

Abstract
The development of metastatic castration resistant prostate cancer (m-CRPC) poses significant challenges for treatment. Specifically, the emergence of elevated levels of cancer stem cells (CSC) coupled with neuroendocrine (NE) carcinoma behavior is likely to play a critical role in m-CRPC and ultimately leads to an aggressive clinical course. Thus, understanding the mechanisms by which CSC and NE phenotypes impact the development of mCRPC will facilitate the identification of biomarkers and development of potentially novel therapeutics. Here, the role of the intracellular chemokine CXCL12γ in CSC induction and NE differentiation impact on m-CRPC was explored. We show that CXCL12γ expression was detected in small cell carcinoma of metastatic tissues and circulating tumor cells (CTCs) from m-CRPC patients and in PCa cells expressing a NE phenotype. CXCL12γ overexpression induces CSC and NE phenotypes through CXCR4-mediated PKCα/NFκB signaling, which promote prostate tumor outgrowth, metastasis, and chemoresistance. Together, these data suggest that CXCL12γ plays a significant role in induction of CSC and NE phenotypic cells leading to the development of m-CRPC and reveals a potential central molecular pathway for targeting of aggressive disease.
Keywords: Prostate cancer, CXCL12γ, Cancer stem cells, Neuroendocrine cells, m-CRPC
Introduction
Bone is the most frequent site of prostate cancer (PCa) metastasis and skeletal lesions are a common and disabling consequence of progressive disease (1,2). Front line therapy for skeletal metastasis includes androgen ablation and chemotherapy. Yet in the vast majority of cases, metastatic castration resistant prostate cancer (m-CRPC) emerges and poses significant challenges for treatment (1–7). Unfortunately, many of the molecular mechanisms which lead to the development of m-CRPC remain unclear.
Growing evidence suggests that cancer stem cells (CSCs) play a crucial role in cancer chemoresistance and recurrence in the development of m-CRPC (8–11). For example, after second generation anti-androgens and chemotherapy, neuroendocrine (NE) carcinoma, potentially derived from CSCs, can accelerate metastatic castration resistant progression, resulting in an aggressive clinical course (12–15). Thus, understanding the mechanisms by which CSCs and NE differentiation impact the development of m-CRPC may facilitate the identification of biomarkers and development of novel therapeutics.
Stromal derived factor-1 (SDF-1 or CXCL12) and CXCR4 signaling is known to play a central role in promoting tumor progression in many cancers and PCa bone metastasis (16–23). The CXCL12-CXCR4 axis also directly promotes tumor cell motility based on chemokine gradients of CXCL12 and CXCR4 expression on tumor cells (21,23). Recently, we demonstrated that enhanced levels of CXCL12 by cancer associated fibroblasts (CAFs) promote an epithelial-mesenchymal transition (EMT), supporting PCa bone metastasis (18,20). Moreover, CXCL12 signaling promotes activation of Protein Kinase C/NFκB signaling, which contributes to cancer stem cell activities (24). These data provide direct evidence that CXCL12 plays a central role in the development of PCa progression. To date, most studies have focused on the secreted isoforms of CXCL12 (CXCL12α, CXCL12β) as they promote metastasis and the survival of tumor cells (16–23). The role of the alternatively transcribed CXCL12γ isoform, an intracellular chemokine, in tumor progression remains unknown (25–30).
Here the role of CXCL12γ expression in induction of CSC and NE phenotypes in the development of m-CRPC was explored. These investigations demonstrate a unique and significant role of CXCL12γ in the reprogramming of CSCs and NE cells leading to the development of m-CRPC and reveal a potential central molecular pathway in the progression of aggressive disease.
Materials and Methods
Animals
Five to seven week-old male and female SCID mice (CB.17. SCID; Taconic, Germantown, NY) were used as transplant recipients. All animal procedures were performed in compliance with the institutional ethical requirements and approved by the University of Michigan Institutional Committee for the Use and Care of Animals (ICUCA).
Cell culture
Human PCa cell lines (PC3, DU145, and LNCaP) were obtained from the American Type Culture Collection (ATCC; Rockville, MD). The metastatic subclone of LNCaP, C4–2B, was originally isolated from a lymph node of a prostate cancer patient with disseminated bony and lymph node involvement. GFP expressing PCa cell lines (PC3GFP and DU145GFP cells) were established by lenti viral transduction. A castration-resistant sub-line of the LNCaP cells, LNCaP95 was kindly provided by Dr. Jun Luo (John Hopkins University). All prostate cancer cell lines were routinely grown in RPMI 1640 (Life Technologies, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS, GEMINI Bio-Products, Sacramento, CA), 1% penicillin-streptomycin (P/S, Life Technologies) and maintained at 37°C, 5% CO2, and 100% humidity. In some cases, PCa cells were cultured in phenol red-free RPMI-1640/DMEM (Hyclone, Logan, UT) supplemented with 10% charcoal/dextran-treated fetal bovine serum (Hyclone). Normal human prostate epithelial RWPE-1 cells (ATCC, CRL-11609) were cultured in Keratinocyte-SFM (Life Technologies) with supplements (17005–042, Life Technologies). Neuroendocrine PCa cells (NCI-H660; ATCC, CRL 5813) was obtained from ATCC and was grown in ATCC formulated RPMI1640 Medium (cat. 30–2001) with supplements. In some cases, the PCa cells were cultured in RPMI with 1% FBS supplemented with 200 ng/ml of rhCXCL12 (cat. 350-NS, R&D Systems, Minneapolis, MN). The human breast cancer (BCa) cell line, MCF7 were kindly provided by Dr. Max Wicha (University of Michigan). MCF7 cell line was cultured in DMEM with supplements (Invitrogen).
CXCL12γ overexpression
A CXCL12γ overexpression plasmid vector, pLV-CXCL12γ and control vector, pLV were kindly provided by Dr. Ramirez (Viral Vector Facility, Technical Unit of Gene Targeting, Fundacion CNIC (National Centre for Cardiovascular Research), Madrid, Spain) (29). pLV-CXCL12γ and pLV were packaged with lenti virus at University of Michigan. Lenti viral pLV-CXCL12γ or pLV were infected into PCa cells (PC3, DU145, LNCaP, C42B) and BCa cells (MCF7). Infected cells were selected for 7 days in media containing 1μg/ml puromycin and analyzed by real-time qPCR or immunofluorescence staining.
RNA Interference
PC3 or DU145 cells at 60% confluence were seeded onto 6-well culture plates. After 24 hours, negative control siRNA (cat. 4390843, Ambion, Foster City, CA) or CXCR4 siRNA (cat. 4390824, Ambion) with OPTI-MEM (cat. 31985–062, Life Technologies, Carlsbad, CA) were transfected into PCa cells using Lipofectamine RNAiMAX (cat. 56532, Life Technologies) according to the manufacturer’s instructions. Transfected cells were incubated at 37°C for 72 hours and the cells were used to various cell assays. Silencing was verified by Western blot.
FACS analysis
For analysis of a cancer stem cell phenotype (CD133+/CD44+), overexpression of CXCL12γ in PCa or control cells (PC3, DU145) (1 × 105) were seeded onto 12-well culture plates and were cultured for 4 days. The cells were incubated with PE-anti-CD133 antibody (cat. 130–080-901, Miltenyi Biotec, San Diego, CA) and APC-anti-CD44 antibody (cat. 559942, BD Biosciences, San Jose, CA) for 20 min at 4°C. For CXCR4 positive cell analysis, the cells were incubated with PE-anti-human CD184 (CXCR4) antibody (cat. 306506, BioLegend, San Diego, CA) or mouse IgG-PE (cat. 130–092-212, Miltenyi Biotec) for 20 min at 4°C. The CD133+/CD44+ or CXCR4 positive fractions were analyzed with a FACS Aria High-Speed Cell Sorter (BD Biosciences). Apoptosis was measured by flow cytometry (FACSAria dual laser flow-cytometer, Becton Dickinson, Mountainview, CA) using PE Annexin V Apoptosis Detection Kit I (cat. 559763, BD Biosciences, San Jose, CA). The PCa cells were pretreated AMD3100 (5μg/ml) or siCXCR4 and treated with of docetaxel (Taxotere; 0.5–1μg/ml, Hospira, Lake Forest, IL). In some cases, the PCa cells were treated with XTANDI® (enzalutamide; 0.5μg/ml) (Selleck Chemicals, Houston, TX).
Prostatosphere culture and assay
PCa cells which overexpress CXCL12γ or control (PC3, DU145) were dissociated to single cells by standard trypsinization and washed three times with PBS. The cells were plated in stem cell culture medium (DMEM:F12 plus 10 ng/mL bFGF, 20 ng/mL EGF, 5 mg/mL insulin, and 0.4% BSA) supplemented with 1% KO serum replacement (Invitrogen/Gibco, p/n 10828–028) at a density of 1,000 cells/ml in low attachment 6 well culture plates. Seven day old spheres are enumerated as size >50 cells.
Quantitative RT-PCR
Total RNA was extracted from cells using the RNeasy mini or micro kit (Qiagen, Valencia, CA) and converted into cDNA using a First-Strand Synthesis Kit (Invitrogen). Quantitative PCR was performed on an ABI 7700 sequence detector (Applied Biosystems) using TaqMan Universal PCR Master Mix Kit (Applied Biosystems) according to the directions of manufacturer. TaqMan MGB probes (Applied Biosystems) were as follows: CXCL12γ (hsSDF-1YJC), CXCR4 (Hs00237052_m1), AR-V7 (Hs04260217_m1), E-Cadherin (CDH1) (Hs010113953_m1), N-Cadherin (CDH2) (Hs00169953_m1), Vimentin (Hs00185584_m1), α-SMA (Hs00426835_g1), CD44 (Hs01075861_m1), Snail1 (Hs00195591_m1), Snail2 (Hs0095344_m1), ZEB1 (Hs00232783_m1), ZEB2 (Hs00207691_m1), TGFBR2 (Hs00234253_m1), Androgen receptor (AR) ( Hs00171172_m1), N-Myc (NYCN) (00232074_m1), NDRG1 ( Hs00608387_m1), ENO2 (Hs00157360_m1), and Synaptophysin (SYP) (Hs00300531_m1). β-actin (Hs99999903_m1) was used as internal controls for the normalization of target gene expression.
For the analysis of gene expression for circulating tumor cells (CTCs) from healthy normal controls (n=2) and m-CRPC patients (n=26), whole blood (5 ml) was drawn from PCa patients (IRB: 00052405, University of Michigan). CTCs were isolated from blood using anti-EpCAM conjugated Dynabeads (Thermo Fisher Scientific) (31–33). After 5 times wash with PBSF (PBS plus 0.1% FBS), CTCs were lysed on beads with the lysis buffer and stored at −80⁰C. mRNA from CTCs was captured using Oligo(dT)25 mRNA Dynabeads (Thermo Fisher Scientific) and reverse transcribed into cDNA. Target gene amplified library was generated with multiplex PCR with a pooled primer set including 150 genes. Real-Time PCR was then used to determined individual gene expression level. Blood from healthy normal controls (no history of cancer) were used to establish baseline gene expression related to WBC contamination.
CXCL12γ expression was also analyzed in normal human tissues and C57BL/6 mouse tissues (n=3 mice). Total human RNA was purchased from Life technologies; brain (cat. AM 7962), heart (cat. AM7966), lung (cat. AM7968), liver (cat. AM7960), spleen (cat. AM7960), kidney (cat. AM7976), prostate (cat. AM7988), testis (cat. AM7972), and skeletal muscle (cat. AM7982). The human or mouse specific CXCL12γ primers were designed through Life technologies (human CXCL12γ primer, cat. SDF-1YJC; mouse CXCL12γ primer, cat. CXCL12C). The human CXCL12γ TaqMan probes are F–5′-CCA AGA GTA CCT GGA GAA AGC TTT AA-3′, R–5′-CGG TCC ATC GGC AGG AA-3′, TaqMan probe–5′-FAM-TTC TCT TCT TCT GTC GCT TCT-TAMRA-3′ (Applied Biosystems) and the mouse CXCL12γ TaqMan probes are F–5′-CCA AGA GTA CCT GGA GAA AGC TTT AA-3′, R–5′-CAC GGA TGT CAG CCT TCC T-3′, TaqMan probe–5′-FAM-AAG TAA GCA CAA CAG CCC-TAMRA-3′ (Applied Biosystems).
Immunohistochemistry
Cells and tumor sections were used for immunostaining. Cells were fixed and permeabilized with Perm/Wash Buffer (cat. 554723, BD Biosciences). Tumor sections were blocked with Image-iT FX signal enhancer for 30 min and incubated for 2 hours at room temperature with primary antibodies combined with reagents of Zenon Alexa Fluor 488 (green) (cat. Z25002, Invitrogen) or 555 (red) (cat. Z25305, Invitrogen) labeling kit. For the CXCL12γ antibody, a rabbit polyclonal antiserum (anti-CXCL12γ) was raised against the peptide KVGKKEKIGKKKRQ, mapping to the specific C-terminal region of CXCL12γ. The N-terminal cysteine enables direct conjugation of the peptide to the protein carrier and is not present in the native sequence. Peptide synthesis, coupling, immunization, ELISA titration and affinity purification were done by BioGenes GmbH (Berlin, Germany) (29). CXCL12α (mouse, cat. ab25117, Abcam), CD133 (cat. 130–090-423, Miltenyi Biotec), CD44 (cat. ab6124, Abcam), CXCR4 (cat. ab181020, Abcam), CD24 (cat. MS-1279-P, ThermoFisher Scientific, Fremont, CA), ENO2 (cat. NB110–58870, Novus), Synaptophysin (cat. ab23754, Abcam), Pan-Cytokeratin (cat. ab86734, Abcam), αTubulin (cat. ab52866, Abcam) antibodies were used as primary antibody. After washing with PBS, tumor sections were mounted with ProLong Gold antifade reagent with DAPI (cat. P36931, Invitrogen). Images were taken with Olympus FV-500 confocal microscope. In some cases, H&E stain, ENO2, Synaptophysin, Pan-Cytokeratin antibodies were used for immunohistochemistry (IHC) staining in s.c. tumors. Images were taken with Olympus 51Amicroscope. Human prostate tissue microarrays (TMAs) were obtained from The Tissue Core of the University of Michigan Comprehensive Cancer Center and US Biomax, Inc. (Rockville, MD). TMAs consist of normal prostate tissues (n=8), benign (n=6), Gleason 6–7 prostate cancer tissues (n=12), Gleason 9–10 prostate cancer tissues (n=25). Tumors were graded using stage progressing system. Human prostate metastatic tissues (lymph node (n=15), lung (n=10), liver (n=12), and bone (N=19)) also obtained from The Tissue Core of the University of Michigan Comprehensive Cancer Center. Staining intensity was ranked on a scale from 1 to 4 (1, negative; 2, weak; 3, moderate; and 4, strong).
Western blots
Whole cell lysates were separated on 4–20% Tris-Glycine gel and transferred to PVDF membranes. The membranes were incubated with 5% milk for 1 hour and incubated with primary antibodies overnight at 4°C. Primary antibodies used included anti-MARCKS (1:1000 dilution, cat. 5607, Cell Signaling), anti-pMARCKS ((Ser159/163) (1:1000 dilution, cat. 11992, Cell Signaling), anti-PKC alpha (1:1000 dilution, cat. 2056, Cell Signaling), anti-p-PKCalpha (1:1000 dilution, cat. 9373, Cell Signaling), monoclonal-anti-NFκBp65, 1:1000 dilution, cat. 8242, Cell Signaling), monoclonal-anti-p-NFκBp65, 1:1000 dilution, cat. 3033, Cell Signaling), anti-CXCR4 (1:1000 dilution, cat. ab181020, Abcam), polyclonal anti-N-Myc (1:1000 dilution, cat. 9405, Cell Signaling), monoclonal NDRG1 (1:1000 dilution, cat. 9485, Cell Signaling), polyclonal ENO2 (1:1000 dilution, cat. 2056, Cell Signaling), polyclonal Synaptophysin (1:1000 dilution, cat. 4329, Cell Signaling. Blots were incubated with peroxidase-coupled anti-mouse IgG secondary antibody (cat. 7076, 1:2,000 dilution, Cell Signaling) or peroxidase-coupled anti-rabbit IgG secondary antibody (cat. 7074, 1:2,000 dilution, Cell Signaling) for 1 hour, and protein expression was detected with SuperSignal West Dura Chemiluminescent Substrate (cat. Prod 34075, Thermo Scientific, Rockford, IL). Membranes were reprobed with monoclonal anti-β-actin antibody (1:1000 dilution; cat. 4970, Cell Signaling) to control for equal loading.
Protein Kinase C (PKC) ELISA
(PC3 or DU145) (1 × 105) were seeded onto 12-well culture plates and cultured for 4 days. Whole cell lysates were prepared to evaluate PKC kinase activity by the following to the directions of manufacturer (cat. ADI-EKS-420A, Enzo Life Sciences, Farmingdale, NY). The levels of PKC kinase activity were normalized to total cell numbers.
Enolase 2, Chromogenin A, and PSA ELISA
For the analysis of serum Enolase 2, Chromogenin A, and PSA levels for circulating tumor cells (CTCs) from PCa patients, serum samples were collected from whole blood, which were drawn from PCa patients (IRB: 00052405, University of Michigan). Serum Enolase 2 and Chromogenin A were evaluated following manufacturer’s directions (Enolase2/NSP, cat. DENL20, R&D System; Chromogenin A, cat. DY9098, R&D System). PSA data were provided by University of Michigan Cancer Center (IRB: 00052405, University of Michigan).
NFκB luciferase reporter assay
Dual-luciferase reporter assays (cat. E1910, Dual-Lucuferase® Reportor Assay System, Promega, Madison, WI) were performed according to the manufacturer’s instructions. Cells plated in 24-well plates were transiently transfected with the NFκB promoter-firefly luciferase plasmid and the internal control plasmid pRL-TK (Promega), which encodes Renilla luciferase and is used to normalize transfection efficacy. The cells were incubated for 48 h. All firefly luciferase activity was normalized to the Renilla luciferase activity or total DNA (cat. P11496, picoQuant-iT™ PicoGreen® dsDNA Assay Kit, Molecular Probes), and data were presented as relative luciferase activity where control values were set to 1 for each cell line.
Subcutaneous tumor growth and in vivo metastasis assay
Tumors were established by injecting PCa cells (CXCL12γ overexpressing PCa cells or controls) or BCa cells (CXCL12γ overexpressing BCa cells or controls) (1 × 106) in growth factor-reduced Collagen Type I gel (cat. 354236, BD Bioscience, Bedford, MA) s.c. into 5–7 week-old male or female SCID mice. The animals were monitored daily. Tumor volumes were calculated using the formula V= (the shortest diameter) x (the longest diameter) x height. After 4 weeks the animals were sacrificed. The tumors were measured and prepared for histology. For in vivo metastasis assays, immunohistochemistry for PCa cells in the marrow was also used for metastasis assays. The numbers of PCa cells were quantified on the endosteal region of the 7 long bones defined as 10 cell diameters from the bone surfaces.
In vivo isolation of disseminated prostate cancer
The PCa cells overexpressing CXCL12γ or control cells (1 × 106 cells) were injected into male CB.17. SCID mice (5–7 weeks of age: Charles River, Wilmington, MA) by intracardiac (i.c.) injection. Groups were PBS (n=2), PC3-control cells (n=7), and PC3 CXCL12γ overexpressing cells (n=8). After 72 hrs, brain, lung, liver, and bone (spine, pelvis, femur, and tibia) tissues were collected. The cells from animal tissues were incubated first with a RBC lysis buffer (Qiagen, cat. 8305333), and then were incubated with a PE/Cy7 anti-mouse H-2Kd (MHC) antibody (cat. 116622, Biolegend) and APC/Cy7 anti-human HLA-ABC antibody (cat. 311426, Biolegend) for 30 min at 4°C. The disseminated prostate tumor cells (PCa DTC) were analyzed with a FACSAria II Cell Sorter by gating on mouse MHC negative and HLA-ABC positive cells. Data are presented as mean ± SD (Student’s t-test).
Statistical analyses
Results are presented as mean ± standard deviation (s.d.) of mean. Significance of the difference between two measurements was determined by unpaired Student’s t-test. Correlation was analyzed with simple linear regression to calculate Pearson’s correlation coefficient (r) and P value. Analyses were conducted in with GraphPad Prism version 7 software. Values of P < 0.05 were considered significant.
Results
CXCL12γ is associated with malignancy in PCa
We previously reported that elevated expression of CXCL12 and its receptor CXCR4 are found in PCa tissues and cell lines, and are associated with increasing malignant potential (22). Here, the role of the alternatively transcribed CXCL12 isoform CXCL12γ, an intracellular chemokine, was explored in relation to prostate tumor progression. Using human prostate tumor and metastatic tissue microarrays (TMAs), CXCL12γ expression was examined using a polyclonal antibody raised against a peptide specific to the C-terminal region of CXCL12γ. Little or no expression of CXCL12γ was detected in normal prostate and benign PCa tissues and relatively low levels of CXCL12γ expression were observed in low-grade PCa tissues and significant expression of CXCL12γ was detected in high-grade PCa tissues dominated by small cell carcinoma (Fig. 1A and 1B). In the metastatic tissues from m-CRPC patients, significant CXCL12γ expressing small cell carcinoma was detected in lymph node, lung, liver, and bone tissues (Fig. 1C and 1D). In contrast, CXCL12γ expression was not detected in regions dominated by luminal type of adenocarcinoma cells from the high-grade PCa tissues (Fig. 1A; Supplementary Fig. S1A). CXCL12γ expression in bone metastases from m-CRPC patients mirrored that of the primary lesions; CXCL12γ expression was observed in epithelial regions (cytokeratin-positive) dominated by a small cell phenotype, whereas it was not observed in regions dominated by luminal type of adenocarcinoma cells (Fig. 1E; Supplementary Fig. S1C).
Figure 1. CXCL12γ expression in PCa patient tissues and m-CRPC cells.
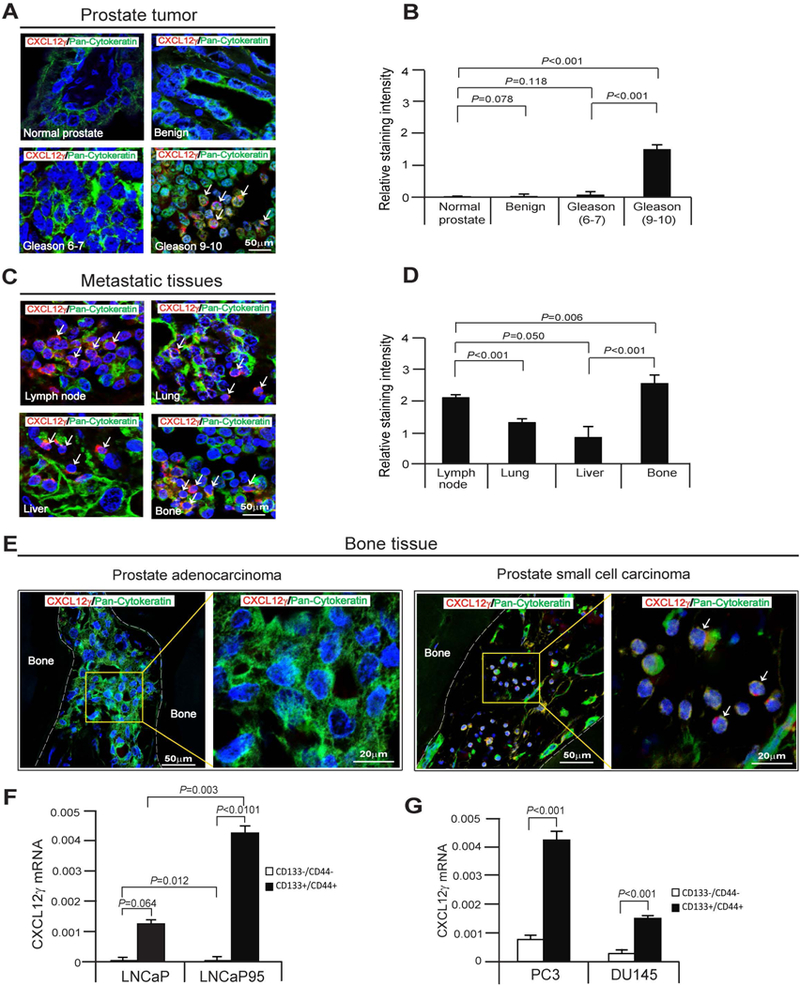
A, Human prostate tissue microarrays (TMAs) from The Tissue Core of the University of Michigan Comprehensive Cancer Center and US Biomax Inc., were stained for CXCL12γ (red) or pan-cytokeratin (green). Cells expressing both markers are indicated with white arrows. DAPI nuclear stain (blue). Bar=50μm. TMAs consist of normal prostate tissues (n=8), benign (n=6), Gleason 6–7 prostate cancer tissues (n=12), Gleason 9–10 prostate cancer tissues (n=25). B, Quantification of Fig. 1A. C, Human prostate metastatic tissues from The Tissue Core of the University of Michigan Comprehensive Cancer Center were stained for CXCL12γ (red) or pan-cytokeratin (green). Metastatic tissues are lymph node (n=15), lung (n=10), liver (n=12), and bone (N=19). D, Quantification of Fig. 1C. E, Left panel: CXCL12γ (red) expression in pan-cytokeratin (green) expressing prostate adenocarcinoma in human bone tissue as detected by immunofluorescence staining, and the magnification of the yellow rectangle from left panel. Bar=20–50μm. Right panel: CXCL12 γ (red) expression in pan-cytokeratin (green) expressing a small cell carcinoma phenotype in human bone tissue as detected by immunofluorescence staining, and the magnification of the yellow rectangle from right panel (white arrows). Bar=20–50μm. F, mRNA expression of CXCL12γ in CSCs (CD133+/CD44+) or non-CSCs (CD133-/CD44-) from LNCaP cells or a castration-resistant sub-line of the LNCaP cells, LNCaP95 as quantified by real-time PCR. G, mRNA expression of CXCL12γ in CSCs (CD133+/CD44+) or non-CSCs (CD133-/CD44-) from the metastatic cell lines PC3 and DU145 as quantified by real-time PCR. Data in Fig. 1B, 1D, 1F, and 1G are representative of mean ± SD (Student’s t-test).
The intriguing observation that CXCL12γ expression is associated with progressive disease and with cellular regions dominated by a small cell phenotype, raises the possibility that CXCL12γ may serve as a marker of CSCs in m-CRPC. To address these possibilities, cells enriched for CSC-like activities were isolated from PCa cell lines based upon their expression of CD133 and CD44 (14) and examined for CXCL12γ expression. CXCL12γ expression in both hormone sensitive and castration resistant CSCs were significantly enhanced relative to non-CSCs (Fig. 1F). Importantly, even amongst the CSC populations, CXCL12γ expression was significantly enhanced in castration resistant cell line LNCaP95 compared to the hormone sensitive cell line LNCaP (Fig. 1F). CXCL12γ expression was also significantly enhanced in CSCs relative to non-CSCs from the metastatic cell lines PC3 and DU145 (Fig. 1G). These data suggest that CXCL12γ expression is associated with the CSC phenotype and m-CRPC.
CXCL12γ induces cancer stem cells (CSCs), which promotes prostate tumor growth and metastasis
Under normal culture conditions, little or no expression of CXCL12γ was detected in human PCa cell lines (Supplementary Fig. S2A) or human breast cancer (BCa) cell line MCF7 (Supplementary Fig. S3A). To further explore the role that CXCL12γ plays in tumor progression, CXCL12γ was overexpressed in PCa (PC3, DU145) and BCa cell lines (MCF7). Overexpression of CXCL12γ was confirmed at the RNA and protein levels (Fig. 2A and 2B; Supplementary S3A and S3B). As higher levels of CXCL12γ expression were observed in the CSC population in castration-resistant PCa cells in Fig. 1F, studies were performed to evaluate the impact of CXCL12γ on the CSC phenotype in vitro. Overexpression of CXCL12γ led to a significant increase in the expression of the CSC phenotype (Fig. 2C) and facilitated the development of spheres in culture (Fig. 2D; Supplementary S3C and S3D).
Figure 2. CXCL12γ induces CSCs, which promotes prostate tumor growth and metastasis.
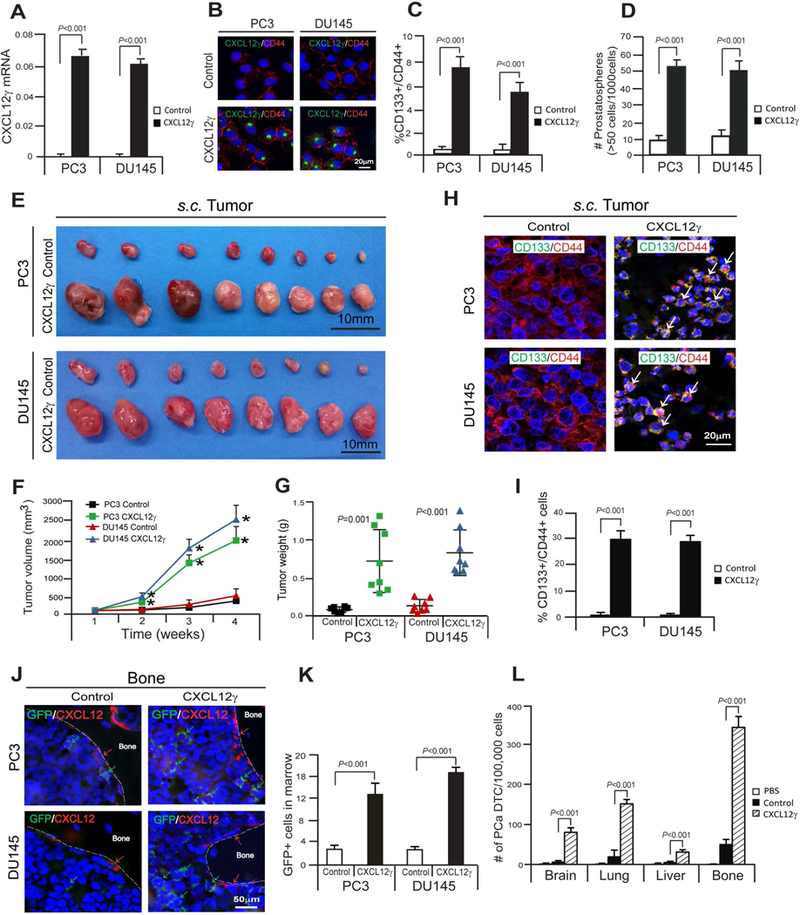
A, Verification of CXCL12γ mRNA expression in CXCL12γ overexpressing PCa cells, PC3 and DU145, as quantified by real-time PCR. B, Verification of CXCL12γ protein expression in CXCL12γ overexpressing PCa cells, PC3 and DU145 as detected by immunofluorescence staining. CXCL12γ (green). CD44 (red). DAPI nuclear stain (blue). Bar=20μm. C, FACS analyses of the CSC (CD133+/CD44+) phenotype in CXCL12γ overexpressing or control PCa cells. D, The formation of prostatospheres in CXCL12γ overexpressing or control PCa cells. Data in Fig. 2A, 2C, and 2D are presented as mean ± SD (Student’s t-test). E-G, The GFP-expressing PCa cells overexpressing CXCL12γ or control cells were implanted s.c. into 5–7 week-old male SCID mice (n=8/group). After 4 weeks, mice were sacrificed and tumor growth was evaluated. *P<0.001. H, Immunostaining of CD133 (green) and CD44 (red) (white arrow) of s.c. PCa tumors generated by CXCL12γ overexpressing PCa cells or control cells. I, Quantification of Fig. 2H. Data are representative of mean ± SD (Student’s t-test). J, GFP-expressing PCa cells were identified in the femur of SCID mice following s.c. injection in Fig. 2E. Green arrows identify GFP-expressing PCa cells. Red arrows identify osteoblast on the bone surface staining positive for CXCL12 expression. Bar=50μm. K, Quantification of Fig. 2J. The numbers of GFP-expressing PCa cells were quantified on the endosteal region of the 10 long bones. Endosteal regions were defined as 12 cell diameters from bone surfaces. Mean ± SD (Student’s t-test) (n=10). L, PCa cells overexpressing CXCL12γ or control cells (1 × 106 cells) were injected into male CB.17. SCID mice (5–7 weeks of age: Charles River, Wilmington, MA) by intracardiac (i.c.) injection. Groups were PBS (n=2), PC3-control cells (n=7), and PC3 CXCL12γ overexpressing cells (n=8). Disseminated prostate tumor cells numbers (PCa DTC) were analyzed on a FACSAria II Cell Sorter by gating on mouse MHC negative and HLA-ABC positive cells from brain, lung, liver, and bone tissues 72 h later. Data are presented as mean ± SD (Student’s t-test).
To validate that CXCL12γ plays an active role in tumor growth, PCa cells or BCa cells were implanted subcutaneously (s.c.) into SCID mice and evaluated for tumor growth. Intriguingly, tumors generated from the CXCL12γ overexpressing PCa or BCa cells grew significantly larger than tumors generated from the control cells (Fig. 2E-2G; Supplementary S3E-S3G). We further confirmed that significant numbers of CSCs (CD133+/CD44+ cells in PCa tumors; CD24-/CD44+ cells in BCa tumors) were detected in tumors generated with CXCL12γ overexpressing cancer cells compared with control cells (Fig. 2H and 2I; Supplementary S3H and S3I). Consistent with more aggressive and larger tumor growth observed following s.c. injection, enhanced numbers of disseminated tumor cells (DTCs) were observed in bone tissues of these animals following injection of CXCL12γ overexpressing PCa cells compared with control cells (Fig. 2J and 2K). The enhanced number of DTCs found in the bone marrows of these animals was likely made possible by the induction of an epithelial to mesenchymal transition (EMT) in cells overexpressing CXCL12γ (Supplementary S2B; Supplementary S3J). To further validate that CXCL12γ plays an active role in tumor metastasis, the PCa cells overexpressing CXCL12γ or control cells were injected intracardially (i.c.) into SCID mice and evaluated for DTCs. Significant numbers of PCa DTC were observed from PC3 CXCL12γ overexpressing cells compared to PC3-control cells in brain, lung, liver, and bone tissues (Fig. 2L; Supplementary S2C). Additionally, we also observed that high levels of CXCL12γ expression are present in normal brain, lung, liver, and heart, adrenal gland, and bone tissues (Supplementary S2D and S2E). Together these data suggest that CXCL12γ induces a CSC phenotype, which promotes PCa tumor outgrowth and ultimately enhanced levels of DTCs in the soft and bone tissues.
CXCL12γ induces neuroendocrine (NE) phenotype, which promotes the development of m-CRPC
NE PCa disease is commonly observed in relapsing individuals particularly after treatment with second generation anti-androgens (e.g., abiraterone and enzalutamide) (10,12,14,34–37). Emerging evidence indicates that the NE cell differentiation and more aggressive disease are associated with enhanced expression of CSC and EMT phenotypes (14,34,35). To explore the extent to which CXCL12γ expression is associated with NE differentiation, we examined the expression of a NE phenotype in CXCL12γ overexpressing PCa cells or BCa cells. In vitro, NE markers were significantly increased in CXCL12γ overexpressing PCa cells or BCa cells compared to control cells (Fig. 3A and 3B; Supplementary S4A). To validate the in vitro studies, enolase-2 (ENO2) and synaptophysin staining were performed on the tumors generated with CXCL12γ overexpressing PCa cells or BCa cells compared to control cells. A high density of NE cells with extensive vascularization (Fig. 3C and 3D; Supplementary S4B, H&E stain), ENO2, and synaptophysin expression were observed in tumors generated from the cells overexpressing CXCL12γ (Fig. 3C and 3D; Supplementary S4B). Pan-cytokeratin expression was used to confirm that these NE PCa cells were of human origin (Fig. 3C and 3D; Supplementary S4B).
Figure 3. CXCL12γ induces a NE phenotype in PCa and CXCL12γ detection in CTCs of PCa patients.
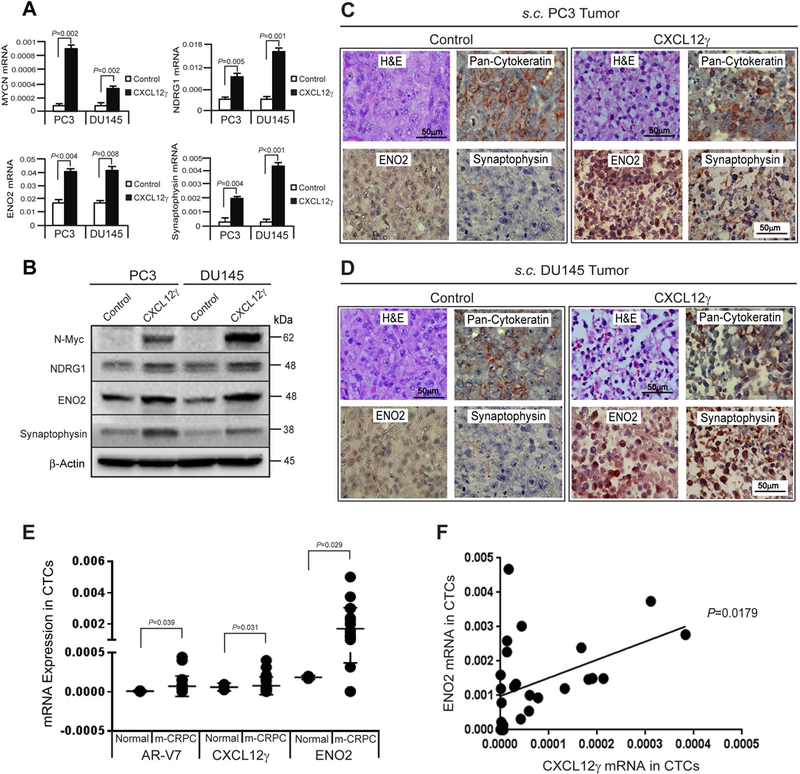
A, mRNA levels of N-Myc (MYCN), N-Myc downstream regulated 1 (NDRG1), enolase-2 (ENO2), and synaptophysin (SYP) expression following CXCL12γ overexpressing or control PCa cells as quantified by real-time PCR. B, Protein levels of N-Myc, NDRG1, ENO2, and synaptophysin in CXCL12γ overexpressing or control PCa cells as quantified by Western blots. Examination of NE markers in s.c. tumors generated by CXCL12γ overexpressing or control PCa cells in SCID mice in Fig. 2E. High density of NE cells with extensive vascularization in C, PC3 tumors and D, DU145 tumors were shown by H&E staining. Bar=50μm. ENO2, synaptophysin, and pan-cytokeratin expressing NE cells as detected by IHC staining. Bar=50μm. E, Gene expression of AR-V7, CXCL12γ, and ENO2 in circulating tumor cells (CTCs) isolated from healthy normal control (n=2) and metastatic castration-resistant PCa (mCRPC) patients (n=26) as quantified by real-time PCR. F, Regression analysis between ENO2 and CXCL12γ. Data are representative of mean ± SD (Student’s t-test).
Detection of circulating tumor cells (CTCs) in blood is a potential prognostic marker in m-CRPC patients and has also been used as a tool to assess gene expression of metastatic tumors (4–6,12). For example, AR-V7-based CTC detection has been shown to be an accurate predictive marker of response to enzalutamide and abiraterone in patients with m-CRPC (4–6). Here, we measured expression of CXCL12γ and the NE marker ENO2 in CTCs of m-CRPC patients in order to assess the association of CXCL12γ with NE disease (Fig. 3E). Critically, we observed that CXCL12γ expression was significantly higher in men with increased ENO2 expression (P=0.018), suggesting that CXCL12γ is associated with a neuroendocrine state (Fig. 3F) (10). Additionally, we observed a trend of positive linear relationship in ENO2 mRNA expression in CTCs and serum enolase and chromogranin A (P=0.129 and P=0.131, respectively) and an inverse linear relationship (P=0.250) in m-CRPC patients (Supplementary S5A-S5C). Together these data suggest that CXCL12γ induces an aggressive NE phenotype, which contributes to the development of m-CRPC.
CXCL12γ contributes activation of CXCR4 signaling in CSC and NE inductions and resistance in PCa
To determine the impact of the major CXCL12 receptor, CXCR4, in CSC activities and resistance, CSCs were examined from hormone sensitive cell line or castration resistant cell line and CXCR4 expression was also examined in CSCs or non-CSCs from hormone sensitive cell line or castration resistant cell line. CSCs were significantly elevated in the castration resistant LNCaP95 cells compared to the parental hormone sensitive LNCaP cells (Fig. 4A) and NE PCa cells (NCI-H660) (Supplementary S6A and S6B). CXCR4 expression in castration resistant CSCs was significantly enhanced relative to hormone sensitive CSCs (Fig. 4B). Total CXCR4 protein levels were enhanced in castration resistant LNCaP95 cells compared to the parental hormone sensitive LNCaP cells (Fig. 4C). These data suggest that CXCL12γ signaling is linked to induction of a CSC phenotype possibly through its receptor, CXCR4. We further verified that the expression levels of CXCL12γ and CXCR4 were dramatically enhanced in cells with a NE phenotype (NCI-H660) compared with PC3 and DU145 cells (Fig. 4D and 4E). Critically, co-localization of CXCL12γ with CXCR4 near the nuclear membrane was observed in NCI-H660 cells (Fig. 4F). We next probed the cells for expression of CXCR4, which is strongly linked to metastasis in solid tumors, including PCa (23). Overexpression of CXCL12γ significantly increased the expression of CXCR4 in PCa cells (Fig. 4G) and BCa cells (Supplementary S7A). Critically, intracellular CXCR4 colocalized with CXCL12γ near the nucleus of CXCL12γ expressing PCa cells in a similar pattern as in neuroendocrine PCa cells (NCI-H660) (Fig. 4H; Supplementary S6C). To further examine CXCR4 involvement in CXCL12γ mediated CSC induction, the prostatosphere formation assays were performed in the presence or absence of the small molecule inhibitor of CXCR4, AMD3100. AMD3100 inhibited the formation of the prostatospheres in control cells, but had minimal impact on the prostatosphere formation in cells overexpressing CXCL12γ, further suggesting that intracellular CXCR4 is more involved in CSC activities than the membrane-binding CXCR4 (Fig. 4I and 4J). Consistent with these observations, blocking CXCR4 signaling with AMD3100 sensitizes PCa cells to docetaxel chemotherapy. However, when cells were induced to overexpress CXCL12γ, AMD3100 did not sensitize PCa cells to docetaxel chemotherapy (Fig. 4K). We also examined the effect of CXCL12γ/CXCR4 signaling on docetaxel-induced microtubule stabilization. CXCL12γ/CXCR4 signaling was significantly countacted docetaxel-induced microtubule stabilization in CXCL12γ overexpressing PCa cells compared to control PCa cells (Fig. 4L). Furthermore, to determine the role of CXCL12γ in AR-dependent PCa cells, CXCL12γ was overexpressed in LNCaP and C42B cells (Supplementary S6D and S6E). The androgen receptor (AR) mRNA was enhanced when CXCL12γ was overexpressed LNCaP and C42B cells (Supplementary S6F). We also observed that less apoptotic cells in CXCL12γ overexpressing LNCaP cells compared to control cells following the enzalutamide treatment and in CXCL12γ overexpressing C42B cells compared to control cells following the docetaxel treatment (Supplementary S6G and S6H). We further found that NE PCa cells (NCI-H660) are highly resistant to docetaxel treatment (Supplementary S6I). Together, these data suggest that the intracellular CXCL12γ and CXCR4 interactions associate with the induction of a CSC phenotype and play a significant role in the regulation of resistance to chemotherapy.
Figure 4. CXCL12γ contributes activation of CXCR4 signaling in CSC and NE inductions and Resistance in PCa.

A, FACS analyses of CSCs (CD133+/CD44+) phenotype in LNCaP cells or LNCaP95 cells. B, mRNA expression of CXCR4 in CSCs (CD133+/CD44+) or non-CSCs (CD133-/CD44-) from LNCaP cells or LNCaP95 cells as quantified by real-time PCR. C, FACS analyses of CXCR4 in LNCaP cells or LNCaP95 cells. Data in Fig. 4A-4C are presented as mean ± SD (Student’s t-test). D, Basal levels of CXCL12γ mRNA expression in neuroendocrine cells (NE; NCI-H660) in comparison to PC3 and DU145 cells as quantified by real-time PCR. E, FACS analyses of CXCR4 in NE cells in comparison to PC3 and DU145 cells. Data in Fig. 4A-4E are presented as mean ± SD (Student’s t-test). F, Immunostaining of CXCL12γ (green) with CXCR4 (red) in NE cells. DAPI nuclear stain (blue). Bar=20μm. G, FACS analyses of % CXCR4 in CXCL12γ overexpressing PCa cells or control cells. H, Immunostaining of CXCL12γ (green) and CXCR4 (red) in CXCL12γ overexpressing or control PCa cells. DAPI nuclear stain (blue). Bar=20μm. I, The formation of prostatospheres in CXCL12γ overexpressing or control PCa cells following with the small molecule inhibitor of CXCR4, AMD3100 treatments as shown by bright-field microscopy. Bar=50μm. J, Quantification of Fig. 4I. K, FACS analyses of % apoptosis of CXCL12γ overexpressing or control PCa cells following docetaxel (DOX) and/or AMD3100 treatments. Data are presented as mean ± SD (Student’s t-test). *P<0.001; +P=0.001–0.01; #P=0.01–0.05. L, Immunostaining analyses of Tubulin stabilization in CXCR4 knock-downed CXCL12γ overexpressing PCa cells (with siControl or siCXCR4 for 72 hrs) or control PCa cells following docetaxel (DOX; 1μg/ml) treatment for 24 hrs. αTubulin (green). DAPI nuclear stain (blue). Bar=20μm.
CXCL12γ contributes activation of CXCR4-mediated PKCα/NFκB signaling in CSC and NE inductions and resistance in PCa
To identify the molecular mechanisms that are functionally associated with CXCL12γ-mediated induction of CSC and NE phenotypes, PKCα/NFκB signaling was explored. We found that PKCα and NFκB signaling were preferentially activated in PCa cells and BCa cells overexpressing CXCL12γ (Fig. 5A-5D; Supplementary S7B and S7C). To prove that PKCα/NFκB activation is responsible for the enhancement of CSC activities, a pan-PKC inhibitor (Go6983) or a NFκB inhibitor (IKK2 inh) was employed. Using each of these inhibitors, significant reductions in CSC phenotype and sphere formation were observed (Fig. 5E-5I). To further prove the role of CXCR4 in the activation of NFκB signaling, we examined the phosphorylation of the NFκB subunit p65 following AMD3100 treatment. AMD3100 inhibited CXCL12-mediated NFκBp65 phosphorylation in parental PCa cells, yet it had little to no impact on NFκBp65 phosphorylation on the CXCL12γ overexpressing PCa cells (Fig. 5J and 5K). To further prove how CXCL12γ/CXCR4-mediated PKCα/NFκB signaling is functionally associated with the development of chemoresistance, myristoylated alanine-rich C-kinase substrate (MARCKS) signaling was explored. Activation of PKC signaling modulates microtubule cytoskeleton and cell morphology (38–40). MARCKS as a downstream target of PKC signaling pathway is known to be critical for regulating multiple pathophysiological processes including chemoresistance (39–41). Recent evidence also shows that docetaxel reduces PKC activity targeting of taxol-mediated suppression of NFκB (40,41). Here, we found that phosphorylation of the MARCKS were preferentially increased in PCa cells overexpressing CXCL12γ (Fig. 5L) and phosphorylation of the MARCKS were significantly reduced along with PKCα and NFκBp65 phosphorylation in CXCR4 knock downed PCa cells overexpressing CXCL12γ (Fig. 5M). These data suggest that the intracellular CXCL12γ and CXCR4 interactions are associated with expression of a CSC phenotype and resistance to chemotherapy through PKCα/NFκB signaling pathways.
Figure 5. CXCL12γ contributes activation of CXCR4-mediated PKCα/NFκB signaling in CSC and NE inductions and resistance in PCa.
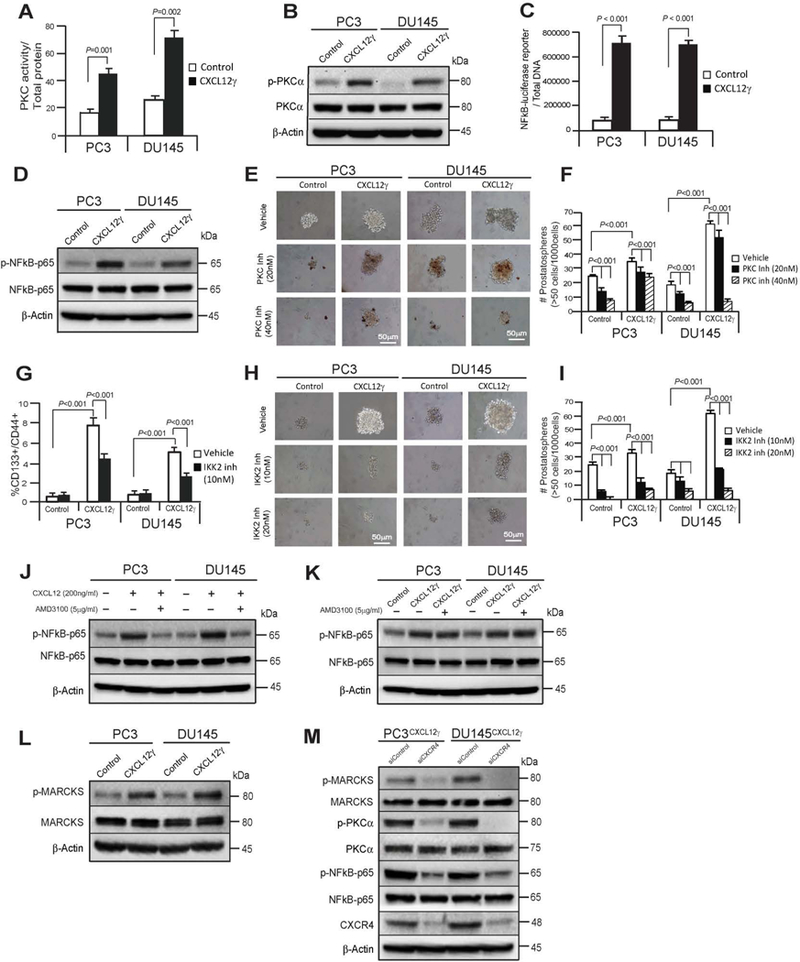
A, Levels of total PKC activity in CXCL12γ overexpressing PCa cells relative to control cells. B, Effect of CXCL12γ on NFκB-reporter luciferase activity. Firefly luciferase was normalized against dsDNA. C, FACS analyses of the CSC (CD133+/CD44+) phenotype in CXCL12γ overexpressing or control PCa cells following NFκB inhibitor (IKK-2 inhibitor VI (IKK2 inh)) treatments. Data in Fig. 5A-5C are representative of mean ± SD (Student’s t-test). D, Immunoblot analysis showing PKCα and its phosphorylation in CXCL12γ overexpressing or control PCa cells as quantified by Western blots. E, The formation of prostatospheres in CXCL12γ overexpressing or control PCa cells following the pan-PKC inhibitor (Go6983) treatments as shown by bright-field microscopy and F, quantification of Fig. 5E. G, Immunoblot analysis showing NFκBp65 and its phosphorylation in CXCL12γ overexpressing or control PCa cells as quantified by Western blots. H, The formation of prostatospheres in CXCL12γ overexpressing or control PCa cells following treatment with an NFκB inhibitor (IKK2 inhibitor VI (IKK2 inh)) as shown by bright-field microscopy and I, quantification of Fig. 5H. Data in Fig. 5F and 5I are representative of mean ± SD (Student’s t-test). J, Immunoblot analysis showing NFκBp65 and its phosphorylation in PCa cells following the CXCL12 and/or AMD3100 treatments as quantified by Western blots and K, in CXCL12γ overexpressing or control PCa cells following AMD3100 treatments. L, Immunoblot analysis showing phosphorylation of myristoylated alanine-rich C-kinase substrate (MARCKS) at Ser159/163 (phospho-MARCKS) in CXCL12γ overexpressing PCa cells or control PCa cells. M, Immunoblot analysis showing reduction of phosphorylation of p-MARCKS, p-PKCα, and p-NFkB-p65 in CXCR4 knock-downed CXCL12γ overexpressing PCa cells.
Discussion
Our studies demonstrate that CXCL12γ expression in PCa regulates induction of CSC and NE phenotypes, promoting PCa tumor outgrowth, metastasis, and chemoresistance, which support the significance impacts of CXCL12γ on the development of m-CRPC.
Despite many advances in therapy for m-CRPC, improvements in survival have generally been on the order of months rather than years (1–7). One possible explanation for this observation is the emergence of metastatic tumor clones or cells with tumor initiating potential or CSCs, which are extraordinarily resistant to conventional therapy (8–11). This biologic process is mediated by therapy-initiated factors and pathways in the tumor. The second-generation anti-androgen therapies alter intrinsic factors and signaling pathways including AR, PI3K/AKT/PTEN, HIF, IGF-1, Wnt, IL-6, SOX2, NANOG, and many others within tumor cells, which facilitate CSC expansion and subsequent m-CRPC development (11). Furthermore, CSCs are associated in the transition from androgen-sensitive to castration-resistant phenotype. For example, PKCα/NFκB signaling is implicated in activating stem-like prostate tumor-initiating cells (TICs) and activation of NFκB signaling, which is thought to contribute to the development of m-CRPC in androgen receptor (AR) independent cells (24,42). These studies support our findings that CXCL12γ induces a CSC phenotype through CXCR4-mediated activation of PKCα/NFκB signaling, which promotes PCa tumor outgrowth and metastasis (43–46).
Additionally, the chemotherapy with docetaxel alters protein targets involved in cell survival, normal physiological functions, and oncogenesis (47–49). Docetaxel also increases circulating cytokines in the castration resistant PCa patients (48). CXCL12-CXCR4 signaling is known to prevent docetaxel-induced microtubule stabilization via p21-activated kinase 4 (PAK4)-dependent activation of LIM domain kinase 1 in PCa cells (47). Activation of PKC signaling modulates microtubule cytoskeleton and cell morphology (38–40) and MARCKS as a downstream target of PKC signaling pathway is known to be critical for regulating multiple pathophysiological processes including chemoresistance (39–41). Recent evidence also shows that docetaxel reduces PKC activity by targeting of taxol-mediated suppression of NF-kB (40,41). These studies support our data demonstrating that intracellular CXCL12γ and CXCR4 interactions associate with the induction of a CSC phenotype and resistance to chemotherapy through PKCα/NFκB signaling pathways. Thus, understanding the biology of CSC expansion by therapy and by what mechanisms CSCs drive resistance to the cytotoxic effects will implicate the development of potential novel therapeutics.
NE PCa disease has an extremely aggressive clinical course with a high mortality rate and commonly observed in the late stages of the PCa coincident with metastatic spread and resistance to treatment (10,12–15,34–37). NE cells may arise by two possible processes, the differentiation of CSCs into NE cells or transdifferentiation of preexisting adenocarcinoma cells. In the first instance, pre-existing CSCs differentiate into cancer cells with three distinct phenotypes (luminal, basal, and NE) or a cellular reprograming of differentiated epithelial cells to stem-like states poised for differentiating to progenitors.
Cells with a NE phenotype highly express the stem cell surface markers PROM1/CD133 and CD44 (10), which suggests that NE cells share features with CSCs (14,34). Recent studies show that the association of a NE phenotype with CSC and EMT phenotypes such that when injected into nude mice, a highly vascularized tumor with a high density of NE cells expressing low levels of E-cadherin and β-catenin and high levels of vimentin are formed (14,34,35).
As the second possible mechanism, the cells with a NE phenotype arise from transdifferentiation of preexisting adenocarcinoma cells, which have accumulated multiple molecular alterations (14,15,50,51). Growing evidence suggests that adenocarcinoma cells can undergo a transdifferentiation process to become NE PCa cells, which acquire a similar phenotype to normal NE cells and express several NE markers (52,53). In this transdifferentiation process, cell autonomous signaling pathways may facilitate acquisition of a NE state, which in conjunction with microenvironmental clues promotes tumor outgrowth, survival, and therapeutic-resistant properties. Recent findings demonstrate that IL-6, epinephrine, and forskolin induce NE differentiation in PCa cells through activation of intracellular cAMP and protein kinase A (PKA) suggests this possibility (54,55). Together, these studies indicate that multiple signaling pathways are likely active in mechanisms associated with NE differentiation. Furthermore, growing evidences also show that deregulation of cellular signaling by genetic or epigenetic alterations are associated with NE differentiation during the therapies. NE PCa is often associated with the accumulation of new genetic alterations such as loss of tumor suppressors (RB1, PTEN, and p53), TMPRSS2-ERG rearrangement, and expression of proto-oncogenes (Aurka, N-Myc, and Bcl 2) (14,29,36,37,51,56,57). NDRG1 expression involves in metastasis and NE differentiation in PCa cells by loss of RB in a hypoxia-dependent fashion and these biologic events are associated with activation of the NFκB signaling pathway, which contributes to metastatic, castrate resistant or NE disease (57). In this investigation, our findings show that CXCL12γ induces N-Myc, NDRG1, and ENO2 expression in NE phenotype of PCa cells (36,58), which contribute to aggressive disease.
Currently there is a continued need to develop biomarkers that predict response to chemotherapy for m-CRPC patients. While most studies have focused on the important role of the androgen receptor (AR) during the transition to m-CRPC, these approaches do not explain the resistance process in conjunction with alternation of signaling pathways, suggesting that AR is not the only pathway involved in the formation of m-CRPC (7,11). Moreover, circulating tumor cell (CTC) numbers have been shown to correlate with patient outcome with overall survival in m-CRPC. In fact, the androgen-receptor splice variant 7 (AR-V7) is associated with resistance to both enzalutamide and abiraterone in PCa cells and has been shown to be one potential marker for CTC (4). Here, we show that CXCL12γ was significantly higher in men with increased ENO2 expression. Thus, CXCL12γ expression may serve as another potential marker of NE PCa disease and the development of m-CRPC.
Finally, our findings show for the first time that expression of the intracellular chemokine CXCL12γ facilitates the emergence of CSC and NE PCa associated with metastatic progression, chemoresistance, and tumor recurrence. That the molecular signatures of cancer stem cells have tumorigenic capability with a large proliferative potential, suggests that CXCL12γ promotes a cellular reprograming of differentiated epithelial cancer cells to stem-like states poised for differentiation into alternative phenotypes.
Supplementary Material
ACKNOWLEDGEMENTS
The authors wish to thank Taocong Jin and Sasha Meshinchi for technical and logistical support in Molecular Core and Microscopy & Image Analysis Laboratory (MIL), Comprehensive Cancer Center Microscopy Core at University of Michigan, Ann Arbor, Michigan, USA. Dr. Juan C. Ramirez (National Centre for Cardiovascular Research, Spain) kindly provided pLV and pLV-CXCL12γ vectors and CXCL12γ antibody. This work is directly supported by the National Cancer Institute (CA093900, CA163124), the Department of Defense (W81XW-15-1-0413 and W81XWH-14-1-0403), and the Prostate Cancer Foundation Challenge Award 16CHAL05. R.T. receives support as the Major McKinley Ash Collegiate Professor. F.C. receives support from a Career Enhancement Award from the NIH / NCI Prostate Cancer Specialized Program in Research Excellence (SPORE) at the University of Michigan.
Footnotes
CONFLICTS OF INTEREST
The authors disclose no potential conflicts of interest.
References
- 1.Hotte SJ, Saad F. Current management of castrate-resistant prostate cancer. Curr Oncol 2010;17 Suppl 2:S72–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pienta KJ, Esper PS. Risk factors for prostate cancer. Ann Intern Med 1993;118:793–803 [DOI] [PubMed] [Google Scholar]
- 3.Morgan TM, Lange PH, Porter MP, Lin DW, Ellis WJ, Gallaher IS, et al. Disseminated tumor cells in prostate cancer patients after radical prostatectomy and without evidence of disease predicts biochemical recurrence. Clinical Cancer Research 2009;15:677–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Qu F, Xie W, Nakabayashi M, Zhang H, Jeong SH, Wang X, et al. Association of AR-V7 and Prostate-Specific Antigen RNA Levels in Blood with Efficacy of Abiraterone Acetate and Enzalutamide Treatment in Men with Prostate Cancer. Clin Cancer Res 2017;23:726–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scher HI, Lu D, Schreiber NA, Louw J, Graf RP, Vargas HA, et al. Association of AR-V7 on Circulating Tumor Cells as a Treatment-Specific Biomarker With Outcomes and Survival in Castration-Resistant Prostate Cancer. JAMA Oncol 2016;2:1441–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med 2014;371:1028–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beltran H, Beer TM, Carducci MA, de Bono J, Gleave M, Hussain M, et al. New therapies for castration-resistant prostate cancer: efficacy and safety. Eur Urol 2011;60:279–90 [DOI] [PubMed] [Google Scholar]
- 8.Cojoc M, Mäbert K, Muders MH, Dubrovska A. A role for cancer stem cells in therapy resistance: cellular and molecular mechanisms 2015. Elsevier; p 16–27. [DOI] [PubMed] [Google Scholar]
- 9.Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nature Reviews Cancer 2005;5:275–84 [DOI] [PubMed] [Google Scholar]
- 10.Borges GT, Vencio EF, Quek SI, Chen A, Salvanha DM, Vencio RZ, et al. Conversion of Prostate Adenocarcinoma to Small Cell Carcinoma-Like by Reprogramming. J Cell Physiol 2016;231:2040–7 [DOI] [PubMed] [Google Scholar]
- 11.Ojo D, Lin X, Wong N, Gu Y, Tang D. Prostate Cancer Stem-like Cells Contribute to the Development of Castration-Resistant Prostate Cancer. Cancers (Basel) 2015;7:2290–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chan F, Goodman O, Fink L, Vogelzang NJ, Pomerantz D, Khoury JD. Dramatically elevated circulating tumor cell numbers in a patient with small cell neuroendocrine carcinoma of the prostate. Arch Pathol Lab Med 2010;134:120–3 [DOI] [PubMed] [Google Scholar]
- 13.Nelson E, Cambio A, Yang J, Ok J, Lara PN, Evans CP. Clinical implications of neuroendocrine differentiation in prostate cancer. Prostate cancer and prostatic diseases 2007;10:6–14 [DOI] [PubMed] [Google Scholar]
- 14.Conteduca V, Aieta M, Amadori D, De Giorgi U. Neuroendocrine differentiation in prostate cancer: current and emerging therapy strategies. Crit Rev Oncol Hematol 2014;92:11–24 [DOI] [PubMed] [Google Scholar]
- 15.Parimi V, Goyal R, Poropatich K, Yang XJ. Neuroendocrine differentiation of prostate cancer: a review. American journal of clinical and experimental urology 2014;2:273. [PMC free article] [PubMed] [Google Scholar]
- 16.Kryczek I, Wei S, Keller E, Liu R, Zou W. Stroma-derived factor (SDF-1/CXCL12) and human tumor pathogenesis. American Journal of Physiology-Cell Physiology 2007;292:C987–C95 [DOI] [PubMed] [Google Scholar]
- 17.Begley LA, MacDonald JW, Day ML, Macoska JA. CXCL12 activates a robust transcriptional response in human prostate epithelial cells. Journal of Biological Chemistry 2007;282:26767–74 [DOI] [PubMed] [Google Scholar]
- 18.Jung Y, Kim JK, Shiozawa Y, Wang J, Mishra A, Joseph J, et al. Recruitment of mesenchymal stem cells into prostate tumours promotes metastasis. Nature communications 2013;4:1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Q, Diao X, Sun J, Chen Z. Stromal cell-derived factor-1 and vascular endothelial growth factor as biomarkers for lymph node metastasis and poor cancer-specific survival in prostate cancer patients after radical prostatectomy. Urol Oncol 2013;31:312–7 [DOI] [PubMed] [Google Scholar]
- 20.Wang J, Ying G, Wang J, Jung Y, Lu J, Zhu J, et al. Characterization of phosphoglycerate kinase-1 expression of stromal cells derived from tumor microenvironment in prostate cancer progression. Cancer research 2010;70:471–80 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 21.Wang J, Loberg R, Taichman RS. The pivotal role of CXCL12 (SDF-1)/CXCR4 axis in bone metastasis. Cancer and Metastasis Reviews 2006;25:573–87 [DOI] [PubMed] [Google Scholar]
- 22.Sun YX, Wang J, Shelburne CE, Lopatin DE, Chinnaiyan AM, Rubin MA, et al. Expression of CXCR4 and CXCL12 (SDF-1) in human prostate cancers (PCa) in vivo. J Cell Biochem 2003;89:462–73 [DOI] [PubMed] [Google Scholar]
- 23.Sun YX, Schneider A, Jung Y, Wang J, Dai J, Wang J, et al. Skeletal localization and neutralization of the SDF-1(CXCL12)/CXCR4 axis blocks prostate cancer metastasis and growth in osseous sites in vivo. J Bone Miner Res 2005;20:318–29 [DOI] [PubMed] [Google Scholar]
- 24.Rajasekhar VK, Studer L, Gerald W, Socci ND, Scher HI. Tumour-initiating stem-like cells in human prostate cancer exhibit increased NF-kappaB signalling. Nat Commun 2011;2:162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cavnar S, Ray P, Moudgil P, Chang S, Luker K, Linderman J, et al. Microfluidic source-sink model reveals effects of biophysically distinct CXCL12 isoforms in breast cancer chemotaxis. Integrative Biology 2014;6:564–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ray P, Stacer AC, Fenner J, Cavnar SP, Meguiar K, Brown M, et al. CXCL12-gamma in primary tumors drives breast cancer metastasis. Oncogene 2015;34:2043–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao S, Chang SL, Linderman JJ, Feng FY, Luker GD. A Comprehensive Analysis of CXCL12 Isoforms in Breast Cancer1,2. Transl Oncol 2014 [DOI] [PMC free article] [PubMed]
- 28.Gahan JC, Gosalbez M, Yates T, Young EE, Escudero DO, Chi A, et al. Chemokine and chemokine receptor expression in kidney tumors: molecular profiling of histological subtypes and association with metastasis. J Urol 2012;187:827–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Torres R, Ramirez JC. A chemokine targets the nucleus: Cxcl12-gamma isoform localizes to the nucleolus in adult mouse heart. PLoS One 2009;4:e7570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu L, Cecil J, Peng SB, Schrementi J, Kovacevic S, Paul D, et al. Identification and expression of novel isoforms of human stromal cell-derived factor 1. Gene 2006;374:174–9 [DOI] [PubMed] [Google Scholar]
- 31.Armstrong AJ, Marengo MS, Oltean S, Kemeny G, Bitting R, Turnbull J, et al. Circulating tumor cells from patients with advanced prostate and breast cancer display both epithelial and mesenchymal markers. Molecular cancer research 2011:molcanres 04902010 [DOI] [PMC free article] [PubMed]
- 32.Reyes EE, Gillard M, Duggan R, Wroblewski K, Kregel S, Isikbay M, et al. Molecular analysis of CD133-positive circulating tumor cells from patients with metastatic castration-resistant prostate cancer. Journal of translational science 2015;1 [PMC free article] [PubMed] [Google Scholar]
- 33.Barriere G, Fici P, Gallerani G, Fabbri F, Zoli W, Rigaud M. Circulating tumor cells and epithelial, mesenchymal and stemness markers: characterization of cell subpopulations. Annals of translational medicine 2014;2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Palapattu GS, Wu C, Silvers CR, Martin HB, Williams K, Salamone L, et al. Selective expression of CD44, a putative prostate cancer stem cell marker, in neuroendocrine tumor cells of human prostate cancer. Prostate 2009;69:787–98 [DOI] [PubMed] [Google Scholar]
- 35.McKeithen D, Graham T, Chung LW, Odero-Marah V. Snail transcription factor regulates neuroendocrine differentiation in LNCaP prostate cancer cells. Prostate 2010;70:982–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee JK, Phillips JW, Smith BA, Park JW, Stoyanova T, McCaffrey EF, et al. N-Myc Drives Neuroendocrine Prostate Cancer Initiated from Human Prostate Epithelial Cells. Cancer Cell 2016;29:536–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med 2016;22:298–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goodnight J, Mischak H, Kolch W, Mushinski JF. Immunocytochemical Localization of Eight Protein Kinase C Isozymes Overexpressed in NIH 3T3 Fibroblasts Isoform-specific association with mircofilaments, golgi, endoplasmic reticulum and nuclear and cell membranes. Journal of Biological Chemistry 1995;270:9991–10001 [DOI] [PubMed] [Google Scholar]
- 39.Yang Y, Chen Y, Saha M, Chen J, Evans K, Qiu L, et al. Targeting phospho-MARCKS overcomes drug-resistance and induces antitumor activity in preclinical models of multiple myeloma. Leukemia 2015;29:715–26 [DOI] [PubMed] [Google Scholar]
- 40.Chen C-H, Cheng C-T, Yuan Y, Zhai J, Arif M, Fong LWR, et al. Elevated MARCKS phosphorylation contributes to unresponsiveness of breast cancer to paclitaxel treatment. Oncotarget 2015;6:15194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Spencer W, Kwon H, Crepieux P, Leclerc N, Lin R, Hiscott J. Taxol selectively blocks microtubule dependent NF-kB activation by phorbol ester via inhibition of IkBa phosphorylation and degradation. Oncogene 1999;18:495–506 [DOI] [PubMed] [Google Scholar]
- 42.Trushin SA, Pennington KN, Carmona EM, Asin S, Savoy DN, Billadeau DD, et al. Protein kinase Calpha (PKCalpha) acts upstream of PKCtheta to activate IkappaB kinase and NF-kappaB in T lymphocytes. Mol Cell Biol 2003;23:7068–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McCall P, Bennett L, Ahmad I, Mackenzie L, Forbes I, Leung H, et al. NFκB signalling is upregulated in a subset of castrate-resistant prostate cancer patients and correlates with disease progression. British journal of cancer 2012;107:1554–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jin R, Yi Y, Yull FE, Blackwell TS, Clark PE, Koyama T, et al. NF-κB gene signature predicts prostate cancer progression. Cancer research 2014;74:2763–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jin R, Yamashita H, Yu X, Wang J, Franco OE, Wang Y, et al. Inhibition of NF-kappa B signaling restores responsiveness of castrate-resistant prostate cancer cells to anti-androgen treatment by decreasing androgen receptor-variant expression. Oncogene 2015;34:3700–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rehman AO, Wang CY. CXCL12/SDF-1 alpha activates NF-kappaB and promotes oral cancer invasion through the Carma3/Bcl10/Malt1 complex. Int J Oral Sci 2009;1:105–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bhardwaj A, Srivastava SK, Singh S, Arora S, Tyagi N, Andrews J, et al. CXCL12/CXCR4 signaling counteracts docetaxel-induced microtubule stabilization via p21-activated kinase 4-dependent activation of LIM domain kinase 1. Oncotarget 2014;5:11490–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mahon K, Lin H, Castillo L, Lee B, Lee-Ng M, Chatfield M, et al. Cytokine profiling of docetaxel-resistant castration-resistant prostate cancer. British journal of cancer 2015;112:1340–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li Y, Li X, Hussain M, Sarkar FH. Regulation of microtubule, apoptosis, and cell cycle-related genes by taxotere in prostate cancer cells analyzed by microarray. Neoplasia (New York, NY) 2004;6:158–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zelivianski S, Verni M, Moore C, Kondrikov D, Taylor R, Lin M-F. Multipathways for transdifferentiation of human prostate cancer cells into neuroendocrine-like phenotype. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research 2001;1539:28–43 [DOI] [PubMed] [Google Scholar]
- 51.Terry S, Beltran H. The many faces of neuroendocrine differentiation in prostate cancer progression. Frontiers in oncology 2014;4:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yuan T-C, Veeramani S, Lin M-F. Neuroendocrine-like prostate cancer cells: neuroendocrine transdifferentiation of prostate adenocarcinoma cells. Endocrine-related cancer 2007;14:531–47 [DOI] [PubMed] [Google Scholar]
- 53.Hu C-D, Choo R, Huang J. Neuroendocrine differentiation in prostate cancer: a mechanism of radioresistance and treatment failure. Frontiers in oncology 2015;5. [DOI] [PMC free article] [PubMed]
- 54.Qiu Y, Robinson D, Pretlow TG, Kung H-J. Etk/Bmx, a tyrosine kinase with a pleckstrin-homology domain, is an effector of phosphatidylinositol 3′-kinase and is involved in interleukin 6-induced neuroendocrine differentiation of prostate cancer cells. Proceedings of the National Academy of Sciences 1998;95:3644–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bang Y, Pirnia F, Fang W, Kang W, Sartor O, Whitesell L, et al. Terminal neuroendocrine differentiation of human prostate carcinoma cells in response to increased intracellular cyclic AMP. Proceedings of the National Academy of Sciences 1994;91:5330–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bandyopadhyay S, Pai SK, Hirota S, Hosobe S, Tsukada T, Miura K, et al. PTEN up-regulates the tumor metastasis suppressor gene Drg-1 in prostate and breast cancer. Cancer research 2004;64:7655–60 [DOI] [PubMed] [Google Scholar]
- 57.Labrecque MP, Takhar MK, Nason R, Santacruz S, Tam KJ, Massah S, et al. The retinoblastoma protein regulates hypoxia-inducible genetic programs, tumor cell invasiveness and neuroendocrine differentiation in prostate cancer cells. Oncotarget 2016;7:24284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee E, Wang J, Yumoto K, Jung Y, Cackowski FC, Decker AM, et al. DNMT1 Regulates Epithelial-Mesenchymal Transition and Cancer Stem Cells, Which Promotes Prostate Cancer Metastasis. Neoplasia (New York, NY) 2016;18:553–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.