

Abstract
Background.
Subcutaneous panniculitis-like T-cell lymphoma (SPTCL) is a rare neoplasm of mature αβ cytotoxic T-cells. Most commonly occurring in young adults, few reports are described in children. A separate analysis of a significant cohort of pediatric patients has not previously been performed.
Procedure.
We analyzed the pathology including molecular results as well as available clinical data from 16 pediatric patients (age 5 months to 21 years) who had a total of 19 biopsies submitted to the National Cancer Institute from 1999 to 2011. This included 6 males and 10 females.
Results.
Most patients (10/16, 62.5%) had multiple skin lesions at the time of biopsy. Histologic features included rimming of adipocytes by atypical lymphocytes, fat necrosis, and karyorrhectic debris. Four biopsies showed only partial involvement by lymphoma; and plasma cells were identified in 14/19 (74%) cases, including three in which they were focally prominent. The neoplastic cells in general were positive for CD3, CD8, TIA-1, and βF1 and were negative for CD4 and CD56. CD5 expression was weak to negative in 5/8 cases (63%). A clonal T-cell receptor gene rearrangement was demonstrated in 11/17 (65%). Patients were treated with a variety of agents. While 5/9 (56%) patients had evidence of recurrent skin lesions, no deaths were attributed to disease for the seven patients with follow-up information.
Conclusions.
Pediatric SPTCL shares many clinical and pathologic features with adult SPTCL. The presence of partial involvement or admixed plasma cells makes the differential diagnosis with reactive conditions challenging in some cases. Pediatr Blood Cancer 2013;60:1165– 1170.
Keywords: lupus panniculitis, pediatric, subcutaneous panniculitis-like T-cell lymphoma
INTRODUCTION
Subcutaneous panniculitis-like T-cell lymphoma (SPTCL) is a neoplasm of mature cytotoxic T-cells with an ab phenotype. This rare lymphoma is most common in young adults, although it can occur at a wide age range. Patients typically present with multiple subcutaneous lesions and may also have systemic symptoms and/or laboratory abnormalities such as cytopenias. The overall 5-year median survival is approximately 80%, although the prognosis is poor if associated with a hemophagocytic syndrome [1,2]. While cases in pediatric patients have been reported as individual case reports or as part of larger series mostly comprised of adult patients, a single study dedicated to pediatric patients has not been reported. We describe the clinical, pathological, and molecular features of SPTCL in a cohort of pediatric patients.
METHODS
The surgical pathology archives of the Laboratory of Pathology, National Cancer Institute (NCI) were queried for all cases of SPTCL diagnosed from January 1999 to December 2011, most of which were reviewed as consultation cases. Only those patients whose age was 21 years or younger at the time of diagnosis were included in this study. All available hematoxylin and eosin-stained slides and immunohisto chemical (IHC) stains were reviewed by two pathologists (ARH, ESJ) for confirmation of the original diagnosis with additional stains performed as necessary. Results of PCR studies for T-cell receptor gene rearrangement, either performed in our laboratory or at an outside institution, were recorded, as well as any clinical information provided. Additional details of clinical data and patient outcome were sought from the submitting pathologist or treating physician in consultation cases. The National Institutes of Health Office of Human Subjects Research and the NCI Institutional Review Board approved this study.
RESULTS
Clinical Presentation
A total of 19 separate biopsies from 16 patients were identified. Clinical information and outcome data are summarized in Table I. Patients’ ages ranged from 5 months to 21 years at the time of biopsy (median 8 years) with a male to female ratio of 1:1.7. The original age of presentation for patients 3, 5, and 6 is unknown, as our review of their pathology was performed on recurrent disease. Genetic/developmental abnormalities included a history of cardiovelofacial abnormalities in patient 3 and trisomy 21 in patient 7 [previously reported [3]]. Autoimmune phenomena include a history of mixed connective tissue disease (MCTD) in patient 8 and a history of a positive antinuclear antibody (ANA) in patient 5 during the early days of her illness (negative ANA at the time of last follow-up).
TABLE I.
Clinical Characteristics and Outcome of Pediatric Patients With SPTCL
| Patient | Age(years, at biopsy), sex |
Extent cutaneous lesions (at presentation) |
Systemic symptoms | Cytopenia(s) | HSM, LAD | Treatment | Recurrence | Last known |
|---|---|---|---|---|---|---|---|---|
| 1 | 9, F | S, T | ND | ND | ND | IS (prednisone, tacrolimus, cyclosporine) | Yes | A, NED (139 mo) |
| 2 | 14, F | S, T | ND | ND | ND | Asparaginase, MTX, adriamycin, vincristine, carmustine, prednisone | No | D, other cause |
| 3 | 3, F | Mul, ND | ND | Pancytopenia | Shotty LAD | Unk | Yesa | Unk |
| 4 | 19, F | Mul, ND | ND | ND | ND | CHOP x 6 cycles | Yes | Unk |
| 5 | 21, F | S, T | Fever, fatigue, weight loss | Anemia, leukopenia | HSM, right axillary LAD | Prior cyclosporine | Yesa | A, NED (> 17 years) |
| 6 | 7, F | S, T | ND | ND | ND | Prior CHOP | Yesa | Unk |
| 7 | 3, F | S, E | ND | ND | ND | CHOP, maintenance with bexarotene | No | A, NED (24+ mo) |
| 8 | 6, F | M, E | ND | ND | HSM | Unk | Unk | |
| 9 | 18, M | Mul, T/E | Recent flu-like illness also in family members | Mild anemia, leukopenia | CT negative | Unk | No | A, NED (38 mo) |
| 10 | 5 Mo, F | Mul, T/E | None | Mild anemia | CT negative | Unk | Unk | Unk |
| 11 | 18, F | Mul, T/E | High fever | Leukopenia (lymphopenia), anemia | CT-no LAD, borderline splenomegaly | Unk | Unk | Unk |
| 12 | 4, M | Mul, E | ND | ND | ND | Steroids alone | Unk | Unk |
| 13 | 20, M | S, T | None | Anemia, leukopenia (neutropenia) | CT- mild splenomegaly, small left axillary LAD | Unk | No | A, NED (28 mo) |
| 14 | 5 Mo, M | Mul, T | ND | ND | ND | Unk | Unk | Unk |
| 15 | 16 Mo, M | Mul, T/E | Mild fever | None | ND | None | Unk | A (2 mo), lost to follow-up |
| 16 | 21, M | Mul, T & head | None | Intermittent leukopenia (for many years) | PET/CT-small retroperitoneal LAD | Unk | Unk | Unk |
Mo, months; F, female; M, male; S, single; Mul, multiple; T, trank; E, extremities; ND, not described; HSM, hepatosplenomegaly; LAD, lymphadenopathy; IS, immunosuppressives; MTX, methotrexate; CHOP, cyclophosphamide, doxorubicin, vincristine, prednisolone; Unk, unknown; A, alive; NED, no evidence of disease; D, deceased, indicates cases in which the recurrent but not original diagnostic specimen was reviewed.
Multiple cutaneous lesions were identified at the time of presentation in 10/16 (62.5%) patients. These lesions were present on the trunk and/or extremities, with the exception of patient 16 who also had lesions on his head. The most common systemic symptom was fever. All except one of the patients on whom data were received had a cytopenia, most commonly anemia. Patient 8 is the only one reported to have a possible hemophagocytic syndrome, with occasional phagocytic histiocytes reported to be seen on a bone marrow biopsy and erythrophagocytosis visualized on a concurrently received liver core biopsy.
Histology and Immunohistochemistry
All tissue examined demonstrated a lobular panniculitis-like infiltrate (Fig. 1). While some slides contained only subcutaneous adipose tissue, the 12 biopsies with skin attached did not display evidence of dermal or epidermal involvement, including lack of epidermal ulceration. While a dermal perivascular or periadnexal lymphocytic infiltrate was present in several cases, these lymphocytes were not cytologically atypical and did not have an abnormal immunophenotype. The neoplastic infiltrate was composed of small to medium-sized lymphocytes with irregular nuclear contours and some with nucleoli; mitotic figures were easily identified in several cases. These neoplastic cells were admixed with histiocytes as well as scattered small lymphocytes without cytologic atypia. Plasma cells were somewhat prominent in 3 cases (patients 1, 14, and 16; Fig. 2) and were less notable although still present in many of the remaining cases, such that only 5 of 19 biopsies had rare to no plasma cells identified. The patient with a history of MCTD had virtually no plasma cells present, while the patient with a history of a positive ANA had a few scattered throughout. Common histologic features included rimming of adipocytes by atypical lymphocytes (18/19 cases, 95%), karyor- rhexis (17/19 cases, 90%), and fat necrosis (18/19 cases, 95%). Four cases had only partial involvement by lymphoma. Some of the non-neoplastic areas showed a vaguely granulomatous reaction or stromal hyalinization, which in one case was associated with previous biopsy site. Of interest, patient 14 also had a focal eosinophilic infiltrate as well as focal benign-appearing lymphoid aggregates.
Fig. 1.
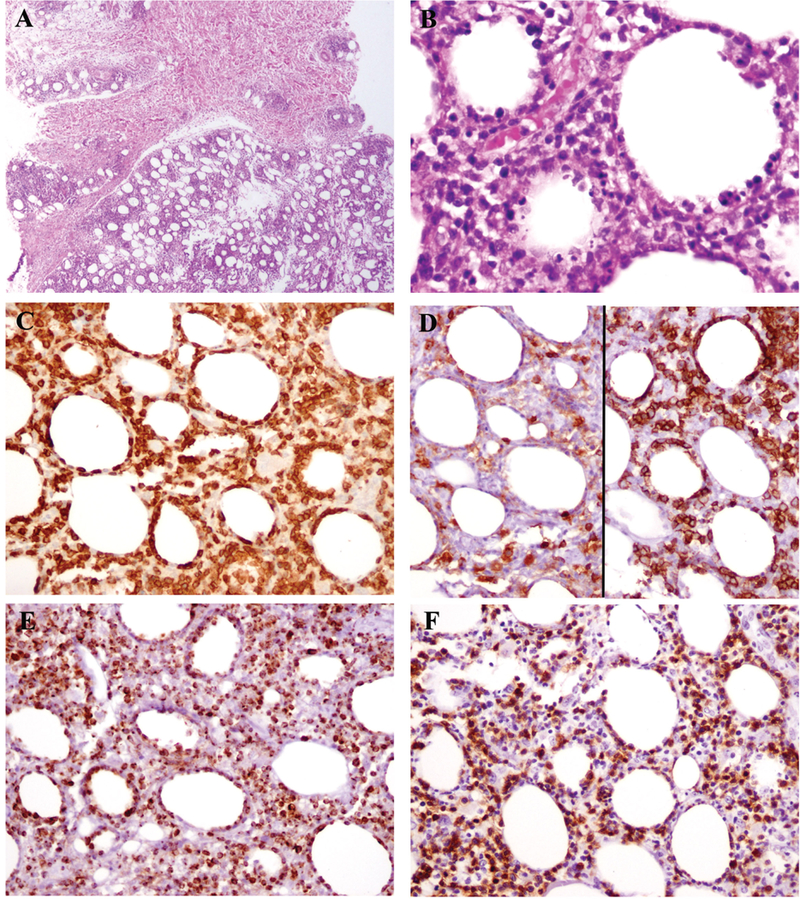
The findings from patient 4 illustrate those typically seen in pediatric SPTCL. The dermis was uninvolved (A, top), while a lobular panniculitic-like infiltrate was present in subcutaneous fat (A, bottom). Atypical lymphoid cells rim adipocytes, and karyorrhectic debris is abundant (B). The atypical lymphocytes express CD3 (C). They are negative for CD4 (D, left) and positive for CD8 (right). Granzyme B (E) and beta F1 (F) are also positive
Fig. 2.
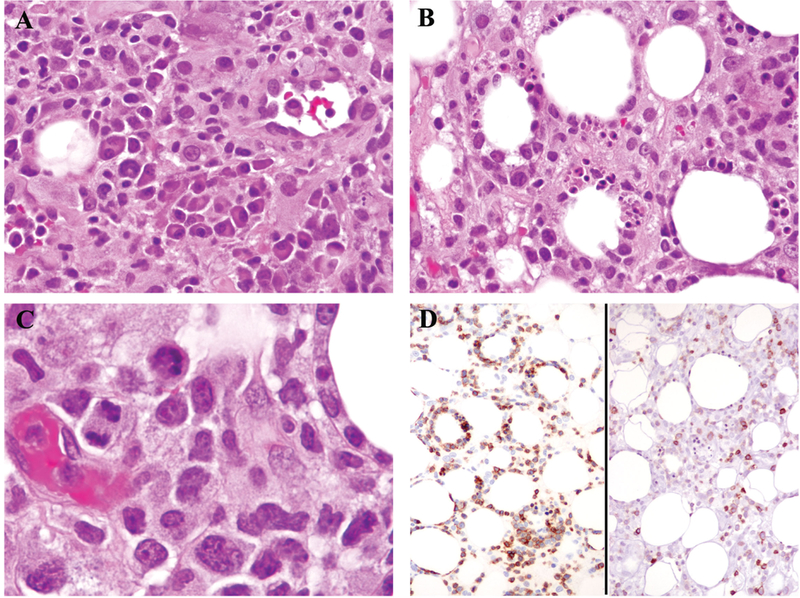
Plasma cells were focally numerous in areas of the tissue from patient 16 (A). However, most of the tissue displayed typical histologic findings of SPTCL (B), including lymphocytes with hyperchromatic nuclei and irregular nuclear contours as well as mitotic figures (C). The neoplastic cells were positive for CD3 (D, left) and negative for CD5 (D, right).
Results of immunohistochemical stains are summarized in Table II. In general, the atypical lymphoid cells had the phenotype of cytotoxic T-cells. Loss of normal antigen expression was demonstrated by weak to negative CD5 expression in 5/8 cases (63%), while CD7 was partially negative in 1/5 (20%). No CD56 expression was identified. Beta F1 staining for T-cell receptor (TCR) αβ was positive in 9/10 (90%). The negative case was from the biopsy of recurrent disease in patient 1. TCR γ and δ stains were attempted on this case but were unsuccessful, as were the same stains and βF1 performed on her original diagnostic material. Testing for Epstein-Barr virus (EBV) by in situ hybridization (EBER) was performed in three cases, all of which were negative.
TABLE II.
Immunohistochemical Findings in Pediatric SPTCL
| Stain | Cases positive/total cases (%) |
|---|---|
| CD3 | 18/18 (100) |
| CD5 | 3/8 (37.5) |
| CD7 | 4/5 (80) |
| CD4 | 0/17 (0) |
| CD8 | 18/18 (0) |
| CD56 | 0/12 (0) |
| TIA-1 | 12/12 (100) |
| Granzyme B | 7/7 (100) |
| Beta F1 | 9/10 (90) |
Molecular Analysis
Seventeen of 19 cases had PCR performed for TCR γ and/or 𝛽 gene rearrangement. Of the 17 tissues tested, 11 demonstrated evidence of a clonal rearrangement. Of interest, the clone detected in case 1 at the time of original diagnosis was different from that seen at a recurrence 7 years later (Fig. 3). No evidence of a clonal rearrangement was identified in four patients, and two more had indeterminate results. Review of these six cases confirmed the diagnosis of SPTCL on the basis of histological features and immunophenotype. One biopsy with indeterminate results was from a patient with two previous diagnostic biopsies, and similar findings were observed. Three additional biopsies with negative or indeterminate results were only focally involved by lymphoma, and additional material was unavailable for repeat testing with macrodissection to enhance the neoplastic population.
Fig. 3.
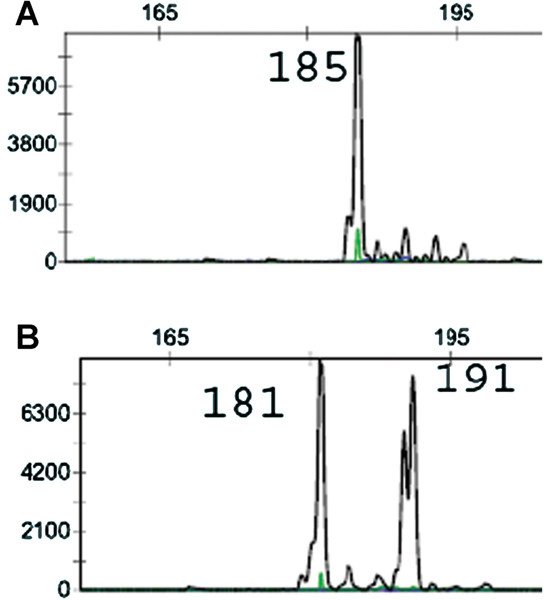
Different clonal populations were detected by TCR-gamma gene rearrangement in the original biopsy (A) and recurrence 7 years later (B).
Treatment and Clinical Course
Patients with known outcome data were subject to a variety of treatments, including cytotoxic chemotherapy [4], immunosuppressive agents [2], steroids [1], or no treatment [1]. Five of nine patients (56%) had evidence of recurrent disease. All patients were alive at the time of last follow-up with the exception of patient 2, who was reported to have died as a complication of accidental medication overdose.
DISCUSSION
Our study establishes the similarities between pediatric SPTCL and published adult cases, including being more common in females, histologic features and good prognosis. Although still in the minority, children account for a notable percentage of patients with SPTCL, with 19% (12/63) of patients studied by Willemze et al. [2] being age 20 years or younger.
Until recently, the diagnosis of SPTCL included neoplastic T cells with either an αβ or γδ phenotype. Accordingly, reports of SPTCL, whether in children or adults, may include cases that would no longer be classified under this heading. The World Health Organization (WHO) and European Organization for Research and Treatment of Cancer (EORTC) have now separated these two entities into SPTCL for malignant αβ T cells and primary cutaneous γδ T-cell lymphoma (PCGD-TCL) [1,4]. This division was based on the difference in prognoses between these two, with SPTCL boasting much better survival rates, a finding that has been confirmed in later studies [2]. Histologic and phenotypic findings may also aid in the distinction. Although both may have a panniculitis-like picture, PCGD-TCL may show involvement of the epidermis and dermis, including possible epidermal ulceration. Immunophenotypic differences include positive CD56 and often lack of expression of both CD4 and CD8 in PCGD-TCL. Although PCGD-TCL is expected to lack CD5 expression, 63% of our SPTCL cases were also weak to negative for CD5 in the neoplastic cells; consequently, a negative CD5 stain may not help to distinguish these two. IHC stains for TCR β (βF1), TCR γ and TCR δ may also be valuable if available.
Patient 1 had negative βF1 staining on a biopsy of her recurrent disease, and a βF1 stain attempted on her original biopsy as well as stains for TCR γ and TCR δ performed on both biopsies were unsuccessful. While absent bF1 expression is concerning for a diagnosis of PCGD-TCL, the histology and remaining phenotype as well as her prolonged survival (>11 years) would argue against that process. This again underscores the need for a complete assessment of all findings without complete reliance on one result.
Another entity within the differential diagnostic spectrum of lobular panniculitis is lupus panniculitis. A useful histologic feature that supports the diagnosis of lupus panniculitis is the presence of plasma cells admixed within the inflammatory infiltrate. However, many of our cases contained plasma cells of varying amounts, including three with areas in which they were focally quite prominent. Two of the three cases were clonal by TCR PCR, supporting a diagnosis of SPTCL. Thus, while a useful part of the evaluation, the presence or absence of plasma cells cannot be used as an absolute criterion. An additional complicating factor is that autoimmune disorders have been associated with SPTCL [2,5], so that a history of one of these conditions does not rule out the possibility of SPTCL as illustrated by the history of MCTD in one of our patients and a reported positive ANA in another.
Molecular studies to demonstrate a monoclonal T-cell population by PCR are diagnostically helpful, but not entirely specific. As with any other suspected T-cell lymphoproliferative disorder, this testing is best used to sustain a diagnosis and not as the sole determining factor as false positive and false negative cases can occur. Clonal populations are well-known to be found in some reactive situations. Also, absence of a clone does not rule out the diagnosis of SPTCL. Willemze et al. [2] were able to detect a clonal population with TCR β in 9/9 (100%) of their cases and for TCR γ in 28/36 (78%). While our overall percentage (11/17, 65%) was lower than either of these, this possibly could have been increased if additional material was available for repeat testing with selective macrodissection in those cases with only partial involvement. This highlights one difficulty of this study, the fact that most of these cases were seen on a consultation basis, thus limiting access to additional material or follow-up data in some cases
In addition to possibly impacting the results of PCR analysis, partial involvement presents a further pitfall to overlooking the diagnosis of SPTCL based on histologic parameters. If the entire tissue is not closely examined, the presence of a neoplastic population of T-cells could easily be missed. While some areas may have granulomas or hyalinization, histologic features characteristic of SPTCL such as rimming of fat cells or cytological atypia may be evident elsewhere. Thorough examination of the biopsy specimen is recommended. As plasma cells do not rule out the diagnosis of SPTCL, cytological atypia of the infiltrating lymphocytes remains the ultimate gold standard for a diagnosis of lymphoma.
The treatment for SPTCL in our patients was variable, ranging from no treatment to steroids or other immunosuppressive agents to multiagent chemotherapy such as CHOP (cyclophosphamide, doxorubicin, vincristine, prednisolone). This inconsistency in therapeutic regimens extends to both pediatric and adult cases reported in the literature. Additional strategies that were not known to have been used in our patients but have been reported include radiation [6] and autologous peripheral blood stem cell transplant [7]. The rarity of this disease has prevented a unified treatment strategy from being developed. Because SPTCL often has a good prognosis, some have suggested that CHOP-bases therapies should not act as the first choice of treatment [2]. That being said, patients with hemophagocytic syndrome or other aggressive features may require more intensive therapy. A number of reports have shown positive results with cyclosporine A, even at the time of relapse [8]. This agent demonstrated activity in two of our patients. Recently, a good response rate in adults and children with either SPTCL or PCGD-TCL was observed with the retinoid bexarotene; this study included patient 7 from our cohort [9]. This provides another option for treatment.
At least five of nine patients on whom information was available had evidence of recurrent disease. This did not show a distinct correlation with the type of treatment received, although it is difficult to draw conclusions due to the small study size. Patient 1 originally presented at the age of 9 and was then diagnosed with recurrent disease at the age of 16. While similar histologic findings were present in both cases, a distinct T-cell clone was identified in each case by TCR gamma testing in our laboratory. We do not have other cases in which PCR results for both the original and recurrent lymphoma identified either the same or dissimilar clones. However, the data for patient 1 suggest a predisposition for the emergence of independent clonal proliferations, rather than true recurrence of the primary disease.
In summary, clinical and pathological features in pediatric SPTCL are and pathologic features are similar to those described in adults, although careful correlation of histologic, immunophenotypic, and molecular results is needed to exclude entities within the differential diagnosis such as lupus panniculitis and PCGD- TCL. Treatments vary among institutions, but based on more recent data emerging from studies in children and adults, an initial conservative approach for patients without evidence of a hemophagocytic syndrome is warranted.
Footnotes
Conflict of interest: Nothing to declare.
REFERENCES
- 1.Swerdlow SH, International Agency for Research on Cancer, World Health Organization. WHO classification of tumours of haematopoietic and lymphoid tissues. Lyon, France: International Agency for Research on Cancer; 2008. [Google Scholar]
- 2.Willemze R, Jansen PM, Cerroni L, et al. Subcutaneous panniculitis-like T-cell lymphoma: Definition, classification, and prognostic factors: An EORTC Cutaneous Lymphoma Group Study of 83 cases. Blood 2008;111:838–845. [DOI] [PubMed] [Google Scholar]
- 3.Mixon B, Drach L, Monforte H, et al. Subcutaneous panniculitis-like T-cell lymphoma in a child with trisomy 21. Fetal Pediatr Pathol 2010;29:380–384. [DOI] [PubMed] [Google Scholar]
- 4.Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood 2005;105:3768–3785. [DOI] [PubMed] [Google Scholar]
- 5.Pincus LB, LeBoit PE, McCalmont TH, et al. Subcutaneous panniculitis-like T-cell lymphoma with overlapping clinicopathologic features of lupus erythematosus: Coexistence of 2 entities? Am J Dermatopathol 2009;31:520–526. [DOI] [PubMed] [Google Scholar]
- 6.Mukai HY, Okoshi Y, Shimizu S, et al. Successful treatment of a patient with subcutaneous panniculitislike T-cell lymphoma with high-dose chemotherapy and total body irradiation. Eur J Haematol 2003;70:413–416. [DOI] [PubMed] [Google Scholar]
- 7.Alaibac M, Berti E, Pigozzi B, et al. High-dose chemotherapy with autologous blood stem cell transplantation for aggressive subcutaneous panniculitis-like T-cell lymphoma. J Am Acad Dermatol 2005;52:S121–S123. [DOI] [PubMed] [Google Scholar]
- 8.Mizutani S, Kuroda J, Shimura Y, et al. Cyclosporine A for chemotherapy-resistant subcutaneous panniculitis-like T cell lymphoma with hemophagocytic syndrome. Acta Haematol 2011;126:8–12. [DOI] [PubMed] [Google Scholar]
- 9.Mehta N, Wayne AS, Kim YH, et al. Bexarotene is active against subcutaneous panniculitis-like T-cell lymphoma in adult and pediatric populations. Clin Lymphoma Myeloma Leuk 2012;12:20–25. [DOI] [PMC free article] [PubMed] [Google Scholar]