


Abstract
Meta-omics approaches have been increasingly used to study the structure and function of the microbial communities. A variety of large-scale collaborative projects are being conducted to encompass samples from diverse environments and habitats. This change has resulted in enormous demands for long-term data maintenance and capacity for data analysis. The Global Catalogue of Metagenomics (gcMeta) is a part of the ‘Chinese Academy of Sciences Initiative of Microbiome (CAS-CMI)’, which focuses on studying the human and environmental microbiome, establishing depositories of samples, strains and data, as well as promoting international collaboration. To accommodate and rationally organize massive datasets derived from several thousands of human and environmental microbiome samples, gcMeta features a database management system for archiving and publishing data in a standardized way. Another main feature is the integration of more than ninety web-based data analysis tools and workflows through a Docker platform which enables data analysis by using various operating systems. This platform has been rapidly expanding, and now hosts data from the CAS-CMI and a number of other ongoing research projects. In conclusion, this platform presents a powerful and user-friendly service to support worldwide collaborative efforts in the field of meta-omics research. This platform is freely accessible at https://gcmeta.wdcm.org/.
INTRODUCTION
‘Meta-omics’ (e.g. metataxonomics, metagenomics and metatranscriptomics) approaches have been increasingly used to study the structure, function and intercellular interactions of the microbial communities and the fundamental mechanisms of microbial life and evolution. Dramatic progress in the next generation sequencing technology has made large-scale sampling and sequencing possible, even for individual laboratory. Meta-omics has also promoted collaborative efforts in a grand vision across the international research community, as exemplified by the Earth Microbiome Project (EMP) (1) and Human Microbiome Project (HMP) (2). These collaborative projects have produced large volume of data and hence generated meaningful interpretations from a full spectrum of sources which are impossible with a single independent study. Along with these changes, microbiome research is becoming a data driven science (3) and rapid advances in this area have brought about significant challenges. Firstly, comparing data from independent research groups becomes difficult if standard operating procedures (SOPs) and reporting standards are not followed. Adhering to universal standards in every step of the study, including sampling, sequencing, data submission, data analysis, and data publication, is necessary to understand the results of a single study within a broader context (4). Recently, significant efforts have been made for the development of universal protocols and standards (5). Although, well-organized and reputable collaborative projects often have built-in standards (http://www.microbiome-standards.org/) and SOPs (6), sometimes it becomes difficult for different projects to implement identical standards. For example, human and environmental microbiota data from different projects are not readily comparable due to inconsistencies in standards, protocols as well as workflows (7). Further developments, and more importantly, adoption of these SOPs and standards by all projects and labs worldwide is crucial for the scientific community. The second challenge is long-term preservation and open access of the data. Integration of all relevant publicly available data is a prerequisite for future cross-studies. A stable and robust data infrastructure is needed that would provide a reliable data archive and rational data organization, thus ensuring data reproducibility and allowing data reinterpretation. The third challenge is to analyze Gigabyte (GB) to Terabyte (TB) scale data on a single computer. Despite the availability of a variety of stand-alone tools, it is almost impossible for any given individual lab to have sufficient infrastructure for data storage and to maintain multi-computer network clusters resources (8). Currently, there are several public resources, including the European Bioinformatics Institute (EBI) metagenomics (9), the Metagenomics RAST server (MG-RAST) (10) and the JGI IMG Integrated Microbial Genomes & Microbiomes (IMG/M) (11). However, considering the rapid increase in data volume and growing demands for data analysis capacity, more public services with the ability to provide data archiving and cloud-based data analysis are required (12).
Because of the wide geographical coverage, rich ecosystem, as well as diverse ethnicity and lifestyles of the people, China harbors enormous diversity of microbial communities. In comparison to developed countries, however, the microbial communities in China are less comprehensively and systematically studied. China also lacks nationwide collaborative projects. The Chinese Academy of Sciences Initiative of Microbiome (CAS-CMI) is one of the leading projects organized at the national level to find solutions to the current challenges in human and environmental health, agriculture and industrial developments. At the same time, it will establish the biobanks (samples, strains and data) of Chinese Microbiome Initiative, and support long term preservation and reuse of data worldwide in a free and open way.
The Global Catalogue of Metagenomics (gcMeta) platform is a part of the CAS-CMI. As a partner database of the World Data Center for Microorganisms (WDCM) (13) as well, gcMeta has two features: firstly, designing and implementing as a standardized and state-of-art database management system to support long-term preservation and integration data from the CAS-CMI project as well as from microbiome research projects worldwide. Secondly, the platform provides web-based tools and pre-defined workflows, along with computing resources for massive data analysis requirements globally.
PLATFORM DESIGN AND IMPLEMENTATION
How to use the platform
The platform supplies management, analysis and publication services for microbiome related data, including genomes, marker genes, metagenomes, metatranscriptomes and their associated metadata (Figure 1). The users can upload the raw data and their metadata into the system via a web submission interface or a data upload web application. After data quality check, the data can be browsed in the system under the user's account. Currently, we provide web-based analysis workflows for marker genes and whole-genome shotgun sequencing (WGS) data. The users can use these workflows or individual tools for data analysis and visualization. The Global Unique Persistent Identifier (GUID) system is used for the open data. To publish the data, users should change the status of their data from ‘private’ to ‘public’, then, the system assigns a persistent identifier (PID) (http://www.pidconsortium.eu/) to the each of the records. The PID is used to cite the data elsewhere and provide a report to the users.
Figure 1.

General pipeline of the gcMeta platform. The functional services of gcMeta can be described in three parts: data management, data analysis and data publication. Users submit the meta-data and primary raw data into the system under their own accounts. Users are allowed to analyze the data by preinstalled tools and workflows. Data and results could be downloaded for further analysis. A unique identifier PID would be assigned to each record before the data is public. If the data is further cited in other resources with the PID, the citation could be traced automatically.
For data protection, login is required before data submission and exploring the full functions. We provide a temporary guest account effective for 24 h along with any submissions, uploaded files and analysis results. All the public available primary raw data or metadata could be downloaded. Access to gcMeta is free at https://gcmeta.wdcm.org/.
Database design
The database hosts information on samples and their associate metadata, and primary ‘raw’ data. A relational database is used to host all relevant data. Schema of the database is shown in Figure 2. The major data record types are ‘Study’, ‘Sample’, ‘Experiment’ and ‘Sequence’. ‘Study’ could include several ‘sub-studies’ and is related to ‘Sample’ by the ‘Study ID’. The samples and their associated metadata are recorded. ‘Sample’ is referenced to ‘Experiment’. ‘Experiment’ is further referenced to sequence information. The sequence information contains the sequencing methods and strategies, as well as the processing of the sequencing results, including data quality control and assembly. The gcMeta platform is implemented by an open source database system PostgreSQL.
Figure 2.

Database schema of gcMeta. Main data structure and relationships between the different tables are illustrated.
Ontologies and data standards are crucial to ensure reusability and interoperability of data. To ensure data comparability and consistency between CAS-CMI and public data resources, gcMeta adopts the Minimum Information about Metagenomic Sequence (MIMS) (14) and Minimum Information about a MARKer gene Sequence (MIMARKS) (15). It also uses the Minimum Information of any(x) Sequence (MIxS), which describes 15 different environmental packages to specify the environmental context of a microbial sample. The Environment Ontology (ENVO) (16) for the three environmental metadata fields including environmental biome, feature and material is used to describe the sampling in the system, using a total of 95 controlled terms.
Data sources
As of August 2018, gcMeta has archived a total of 3053 studies and 124 052 samples, hosting more than 120 TB of sequencing data. We have two major data sources. The first is publicly available data (e.g. MG-RAST, EBI metagenomics and HMP). Publicly available data are integrated based on isolation sources, environmental features and experiment types, and thus allowing data comparison across different data resources. Efforts were made to overcome varying data quality level, format and (lack of) metadata description. Expression was unified according to environmental ontologies. Secondly, gcMeta serves as a data catalogue for the CAS-CMI project and some other ongoing projects in China. This platform has been rapidly expanding, and now hosts CAS-CMI project sample data from waste water, human gut, characteristic Chinese fermented food and so on. The number of samples from these projects is more than 2000 up to now.
Data management services
The system allows two routes of data submission. Users can establish a record of their studies and associated samples and experiments online one by one through a web form as indicated in Figure 3. For raw sequence data, users can upload data to the system using a web application. To submit high volume of data in a single session, users can choose a simple tab-delimited file format such as Microsoft Excel. Implementation of data standards occurs during database design as well as prior to the generation of the data in the sampling stage. We also use data validation tools during the on-batch data submission. The system is able to accept data submission from all over the world and offers standardized quality control for the submitted data.
Figure 3.

Screenshots and examples of user cases in gcMeta. (A) Homepage of the gcMeta. Statistic number of public and private studies, samples, experiments and runs are showed in the homepage. (B) A screenshot of data submission by web table. Each entry could be set ‘private’ or ‘public’ as highlighted in the red box. (C) A screenshot of database browser. In the search interface, search results could be filtered by ‘experiment type’, ‘sample environment’ and ‘data sources’.
The platform under the CAS-CMI project coordinates with other research institutes, universities, and hospital across China for data repository. Since the ongoing projects are one of the forms of data sources, some data are currently not available to the general public at this stage; data can be accessed via project members only, but it will eventually be publicly available. Data submitters can upload and share their pre-publication data with their research collaborators. Only controlled-access is available for pre-publication data. When required by journals, the data status could be seamlessly switched from ‘private’ to ‘public’ in the system. In this way, we encourage data sharing while protecting data privacy and security. We limit the number of mandatory fields to keep a balance between the burden on contributing scientists and reusability of the data for future analysis. The data could be searched and browsed online after it is submitted.
After the data is set public, PID will be assigned to each ‘Study’ ‘Sample’ ‘Experiment’ and ‘Sequence’ record. The PID is a Global Unique Persistent Identifier system that provides long-term identifying service similar to Digital Object Identifier (DOI) (http://www.doi.org/). An example of PID in gcMeta for ‘Study’ is 21.86101/gcm.study.88c292e8f67c11e7b172b49691092464, where ‘21’ is the handle prefix for PID. ‘gcm.study’ is identifier for ‘Study’ records in the gcMeta database and ‘88c292e8f67c11e7b172b49691092464’ is a randomly assigned series code for the record. The PID can be used to search the Handle (http://hdl.handle.net) site.
Data analysis and visualization workflows
Metagenomics data are often referred as ‘marker gene amplification metagenomics’ and ‘full shotgun metagenomics’. Depending on the data types, general workflows for data analysis include two different categories. A wide array of tools are currently available to carry out each step of the workflows (17). In gcMeta, we supply analysis tools and workflows which are installed based on Docker technologies, and thus allow users with limited computational resources to perform analysis of metagenomic samples.
Dockerized analysis tools
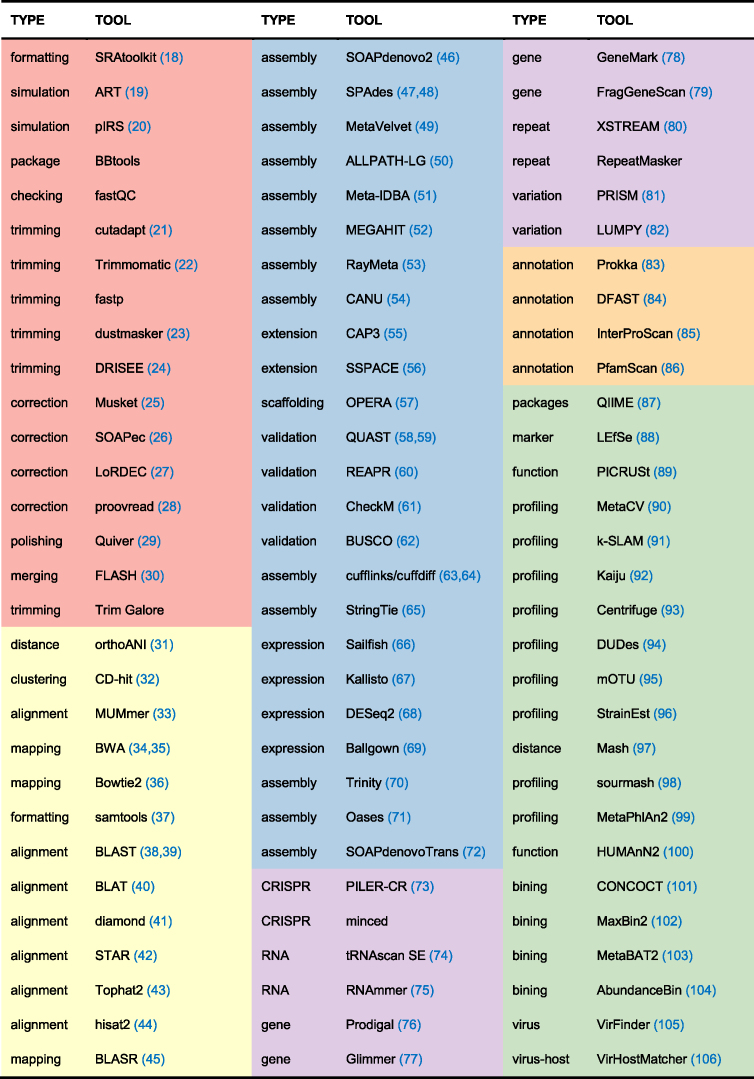
More than 90 tools could be used for microbial genomic and metagenomic analysis in web-based interactive mode. These tools are grouped into 6 different categories according to their functions: raw reads preprocessing, sequence assembly, genome structural analysis, database annotation, community profiling and sequence alignment (shown in Table 1). (1) Raw reads preprocessing contains formatting, trimming, filtering, error-correcting and other tools, which are used to reformat or filter the raw data for downstream analysis. (2) Sequence assembly includes assembly, extension and validation tools for both DNA and RNA sequences. Short or long reads become contigs, scaffolds, draft genomes or even complete genomes after this process. (3) Genome structural analysis contains tools for gene prediction, tandem or interspersed repeat detection, CRISPR array detection, etc. The outputs of some tools can be used for further annotation with various databases. (4) Database annotation groups some of the automatic gene annotation tools such as Prokka, DFAST and InterProScan. Formatted databases for annotation are stored in gcMeta for sequence alignment. (5) Community profiling includes tools for classification and quantification, de novo binning, community function prediction and downstream analysis both from short reads and contigs of metagenomic data. (6) Sequence alignment contains mapping and alignment, phylogenetic and evolution analysis tools.
Table 1.
Tools embedded in the gcMeta platform. The tools belong to the group raw reads preprocessing, sequence assembly, genome structural analysis, database annotation, community profiling and sequence alignment are set as red, blue, purple, orange green and yellow respectively. BBtools software suite (http://jgi.doe.gov/data-and-tools/bbtools/), FastQC (http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc/), fastp (https://github.com/OpenGene/fastp/), Trim Galore (http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/), minced (https://github.com/ctSkennerton/minced/tree/master) and RepeatMasker (http://ftp.genome.washington.edu/cgi-bin/RepeatMasker) are all referenced to their websites
|
Integrated analysis workflows
Since outputs of upstream tools can be severed as the inputs of downstream tools, tools can be easily concatenated to achieve a predefined workflow in this platform. Till date, there has been no generally accepted ‘analysis standard’ for a metagenomic analysis workflow. Most workflows involve aspects such as quality control, assembly, binning, taxonomic assignment and functional annotation. However, each workflow is tailored for specific computing resources, aims of analysis and characteristics of the data. In gcMeta, we provide five main workflows for genomes, marker genes, metagenomes analysis. All merged in the workflow overview in Figure 4, and they are:
Metagenome assembly and annotation (microbiome sample - NGS reads—quality control—assembly and validation—binning—genome structural analysis—database annotation): In this workflow, we assemble the short reads into contigs. These contigs can be further sorted or binned by similarity to assemble partial to full genomes of microorganisms. The assembled sequences are used for subsequent structural and functional analysis. Firstly, NGS reads, as input, are trimmed into clean reads after performing quality control (host genome contamination removal with Bowtie using parameter ‘very-sensitive’, quality viewing with FastQC and trimming with cutadapt and Trimmomatic). During the host contamination removal process, users can upload the host reference genome or use the index file we provide. Next, clean reads are assembled into contig and scaffold (MEGAHIT). After assembly, contigs and scaffolds are clustered into different bins (MaxBin) and used to perform genome structural analysis (CRISPR detection with PILER-CR, gene prediction with Prodigal, RNA identification with tRNAscan). Then, genes are used to perform annotation (annotation with all referred annotation and alignment tools).
Metagenomic 16S rRNA sequencing amplicon taxonomic assignment (microbiome sample—NGS reads—quality control—taxonomic assignment—downstream analysis): As shown in Figure 4, NGS reads, as input, are trimmed and demultiplexed (cutadapt, dada2, demux plugins) with QIIME2 (https://qiime2.org/), and used to perform taxonomic assignment, diversity analysis (diversity analysis, feature-classifier, feature-table, taxa, composition plugins) and phylogenic analysis (phylogeny plugins) with QIIME2 and other downstream analysis (function prediction with PICRUSt and biomarker discovery with LEfSe).
Reference based metagenome taxonomic assignment (microbiome sample—NGS reads—quality control—taxonomic assignment—downstream analysis): Read-based taxonomic assignment uses the unassembled DNA or mRNA sequence reads directly and compares them against reference databases to assign taxonomy name to the sequence. NGS reads, as input, are trimmed into clean reads as depicted in workflow 1. Clean reads are then used to perform taxonomic (MetaPhlAn2) and functional assignment (HUMAnN2).
Genome assembly and annotation (isolated sample—NGS/TGS reads—quality control—assembly and validation—Genome structural analysis - database annotation): For NGS reads, the workflow is the same as workflow 1, except that there is no contamination removal step in quality control process and contig binning step in assembly process in workflow 4. For TGS reads, as input, are trimmed into clean reads and assembled into contigs and scaffolds with CANU. The draft genomes are then polished with Quiver. Structure analysis and annotation process are the same as in workflow 1.
RNA-seq analysis (isolated sample - NGS reads—quality control—alignment—assembly and differential expression analysis): This workflow allows users to identify differentially expressed genes and transcripts by comparing each sample with RNA-seq data. Firstly, NGS reads, as input, are trimmed into clean reads after quality control (quality viewing with FastQC and trimming with TrimGalore). Next, cleaned reads are aligned to the reference genomes with Hisat2. Then, the alignment result is used as an input to assemble transcripts. After assembly, differential expression analysis based on the assembled transcripts will be executed with DESeq2.
Figure 4.

Integrated workflows on gcMeta. The tools can be grouped into 6 clusters shown in different colors (metagenome binning, taxonomic assignment and downstream analysis are all belong to the group community profiling shown in green color). Tools from different functional groups are connected in proper sequence to create workflows. Five main workflows covering different tools according to analysis aims are accessible from a unified user interface exemplified. Comparative analysis tools (shown in yellow) are widely involved in all the workflows. NGS and TGS stands for next-generation sequencing and third-generation sequencing, respectively.
Implementation and utility of the tools and workflows
The currently available tools and workflows are developed for different server systems, and often difficult to install, configure and deploy by the microbiologists. Since software typically depends on libraries and other components of the installed environment, a workflow implemented in one environment may not work in another environment without extra configuration. To solve this problem, we use the Docker-based technologies. Docker is a Linux-based container technology that allows tools to run across a wide range of operating systems, and helps users to deploy and reproduce tools and workflows without undue efforts (107).
The tools and workflows provide a web-based interface for the users as indicated in Figure 5. The job can be submitted into the tasks queue after the users submit their files into the system and select a specific tool or workflow and set the essential parameters. If sufficient computing resources are not available, the job is put on waiting schedule. Job status is refreshed in the task page for user view. When the job is completed, results could be downloaded and visualized online.
Figure 5.

Screenshots of the utility of the tool and workflow. (A, B) The ANI (average nucleotide identity) and dDDH (digital DNA-DNA hybridization) calculation tool which can be used by guest users. (A) Screenshots of the job submission including file upload module and necessary arguments setting. (B) The results of the job. (C–F) Metagenomic 16S rRNA sequencing taxonomic assignment workflow. (C) A screenshot of the sketch of the workflow. (D) The screenshots of the inputs, ouputs and arguments settings. (E) The result of the workflow. (F) The screenshots of the visualization of the analysis result. The example shows PCoA plot generated by ggplot package.
Details of all the tools and workflows, including input format requirements, arguments setting and examples, are described in the Supplementary file and online manual https://gcmeta.wdcm.org/im/manual/index.jsp.
System design and implementation
The system as indicated in Figure 6 is based on centralized computing and storage resources. The database management system is divided into metadata management, sequencing raw data management and user information management. The current version of the gcMeta database is constructed on the basis of PostgreSQL for metadata and user information, and MongoDB for sequencing raw data index information. The system is operated on Linux servers. The web interface was developed using Python. Tools and workflows were installed with Docker technologies.
Figure 6.

System structure of gcMeta. The platform integrates storage cluster and computing cluster resources with database management system and Docker based tools and workflows to supply comprehensive data archive, publication and analysis service to users.
DISCUSSION
With the vast diversity of microbial communities and exponentially increasing amount of meta-omics data, we are facing great challenges in organized data management and deep data mining. As a part of the efforts of CAS-CMI, gcMeta provides long-term data preservation and management following the internationally used standards and hence serves as a public data repository. We provide data protection for pre-publication data and GUID for public data, which ensures the reuse of data as required by the scientific community. The platform houses and incorporates data from public and ongoing microbiome projects, and supports comparative analysis of the data collected from distinct projects.
Another key feature of the platform is we offer a set of data analysis and visualization tools and predefined workflows by web interface which facilitates data analysis by microbiologists in an easy way. As the system is based on the Docker technology, it can be run by a variety of operational systems. The analysis application is integrated with the in-house computing resources which provide scientists a robust site for powerful data analysis.
We will keep updating the database and workflows to support the rapidly increasing datasets and complicated studies. gcMeta accepts data submission for single genome, microbiome and transcriptome data. Meanwhile, we will establish connection with other data portals to ensure data properly deposited and preserved within the International Nucleotide Sequence Database Consortium (INSDC). On the other hand, the meta-omics data analysis is becoming more and more diverse. Predefined workflows could not fit for all the analysis aims. Therefore, the future version of gcMeta will provide customized workflows for professional bioinformatics who are interested in the data and computing resources while hope to develop their own analysis workflows.
Additionally, current meta-omics data and associated analysis results are widely dispersed in different kinds of resources from public data archives to specialized databases. Integration of various types of meta-omics data is essential for a comprehensive understanding of the structure, functions and expressions of a specific community, species, strain or gene. Updating our database schema to accommodate diverse data and providing rational links among those data through semantic web technologies are future planned directions as well.
Moreover, with increasing data from microbiome projects in China and worldwide, high quality reference data are needed for accurate data annotation. However, the current type strain genomic data resources in the public database are still unable to fullfill the requirements. The Global Catalogue of Microorganisms (GCM) 2.0 type strains (108) sequencing project and The Genomic Encyclopedia of Bacteria and Archaea (GEBA) project (109) are the ongoing efforts to fill in this gap. We plan to integrate the GCM 2.0 sequencing outputs into our system to provide more accurate annotation of metagenomics data. In conclusion, gcMeta will continuously improve to accommodate the evolving meta-omics researches.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank the former director of WDCM, Hideaki Sugawara for his thoughtful suggestions and continuous supports. We would like to thank the support from GeneDock Co., Ltd., Beijing, China for Docker container technologies. We would like to thank the technical support staff from BGI Shenzhen for their suggestions on data curation.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National key Research Program of China [2017YFD0400302, 2017YFC1201202, 2017YFC0907505, 2016YFC1200801, 2016YFB1000605, 2016YFC0901702]; Key Research Program of the Chinese Academy of Sciences [KFZD-SW-219]; Strategic Priority Research Program of the Chinese Academy of Sciences [XDA19050301]; 13th Five-year Informatization Plan of Chinese Academy of Sciences [XXH13506, XXH13505]; National Science Foundation for Young Scientists of China [31701157]; Supporting Program for the Developing Countries Around China [KY201701011]; Major State Basic Research Development Program [2015CB554200]. Funding for open access charge: National key Research Program of China [2016YFC0901702].
Conflict of interest statement. None declared.
REFERENCES
- 1. Thompson L.R., Sanders J.G., McDonald D., Amir A., Ladau J., Locey K.J., Prill R.J., Tripathi A., Gibbons S.M., Ackermann G. et al. A communal catalogue reveals Earth's multiscale microbial diversity. Nature. 2017; 551:457–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lloyd-Price J., Mahurkar A., Rahnavard G., Crabtree J., Orvis J., Hall A.B., Brady A., Creasy H.H., McCracken C., Giglio M.G. et al. Strains, functions and dynamics in the expanded Human Microbiome Project. Nature. 2017; 550:61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kyrpides N.C., Eloe-Fadrosh E.A., Ivanova N.N.. Microbiome Data Science: understanding our microbial planet. Trends Microbiol. 2016; 24:425–427. [DOI] [PubMed] [Google Scholar]
- 4. Hoopen P.T. P., Finn R.D., Bongo L.A., Corre E., Fosso B., Meyer F., Mitchell A., Pelletier E., Pesole G., Santamaria M. et al. The metagenomic data life-cycle: standards and best practices. Gigascience. 2017; 6:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Field D., Amaral-Zettler L., Cochrane G., Cole J.R., Dawyndt P., Garrity G.M., Gilbert J., Glockner F.O., Hirschman L., Karsch-Mizrachi I. et al. The Genomic Standards Consortium. PLoS Biol. 2011; 9:e1001088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ten Hoopen P., Pesant S., Kottmann R., Kopf A., Bicak M., Claus S., Deneudt K., Borremans C., Thijsse P., Dekeyzer S. et al. Marine microbial biodiversity, bioinformatics and biotechnology (M2B3) data reporting and service standards. Stand. Genomic Sci. 2015; 10:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dubilier N., McFall-Ngai M., Zhao L.. Microbiology: create a global microbiome effort. Nature. 2015; 526:631–634. [DOI] [PubMed] [Google Scholar]
- 8. Niu S.Y., Yang J., McDermaid A., Zhao J., Kang Y., Ma Q.. Bioinformatics tools for quantitative and functional metagenome and metatranscriptome data analysis in microbes. Brief Bioinform. 2017; 19:360. [DOI] [PubMed] [Google Scholar]
- 9. Mitchell A.L., Scheremetjew M., Denise H., Potter S., Tarkowska A., Qureshi M., Salazar G.A., Pesseat S., Boland M.A., Hunter F.M.I. et al. EBI Metagenomics in 2017: enriching the analysis of microbial communities, from sequence reads to assemblies. Nucleic Acids Res. 2018; 46:D726–D735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wilke A., Bischof J., Gerlach W., Glass E., Harrison T., Keegan K.P., Paczian T., Trimble W.L., Bagchi S., Grama A. et al. The MG-RAST metagenomics database and portal in 2015. Nucleic Acids Res. 2016; 44:D590–D594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen I.M.A., Markowitz V.M., Chu K., Palaniappan K., Szeto E., Pillay M., Ratner A., Huang J.H., Andersen E., Huntemann M. et al. IMG/M: integrated genome and metagenome comparative data analysis system. Nucleic Acids Res. 2017; 45:D507–D516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Huang L.R., Kruger J., Sczyrba A.. Analyzing large scale genomic data on the cloud with Sparkhit. Bioinformatics. 2018; 34:1457–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wu L., Sun Q., Desmeth P., Sugawara H., Xu Z., McCluskey K., Smith D., Alexander V., Lima N., Ohkuma M.. World data centre for microorganisms: an information infrastructure to explore and utilize preserved microbial strains worldwide. Nucleic Acids Res. 2016; 45:D611–D618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Field D., Garrity G., Gray T., Morrison N., Selengut J., Sterk P., Tatusova T., Thomson N., Allen M.J., Angiuoli S.V. et al. The minimum information about a genome sequence (MIGS) specification. Nat. Biotechnol. 2008; 26:541–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yilmaz P., Kottmann R., Field D., Knight R., Cole J.R., Amaral-Zettler L., Gilbert J.A., Karsch-Mizrachi I., Johnston A., Cochrane G. et al. Minimum information about a marker gene sequence (MIMARKS) and minimum information about any (x) sequence (MIxS) specifications. Nat. Biotechnol. 2011; 29:415–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Buttigieg P.L., Pafilis E., Lewis S.E., Schildhauer M.P., Walls R.L., Mungall C.J.. The environment ontology in 2016: bridging domains with increased scope, semantic density, and interoperation. J. Biomed. Semant. 2016; 7:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Oulas A., Pavloudi C., Polymenakou P., Pavlopoulos G.A., Papanikolaou N., Kotoulas G., Arvanitidis C., Iliopoulos I.. Metagenomics: tools and insights for analyzing next-generation sequencing data derived from biodiversity studies. Bioinform. Biol. Insights. 2015; 9:75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Leinonen R., Sugawara H., Shumway M., Collaboration I.N.S.D.. The sequence read archive. Nucleic Acids Res. 2010; 39:D19–D21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Huang W., Li L., Myers J.R., Marth G.T.. ART: a next-generation sequencing read simulator. Bioinformatics. 2012; 28:593–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hu X., Yuan J., Shi Y., Lu J., Liu B., Li Z., Chen Y., Mu D., Zhang H., Li N. et al. pIRS: Profile-based Illumina pair-end reads simulator. Bioinformatics. 2012; 28:1533–1535. [DOI] [PubMed] [Google Scholar]
- 21. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011; 17:10–12. [Google Scholar]
- 22. Bolger A.M., Lohse M., Usadel B.. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014; 30:2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Morgulis A., Gertz E.M., Schaffer A.A., Agarwala R.. A fast and symmetric DUST implementation to mask low-complexity DNA sequences. J. Comput. Biol. 2006; 13:1028–1040. [DOI] [PubMed] [Google Scholar]
- 24. Keegan K.P., Trimble W.L., Wilkening J., Wilke A., Harrison T., D'Souza M., Meyer F.. A platform-independent method for detecting errors in metagenomic sequencing data: DRISEE. PLoS Comput. Biol. 2012; 8:e1002541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu Y., Schröder J., Schmidt B.. Musket: a multistage k-mer spectrum-based error corrector for Illumina sequence data. Bioinformatics. 2012; 29:308–315. [DOI] [PubMed] [Google Scholar]
- 26. Li R., Zhu H., Ruan J., Qian W., Fang X., Shi Z., Li Y., Li S., Shan G., Kristiansen K.. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010; 20:265–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Salmela L., Rivals E.. LoRDEC: accurate and efficient long read error correction. Bioinformatics. 2014; 30:3506–3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hackl T., Hedrich R., Schultz J., Förster F.. proovread: large-scale high-accuracy PacBio correction through iterative short read consensus. Bioinformatics. 2014; 30:3004–3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chin C.-S., Alexander D.H., Marks P., Klammer A.A., Drake J., Heiner C., Clum A., Copeland A., Huddleston J., Eichler E.E. et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods. 2013; 10:563–569. [DOI] [PubMed] [Google Scholar]
- 30. Magoč T., Salzberg S.L.. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011; 27:2957–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lee I., Kim Y.O., Park S.-C., Chun J.. OrthoANI: an improved algorithm and software for calculating average nucleotide identity. Int. J. Syst. Evol. Microbiol. 2016; 66:1100–1103. [DOI] [PubMed] [Google Scholar]
- 32. Li W., Godzik A.. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006; 22:1658–1659. [DOI] [PubMed] [Google Scholar]
- 33. Kurtz S., Phillippy A., Delcher A.L., Smoot M., Shumway M., Antonescu C., Salzberg S.L.. Versatile and open software for comparing large genomes. Genome Biol. 2004; 5:R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li H., Durbin R.. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009; 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li H., Durbin R.. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010; 26:589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Langmead B., Salzberg S.L.. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J.. Basic local alignment search tool. J. Mol. Biol. 1990; 215:403–410. [DOI] [PubMed] [Google Scholar]
- 39. Camacho C., Coulouris G., Avagyan V., Ma N., Papadopoulos J., Bealer K., Madden T.L.. BLAST+: architecture and applications. BMC Bioinformatics. 2009; 10:421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kent W.J. BLAT—the BLAST-like alignment tool. Genome Res. 2002; 12:656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Buchfink B., Xie C., Huson D.H.. Fast and sensitive protein alignment using DIAMOND. Nat. Methods. 2014; 12:59–60. [DOI] [PubMed] [Google Scholar]
- 42. Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R.. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L.. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013; 14:R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kim D., Langmead B., Salzberg S.L.. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods. 2015; 12:357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chaisson M.J., Tesler G. Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): application and theory. BMC Bioinformatics. 2012; 13:238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Luo R., Liu B., Xie Y., Li Z., Huang W., Yuan J., He G., Chen Y., Pan Q., Liu Y. et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience. 2012; 1:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bankevich A., Nurk S., Antipov D., Gurevich A.A., Dvorkin M., Kulikov A.S., Lesin V.M., Nikolenko S.I., Pham S., Prjibelski A.D.. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012; 19:455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nurk S., Meleshko D., Korobeynikov A., Pevzner P.A.. metaSPAdes: a new versatile metagenomic assembler. Genome Res. 2017; 27:824–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Namiki T., Hachiya T., Tanaka H., Sakakibara Y.. MetaVelvet: an extension of Velvet assembler to de novo metagenome assembly from short sequence reads. Nucleic Acids Res. 2012; 40:e155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gnerre S., Maccallum I., Przybylski D., Ribeiro F.J., Burton J.N., Walker B.J., Sharpe T., Hall G., Shea T.P., Sykes S. et al. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc. Natl Acad. Sci. U.S.A. 2011; 108:1513–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Peng Y., Leung H.C., Yiu S.M., Chin F.Y.. Meta-IDBA: a de novo assembler for metagenomic data. Bioinformatics. 2011; 27:i94–i101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Li D., Liu C.-M., Luo R., Sadakane K., Lam T.-W.. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics. 2015; 31:1674–1676. [DOI] [PubMed] [Google Scholar]
- 53. Boisvert S., Raymond F., Godzaridis E., Laviolette F., Corbeil J.. Ray Meta: scalable de novo metagenome assembly and profiling. Genome Biol. 2012; 13:R122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Koren S., Walenz B.P., Berlin K., Miller J.R., Bergman N.H., Phillippy A.M.. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017; 27:722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Huang X., Madan A.. CAP3: a DNA sequence assembly program. Genome Res. 1999; 9:868–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Boetzer M., Henkel C.V., Jansen H.J., Butler D., Pirovano W.. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics. 2011; 27:578–579. [DOI] [PubMed] [Google Scholar]
- 57. Gao S., Sung W.K., Nagarajan N.. Opera: reconstructing optimal genomic scaffolds with high-throughput paired-end sequences. J. Comput. Biol. 2011; 18:1681–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gurevich A., Saveliev V., Vyahhi N., Tesler G.. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013; 29:1072–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mikheenko A., Saveliev V., Gurevich A.. MetaQUAST: evaluation of metagenome assemblies. Bioinformatics. 2015; 32:1088–1090. [DOI] [PubMed] [Google Scholar]
- 60. Hunt M., Kikuchi T., Sanders M., Newbold C., Berriman M., Otto T.D.. REAPR: a universal tool for genome assembly evaluation. Genome Biol. 2013; 14:R47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Parks D.H., Imelfort M., Skennerton C.T., Hugenholtz P., Tyson G.W.. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015; 25:1043–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Simão F.A., Waterhouse R.M., Ioannidis P., Kriventseva E.V., Zdobnov E.M.. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015; 31:3210–3212. [DOI] [PubMed] [Google Scholar]
- 63. Trapnell C., Williams B.A., Pertea G., Mortazavi A., Kwan G., van Baren M.J., Salzberg S.L., Wold B.J., Pachter L.. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010; 28:511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Trapnell C., Roberts A., Goff L., Pertea G., Kim D., Kelley D.R., Pimentel H., Salzberg S.L., Rinn J.L., Pachter L.. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012; 7:562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Pertea M., Pertea G.M., Antonescu C.M., Chang T.-C., Mendell J.T., Salzberg S.L.. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015; 33:290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Patro R., Mount S.M., Kingsford C.. Sailfish enables alignment-free isoform quantification from RNA-seq reads using lightweight algorithms. Nat. Biotechnol. 2014; 32:462–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bray N.L., Pimentel H., Melsted P., Pachter L.. Erratum: near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016; 34:525–527. [DOI] [PubMed] [Google Scholar]
- 68. Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Frazee A.C., Pertea G., Jaffe A.E., Langmead B., Salzberg S.L., Leek J.T.. Ballgown bridges the gap between transcriptome assembly and expression analysis. Nat. Biotechnol. 2015; 33:243–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Grabherr M.G., Haas B.J., Yassour M., Levin J.Z., Thompson D.A., Amit I., Adiconis X., Fan L., Raychowdhury R., Zeng Q.. Trinity: reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 2011; 29:644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Schulz M.H., Zerbino D.R., Vingron M., Birney E.. Oases: Robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics. 2012; 28:1086–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Xie Y., Wu G., Tang J., Luo R., Patterson J., Liu S., Huang W., He G., Gu S., Li S. et al. SOAPdenovo-Trans: de novo transcriptome assembly with short RNA-Seq reads. Bioinformatics. 2014; 30:1660–1666. [DOI] [PubMed] [Google Scholar]
- 73. Edgar R.C. PILER-CR: fast and accurate identification of CRISPR repeats. BMC Bioinformatics. 2007; 8:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Lowe T.M., Chan P.P.. tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016; 44:W54–W57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lagesen K., Hallin P., Rodland E.A., Staerfeldt H.H., Rognes T., Ussery D.W.. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007; 35:3100–3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Hyatt D., Chen G.L., Locascio P.F., Land M.L., Larimer F.W., Hauser L.J.. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010; 11:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Delcher A.L., Harmon D., Kasif S., White O., Salzberg S.L.. Improved microbial gene identification with GLIMMER. Nucleic. Acids. Res. 1999; 27:4636–4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Borodovsky M., Mcininch J.. GENMARK: Parallel gene recognition for both DNA strands. Comput. Chem. 1993; 17:123–133. [Google Scholar]
- 79. Rho M., Tang H., Ye Y.. FragGeneScan: predicting genes in short and error-prone reads. Nucleic Acids Res. 2010; 38:e191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Newman A.M., Cooper J.B.. XSTREAM: a practical algorithm for identification and architecture modeling of tandem repeats in protein sequences. BMC Bioinformatics. 2007; 8:382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Jiang Y., Wang Y., Brudno M.. PRISM: pair-read informed split-read mapping for base-pair level detection of insertion, deletion and structural variants. Bioinformatics. 2012; 28:2576–2583. [DOI] [PubMed] [Google Scholar]
- 82. Layer R.M., Chiang C., Quinlan A.R., Hall I.M.. LUMPY: a probabilistic framework for structural variant discovery. Genome Biol. 2014; 15:R84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014; 30:2068–2069. [DOI] [PubMed] [Google Scholar]
- 84. Tanizawa Y., Fujisawa T., Nakamura Y.. DFAST: a flexible prokaryotic genome annotation pipeline for faster genome publication. Bioinformatics. 2018; 34:1037–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Zdobnov E.M., Apweiler R.. InterProScan–an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 2001; 17:847–848. [DOI] [PubMed] [Google Scholar]
- 86. Bateman A., Coin L., Durbin R., Finn R.D., Hollich V., Griffiths‐Jones S., Khanna A., Marshall M., Moxon S., Sonnhammer E.L. et al. The Pfam protein families database. Nucleic Acids Res. 2004; 32:D138–D141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Caporaso J.G., Kuczynski J., Stombaugh J., Bittinger K., Bushman F.D., Costello E.K., Fierer N., Pena A.G., Goodrich J.K., Gordon J.I. et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods. 2010; 7:335–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Segata N., Izard J., Waldron L., Gevers D., Miropolsky L., Garrett W.S., Huttenhower C.. Metagenomic biomarker discovery and explanation. Genome Biol. 2011; 12:R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Langille M.G.I., Zaneveld J., Caporaso J.G., McDonald D., Knights D., Reyes J.A., Clemente J.C., Burkepile D.E., Vega Thurber R.L.V., Knight R. et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013; 31:814–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Liu J.M., Wang H.F., Yang H.X., Zhang Y.Z., Wang J.F., Zhao F.Q., Qi J.. Composition-based classification of short metagenomic sequences elucidates the landscapes of taxonomic and functional enrichment of microorganisms. Nucleic Acids Res. 2012; 41:e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Ainsworth D., Sternberg M.J.E., Raczy C., Butcher S.A.. k-SLAM: accurate and ultra-fast taxonomic classification and gene identification for large metagenomic data sets. Nucleic Acids Res. 2017; 45:1649–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Menzel P., Ng K.L., Krogh A.. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 2016; 7:11257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kim D., Song L., Breitwieser F.P., Salzberg S.L.. Centrifuge: rapid and sensitive classification of metagenomic sequences. Genome Res. 2016; 26:1721–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Piro V.C., Lindner M.S., Renard B.Y.. DUDes: a top-down taxonomic profiler for metagenomics. Bioinformatics. 2016; 32:2272–2280. [DOI] [PubMed] [Google Scholar]
- 95. Sunagawa S., Mende D.R., Zeller G., Izquierdo-Carrasco F., Berger S.A., Kultima J.R., Coelho L.P., Arumugam M., Tap J., Nielsen H.B. et al. Metagenomic species profiling using universal phylogenetic marker genes. Nat. Methods. 2013; 10:1196–1199. [DOI] [PubMed] [Google Scholar]
- 96. Albanese D., Donati C.. Strain profiling and epidemiology of bacterial species from metagenomic sequencing. Nat. Commun. 2017; 8:2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Ondov B.D., Treangen T.J., Melsted P., Mallonee A.B., Bergman N.H., Koren S., Phillippy A.M., Altschul S., Gish W., Miller W. et al. Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016; 17:132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Brown C.T., Irber L.. sourmash: a library for MinHash sketching of DNA. J. Open Source Softw. 2016; 1:27. [Google Scholar]
- 99. Segata N., Waldron L., Ballarini A., Narasimhan V., Jousson O., Huttenhower C.. Metagenomic microbial community profiling using unique clade-specific marker genes. Nat. Methods. 2012; 9:811–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Abubucker S., Segata N., Goll J., Schubert A.M., Izard J., Cantarel B.L., Rodriguez-Mueller B., Zucker J., Thiagarajan M., Henrissat B. et al. Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS Comput. Biol. 2012; 8:e1002358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Alneberg J., Bjarnason B.S., de Bruijn I., Schirmer M., Quick J., Ijaz U.Z., Lahti L., Loman N.J., Andersson A.F., Quince C.. Binning metagenomic contigs by coverage and composition. Nat. Methods. 2014; 11:1144–1146. [DOI] [PubMed] [Google Scholar]
- 102. Wu Y.W., Simmons B.A., Singer S.W.. MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics. 2015; 32:605–607. [DOI] [PubMed] [Google Scholar]
- 103. Kang D.D., Froula J., Egan R., Wang Z.. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ. 2015; 3:e1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Wu Y.W., Ye Y.. A Novel Abundance-Based Algorithm for Binning Metagenomic Sequences Using l-tuples. J. Comput. Biol. 2011; 18:523–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Ren J., Ahlgren N.A., Lu Y.Y., Fuhrman J.A., Sun F.. VirFinder: a novel k-mer based tool for identifying viral sequences from assembled metagenomic data. Microbiome. 2017; 5:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Ahlgren N.A., Ren J., Young L.Y., Fuhrman J.A., Sun F.. Alignment-free d2* oligonucleotide frequency dissimilarity measure improves prediction of hosts from metagenomically-derived viral sequences. Nucleic Acids Res. 2017; 45:39–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Boettiger C. An introduction to Docker for reproducible research. ACM SIGOPS Oper. Syst. Rev. 2015; 49:71–79. [Google Scholar]
- 108. Wu L., McCluskey K., Desmeth P., Liu S., Hideaki S., Yin Y., Moriya O., Itoh T., Kim C.Y., Lee J.-S. et al. The global catalogue of microorganisms 10K type strain sequencing project: closing the genomic gaps for the validly published prokaryotic and fungi species. GigaScience. 2018; 7:giy026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Mukherjee S., Seshadri R., Varghese N.J., Eloe-Fadrosh E.A., Meier-Kolthoff J.P., Göker M., Coates R.C., Hadjithomas M., Pavlopoulos G.A., Paez-Espino D.. 1,003 reference genomes of bacterial and archaeal isolates expand coverage of the tree of life. Nat. Biotechnol. 2017; 35:676–683. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.