peak pairs (FDR<0.1) for three histone markers in LCLs, as hQTLs enable the identification of putative target genes of disease-associated variants from genome-wide association studies (79). The statistical test to measure significance is similar to that used for GTEx eQTLs.
Gene expression in human cancers and normal tissues
The gene expression profiles in human cancers were obtained from the TCGA data portal (https://gdc-portal.nci.nih.gov/) (46), which contains 20 531 genes for each sample. In total, we collected expression data from 13 250 tumor samples in 36 cancer types from TCGA. For each cancer type, weighted gene co-expression network analysis was performed as described below. As the normal control, we collected gene expression data for 53 normal human tissues from the genotype-tissue expression project (GTEx) (72). Both the gene expression of human cancers and normal tissues were displayed in bar plot figures from the searching results.
Weighted gene co-expression network analysis of 36 tumor types
Co-expression analysis is a powerful method for the identification of genes involved in the same molecular processes and regulating relationships. Weighted gene co-expression network analysis (WGCNA) (80) was performed to understand the co-expression relationships between genes for 36 tumor types by using the pipeline we previously employed (81). Genes with a null expression <80% in all samples in each tumor type were selected for WGCNA analysis. A step-by-step network construction workflow was employed with a soft-thresholding power value of 10 for each tumor type. A kME >0.1 was assigned to an eigengene module for co-expression networks in each tumor type. The co-expression networks were present as an interactive networks and ranked by Google PageRank algorithm. The size of the circle of each gene is positively correlated with the PageRank score.
Network diffusion system to prioritize mutations and target genes
Google PageRank, a network-based diffusion algorithm, has emerged as the leading method to rank web content, ecological species and biology scientists (80). PageRank computes the ranking of nodes in graph G based on the structures of incoming connections. It was originally designed as an algorithm to rank web pages. But, here PageRank expresses each gene or mutation as a single item node, and an item containing one or more connections pointing to another item B indicates that item A approves the importance of item B and casts a vote for item B. This relationship can be abstracted as a directed edge in a graph structure. From the point of view of energy passing, each node will distribute its own weight to the nodes to which it points. After several rounds of iterations, diffusion system will complete its convergence and obtain each respective PageRank score. In mathematical terms, the general form of PageRank is expressed as follows:
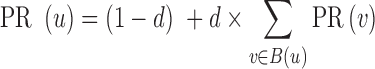 |
PR(u) represents the PageRank score of node u and B(u) represents the set of nodes that point to u. The parameter d is used to solve the situation in which no node points to u. If it is not set, then the converging value will be 0 at the end. Here, we set d = 0.85 for its best practice (82).
Here, the regulatory networks were considered as a graph structure by PageRank. For example, if there are 10 connections in item A, 9-point to item B, and 1 points to item C, then A should assign more weight to B than A. Therefore, a slightly improved version of the PageRank form is as follows:
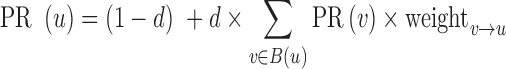 |
Weightv→u is used to measure the weight of the edge. For the edge derived from chromatin interaction determined by EpiTensor, we assigned the weight value as 1 while we used the enhancer–target scores ([0, 1]) as the weight value from the interaction connections predicted by JEME. Finally, we provided an interactive view of periodization of genes related to certain somatic mutation in two layers’ network ranked by score of PageRank in section ‘Regulatory_Network’. PageRank ranking were applied to all genes regulated by certain somatic mutation and all of other somatic mutations regulating these genes. The network view only shows the first layer networks and the somatic mutations or genes related to second layer networks are showed in a table.
DATABASE FEATURES AND APPLICATIONS
Architecture and statistics
The user-friendly web interface OncoBase (http://www.oncobase.biols.ac.cn/ or http://159.226.67.237/sun/oncobase) was developed by combining jQuery with the PHP-based web framework CodeIgniter, supported by versatile browsing and searching functionalities similar to our previous databases and webservers (71,81,83–87). Annotation information was stored in either the MySQL database or flat files. Academic users can access genetic data or extended analysis results freely via the web interface with no requirement permissions. OncoBase stored 81 385 242 somatic mutations and 345 756 clinical mutations collected from database ICGC or TCGA or COSMIC, and ClinVar, respectively. More than 90% mutations were located in intergenic or intronic regions (Figure 2A), and the majority of mutations were located in intergenic and intronic regions of both three dimension (3D) spatial long-range interactions regions and one dimension (1D) epigenomic regulatory regions (Figure 2B, C).
Figure 2.

Distribution of mutations in OncoBase. (A). Total mutations located in eight elements annotated by software ANNOVAR. (B, C). Distribution of mutations located in 3D interaction regions and 1D regulatory regions.
Website interface
The data retrieved in OncoBase can be searched in three formats: ‘gene symbols’, ‘somatic mutations’, ‘dbSNP ID’ and ‘regulatory regions’ (Figure 3). A ‘gene symbols’ search is very useful in terms of searching for gene expression and epigenetic regulation of genes of interest in human cancers and candidate regulatory regions or somatic mutations that are based on genes. ‘Somatic mutation’ retrieval is appropriate for analyzing the results of genetic studies for human cancers and is especially useful for the results of high-throughput studies. ‘Somatic mutation’ retrieval gives support for further functional studies to identify regulatory somatic mutations and sheds light on the underlying molecular mechanisms of human carcinogenesis. In addition, OncoBase allows ‘regulatory region’ retrieval, which could elucidate the potential roles of regulatory regions by providing genomic regions to search somatic mutations that were deposited in the curated database. Furthermore, the JBrowse Genome Browser (http://jbrowse.org) was applied to establish a well-organized ‘JBrowse’ page for visualizing genome-wide signals of epigenetic data sets from 127 Roadmap epigenomes, including signals of fold change compared with input for H2A ChIP-seq, DNase-seq and 30 kinds of histone modifications measured by ChIP-seq. Users can select and browse sequencing signals from any epigenetic type and any cell or tissue across a specific genomic region or mutation of interest. Genetic mutations located in the regulatory region may provide a clue that this mutation may affect the local epigenetic status and result in dysregulation of gene expression.
Figure 3.

Web interface of OncoBase.
Case study
To illustrate the usage of OncoBase, we search the database with a well-studied 3′ UTR’s somatic mutation at the chromosome 1p33 locus, rs977747 (‘chr1:47684677-47684677:T-G’), associated with T-cell acute lymphoblastic leukemia (T-ALL) (88). From the ‘BioCircos_View’ summary section, we can see that somatic mutation rs977747 is located in an endogenous super-enhancer ‘chr1:47675704-47707659’ upstream of the TAL1 oncogene (Figure 4A). In the ‘Mutation_Target_Pair’ information section, we can see five potential target genes of rs977747 based on the targets of enhancers: CMPK1, TAL1, STIL, PDZK1IP1 and CYP4A22 (Figure 4B). The prioritization of the target genes was performed by Google PageRank and showed in ‘Regulatory_Network’ section, which displays the size of genes based on the significance in the regulatory network centered on a functional mutation. According to the annotation of ‘TF_binding’ section, ESR1 binds to this position in invasive ductal breast carcinoma. More interestingly, we find that somatic mutation rs977747 is also an mQTL in the blood of different developmental stages showed in ‘Quantitative_Trait_Locus’ section. It suggests that rs977747 may regulate gene expression through affecting DNA methylation. In addition, somatic mutation rs977747 is also eQTL in blood and cerebellum with the target genes CMPK1 and STIL, respectively. Thus, this case study redisplayed a genetic mechanism responsible for the generation of oncogenic super-enhancers in malignant cells (88) and provided additional insight into the molecular functions of the noncoding somatic mutation rs977747. In addition, we also present a recurrent mutation (chr19:49990694:49990694:G-A) in the promoter of gene RPL13A and RPL13AP5 in melanoma. In according with the article reported, this mutation overlapped with ETS family protein (ELF1, ELK1, ETS1 and GABPA) from ENCODE data in our ‘TF_Binding’ part. Further, the ‘Motif_Break’ prediction results show this mutation can strong affect the binding of that family protein. In addition, ‘Mutation_Target_Pair’ show this mutation may interact with several genes such as RPL13A, ALDH16A1, RCN3 and FLT3LG in long-range distance (Figure 4C, D) (89). These information provide a hypothesis: this G/A mutation may affect TFs binding and result in differential gene expression of its target genes. Users can validate their own hypothesis by molecular and functional experiments with lots of valuable clues provided by our platform.
Figure 4.

Two case studies of mutation in OncoBase. (A). Circos plot of 3D and 1D information related to rs977747 and chr19:49990694:49990694:G:A. (B). Regulatory network of mutation and its target genes produced by Google PageRank method for rs977747. (C) Recurrent mutation in promoter can affect binding of ETS family protein. (D) Regulatory network of mutation and its target genes produced by Google PageRank method for chr19:49990694:49990694:G:A.
DISCUSSION AND PERSPECTIVES
The expansion of functional data sets across a wide range of cell types will improve the functional predictions of noncoding variants for tissue-specific phenotypes (90). 3D chromatin interactions are crucial for deciphering the roles of regulatory elements and embedded variants (21). Recently, several data-driven methods (15,18,21–23,81,91) and sequence-based tools (92–97) have been developed to decode noncoding germline mutations from the GWASdb (65) or dbSNP (98) databases, but none of these databases were designed to investigate functional somatic mutations in noncoding elements for human cancers. And the scientific evidence on noncoding mutations being driver events in cancer remains limited. Compared with previous tools, the principal advantages of OncoBase for the annotation of regulatory somatic mutations are as follows:
Collected all of the somatic mutations identified by TCGA, ICGC and other somatic or clinical mutations deposited in COSMIC and ClinVar. These somatic mutations comprise noncoding variants as well as coding variants with comprehensive annotations by ANNOVAR.
Constructed more than 49 million enhancer–target interactions by multiple predictions from multiple resources.
Incorporated 127 tissue/cell type-specific epigenomes data from the ENCODE and Roadmap epigenomics project.
Integrated the motifs of 2817 transcriptional regulators from four public resources and predicted the effects of mutations on binding motifs.
Uniformly processed Hi-C sequencing data and reconstructed 25 million chromatin interactions at a high resolution across 127 tissues/cell types.
Provided comprehensive functional annotations and predictions of regulatory somatic mutations.
Equipped a highly interactive visualization function for mutation-target interactions.
Included multiple concepts of QTLs, including eQTLs, mQTLs, dsQTLs and hQTLs.
Prioritized regulatory mutations and target genes by network diffusion.
Established weighted gene co-expression networks for 36 tumor types.
It is still a challenge to identify noncoding driver mutations though several studies pointed out dysregulation of enhancer-promoter interaction due to somatic mutations could constitute a general mechanism of carcinogenesis (88,99–101). The widespread implementation of noncoding variant annotation methods will help predict the effects of genomic variation, elucidate the mechanisms and pathways of human cancers, and understand the full complexity of the human genome. With the discovery of clustered regularly interspaced short palindromic repeat (CRISPR) editing, the functions of noncoding variants can now be investigated more easily with these experiment-based systems (102). Currently, owe to the limited number of 1D and 3D sequencing data sets, the 1D epigenomic profiles were mainly collected from healthy cell lines/tissues. Those data sets can easily provide loss of long-range interaction resulted from somatic mutation. Whereas, more long-range interaction data sets in cancer cells are required to identify gain of long-range interaction. In addition, genome sequencing may be changing after many times of passage in the laboratory and long-range interaction may also change. Taken together, more 1D and 3D sequencing data sets are required to provide more accurate regulatory role prediction in long distance. In the future, OncoBase will be frequently updated with new Hi-C datasets and extended to other functional somatic mutations validated by experimental methods, such as CRISPR editing. We are dedicated to maintaining and improving OncoBase since it is a valuable resource for the research community. Finally, incorporation of 3D chromatin interactions will likely improve our ability to assign regulatory somatic mutations to their target genes, thus providing additional improvements to our ability to discern their functions and place them in their biological context, a necessary step for critical pharmacogenetic advancement.
ACKNOWLEDGEMENTS
We thank Dr Chenghang Du in the Beijing Institutes of Life Science, Chinese Academy of Sciences for his help in maintaining the high performance computing systems. It is greatly appreciated for the initial discussion with Dr Wenyu Shi in the Beijing Institutes of Life Science, Chinese Academy of Sciences (CAS). We also thank for OncoBase's logo designer Zehui Chen in Key Laboratory of Animal Ecology and Conservation Biology, CAS.
Authors’ contributions: F.B.M., Q.M.Z. and Z.S.S. conceived and designed the database; F.B.M., Y.W. and X.L.Z. collected the 1D chromatin signatures and profiles from ENCODE and Roadmap projects. X.F.L. and M.K.L. collected somatic mutations from TCGA, ICGC, COSMIC and ClinVar, and Hi-C sequencing data from NCBI database GEO. F.B.M., H.J.T. and Y.W. collected the multiple concepts of QTLs and enhancers from multiple sources. X.F.L and J.N.Z. performed the prediction of the TFBS gain/loss by motifbreakR. F.B.M. and X.L.Z. calculated the gene co-expression by R package WGCNA. X.F.L. and J.N.Z. reconstructed 3D interaction maps with 200 bp resolution by using the EpiTensor algorithm. X.H.S. collected predictions of enhancer–promoter pairs from JEME and GeneHancer. H.N.Y. and L.S.S. constructed the diffusion networks by Google PageRank. X.F.L. and L.S.S. generated the interactive view of chromosome interactions by BioCircos software. L.S.S., X.F.L. and F.B.M. established and maintained the full functional database; X.H.S., Y.W. and S.S.R. tested and debugged the database. F.B.M., Q.M.Z., Y.W. and H.J.T. wrote and revised the manuscript.
FUNDING
National Key R&D Program of China [2016YFC0900400 to Z.S.S.]. Funding for open access charge: National Key R&D Program of China [2016YFC0900400 to Z.S.S.]; Beijing Municipal Commission of Health and Family Planning Project [PXM2018_026279_000005 to Q.M.Z.]; National Natural Science Foundation of China [81490753, 81502150 to Q.M.Z.]; National 973 Program [2015CB553904 to Q.M.Z.]; China Postdoctoral Science Foundation (2018M641117 to X.F.L.).
Conflict of interest statement. None declared.
REFERENCES
- 1. Whalen S., Truty R.M., Pollard K.S.. Enhancer-promoter interactions are encoded by complex genomic signatures on looping chromatin. Nat. Genet. 2016; 48:488–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Li J., Zhao T., Zhang Y., Zhang K., Shi L., Chen Y., Wang X., Sun Z.. Performance evaluation of pathogenicity-computation methods for missense variants. Nucleic Acids Res. 2018; 46:7793–7804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Song Y., Li L., Ou Y., Gao Z., Li E., Li X., Zhang W., Wang J., Xu L., Zhou Y. et al. Identification of genomic alterations in oesophageal squamous cell cancer. Nature. 2014; 509:91–95. [DOI] [PubMed] [Google Scholar]
- 4. Zhang L., Jia Z., Mao F., Shi Y., Bu R.F., Zhang B.. Whole-exome sequencing identifies a somatic missense mutation of NBN in clear cell sarcoma of the salivary gland. Oncol. Rep. 2016; 35:3349–3356. [DOI] [PubMed] [Google Scholar]
- 5. Weinhold N., Jacobsen A., Schultz N., Sander C., Lee W.. Genome-wide analysis of noncoding regulatory mutations in cancer. Nat. Genet. 2014; 46:1160–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ernst J., Kheradpour P., Mikkelsen T.S., Shoresh N., Ward L.D., Epstein C.B., Zhang X.L., Wang L., Issner R., Coyne M. et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011; 473:43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Finucane H.K., Bulik-Sullivan B., Gusev A., Trynka G., Reshef Y., Loh P.R., Anttila V., Xu H., Zang C.Z., Farh K. et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 2015; 47:1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Huang F.W., Hodis E., Xu M.J., Kryukov G.V., Chin L., Garraway L.A.. Highly recurrent TERT promoter mutations in human melanoma. Science. 2013; 339:957–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Horn S., Figl A., Rachakonda P.S., Fischer C., Sucker A., Gast A., Kadel S., Moll I., Nagore E., Hemminki K. et al. TERT promoter mutations in familial and sporadic melanoma. Science. 2013; 339:959–961. [DOI] [PubMed] [Google Scholar]
- 10. Huang D.S., Wang Z., He X.J., Diplas B.H., Yang R., Killela P.J., Meng Q., Ye Z.Y., Wang W., Jiang X.T. et al. Recurrent TERT promoter mutations identified in a large-scale study of multiple tumour types are associated with increased TERT expression and telomerase activation. Eur. J. Cancer. 2015; 51:969–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liang J., Cai W., Feng D., Teng H., Mao F., Jiang Y., Hu S., Li X., Zhang Y., Liu B. et al. Genetic landscape of papillary thyroid carcinoma in the Chinese population. J. Pathol. 2018; 244:215–226. [DOI] [PubMed] [Google Scholar]
- 12. Fredriksson N.J., Ny L., Nilsson J.A., Larsson E.. Systematic analysis of noncoding somatic mutations and gene expression alterations across 14 tumor types. Nat. Genet. 2014; 46:1258–1263. [DOI] [PubMed] [Google Scholar]
- 13. Piraino S.W., Furney S.J.. Beyond the exome: the role of non-coding somatic mutations in cancer. Ann. Oncol. 2016; 27:240–248. [DOI] [PubMed] [Google Scholar]
- 14. Khurana E., Fu Y., Chakravarty D., Demichelis F., Rubin M.A., Gerstein M.. Role of non-coding sequence variants in cancer. Nat. Rev. Genet. 2016; 17:93–108. [DOI] [PubMed] [Google Scholar]
- 15. Boyle A.P., Hong E.L., Hariharan M., Cheng Y., Schaub M.A., Kasowski M., Karczewski K.J., Park J., Hitz B.C., Weng S. et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012; 22:1790–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ward L.D., Kellis M.. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012; 40:D930–D934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ward L.D., Kellis M.. HaploReg v4: systematic mining of putative causal variants, cell types, regulators and target genes for human complex traits and disease. Nucleic Acids Res. 2016; 44:D877–D881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhou L., Zhao F.. Prioritization and functional assessment of noncoding variants associated with complex diseases. Genome Med. 2018; 10:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fu Y., Liu Z., Lou S., Bedford J., Mu X.J., Yip K.Y., Khurana E., Gerstein M.. FunSeq2: a framework for prioritizing noncoding regulatory variants in cancer. Genome Biol. 2014; 15:480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xie X., Ma W., Songyang Z., Luo Z., Huang J., Dai Z., Xiong Y.. CCSI: a database providing chromatin-chromatin spatial interaction information. Database (Oxford). 2016; 2016:bav124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lu Y., Quan C., Chen H., Bo X., Zhang C.. 3DSNP: a database for linking human noncoding SNPs to their three-dimensional interacting genes. Nucleic Acids Res. 2017; 45:D643–D649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li M.J., Wang L.Y., Xia Z., Sham P.C., Wang J.. GWAS3D: Detecting human regulatory variants by integrative analysis of genome-wide associations, chromosome interactions and histone modifications. Nucleic Acids Res. 2013; 41:W150–W158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Huang D., Yi X., Zhang S., Zheng Z., Wang P., Xuan C., Sham P.C., Wang J., Li M.J.. GWAS4D: multidimensional analysis of context-specific regulatory variant for human complex diseases and traits. Nucleic Acids Res. 2018; 46:W114–W120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yang D., Jang I., Choi J., Kim M.S., Lee A.J., Kim H., Eom J., Kim D., Jung I., Lee B.. 3DIV: A 3D-genome Interaction Viewer and database. Nucleic Acids Res. 2018; 46:D52–D57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McGranahan N., Swanton C.. Clonal heterogeneity and tumor evolution: past, present, and the future. Cell. 2017; 168:613–628. [DOI] [PubMed] [Google Scholar]
- 26. Hornshoj H., Nielsen M.M., Sinnott-Armstrong N.A., Switnicki M.P., Juul M., Madsen T., Sallari R., Kellis M., Orntoft T., Hobolth A. et al. Pan-cancer screen for mutations in non-coding elements with conservation and cancer specificity reveals correlations with expression and survival. NPJ Genomic Med. 2018; 3:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Consortium, E.P. The ENCODE (ENCyclopedia Of DNA Elements) Project. Science. 2004; 306:636–640. [DOI] [PubMed] [Google Scholar]
- 28. Bernstein B.E., Stamatoyannopoulos J.A., Costello J.F., Ren B., Milosavljevic A., Meissner A., Kellis M., Marra M.A., Beaudet A.L., Ecker J.R. et al. The NIH roadmap epigenomics mapping consortium. Nat. Biotechnol. 2010; 28:1045–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Heintzman N.D., Stuart R.K., Hon G., Fu Y., Ching C.W., Hawkins R.D., Barrera L.O., Van Calcar S., Qu C., Ching K.A. et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007; 39:311–318. [DOI] [PubMed] [Google Scholar]
- 30. Visel A., Blow M.J., Li Z., Zhang T., Akiyama J.A., Holt A., Plajzer-Frick I., Shoukry M., Wright C., Chen F. et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009; 457:854–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Heintzman N.D., Hon G.C., Hawkins R.D., Kheradpour P., Stark A., Harp L.F., Ye Z., Lee L.K., Stuart R.K., Ching C.W. et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009; 459:108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fullwood M.J., Ruan Y.. ChIP-based methods for the identification of long-range chromatin interactions. J. Cell. Biochem. 2009; 107:30–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fullwood M.J., Wei C.L., Liu E.T., Ruan Y.. Next-generation DNA sequencing of paired-end tags (PET) for transcriptome and genome analyses. Genome Res. 2009; 19:521–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sanyal A., Lajoie B.R., Jain G., Dekker J.. The long-range interaction landscape of gene promoters. Nature. 2012; 489:109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lieberman-Aiden E., van Berkum N.L., Williams L., Imakaev M., Ragoczy T., Telling A., Amit I., Lajoie B.R., Sabo P.J., Dorschner M.O. et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009; 326:289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rao S.S., Huntley M.H., Durand N.C., Stamenova E.K., Bochkov I.D., Robinson J.T., Sanborn A.L., Machol I., Omer A.D., Lander E.S. et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014; 159:1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dixon J.R., Selvaraj S., Yue F., Kim A., Li Y., Shen Y., Hu M., Liu J.S., Ren B.. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012; 485:376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schmitt A.D., Hu M., Jung I., Xu Z., Qiu Y., Tan C.L., Li Y., Lin S., Lin Y., Barr C.L. et al. A Compendium of Chromatin Contact Maps Reveals Spatially Active Regions in the Human Genome. Cell Rep. 2016; 17:2042–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mifsud B., Tavares-Cadete F., Young A.N., Sugar R., Schoenfelder S., Ferreira L., Wingett S.W., Andrews S., Grey W., Ewels P.A. et al. Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nat. Genet. 2015; 47:598–606. [DOI] [PubMed] [Google Scholar]
- 40. Dryden N.H., Broome L.R., Dudbridge F., Johnson N., Orr N., Schoenfelder S., Nagano T., Andrews S., Wingett S., Kozarewa I. et al. Unbiased analysis of potential targets of breast cancer susceptibility loci by Capture Hi-C. Genome Res. 2014; 24:1854–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Martin P., McGovern A., Orozco G., Duffus K., Yarwood A., Schoenfelder S., Cooper N.J., Barton A., Wallace C., Fraser P. et al. Capture Hi-C reveals novel candidate genes and complex long-range interactions with related autoimmune risk loci. Nat. Commun. 2015; 6:10069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Smemo S., Tena J.J., Kim K.H., Gamazon E.R., Sakabe N.J., Gomez-Marin C., Aneas I., Credidio F.L., Sobreira D.R., Wasserman N.F. et al. Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature. 2014; 507:371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Javierre B.M., Burren O.S., Wilder S.P., Kreuzhuber R., Hill S.M., Sewitz S., Cairns J., Wingett S.W., Varnai C., Thiecke M.J. et al. Lineage-Specific genome architecture links enhancers and non-coding disease variants to target gene promoters. Cell. 2016; 167:1369–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sahni N., Yi S., Taipale M., Fuxman Bass J.I., Coulombe-Huntington J., Yang F., Peng J., Weile J., Karras G.I., Wang Y. et al. Widespread macromolecular interaction perturbations in human genetic disorders. Cell. 2015; 161:647–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hofree M., Shen J.P., Carter H., Gross A., Ideker T.. Network-based stratification of tumor mutations. Nat. Methods. 2013; 10:1108–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cancer Genome Atlas Research, N. Weinstein J.N., Collisson E.A., Mills G.B., Shaw K.R., Ozenberger B.A., Ellrott K., Shmulevich I., Sander C., Stuart J.M.. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013; 45:1113–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. International Cancer Genome, C. Hudson T.J., Anderson W., Artez A., Barker A.D., Bell C., Bernabe R.R., Bhan M.K., Calvo F., Eerola I. et al. International network of cancer genome projects. Nature. 2010; 464:993–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Forbes S.A., Bindal N., Bamford S., Cole C., Kok C.Y., Beare D., Jia M., Shepherd R., Leung K., Menzies A. et al. COSMIC: mining complete cancer genomes in the catalogue of somatic mutations in Cancer. Nucleic Acids Res. 2011; 39:D945–D950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Landrum M.J., Lee J.M., Benson M., Brown G., Chao C., Chitipiralla S., Gu B., Hart J., Hoffman D., Hoover J. et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016; 44:D862–D868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Landrum M.J., Lee J.M., Riley G.R., Jang W., Rubinstein W.S., Church D.M., Maglott D.R.. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014; 42:D980–D985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang K., Li M., Hakonarson H.. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010; 38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ernst J., Kellis M.. ChromHMM: automating chromatin-state discovery and characterization. Nat. Methods. 2012; 9:215–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ernst J., Kellis M.. Chromatin-state discovery and genome annotation with ChromHMM. Nat. Protoc. 2017; 12:2478–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kulakovskiy I.V., Medvedeva Y.A., Schaefer U., Kasianov A.S., Vorontsov I.E., Bajic V.B., Makeev V.J.. HOCOMOCO: a comprehensive collection of human transcription factor binding sites models. Nucleic Acids Res. 2013; 41:D195–D202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wang J., Zhuang J., Iyer S., Lin X.Y., Greven M.C., Kim B.H., Moore J., Pierce B.G., Dong X., Virgil D. et al. Factorbook.org: a Wiki-based database for transcription factor-binding data generated by the ENCODE consortium. Nucleic Acids Res. 2013; 41:D171–D176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K.. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010; 38:576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kheradpour P., Kellis M.. Systematic discovery and characterization of regulatory motifs in ENCODE TF binding experiments. Nucleic Acids Res. 2014; 42:2976–2987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Coetzee S.G., Coetzee G.A., Hazelett D.J.. motifbreakR: an R/Bioconductor package for predicting variant effects at transcription factor binding sites. Bioinformatics. 2015; 31:3847–3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rhead B., Karolchik D., Kuhn R.M., Hinrichs A.S., Zweig A.S., Fujita P.A., Diekhans M., Smith K.E., Rosenbloom K.R., Raney B.J. et al. The UCSC Genome Browser database: update 2010. Nucleic Acids Res. 2010; 38:D613–D619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zhang Y., An L., Xu J., Zhang B., Zheng W.J., Hu M., Tang J., Yue F.. Enhancing Hi-C data resolution with deep convolutional neural network HiCPlus. Nat. Commun. 2018; 9:750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zhu Y., Chen Z., Zhang K., Wang M., Medovoy D., Whitaker J.W., Ding B., Li N., Zheng L., Wang W.. Constructing 3D interaction maps from 1D epigenomes. Nat. Commun. 2016; 7:10812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Teng L., He B., Wang J., Tan K.. 4DGenome: a comprehensive database of chromatin interactions. Bioinformatics. 2016; 32:2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gao T., He B., Liu S., Zhu H., Tan K., Qian J.. EnhancerAtlas: a resource for enhancer annotation and analysis in 105 human cell/tissue types. Bioinformatics. 2016; 32:3543–3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Khan A., Zhang X.. dbSUPER: a database of super-enhancers in mouse and human genome. Nucleic Acids Res. 2016; 44:D164–D171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wei Y., Zhang S., Shang S., Zhang B., Li S., Wang X., Wang F., Su J., Wu Q., Liu H. et al. SEA: a super-enhancer archive. Nucleic Acids Res. 2016; 44:D172–D179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Polychronopoulos D., King J.W.D., Nash A.J., Tan G., Lenhard B.. Conserved non-coding elements: developmental gene regulation meets genome organization. Nucleic Acids Res. 2017; 45:12611–12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Dimitrieva S., Bucher P.. UCNEbase–a database of ultraconserved non-coding elements and genomic regulatory blocks. Nucleic Acids Res. 2013; 41:D101–D109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Cao Q., Anyansi C., Hu X., Xu L., Xiong L., Tang W., Mok M.T.S., Cheng C., Fan X., Gerstein M. et al. Reconstruction of enhancer-target networks in 935 samples of human primary cells, tissues and cell lines. Nat. Genet. 2017; 49:1428–1436. [DOI] [PubMed] [Google Scholar]
- 69. Fishilevich S., Nudel R., Rappaport N., Hadar R., Plaschkes I., Iny Stein T., Rosen N., Kohn A., Twik M., Safran M. et al. GeneHancer: genome-wide integration of enhancers and target genes in GeneCards. Database (Oxford). 2017; 2017:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Cui Y., Chen X., Luo H., Fan Z., Luo J., He S., Yue H., Zhang P., Chen R.. BioCircos.js: an interactive Circos JavaScript library for biological data visualization on web applications. Bioinformatics. 2016; 32:1740–1742. [DOI] [PubMed] [Google Scholar]
- 71. Mao F., Xiao L., Li X., Liang J., Teng H., Cai W., Sun Z.S.. RBP-Var: a database of functional variants involved in regulation mediated by RNA-binding proteins. Nucleic Acids Res. 2016; 44:D154–D163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Genetic effects on gene expression across human tissues. Nature. 2017; 550:204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Gong J., Mei S., Liu C., Xiang Y., Ye Y., Zhang Z., Feng J., Liu R., Diao L., Guo A.Y. et al. PancanQTL: systematic identification of cis-eQTLs and trans-eQTLs in 33 cancer types. Nucleic Acids Res. 2018; 46:D971–D976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Xia K., Shabalin A.A., Huang S., Madar V., Zhou Y.H., Wang W., Zou F., Sun W., Sullivan P.F., Wright F.A.. seeQTL: a searchable database for human eQTLs. Bioinformatics. 2012; 28:451–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Gamazon E.R., Zhang W., Konkashbaev A., Duan S., Kistner E.O., Nicolae D.L., Dolan M.E., Cox N.J.. SCAN: SNP and copy number annotation. Bioinformatics. 2010; 26:259–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Pickrell J.K., Marioni J.C., Pai A.A., Degner J.F., Engelhardt B.E., Nkadori E., Veyrieras J.B., Stephens M., Gilad Y., Pritchard J.K.. Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature. 2010; 464:768–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Gaunt T.R., Shihab H.A., Hemani G., Min J.L., Woodward G., Lyttleton O., Zheng J., Duggirala A., McArdle W.L., Ho K. et al. Systematic identification of genetic influences on methylation across the human life course. Genome Biol. 2016; 17:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Degner J.F., Pai A.A., Pique-Regi R., Veyrieras J.B., Gaffney D.J., Pickrell J.K., De Leon S., Michelini K., Lewellen N., Crawford G.E. et al. DNase I sensitivity QTLs are a major determinant of human expression variation. Nature. 2012; 482:390–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Grubert F., Zaugg J.B., Kasowski M., Ursu O., Spacek D.V., Martin A.R., Greenside P., Srivas R., Phanstiel D.H., Pekowska A. et al. Genetic Control of Chromatin States in Humans Involves Local and Distal Chromosomal Interactions. Cell. 2015; 162:1051–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ghoshal G., Barabasi A.L.. Ranking stability and super-stable nodes in complex networks. Nat. Commun. 2011; 2:394. [DOI] [PubMed] [Google Scholar]
- 81. Mao F., Liu Q., Zhao X., Yang H., Guo S., Xiao L., Li X., Teng H., Sun Z., Dou Y.. EpiDenovo: a platform for linking regulatory de novo mutations to developmental epigenetics and diseases. Nucleic Acids Res. 2018; 46:D92–D99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Paolo Boldi M.S., Sebastiano Vigna. PageRank as a function of the damping factor. Proceedings of the 14th International Conference on World Wide Web. 2005; 557–566. [Google Scholar]
- 83. Ran X., Li J., Shao Q., Chen H., Lin Z., Sun Z.S., Wu J.. EpilepsyGene: a genetic resource for genes and mutations related to epilepsy. Nucleic Acids Res. 2015; 43:D893–D899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Li J., Jiang Y., Wang T., Chen H., Xie Q., Shao Q., Ran X., Xia K., Sun Z.S., Wu J.. mirTrios: an integrated pipeline for detection of de novo and rare inherited mutations from trios-based next-generation sequencing. J. Med. Genet. 2015; 52:275–281. [DOI] [PubMed] [Google Scholar]
- 85. Li J., Cai T., Jiang Y., Chen H., He X., Chen C., Li X., Shao Q., Ran X., Li Z. et al. Genes with de novo mutations are shared by four neuropsychiatric disorders discovered from NPdenovo database. Mol. Psychiatry. 2016; 21:298. [DOI] [PubMed] [Google Scholar]
- 86. Li X., Shi L., Zhang K., Wei W., Liu Q., Mao F., Li J., Cai W., Chen H., Teng H. et al. CirGRDB: a database for the genome-wide deciphering circadian genes and regulators. Nucleic Acids Res. 2018; 46:D64–D70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Li J., Shi L., Zhang K., Zhang Y., Hu S., Zhao T., Teng H., Li X., Jiang Y., Ji L. et al. VarCards: an integrated genetic and clinical database for coding variants in the human genome. Nucleic Acids Res. 2018; 46:D1039–D1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Mansour M.R., Abraham B.J., Anders L., Berezovskaya A., Gutierrez A., Durbin A.D., Etchin J., Lawton L., Sallan S.E., Silverman L.B. et al. Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science. 2014; 346:1373–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Fredriksson N.J., Elliott K., Filges S., Van den Eynden J., Stahlberg A., Larsson E.. Recurrent promoter mutations in melanoma are defined by an extended context-specific mutational signature. PLos Genet. 2017; 13:e1006773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Nishizaki S.S., Boyle A.P.. Mining the unknown: assigning function to noncoding single nucleotide polymorphisms. Trends Genet. 2017; 33:34–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Ritchie G.R., Dunham I., Zeggini E., Flicek P.. Functional annotation of noncoding sequence variants. Nat. Methods. 2014; 11:294–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Zhou J., Troyanskaya O.G.. Predicting effects of noncoding variants with deep learning-based sequence model. Nat. Methods. 2015; 12:931–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Lee D., Gorkin D.U., Baker M., Strober B.J., Asoni A.L., McCallion A.S., Beer M.A.. A method to predict the impact of regulatory variants from DNA sequence. Nat. Genet. 2015; 47:955–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Alipanahi B., Delong A., Weirauch M.T., Frey B.J.. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat. Biotechnol. 2015; 33:831–838. [DOI] [PubMed] [Google Scholar]
- 95. Whitaker J.W., Chen Z., Wang W.. Predicting the human epigenome from DNA motifs. Nat. Methods. 2015; 12:265–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Huang Y.F., Gulko B., Siepel A.. Fast, scalable prediction of deleterious noncoding variants from functional and population genomic data. Nat. Genet. 2017; 49:618–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Zhou J., Theesfeld C.L., Yao K., Chen K.M., Wong A.K., Troyanskaya O.G.. Deep learning sequence-based ab initio prediction of variant effects on expression and disease risk. Nat. Genet. 2018; 50:1171–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Sherry S.T., Ward M.H., Kholodov M., Baker J., Phan L., Smigielski E.M., Sirotkin K.. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001; 29:308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Zhang W., Bojorquez-Gomez A., Velez D.O., Xu G., Sanchez K.S., Shen J.P., Chen K., Licon K., Melton C., Olson K.M. et al. A global transcriptional network connecting noncoding mutations to changes in tumor gene expression. Nat. Genet. 2018; 50:613–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Gao P., Xia J.H., Sipeky C., Dong X.M., Zhang Q., Yang Y., Zhang P., Cruz S.P., Zhang K., Zhu J. et al. Biology and clinical implications of the 19q13 aggressive prostate cancer susceptibility locus. Cell. 2018; 174:576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Zhang X., Choi P.S., Francis J.M., Gao G.F., Campbell J.D., Ramachandran A., Mitsuishi Y., Ha G., Shih J., Vazquez F. et al. Somatic superenhancer duplications and hotspot mutations lead to oncogenic activation of the KLF5 transcription factor. Cancer Discov. 2018; 8:108–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Sanjana N.E., Wright J., Zheng K., Shalem O., Fontanillas P., Joung J., Cheng C., Regev A., Zhang F.. High-resolution interrogation of functional elements in the noncoding genome. Science. 2016; 353:1545–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]