

Abstract
Efavirenz (EFV), a widely used antiretroviral drug, is associated with idiosyncratic hepatotoxicity and dyslipidemia. Here we demonstrate that EFV stimulates the activation in primary hepatocytes of key cell stress regulators: inositol-requiring 1α (IRE1α) and X-box binding protein 1 (XBP1). Following EFV exposure, XBP1 splicing (indicating activation) was increased 35.7-fold in primary human hepatocytes. In parallel, XBP1 splicing and IRE1α phosphorylation (p-IRE1α, active IRE1α) were elevated 36.4-fold and 4.9-fold, respectively, in primary mouse hepatocytes. Of note, with EFV treatment, 47.2% of mouse hepatocytes were apoptotic; which was decreased to 23.9% in the presence of STF 083010, an inhibitor of XBP1 splicing. Experiments performed using pregnane X receptor (PXR)-null mouse hepatocytes revealed that EFV-mediated XBP1 splicing and hepatocyte death were not dependent on PXR, which is a nuclear receptor transcription factor that plays a crucial role in the cellular response to xenobiotics. Interestingly, incubation with the primary metabolite of EFV, 8-hydroxyefavirenz (8-OHEFV), only resulted in 10.3- and 2.9-fold increased XBP1 splicing in human and mouse hepatocytes and no change in levels of p-IRE1α in mouse hepatocytes. To further probe the structure-activity relationship of IRE1α-XBP1 activation by EFV, 16 EFV analogs were employed. Of these, an analog in which the EFV alkyne is replaced with an alkene and an analog in which the oxazinone oxygen is replaced by a carbon stimulated XBP1 splicing in human, mouse, and macaque hepatocytes. These data demonstrate that EFV and compounds sharing the EFV scaffold can activate IRE1α-XBP1 across human, mouse, and macaque species.
Introduction
Efavirenz (EFV) is a non-nucleoside reverse transcriptase inhibitor widely used for treatment of HIV. Both EFV and EFV-containing multidrug regimens are classified as essential therapies by the World Health Organization (http://www.who.int/medicines/publications/essentialmedicines/en/). However, EFV causes dyslipidemia (Feeney and Mallon, 2011) and hepatotoxicity (Shubber et al., 2013) in some patients, and elevated plasma EFV concentrations have been linked to EFV-induced liver injury (Yimer et al., 2012). Target therapeutic plasma concentration range for EFV is 3–12 μM; however, concentrations can climb dramatically above this, requiring dose adjustment (Fayet Mello et al., 2011). In some instances, plasma EFV concentrations have been reported to meet or exceed 50 μM (Marzolini et al., 2001; Gounden et al., 2010).
EFV is extensively metabolized in the liver by cytochrome P450 (P450) mono-oxygenases (Avery et al., 2013), with the primary mono-oxygenated metabolite being 8-hydroxyefavirenz (8-OHEFV) (Ward et al., 2003). Concentrations of 8-OHEFV can become nearly equal to EFV in plasma during EFV dosing (Ngaimisi et al., 2010; Grilo et al., 2016). One study measured average plasma EFV and 8-OHEFV concentrations of 4.61 and 3.14 μM, respectively, in patients receiving 600 mg of EFV daily for at least 1 month (Grilo et al., 2016). In addition, EFV induces its own metabolism via activation of the pregnane X receptor (PXR) (Ngaimisi et al., 2010; Swart et al., 2012; Sharma et al., 2013; Narayanan et al., 2018), which is a nuclear receptor transcription factor that modulates the cellular response to xenobiotics via altering expression of drug-metabolizing enzymes (Hedrich et al., 2016; Buchman et al., 2018). Previously, we demonstrated that 8-OHEFV causes hepatocyte death in vitro, observing that incubation with either EFV or 8-OHEFV is able to activate cell death in a c-Jun N-terminal kinase (JNK)-dependent fashion in primary human hepatocytes (Bumpus, 2011).
One highly conserved signaling pathway that results in JNK-dependent cell death activation is the inositol-requiring enzyme 1α (IRE1α) arm of the endoplasmic reticulum (ER) stress response (Urano et al., 2000). During ER stress, misfolded proteins accumulate in the ER, stimulating the release of deactivating chaperone binding immunoglobulin protein from IRE1α, allowing this membrane protein to dimerize and autophosphorylate, at which point IRE1α is considered to be activated (Bertolotti et al., 2000; Lee et al., 2008b; Korennykh et al., 2009). When activated, IRE1α can recruit tumor necrosis factor receptor-associated factor 2 and subsequently activate JNK-dependent cell death (Urano et al., 2000). In addition, dimerized IRE1α possess endoribonuclease activity, allowing for targeted cleavage of the mRNA transcript of the transcription factor X-box-binding protein 1 (XBP1), resulting in the loss of an internal 26-nucleotide sequence, which converts this transcript from its unspliced, inactive form (uXBP1) to its spliced, active form (sXBP1) (Lee et al., 2002). As such, the levels of phosphorylated IRE1α and XBP1 splicing are routinely measured experimentally as hallmarks of the activation of this pathway (Sha et al., 2009; Ning et al., 2011; Hur et al., 2012).
It is generally assumed that sXBP1 transcriptional regulation serves a cytoprotective function, as many of its known gene targets consist of membrane biogenesis and ER-associated protein degradation components that work to reduce the build-up of potentially harmful misfolded proteins, as well as to expand the capacity of the ER (Jiang et al., 2015). However, in the instance of hepatotoxicity and hepatic dyslipidemia, sXBP1 has been shown both to prevent and promote these occurrences, depending on the stimuli. Inhibition of XBP1 splicing diminished the negative effects of high-fat diet in mice, which exhibited decreased steatosis, decreased hepatic lipid droplet number, and decreased serum alanine and aspartate transaminases (Lebeaupin et al., 2018). Conversely, hepatocyte-specific IRE1α-null mice, which lack the ability to activate XBP1, show increased hepatic steatosis and lipid content over wild-type (WT) mice both basally and during ER stress activation (Zhang et al., 2011).
With this previous work in mind, we sought to gain a deeper understanding of the impact of EFV and 8-OHEFV on hepatocytes. In doing so, we tested whether EFV and 8-OHEFV activate IRE1α-XBP1 signaling and, on a broader level, the relationship between EFV structure and its ability to activate IRE1α-XBP1. We observed the conservation of this response across genders and between humans versus model organisms. Because EFV causes patient hepatic dyslipidemia and XBP1 regulates lipid homeostasis, we probed for XBP1-dependency in EFV-mediated hepatic lipid accumulation. And finally, we investigated whether activation of either IRE1α-XBP1 signaling plays a role in EFV- and 8-OHEFV-induced hepatotoxicity. Using EFV, 8-OHEFV, and structurally-related compounds to EFV, we aimed to contribute to the growing body of work on the stimuli-specific role of XBP1s in liver disease progression.
Materials and Methods
Chemical Reagents.
Efavirenz (EFV) was obtained from the National Institutes of Health AIDS Research and Reference Reagent Program (Germantown, MD), and had a purity of 99.4% as assessed by the manufacturer using HPLC in comparison with a USP reference standard. EFV analogs and 8-OHEFV were synthesized by Toronto Research Chemicals (Toronto, Canada). Analogs used in this study were (S)-1-(2-amino-5-chlorophenyl)-1-(trifluoromethyl)-3-cyclopropyl-2-propyn-1-ol (1), (R)-5-chloro-α-(cyclopropylethynyl)-2-amino-α-(trifluoromethyl) benzenemethanol (2), (4S)-6-chloro-4-[(1E)-2-cyclopropylethenyl]-1,4-dihydro-4-(trifluoromethyl)-2H-3,1-benzoxazin-2-one (3), rac 6-chloro-1,4-dihydro-4-(1-pentynyl)-4-(trifluoromethyl)-2H-3,1-benzoxazin-2-one (4), (R)-5-chloro-α-(cyclopropylethynyl)-2-[[(4-methoxyphenyl)methyl]amino]-α-(trifluoromethyl)benzenemethanol (5), (S)-5-chloro-α-(cyclopropylethynyl)-2-[[(4-methoxyphenyl)methyl]amino]-α-(trifluoromethyl)benzenemethanol (6), rac N-[4-chloro-2-[3-cyclopropyl-1-hydroxy-1-(trifluoromethyl) -2-propynyl]phenyl]-4-methoxybenzamide (7), (4S)-6-chloro-4-(2-cyclopropylethynyl)-1,4-dihydro-4-(trifluoromethyl)-2H-3,1- benzoxazine (8), (4S)-6-chloro-4-(cyclopropylethynyl)-1,4-dihydro-2-(4-methoxyphenyl)-4-(trifluoromethyl)-2H-3,1-benzoxazine (9), 1-(2-amino-5-chloro-3-methoxyphenyl)-2,2,2-trifluoro Ethanone (10), 6-chloro-4-(2-cyclopropylethynyl)-1,4-dihydro-2-methyl-4-(trifluoromethyl)-2H-3,1-benzoxazine (11), (4S)-6-chloro-4-[(1E)-2-cyclopropylethenyl]-3,4-dihydro-4-(trifluoromethyl)-,2(1H)-quinazolinone (12), (4S)-6-chloro-4-(cyclopropylethynyl)-3,4-dihydro-4-(trifluoromethyl)-2(1H)-Quinazolinone (13), 6-chloro-4-(cyclopropylethynyl)-3,4-dihydro-4-(trifluoromethyl)-2(1H)-quinolinone (14), 6-chloro-4-(cyclopropylethynyl)-4-(trifluoromethyl)-4H-benzo[d][1,3]dioxin-2-one (15), and 7-chloro-1-(cyclopropylethynyl)-1-(trifluoromethyl)isochroman-3-one (16). EFV analogs and 8-OHEFV had a purity of ≥97%, with the exception of analog 9, which had a purity of 95%, as assessed by the manufacturer (using 1H nuclear magnetic resonance and mass spectrometry). Tunicamycin (TM) was purchased from MilliporeSigma (St. Louis, MO), STF 083010 from Tocris Bioscience/Bio-Techne (Minneapolis, MN), staurosporine from Cell Signaling Technology (Danvers, MA), and oleic acid and hydrogen peroxide from MilliporeSigma.
Primary Hepatocyte Isolation and Culture.
Experiments using mice were approved by the Johns Hopkins Animal Care and Use Committee and performed in accordance with Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the US National Institutes of Health. Primary mouse hepatocytes were isolated from C57BL/6J male and female wild-type mice, aged 8–12 weeks (The Jackson Laboratory, Bar Harbor, ME), and from male and female PXR-null mice, aged 8–12 weeks (Taconic Biosciences, Inc., Rensselaer, NY). Hepatocytes were isolated as previously described (Lee et al., 2004). All hepatocyte preparations used for experiments were ≥80% viable upon plating. For mRNA isolation, cells were plated onto 12-well collagen-coated plates (Corning, Corning, NY) at a density of 100,000 viable cells/well. For protein isolation, cells were plated onto six-well collagen-coated plates (Corning) at a density of 240,000 viable cells/well. For cell-staining experiments, cells were plated on rat tail collagen-1 (Thermo Fisher Scientific, Waltham, MA) -coated 18-mm cover slips and placed in noncoated 12-well plates (Falcon/Corning), at a density of 100,000 cells/well. Cells were allowed to adhere overnight following isolation, and new medium was added prior to treatment.
Primary human (male and female) and cynomolgus macaque (male) hepatocytes were purchased from BioIVT (Baltimore, MD). Cells were plated at a density of 700,000 viable cells/well for EFV versus analog incubations and at 100,000 viable cells/well for EFV versus 8-OHEFV with a viability of ≥90%. Cells were plated in 12-well collagen-coated plates. Upon receipt of the cells, the medium was changed to the above described and cells were allowed to acclimate to medium for 4 hours prior to addition of drug. Hepatocytes from all species were cultured in Williams’ E Medium (Gibco/Thermo Fisher Scientific) supplemented with 5% fetal bovine serum (Gibco), 2 mM l-glutamine (Gibco), 100 IU/ml penicillin, and 100 μg/ml streptomycin (Gibco).
Reverse Transcriptase–Polymerase Chain Reaction Measurement of XBP1 Splicing.
EFV, 8-OHEFV, and analogs dissolved in 100% dimethyl sulfoxide (DMSO) were added directly to culture wells (containing 1 ml of medium) for final concentrations of 10, 20, 30, 40, or 50 μM and 0.1% DMSO. For comparison of EFV and 8-OHEFV, cells were incubated with 10 or 50 μM EFV or 8-OHEFV for 4 hours. For time- and concentration-dependency experiments, cells were exposed to concentrations ranging from 10 to 50 μM EFV for 4 hours or 50 μM EFV for 5, 15, 30 minutes, 1, 2, or 4 hours. For comparison of analogs, a final concentrations of 50 μM EFV or analogs of EFV were tested for 4 hours. For PXR-dependency experiments, WT and PXR-null cells were treated with 50 μM EFV for 4 hours. For cotreatments with STF 083010, STF 083010 was added at the same time as EFV for final concentrations of 100 μM inhibitor, 50 μM EFV or analogs, and 0.2% DMSO, with which cells were incubated for 8 hours. Positive control incubations with tunicamycin at 1 μg/ml were performed. After incubations, medium was aspirated, cells were harvested in 0.5 ml TRIzol reagent (Life Technologies/Thermo Fisher Scientific). RNA was isolated following the manufacturer’s protocol for TRIzol reagent. RNA was then quantified (A260) and reverse-transcribed at 50 ng/μl into cDNA (Maxima enzyme; Thermo Fisher Scientific). Polymerase chain reaction (PCR) (Phusion HF enzyme; New England Biolabs, Ipswich, MA) was then performed using primers (purchased from Integrated DNA Technologies, Coralville, IA) that simultaneously amplified both sXBP1 and uXBP1, which was confirmed by Sanger sequencing. The primer sequences used were:
5′-TACGGGAGAAAACTCACGG-3′ (mouse XBP1 forward),
5′-TTCCAGCTTGGCTGATGAGG-3′ (mouse XBP1 reverse),
5′-GCTCGAATGAGTGAGCTGGA-3′ (human and cynomolgus macaque XBP1 forward), and
5′-GGTGGTAAGGAACTGGGTC-3′ (human and cynomolgus macaque XBP1 reverse).
PCR products were resolved using 7.5% polyacrylamide (Bio-Rad, Hercules, CA) Tris-borate EDTA (Quality Biologicals, Gaithersburg, MD) gel electrophoresis. Gels were stained with SYBR safe DNA stain (Invitrogen/Thermo Fisher Scientific) and visualized using UV light with a Bio-Rad ChemiDoc gel imager. Image Laboratory software by Bio-Rad (version 5.2.1) was used for densitometric PCR band quantitation. Densitometric signal for sXBP1 was divided by that for uXBP1 to obtain a semiquantitative measure of XBP1 splicing activation.
Immunoblotting.
For comparison of IRE1α phosphorylation activation by EFV and 8-OHEFV, cells were incubated with EFV at 10, 20, 30, 40, or 50 μM, or 8-OHEFV at 50 μM for 2 hours. For PXR-dependency of IRE1α phosphorylation experiments, cells were treated with EFV at 50 μM for 2 hours. For 4-hydroxynonenal measurements, cell were treated with EFV at 20 μM for 8 hours. DMSO (0.1%) was used for vehicle. Hepatocytes were washed and harvested in phosphate-buffered saline (PBS) (Gibco), lysed in 1× cell lysis buffer (Cell Signaling Technology) with 0.5 mM phenylmethylsulfonyl fluoride (MilliporeSigma) and 1× Halt protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific) by passage through a 27-gauge syringe tip 30 times. Insoluble debris was removed by centrifugation at 3000g for 10 minutes at 4°C. Protein concentration was quantified using a bicinchoninic acid assay (Pierce Protein Biology/Thermo Fisher Scientific), 50 μg of protein per sample was resolved using 10% SDS-PAGE (Bio-Rad), and then transferred to a nitrocellulose membrane (Life Technologies). Blots were then probed using antibodies specific to the following target proteins: phospho-S724-IRE1α (ab48187; Abcam, Cambridge, UK), IRE1α (14C10; Cell Signaling Technology), 4-hydroxynonenal (ab46545; Abcam), β-actin (13E5; Cell Signaling Technology). Probe binding was detected with horseradish peroxidase-conjugated goat anti-rabbit IgG secondary antibody (Cell Signaling Technology) followed by development with SuperSignal West Femto Maximum Sensitivity Substrate (p-IRE1α, and IRE1α) or SuperSignal West Dura Extended Duration Substrate (β-actin) (Thermo Fisher Scientific). Light release was imaged using a Bio-Rad ChemiDoc gel imager. Densitometry quantitation was performed as described above and p-IRE1α signal intensity divided by IRE1α signal intensity.
Lipid Droplet Staining.
Hepatocytes were incubated for 8 hours with EFV at 20 μM (with 0.1% DMSO as a final vehicle concentration) or with EFV at 20 μM in the presence or absence of 100 μM STF083010 (with 0.2% DMSO as a final vehicle concentration). Positive control incubations were with oleic acid at 500 μM. Cells were washed in PBS, fixed in 4% paraformaldehyde in PBS (Affymetrix, Santa Clara, CA) for 7 minutes, permeabilized in 60% isopropanol (Fisher Chemical, Waltham, MA) for 5 minutes, stained with 0.3% Oil Red O (MilliporeSigma) in 60% isopropanol, and counterstained with hematoxylin (Cell Signaling Technology). Cover slips were then attached to slides with SignalStain mounting media (Cell Signaling Technology) and allowed to dry overnight. Images were taken using Olympus BX51TF brightfield microscope equipped with a DP70 color camera, maintained by the Johns Hopkins Medicine Institute for Basic Biomedical Sciences Microscope Facility. Oil Red O stain was quantified using Nikon NIS-Elements, Advanced Research Version 3.22.00, and normalized to cell count for each image.
8-Hydroxydeoxyguanosine Staining.
Following 4 hour exposure to 20 μM EFV, 50 μM EFV, or 200 μM of the positive control hydrogen peroxide (all treatments were in a final vehicle concentration of 0.1% DMSO) cells were washed in PBS, then fixed and permeabilized by incubation at room temperature in pre-chilled 100% methanol (Fisher Chemical) for 5 minutes. Cover slips were blocked using 1% bovine serum albumin (MilliporeSigma), 300 mM glycine (Affymetrix), and 0.1% Triton Xte-100 (MilliporeSigma) in PBS, probed with an antibody specific to 8-hydroxydeoxyguanosine (8-OHdG; Thermo Fisher Scientific, PA1-84172), and probe binding detected with an Alexa Fluor 488-conjugated rabbit anti-goat IgG Superclonal secondary antibody (Thermo Fisher Scientific). Nuclei were counterstained with 4′,6-diamidino-2-phenylindole, dihydrochloride (DAPI; Cell Signaling Technology). Cover slips were mounted as described above and images were obtained using an Olympus BX51 Fluorescence Microscope equipped with a Roper Photometrics Coolsnap HQ CCD camera, maintained by the Johns Hopkins Medicine Institute for Basic Biomedical Sciences Microscope Facility. DAPI was detected using a dichroic 400-nm filter, and for Alexa Fluor 488 a dichroic filter of 495-nm for excitation and a 525/50-nm filter for emission. Images were processed using ImageJ software (NIH, Bethesda, MD). Staining was assessed by cell counting, with cell nuclei costaining for both DAPI and 8-hydroxydeoxyguanosine identified as positive.
Cell Viability Staining.
Cells were treated for 8 hours with EFV, analog 3, or analog 14 at 20 μM in the presence or absence of 100 μM STF083010, or staurosporine alone at 10 μM (all treatments were in a final vehicle concentration of 0.2% DMSO). Cells were then washed in PBS and incubated in 100 μg/ml of ethidium bromide (EtBr; MilliporeSigma) and 100 μg/ml of acridine orange (AcrO; Life Technologies/Thermo Fisher Scientific) diluted in PBS for 15 minutes at room temperature. Cover slips were mounted, fluorescently imaged, and images processed in ImageJ. EtBr was detected using dichroic filter of 593 nm for excitation and a 642/40 nm filter for emission, and for AcrO, a dichroic filter of 495 nm for excitation and a 525/50 nm filter for emission. Viability was assessed by cell counting, with cell nuclei costaining with both AcrO and EtBr identified as apoptotic.
Quantitative Real-Time PCR Measurement of DNJB9 and CYP2B10 Gene Expression.
Hepatocytes were incubated for 8 hours with 20 μM EFV, 20 μM analog 3, 20 μM analog 14, or 1 μg/mL TM for DnaJ heat-shock protein family member 9 (DNAJB9) expression analysis or with 50 μM EFV for 4 hours for CYP2B10 expression analysis (treatments were performed with final vehicle concentrations of 0.1% DMSO). RNA was isolated and reverse-transcribed as described above. Quantitative real-time PCR (qPCR) was performed for DNAJB9 and GAPDH using Maxima SYBR Green qPCR Master Mix (Thermo Fisher Scientific). The primers used during quantitative real-time PCR analyses of mouse DNAJB9, CYP2B10, and GAPDH were as follows:
DNJB9 forward 5′-GGGCGCACAGGTTATTAGAA-3′,
DNAJB9 reverse 5′-ACGCTTCTGCAATCTCTCTGA-3′,
CYP2B10 forward 5′-GCCCAATGTTTAGTGGAGGA-3′,
CYP2B10 reverse 5′-GACTTCTCCTTCTCCATGCG-3′,
GAPDH forward 5′-GAAGGCCGGGGCCCACTTGA-3′, and
GAPDH reverse 5′-TCTCCAGGCGGCACGTCAGA-3′.
The GAPDH PCR product from mouse cDNA was isolated and ligated into a pJET1.2 plasmid using a CloneJET PCR cloning kit (Thermo Fisher Scientific). This plasmid was transformed into and purified from DH5α-competent cells (Invitrogen). Purified plasmid was used to generate a gene-copy-number standard curve used in qPCR analyses. Normalized DNAJB9 expression levels were calculated by dividing DNAJB9 copy number by GAPDH copy number.
Data Analysis.
Statistical analysis was performed in GraphPad Prism 7. For any measures involving ratios (sXBP1/uXBP1, p-IRE1α/IRE1α, qPCR gene expression analysis, and fold change in Oil Red O staining), data were log-transformed, and the data analysis performed on the log-transformed data. For these data sets, fold changes throughout the text are reported as ratios of geometric means. For XBP1 splicing, Western blotting, qPCR, and Oil Red O staining data, statistical significance was determined using an unpaired t test (without assuming consistent S.D., without corrections for multiple comparisons, generating two-tailed P values). For comparison of XBP1 splicing levels across sex, statistical significance from vehicle was determined for treatments in both male and female mouse primary hepatocytes, and for any compounds that showed statistically significant splicing in either group, statistical significance of splicing differences between male and female mouse hepatocytes was assessed. For cell imaging with EtBr/AcrO and 8-OHdG staining a paired-ratio t test was performed to assess statistical significance of fold changes between control and treated values, generating two-tailed P values. P values are shown as: * or # P < 0.05, ** or ## P < 0.01, *** or ### P < 0.001, and n.s. highlighting select nonstatistically significant differences. For values reported in the text, 95% confidence intervals have been provided, and were calculated assuming normal distribution.
Results
Stimulation of IRE1α-XBP1 Activation by EFV in Human and Mouse Primary Hepatocytes.
To investigate whether IRE1α -XBP1 is activated by EFV and 8-OHEFV, XBP1 splicing was quantified following incubation of primary hepatocytes with these two compounds. In primary human hepatocytes, a 35.7-fold (95% CI [10.6, 120.3]) increase in the ratio of unspliced to spliced XBP1 was measured following 4-hour exposure of cells to 50 μM EFV compared with vehicle control, whereas parallel treatments performed using 8-OHEFV only rendered a 10.3-fold increase (95% CI [4.9, 21.7]). Neither compound stimulated splicing at 10 μM in primary human hepatocytes. To test whether the impact of EFV and 8-OHEFV on hepatic XBP1 splicing is conserved in mice, mouse primary hepatocytes were employed. Commensurate with what was observed using human hepatocytes, XBP1 splicing was increased 36.4-fold (95% CI [21.0, 63.3]) in mouse hepatocytes in response to 50 μM EFV. In addition, primary mouse hepatocytes incubated with 10 μM EFV showed a 1.9-fold (95% CI [1.3, 2.9]) increase in XBP1 splicing. In contrast to what was observed using primary human hepatocytes, the presence of 50 μM 8-OHEFV only resulted in a 2.9-fold increase (95% CI [1.5, 6.0]) in spliced XBP1 in mouse hepatocytes. Tunicamycin, a known activator of IRE1α -XBP1 signaling (Yoshida et al., 2001), was used as a positive control in this study, and exhibited an increase in splicing in both human and mouse primary hepatocytes (Fig. 1). To further characterize this response in primary mouse hepatocytes, we examined time- and dose-dependence of EFV activation of XBP1 splicing. When primary mouse hepatocytes were incubated for 4 hours with a range of concentrations of EFV (10–50 μM), XBP1 splicing was elevated in the presence of 30, 40, and 50 μM EFV (Fig. 2A). Following primary mouse hepatocyte treatment with 50 μM EFV for a range of time points (5 minutes to 4 hours), XBP1 splicing was first elevated at 1 hour and this was sustained through 4 hours. The time profile determined for EFV paralleled that of TM (Fig. 2B).
Fig. 1.

XBP1 splicing during incubation of primary human and mouse hepatocytes with EFV and 8-OHEFV. Representative gel images and sXBP1/uXBP1 densitometry quantification ratios of XBP1 mRNA splicing from semiquantitative reverse transcriptase PCR in (A) human primary hepatocytes (n = 6) and (B) male mouse primary hepatocytes (n = 6) in the presence of EFV or 8OH-EFV at 10 or 50 μM for 4 hours. Data are the mean ± S.D. Ratios of sXBP1/uXBP1 densitometry were log-transformed and the statistical significance of TM, EFV, or 8-OHEFV treatments from DMSO (0.1%) control was determined using an unpaired t test on the transformed data, generating two-tailed P values (*P < 0.05; ***P < 0.001).
Fig. 2.

IRE1α-XBP1 signaling during exposure of primary mouse hepatocytes to EFV or 8-OHEFV. Representative gel images and sXBP1/uXBP1 densitometry quantification ratios of XBP1 mRNA splicing from semiquantitative reverse transcriptase PCR in (A) male mouse primary hepatocytes incubated with 10, 20, 30, 40, or 50 μM EFV for 4 hours (mean ± S.D, n = 5), and (B) male mouse primary hepatocytes with 50 μM EFV for 5, 15, 30 minutes, 1, 2, or 4 hours (mean ± S.D., n = 4). (C) Representative images for Western blotting of p-IRE1α, total IRE1α, and β-Actin and densitometry quantification ratios of p-IRE1α/total IRE1α for male mouse primary hepatocytes treated with 10, 20, 30, 40, 50 μM EFV or 50 μM 8-OH EFV for 2 hours (mean ± S.D., n = 3). Ratios of sXBP1/uXBP1 and p-IRE1α/total IRE1α densitometry were log-transformed and the statistical significance of TM, EFV, or 8-OHEFV treatments from DMSO (0.1%) control was determined using an unpaired t test on the transformed data. For (B) treatments were compared with the DMSO control from the same time point. Two-tailed P values were generated for all analyses (*P < 0.05; **P < 0.01; ***P < 0.001).
Since IRE1α is the endoribonuclease responsible for splicing XBP1 mRNA, and phosphorylation of IRE1α is a marker for activation of this enzyme (Lee et al., 2008b), we tested whether IRE1α is phosphorylated in response to EFV or 8-OHEFV. Following 2 hours of treatment, the levels of phosphorylated IRE1α were increased in a dose-responsive manner from 1.5-fold (95% CI [1.2, 1.9]) with 20 μM EFV to 4.9-fold (95% CI [4.1, 5.8]) with 50 μM EFV, compared with vehicle control. Phosphorylation of IRE1α was not elevated above vehicle controls in the presence of 10 μM EFV or 50 μM 8-OHEFV (Fig. 2C).
Impact of EFV Structure on XBP1 Splicing Activation in Primary Mouse Hepatocytes.
The single oxygen insertion changing EFV to 8-OHEFV markedly decreased IRE1α-XBP1 activation. Because of this, we were interested in investigating how other structural changes to EFV impact its ability to activate this signaling event, and whether the structure-activity relationship between EFV and IRE1α-XBP1 activation is conserved across species. A panel of 16 EFV analogs was employed (Fig. 3). The structural changes in these analogs have been previously observed by our group to impact both PXR activation (Narayanan et al., 2018) and metabolism by CYP2B6 (Cox and Bumpus, 2014, 2016). They have been designed to probe specific sections and features of the EFV molecule: the integrity of the oxazinone ring (analogs 1 and 2), the alkyne-cyclopropyl arm (analogs 3 and 4), increases in EFV size (analogs 5–7, and 9), decrease in size (analog 10), and discreet changes to the constituents of the oxazinone ring (analogs 8 and 11–16).
Fig. 3.

EFV, 8-OHEFV, and the structural analogs of EFV used in this study, with differences from EFV highlighted in blue.
In addition to EFV, treatment with several of the 16 analogs tested consistently resulted in XBP1-splicing activation across species (Fig. 4): analog 3, in which the EFV alkyne is changed to a trans-alkene; analog 14, in which the oxazinone ring oxygen has been replaced with a carbon atom; and analogs 12, 13, 14, and 15. Macaque hepatocytes were least sensitive to XBP1-splicing activation in response to both EFV and analogs of EFV (Fig. 4, A and D). TM incubations resulted in XBP1 splicing for all primary hepatocytes tested (Fig. 4, A and C).
Fig. 4.

XBP1 splicing activation in primary hepatocytes in the presence of EFV and structural analogs of EFV. (A) Representative XBP1 mRNA splicing PAGE gel images for male mouse (n = 5 for all but 3 and 14, for which n = 8), female mouse (n = 4), human (n = 4, for all but 14, in which n = 3), and cynomolgus macaque (n = 4 for all but 8, in which n = 3) primary hepatocytes incubated with EFV and analogs of EFV at 50 μM for 4 hours. (B) sXBP1/uXBP1 densitometry quantification ratios of XBP1 mRNA splicing for the above described treatments in male and female mouse primary hepatocytes; (C) in human hepatocytes; and (D) in primary cynomolgus macaque hepatocytes. In male primary mouse hepatocytes, a DMSO (0.1%) control and TM-positive control were run for all replicates, including additional replicates measuring splicing with only analog 3 and analog 14. For statistical analysis of this data from male mouse hepatocytes, each treatment was compared with the DMSO controls that were run in the same preparations of primary hepatocytes. For all data sets, ratios of sXBP1/uXBP1 densitometry were log-transformed and the statistical significance of TM, EFV, or analog treatments from DMSO (0.1%) control was determined using an unpaired t test on the transformed data, generating two-tailed P values (*P < 0.05; **P < 0.01; ***P < 0.001). Ratios were compared between male and female hepatocytes for any treatment that showed significant splicing in either group using an unpaired t test, generating two-tailed P values (#P < 0.05; ##P < 0.01).
To investigate potential sexual dimorphisms in the activation of IRE1α-XBP1 by EFV and EFV analogs, XBP1 splicing was quantified in male and female primary mouse hepatocytes separately (Fig. 4B). XBP1 splicing in vehicle control-treated hepatocytes was not statistically different between the two groups. In the presence of EFV, as well as analogs 3, 5, 6, 7, 12, 14, and 15, similar magnitudes of XBP1 splicing were observed in male and female primary mouse hepatocytes. Of note, analogs 3 and 14 exhibited higher levels of XBP1 splicing than that of EFV in both groups. Using analog 3, XBP1 splicing was 2.4-fold (male) and 1.8-fold (female) higher (95% CI are [0.8, 7.0] and [0.67, 4.9], respectively) than splicing in EFV cells, and using analog 14, splicing was 3.7-fold (male) and 2.7-fold (female) greater (95% CI are [1.2, 11.5] and [1.0, 6.9], respectively) than that measured following treatment with EFV. Analog 11 stimulated splicing to a level that reached statistical significance only in male mouse hepatocytes, although XBP1 splicing was also elevated, albeit not to a statistically significant degree, by this compound in female mouse hepatocytes as well. XBP1 splicing in the presence of analog 13 was measured in both male and female primary hepatocytes but was higher in female (4.1-fold above that in male, 95% CI [2.1, 8.2]). Following incubation with analog 1 or 2, both of which have broken oxazinone rings with missing carbonyl moieties, splicing was detectable over vehicle, demonstrating a 3.5-fold increase for analog 1 and 3.2-fold with analog 2 (95% CI are [2.0, 6.2] and [1.7, 5.9], respectively) in female mouse hepatocytes only (Fig. 4B).
IRE1α-XBP1 Activation with EFV in PXR-Null Mouse Primary Hepatocytes.
EFV is a well known activator of the xenobiotic sensing nuclear receptor PXR (Sharma et al., 2013). We therefore sought to determine whether PXR might play a role in the activation of IRE1α-XBP1 in response to EFV. Of note, EFV activation of XBP1 splicing was not dependent on PXR, since in fact there was a trend toward increased XBP1 splicing in hepatocytes isolated from PXR-null mice versus WT mice, demonstrating 4.2-fold (95% CI [1.1, 18.7]) greater splicing in male PXR-null mouse hepatocytes compared with male WT, and 2.0-fold greater (95% CI [0.64, 6.3]) in female PXR-null mouse compared with female WT, although this difference did not rise to the level of statistical significance (Fig. 5A). Still, in consideration of this increase, we moved forward by probing for differences in the levels of IRE1α phosphorylation between WT and male PXR-null mouse hepatocytes in response to EFV. In line with the above XBP1 splicing data, the levels of phosphorylated IRE1α in PXR-null mouse hepatocytes were commensurate with those of WT mice (Fig. 5B).
Fig. 5.

IRE1α-XBP1 signaling in WT and PXR-null primary hepatocytes during exposure to EFV. (A) Representative XBP1 mRNA splicing PAGE images and densitometry quantification (mean ± S.D.) of XBP1 mRNA splicing from semiquantitative reverse transcriptase PCR for sXBP1 and uXBP1 in WT (n = 5) and PXR-null male (n = 5) mouse primary hepatocytes, as well as WT (n = 5) and PXR-null (n = 5) female hepatocytes treated with EFV and analogs of EFV at 50 μM for 4 hours. (B) Representative images for Western blotting of p-IRE1α, total IRE1α, and β-Actin and densitometry quantification ratios of p-IRE1α/total IRE1α for WT (n = 3) and PXR-null (n = 3) male mouse primary hepatocytes incubated with 50 μM EFV for 2 hours (mean ± S.D.). Ratios of sXBP1/uXBP1 and p-IRE1α/total IRE1α densitometry were log-transformed and the statistical significance of EFV treatments from DMSO (0.1%) control determined using an unpaired t test on transformed data, generating two-tailed P values (**P < 0.01; ***P < 0.001). n.s., not significant.
Given that EFV activates PXR, resulting in the increased expression of P450 drug-metabolizing enzymes, we wanted to investigate whether a change in the expression of EFV-metabolizing P450 enzymes occurs during EFV treatment in these hepatocytes. Though it is unknown which P450 metabolizes EFV in mouse, EFV is primarily metabolized by CYP2B6 in humans (Ward et al., 2003), and because of this we measured expression of mouse CYP2B10, which shares 74% amino acid sequence identity with human CYP2B6 (Davies et al., 2005). The normalized expression of CYP2B10 did not change with EFV treatment (50 μM, 4 hours) compared with the DMSO control in both WT and PXR-null primary mouse hepatocytes. However, the overall expression of CYP2B10 was on average 2.9-fold higher in WT cells compared with PXR-null cells (n = 3, 95% CI [2.1, 4.0], P < 0.0001, unpaired t test).
Effects of Inhibition of XBP1 Splicing on Lipid Droplet Formation with EFV.
Because XBP1’s involvement in lipid biogenesis has been previously demonstrated (Sriburi et al., 2004), and hepatic dyslipidemia is commonly indicated clinically along with EFV toxicity (Feeney and Mallon, 2011), we tested whether we could detect lipid accumulation in mouse hepatocytes following treatment with EFV and, subsequently, if this accumulation was XBP1-dependent. Following 8 hours in the presence of 20 μM EFV, an approximate 7.7-fold (95% CI [3.6, 16.4] increase in lipid droplet formation was measured compared with vehicle in primary mouse hepatocytes (Fig. 6). Since lipid homeostasis has been shown previously to be differentially regulated in WT versus PXR-null mice (He et al., 2013; Choi et al., 2018), we also performed experiments to probe the impact of PXR on EFV-mediated lipid droplet formation. We observed no difference in lipid droplet formation in response to EFV between PXR-null and WT mouse hepatocytes (Fig. 6).
Fig. 6.

Lipid droplet formation in WT and PXR-null primary hepatocytes treated with EFV. Representative images (40×) of Oil Red O (red) and hematoxylin (blue) stained primary mouse hepatocytes and quantitation of Oil Red O signal intensity in WT (n = 4) and PXR-null (n = 4) primary mouse hepatocytes incubated with 20 μM EFV for 8 hours (mean ± S.D.). Fold change was calculated with respect to the mean of the DMSO (0.1%) control within each group, and fold changes were log-transformed. The statistical significance of EFV treatments from DMSO (0.1%) control was determined using an unpaired t test on transformed data, generating two-tailed P values (*P < 0.05; **P < 0.01; ***P < 0.001). n.s., not significant.
To investigate whether EFV increases lipid droplet formation in an XBP1-dependent manner, STF 083010, a chemical inhibitor of IRE1α endoribonuclease activity (Papandreou et al., 2011), was employed. Following 8 hours with 20 μM EFV ± 100 μM STF 083010, there was no observed impact of XBP1 splicing inhibition on EFV-mediated lipid droplet accumulation in primary mouse hepatocytes (Fig. 7A). We did however confirm that cotreatment with STF 083010 at 100 μM decreased the activation of XBP1 splicing by both EFV at 50 μM and TM after 8 hours (Fig. 7B): Though splicing levels were still higher than vehicle with both compounds, XBP1 splicing with EFV was 9.9-fold lower (95% CI [6.1, 16.0]) in the presence of STF 083010.
Fig. 7.

XBP1 splicing and lipid droplet formation in male mouse primary hepatocytes during EFV exposure. (A) Representative images (20×) of Oil Red O (red)- and hematoxylin (blue)-stained primary mouse hepatocytes and quantitation of Oil Red O signal intensity in WT (n = 4) primary mouse hepatocytes in presence of 20 μM EFV for 8 hours ± STF 083010 at 100 μM (mean ± S.D.). Fold change in Oil Red O intensity was calculated with respect to the mean of the DMSO (0.1%) control and fold changes were log-transformed. (B) Representative XBP1 mRNA splicing PAGE images and densitometry quantification (mean ± S.D.) of XBP1 mRNA splicing from semiquantitative reverse transcriptase PCR for sXBP1 and uXBP1 in male mouse primary hepatocytes (n = 4) treated with EFV and analogs of EFV at 50 μM for 8 hours ± STF 083010 at 100 μM. Ratios of sXBP1/uXBP1 and p-IRE1α/total IRE1α densitometry were log-transformed. The statistical significance in both (A and B) was determined for all treatments with respect to DMSO (0.2%) control using an unpaired t test on transformed data, generating two-tailed P values (*P < 0.05; **P < 0.01; ***P < 0.001). Additional un-paired t tests were performed to compare treatments indicated by brackets generating two-tailed P values (##P < 0.01; ###P < 0.001). n.s., not significant.
Primary Hepatocyte Death in Response to EFV and the Role of XBP1 Splicing.
Reactive oxygen species accumulation is one example of a cellular stressor that can result in IRE1α-XBP1 activation (Hanada et al., 2007). Formation of 8-OHdG DNA adducts, which are formed when reactive oxygen species build up in the cell (Kasai et al., 1986), was measured during incubation with EFV. In primary mouse hepatocytes treated for 4 hours, positive 8-OHdG staining increased from 25.3% (95% CI [13.2, 37.3]) with vehicle to 52.2% (95% CI [41.1, 73.9]) with 20 μM EFV, and increased from 20.9% (95% CI [7.7, 34.1]) with vehicle to 52.2% (95% CI [39.9, 64.5]) in the presence of 50 μM EFV (Fig. 8A). The pairing of treated and untreated samples for ratio-paired t test analysis of this data is provided in Supplemental Fig. 1. Given this observed increase in 8-OHdG with EFV, as well as the increase in lipid droplet formation with EFV, we also probed for formation of 4-hydroxynonenal protein adducts, a result of increased lipid oxidation during cellular oxidative stress (Comporti, 1998). After an 8-hour incubation with EFV at 20 μM, no 4-hydroxynonenal protein adducts were observed (data not shown).
Fig. 8.

8-OHdG and EtBr/AcrO viability staining in primary mouse hepatocytes during EFV incubations. (A) Representative images (20×) of 8-OHdG (red)- and DAPI (blue)-stained primary mouse hepatocytes and quantitation (mean ± S.D., n = 4) of 8-OhdG-stained cells after incubations with EFV at either 20 or 50 μM for 4 hours. Positively stained 8-OHdG nuclei are purple. (B) Representative images (20×) of EtBr (red)- and AcrO (green)-stained primary mouse hepatocytes and quantitation (mean ± S.D., n = 5) after treatment with EFV, analog 3 or analog 14 for 8 hours ± cotreatment with STF 083010 at 100 μM. Hepatocytes positive for cell death have yellow/orange nuclei. Ratio-paired t tests were performed to determine significance from DMSO controls or for bracketed comparisons. For (A) five separate isolations of mouse hepatocytes were used to generate these results, and each treatment EFV (20 μM) and EFV (50 μM) was performed four times among these five batches of cells. The pairing of these treatments for statistical analysis is indicated in Supplemental Fig. 1. For (B) STF 083010 (100 μM), EFV (20 μM), and EFV (20 μM) + STF 083010 (100 μM) were performed in the same group of five hepatocyte isolations, and treatments with 3 (20 μM), 3 (20 μM) + STF 083010 (100 μM), 14 (20 μM), and 14 (20 μM) + STF 083010 (100 μM) were performed in a separate set of five isolations. DMSO (0.2%) treatments were performed for each separate isolation, and the pairing of the experimental treatments to their respective DMSO control is shown in Supplemental Fig. 2. A ratio paired t test was used to test for significance from DMSO controls or for bracketed comparisons in both (A and B), generating two-tailed P values (*P < 0.05; **P < 0.01 for significance from DMSO control and ##P < 0.01 for indicated comparisons). n.s., not significant.
To assay the impact of EFV exposure on primary hepatocyte viability, AcrO/EtBr co-staining was used in primary male mouse hepatocytes treated with 20 μM EFV for 8 hours (Figure 8B). In the presence of EFV, a marked increase in EtBr-positive cells was observed, with 47.2% (95% CI [32.3, 62.0]) of cells positive, compared with 10.8% (95% CI [1.59, 20.0]) for vehicle. Because we saw higher levels of XBP1 splicing with analog 3 and analog 14, we also performed EtBr/AcrO viability measurements during incubation with these compounds at 20 μM for 8 hours. Interestingly, analog 3 also resulted in an increase in EtBr positive cells, with 49.7% (95% CI [30.8, 68.6]) positive, as compared to 7.3% (95% CI [4.0, 10.5]) for vehicle. Analog 14 showed no increase in positive EtBr/AcrO staining (Fig. 8B). To assay whether this cell death was dependent on IRE1α endoribonuclease activity, we also performed EtBr/AcrO staining on cells cotreated with 100 μM of the splicing inhibitor STF 083010 and either EFV, analog 3, or analog 14. Cotreatment with this XBP1 splicing inhibitor decreased EFV-mediated hepatocyte death to 23.9% (95% CI [15.9, 32.0]) of cells positive for apoptosis but did not decrease cell death with analog 3 and had no impact on cell death with analog 14 (Fig. 8B). The pairing of treated and untreated samples for ratio-paired t test analysis of this data has been provided (Supplemental Fig. 2).
Because the cell death levels with EFV, analog 3, and analog 14 showed differential dependence on IRE1α-XBP1 signaling, we hypothesized that the activity of XBP1 as a transcription factor may differ across these stimuli. Because of this, we measured the expression of the XBP1 target gene DNAJB9 (Lee et al., 2003). During incubations with EFV, analog 3, and analog 14, 2.4- (n = 4, 95% CI [1.7, 3.4], P < 0.01), 1.9- (n = 4, 95% CI [1.2, 2.9], P < 0.05), and 1.9-fold (n = 4, 95% CI [1.2, 3.2], P < 0.05) increases were measured, respectively, in DNAJB9 expression (P values generated using an un-paired t test).
Discussion
In this work we demonstrated that EFV and 8-OHEFV, despite being very structurally similar compounds, differ in their activation of IRE1α-XBP1signaling. EFV treatment resulted in greater XBP1 splicing in human and mouse primary hepatocytes than did 8-OHEFV. Previously, our group has shown that both compounds activate cell death in a JNK- and Bcl-2 interacting mediator of cell death, extra long isoform (BimEL)-dependent mechanism, with 8-OHEFV exposure resulting in greater activation than EFV (Bumpus, 2011). Taken together, these results indicate that EFV and 8-OHEFV have diverging pharmacological effects, via activation of different cell-signaling events. It is especially important to consider the effects of 8-OHEFV because the levels of this metabolite in the blood stream can reach levels nearly equal to EFV (Ngaimisi et al., 2010; Grilo et al., 2016).
Although it has been previously shown that EFV activates XBP1 splicing in HEP3B cells and increases XBP1 expression in both Hep3B cells and primary human hepatocytes (Apostolova et al., 2013), the role of XBP1 splicing activation in EFV-mediated hepatocyte death has not been explored. In addition, we expanded on this by directly monitoring EFV activation of XBP1 splicing in primary human, male and female mouse, and cynomolgus macaque primary hepatocytes. Importantly, we have examined the impact of structural changes to EFV on IRE1α-XPB1 activation, and to the best of our knowledge this is the first study to perform this structure-activity analysis. As such, these data can be leveraged more broadly to improve understanding of the activation of these signaling molecules. Several changes to the structure of EFV, including breaking of the oxazinone ring (analogs 1 and 2), the cyclopropyl group (analog 4), removal of the oxazinone carbonyl (analog 8), or replacement of the oxazinone ring oxygen with carbon (analog 16) all decreased or abrogate activation of XBP1 splicing. Conversely, a change of the EFV alkyne to an alkene (analog 3) and replacement of the oxazinone ring nitrogen with carbon (analog 14) resulted in augmented XBP1 splicing. These structural changes to EFV could modulate IRE1α-XBP1 activation in several ways: through their impact on compound reactivity and therefore stimulation of reactive oxygen species formation, or by changing certain physiochemical properties (such as Log P and protein binding) that affect free intracellular concentration of the compound. Future work to directly monitor reactive oxygen species formation with these compounds and the free intracellular concentration would help to elucidate how each structural change impacts IRE1α-XBP1 activation. Though few of the analogs in this study have been tested for antiretroviral activity, two compounds, analog 12 and 13, have previously demonstrated IC90s against viral replication nearly identical to that of EFV (Corbett et al., 2000). Here we observed that, like EFV, both of these compounds activate IRE1α-XBP1.
Here we observed a trend toward increased activation of XBP1 splicing in primary hepatocytes from female mice, compared with those from male mice, though for most compounds tested this change was not statistically significant. This difference could be suggestive of increased sensitivity to IRE1α-XBP1 activation in females, which could be cytoprotective or cytotoxic. It would also be interesting to test whether all ER stress machinery is more readily activated in females. Further research is necessary to investigate this sex difference and its potential biologic implications.
We show similar impacts of EFV structure on activation of XBP1 splicing across model organisms. Of note, although IRE1α-XBP1 activation has been previously measured in rhesus macaque primary retinal pigment epithelium cells (Ma et al., 2016), it has not to our knowledge been measured before in cynomolgus macaque. In fact, previous work regarding ER stress in this biologic model is limited to one study that utilized a cynomolgus macaque glaucoma model (Ito et al., 2011). Considering the growing knowledge of this pathway’s role in drug toxicities (Cao et al., 2010; Van Summeren et al., 2011; Hur et al., 2012; Uzi et al., 2013; McConkey, 2017), and the fact that cynomolgus macaques are often used as models in drug development studies, during which early indications of toxicities may be observed, we feel our comparison is especially important. Here we demonstrate for EFV and this panel of structures that similar XBP1 splicing activation results can be seen across nonhuman models (cynomolgus macaque and mouse) used in various stages of clinical development. In addition, we have expanded on the characterization of EFV hepatotoxicity in vitro by demonstrating lipid droplet formation, 8-OHdG formation, and positive EtBr/AcrO staining in primary mouse hepatocytes treated with EFV.
PXR ablation has been shown to reduce high-fat diet–induced JNK-dependent cell death and lower levels of hepatic lipid accumulation in mice receiving a high-fat diet (He et al., 2013). On the other hand, PXR ablation shows no protective effect against ethanol-induced triglyceride formation and lipid droplet staining in mice, and higher basal levels of the lipid biosynthesis-activating transcription factor sterol regulatory element binding protein 1c (Srebp1c) (Choi et al., 2018). From this, it seems that PXR’s involvement in lipid homeostasis and hepatotoxicity varies across stimuli. With this work we showed that there is no measurable effect of PXR-ablation on EFV-mediated lipid droplet formation or IRE1α- XBP1 activation.
Though the targets of XBP1, such as protein degradation and membrane biogenesis machinery, are often considered to be cytoprotective (Iurlaro and Muñoz-Pinedo, 2016), the role of XBP1 in hepatocyte cell death and dyslipidemia is not well understood. Increased sXBP1 levels have been shown to decrease triglyceride levels, and lipogenic gene expression in diet-induced obesity mouse models (Herrema et al., 2016) and hepatocyte-specific IRE1α-null mice have increased steatosis over WT mice (Zhang et al., 2011). On the other hand, XBP1 ablation has been shown to decrease hepatic lipogenesis associated with high-carbohydrate diet in mice (Lee et al., 2008a). In addition, cotreatment with the IRE1α inhibitor STF 083010 has been demonstrated to decrease serum transaminase levels, hepatic lipid accumulation, steatosis, and apoptosis associated with high-fat diet in mice (Lebeaupin et al., 2018). As with PXR, the contribution of XBP1 splicing to hepatocyte cell death and lipid formation appears to be stimuli-dependent.
In this study we determined that cotreatment of cells with the IRE1α endoribonuclease inhibitor STF 083010, which inhibited XBP1 splicing activated by EFV in primary mouse hepatocytes, lessened EFV-induced primary hepatocyte death but not lipid droplet formation. These results suggest that for EFV, IRE1α-XBP1 signaling promotes cell death in response to EFV (Fig. 9) but does not play a role in EFV-mediated hepatocyte lipid accumulation. Interestingly, STF 083010 did not block primary hepatocyte cell death caused by the EFV analog 3, which stimulated splicing to a greater extent than did EFV. Further, analog 14, despite resulting in the highest levels of XBP1 splicing activation, caused no measureable primary hepatocyte cell death. Yet, we found that EFV, analog 3, and analog 14 were all able to increase to a similar extent the mRNA abundance of DNAJB9, an XBP1 target gene. Taken together, these data reveal that the consequences of IRE1α-XBP1 are stimuli-dependent, and although beyond the scope of the present study, this warrants future investigation.
Fig. 9.
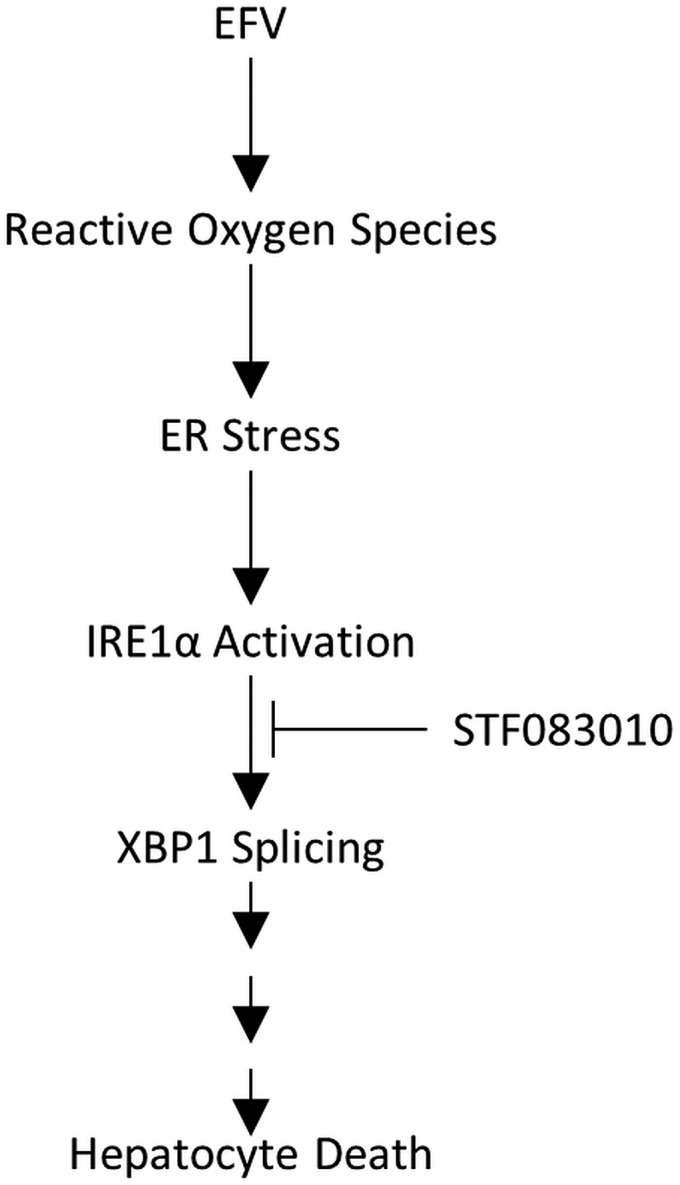
EFV activation of IRE1α-XBP1. Treatment with EFV results in the activation of IRE1α and subsequently IRE1α-catalyzed mRNA splicing of XBP1. In addition, EFV exposure causes formation of reactive oxygen species, an event that can lead to the activation of ER stress response, including the IRE1α-XBP1 signaling axis (Hanada et al., 2007). Reactive oxygen species production may drive the activation IRE1α-XBP1 by EFV, though a direct link between these two events remains to be established. STF 083010 inhibition of XBP1 splicing by IRE1α during incubation with EFV results in decreased EFV-mediated hepatocyte death.
In summary, our results demonstrate that EFV and several EFV-like compounds activate XBP1, and this is conserved among humans, mice, and macaques. Further, activation of XBP1 by EFV may contribute to EFV-mediated hepatocyte death. Of broader interest, our data indicate that even within a class of structurally similar compounds discreet changes in structure can impact the magnitude and outcome of XBP1 splicing. These findings provide insight into the impact of EFV on cellular signaling within hepatocytes while also identifying structural features that may contribute to IRE1α-XBP1 activation.
Acknowledgments
We acknowledge Dr. Dionna Williams for training in and use of Nikon NIS- Elements image analysis software.
Abbreviations
- 8-OhdG
8-hydroxydeoxyguanosine
- 8-OHEFV
8-hydroxyefavirenz
- AcrO
acridine orange
- DAPI
4′,6-diamidino-2-phenylindole, dihydrochloride
- DMSO
dimethyl sulfoxide
- DNAJB9
DnaJ heat-shock protein family member 9
- EFV
efavirenz
- ER
endoplasmic reticulum
- EtBr
ethidium bromide
- IRE1α
inositol-requiring enzyme 1α
- JNK
c-Jun N-terminal kinase
- P450
cytochrome P450
- PBS
phosphate-buffered saline
- PCR
polymerase chain reaction
- PXR
pregnane X receptor
- qPCR
quantitative real-time PCR
- STF 083010
N-[(2-hydroxy-1-naphthalenyl)methylene]-2-thiophenesulfonamide
- sXBP1
spliced XBP1
- TM
tunicamycin
- uXBP1
unspliced XBP1
- WT
wild-type
- XBP1
X-box-binding protein 1
Authorship Contributions
Participated in research design: Heck, Hamlin, Bumpus.
Conducted experiments: Heck, Hamlin.
Performed data analysis: Heck, Hamlin, Bumpus.
Wrote or contributed to the writing of the manuscript: Heck, Hamlin, Bumpus.
Footnotes
This work was supported by the National Institutes of Health [Grant R01 GM103853 awarded to N.N.B. and Grant T32 GM008763 awarded to the Department of Pharmacology and Molecular Sciences at the Johns Hopkins School of Medicine], and a National Science Foundation Graduate Research Fellowship [DGE-1232825 awarded to C.J.S.H.].
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Apostolova N, Gomez-Sucerquia LJ, Alegre F, Funes HA, Victor VM, Barrachina MD, Blas-Garcia A, Esplugues JV. (2013) ER stress in human hepatic cells treated with efavirenz: mitochondria again. J Hepatol 59:780–789. [DOI] [PubMed] [Google Scholar]
- Avery LB, VanAusdall JL, Hendrix CW, Bumpus NN. (2013) Compartmentalization and antiviral effect of efavirenz metabolites in blood plasma, seminal plasma, and cerebrospinal fluid. Drug Metab Dispos 41:422–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. (2000) Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol 2:326–332. [DOI] [PubMed] [Google Scholar]
- Buchman CD, Chai SC, Chen T. (2018) A current structural perspective on PXR and CAR in drug metabolism. Expert Opin Drug Metab Toxicol 14:635–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bumpus NN. (2011) Efavirenz and 8-hydroxyefavirenz induce cell death via a JNK- and BimEL-dependent mechanism in primary human hepatocytes. Toxicol Appl Pharmacol 257:227–234. [DOI] [PubMed] [Google Scholar]
- Cao R, Hu Y, Wang Y, Gurley EC, Studer EJ, Wang X, Hylemon PB, Pandak WM, Sanyal AJ, Zhang L, et al. (2010) Prevention of HIV protease inhibitor-induced dysregulation of hepatic lipid metabolism by raltegravir via endoplasmic reticulum stress signaling pathways. J Pharmacol Exp Ther 334:530–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S, Gyamfi AA, Neequaye P, Addo S, Gonzalez FJ, Gyamfi MA. (2018) Role of the pregnane X receptor in binge ethanol-induced steatosis and hepatotoxicity. J Pharmacol Exp Ther DOI: 10.1124/jpet.117.244665 [published ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comporti M. (1998) Lipid peroxidation and biogenic aldehydes: from the identification of 4-hydroxynonenal to further achievements in biopathology. Free Radic Res 28:623–635. [DOI] [PubMed] [Google Scholar]
- Corbett JW, Ko SS, Rodgers JD, Gearhart LA, Magnus NA, Bacheler LT, Diamond S, Jeffrey S, Klabe RM, Cordova BC, et al. (2000) Inhibition of clinically relevant mutant variants of HIV-1 by quinazolinone non-nucleoside reverse transcriptase inhibitors. J Med Chem 43:2019–2030. [DOI] [PubMed] [Google Scholar]
- Cox PM, Bumpus NN. (2014) Structure-activity studies reveal the oxazinone ring is a determinant of cytochrome P450 2B6 activity toward efavirenz. ACS Med Chem Lett 5:1156–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox PM, Bumpus NN. (2016) Single heteroatom substitutions in the efavirenz oxazinone ring impact metabolism by CYP2B6. ChemMedChem 11:2630–2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies R, Schuurman A, Barker CR, Clothier B, Chernova T, Higginson FM, Judah DJ, Dinsdale D, Edwards RE, Greaves P, et al. (2005) Hepatic gene expression in protoporphyic Fech mice is associated with cholestatic injury but not a marked depletion of the heme regulatory pool. Am J Pathol 166:1041–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fayet Mello A, Buclin T, Decosterd LA, Delhumeau C, di Iulio J, Fleurent A, Schneider MP, Cavassini M, Telenti A, Hirschel B, et al. (2011) Successful efavirenz dose reduction guided by therapeutic drug monitoring. Antivir Ther 16:189–197. [DOI] [PubMed] [Google Scholar]
- Feeney ER, Mallon PW. (2011) HIV and HAART-associated dyslipidemia. Open Cardiovasc Med J 5:49–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gounden V, van Niekerk C, Snyman T, George JA. (2010) Presence of the CYP2B6 516G> T polymorphism, increased plasma efavirenz concentrations and early neuropsychiatric side effects in South African HIV-infected patients. AIDS Res Ther 7:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grilo NM, Correia MJ, Sequeira C, Harjivan SG, Caixas U, Diogo LN, Marques MM, Monteiro EC, Antunes AM, Pereira SA. (2016) Efavirenz biotransformation as an up-stream event of mood changes in HIV-infected patients. Toxicol Lett 260:28–35. [DOI] [PubMed] [Google Scholar]
- Hanada S, Harada M, Kumemura H, Bishr Omary M, Koga H, Kawaguchi T, Taniguchi E, Yoshida T, Hisamoto T, Yanagimoto C, et al. (2007) Oxidative stress induces the endoplasmic reticulum stress and facilitates inclusion formation in cultured cells. J Hepatol 47:93–102. [DOI] [PubMed] [Google Scholar]
- He J, Gao J, Xu M, Ren S, Stefanovic-Racic M, O’Doherty RM, Xie W. (2013) PXR ablation alleviates diet-induced and genetic obesity and insulin resistance in mice. Diabetes 62:1876–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrich WD, Hassan HE, Wang H. (2016) Insights into CYP2B6-mediated drug-drug interactions. Acta Pharm Sin B 6:413–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrema H, Zhou Y, Zhang D, Lee J, Salazar Hernandez MA, Shulman GI, Ozcan U. (2016) XBP1s is an anti-lipogenic protein. J Biol Chem 291:17394–17404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur KY, So JS, Ruda V, Frank-Kamenetsky M, Fitzgerald K, Koteliansky V, Iwawaki T, Glimcher LH, Lee AH. (2012) IRE1α activation protects mice against acetaminophen-induced hepatotoxicity. J Exp Med 209:307–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito Y, Shimazawa M, Inokuchi Y, Yamanaka H, Tsuruma K, Imamura K, Onoe H, Watanabe Y, Aihara M, Araie M, et al. (2011) Involvement of endoplasmic reticulum stress on neuronal cell death in the lateral geniculate nucleus in the monkey glaucoma model. Eur J Neurosci 33:843–855. [DOI] [PubMed] [Google Scholar]
- Iurlaro R, Muñoz-Pinedo C. (2016) Cell death induced by endoplasmic reticulum stress. FEBS J 283:2640–2652. [DOI] [PubMed] [Google Scholar]
- Jiang D, Niwa M, Koong AC. (2015) Targeting the IRE1α-XBP1 branch of the unfolded protein response in human diseases. Semin Cancer Biol 33:48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai H, Crain PF, Kuchino Y, Nishimura S, Ootsuyama A, Tanooka H. (1986) Formation of 8-hydroxyguanine moiety in cellular DNA by agents producing oxygen radicals and evidence for its repair. Carcinogenesis 7:1849–1851. [DOI] [PubMed] [Google Scholar]
- Korennykh AV, Egea PF, Korostelev AA, Finer-Moore J, Zhang C, Shokat KM, Stroud RM, Walter P. (2009) The unfolded protein response signals through high-order assembly of Ire1. Nature 457:687–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebeaupin C, Vallée D, Rousseau D, Patouraux S, Bonnafous S, Adam G, Luciano F, Luci C, Anty R, Iannelli A, et al. (2018) Bax inhibitor-1 protects from nonalcoholic steatohepatitis by limiting inositol-requiring enzyme 1 alpha signaling in mice. Hepatology 68:515–532. [DOI] [PubMed] [Google Scholar]
- Lee AH, Iwakoshi NN, Glimcher LH. (2003) XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol 23:7448–7459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AH, Scapa EF, Cohen DE, Glimcher LH. (2008a) Regulation of hepatic lipogenesis by the transcription factor XBP1. Science 320:1492–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Tirasophon W, Shen X, Michalak M, Prywes R, Okada T, Yoshida H, Mori K, Kaufman RJ. (2002) IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev 16:452–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KP, Dey M, Neculai D, Cao C, Dever TE, Sicheri F. (2008b) Structure of the dual enzyme Ire1 reveals the basis for catalysis and regulation in nonconventional RNA splicing. Cell 132:89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee P, Peng H, Gelbart T, Beutler E. (2004) The IL-6- and lipopolysaccharide-induced transcription of hepcidin in HFE-, transferrin receptor 2-, and beta 2-microglobulin-deficient hepatocytes. Proc Natl Acad Sci USA 101:9263–9265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma JH, Wang JJ, Li J, Pfeffer BA, Zhong Y, Zhang SX. (2016) The role of IRE-XBP1 pathway in regulation of retinal pigment epithelium tight junctions. Invest Ophthalmol Vis Sci 57:5244–5252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzolini C, Telenti A, Decosterd LA, Greub G, Biollaz J, Buclin T. (2001) Efavirenz plasma levels can predict treatment failure and central nervous system side effects in HIV-1-infected patients. AIDS 15:71–75. [DOI] [PubMed] [Google Scholar]
- McConkey DJ. (2017) The integrated stress response and proteotoxicity in cancer therapy. Biochem Biophys Res Commun 482:450–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan B, Lade JM, Heck CJS, Dietz KD, Wade H, Bumpus NN. (2018) Probing ligand structure-activity relationships in pregnane X receptor (PXR): efavirenz and 8-hydroxyefavirenz exhibit divergence in activation. ChemMedChem 13:736–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngaimisi E, Mugusi S, Minzi OM, Sasi P, Riedel KD, Suda A, Ueda N, Janabi M, Mugusi F, Haefeli WE, et al. (2010) Long-term efavirenz autoinduction and its effect on plasma exposure in HIV patients. Clin Pharmacol Ther 88:676–684. [DOI] [PubMed] [Google Scholar]
- Ning J, Hong T, Ward A, Pi J, Liu Z, Liu HY, Cao W. (2011) Constitutive role for IRE1α-XBP1 signaling pathway in the insulin-mediated hepatic lipogenic program. Endocrinology 152:2247–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papandreou I, Denko NC, Olson M, Van Melckebeke H, Lust S, Tam A, Solow-Cordero DE, Bouley DM, Offner F, Niwa M, et al. (2011) Identification of an Ire1alpha endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. Blood 117:1311–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha H, He Y, Chen H, Wang C, Zenno A, Shi H, Yang X, Zhang X, Qi L. (2009) The IRE1alpha-XBP1 pathway of the unfolded protein response is required for adipogenesis. Cell Metab 9:556–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma D, Lau AJ, Sherman MA, Chang TK. (2013) Agonism of human pregnane X receptor by rilpivirine and etravirine: comparison with first generation non-nucleoside reverse transcriptase inhibitors. Biochem Pharmacol 85:1700–1711. [DOI] [PubMed] [Google Scholar]
- Shubber Z, Calmy A, Andrieux-Meyer I, Vitoria M, Renaud-Théry F, Shaffer N, Hargreaves S, Mills EJ, Ford N. (2013) Adverse events associated with nevirapine and efavirenz-based first-line antiretroviral therapy: a systematic review and meta-analysis. AIDS 27:1403–1412. [DOI] [PubMed] [Google Scholar]
- Sriburi R, Jackowski S, Mori K, Brewer JW. (2004) XBP1: a link between the unfolded protein response, lipid biosynthesis, and biogenesis of the endoplasmic reticulum. J Cell Biol 167:35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swart M, Whitehorn H, Ren Y, Smith P, Ramesar RS, Dandara C. (2012) PXR and CAR single nucleotide polymorphisms influence plasma efavirenz levels in South African HIV/AIDS patients. BMC Med Genet 13:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D. (2000) Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287:664–666. [DOI] [PubMed] [Google Scholar]
- Uzi D, Barda L, Scaiewicz V, Mills M, Mueller T, Gonzalez-Rodriguez A, Valverde AM, Iwawaki T, Nahmias Y, Xavier R, et al. (2013) CHOP is a critical regulator of acetaminophen-induced hepatotoxicity. J Hepatol 59:495–503. [DOI] [PubMed] [Google Scholar]
- Van Summeren A, Renes J, Bouwman FG, Noben JP, van Delft JH, Kleinjans JC, Mariman EC. (2011) Proteomics investigations of drug-induced hepatotoxicity in HepG2 cells. Toxicol Sci 120:109–122. [DOI] [PubMed] [Google Scholar]
- Ward BA, Gorski JC, Jones DR, Hall SD, Flockhart DA, Desta Z. (2003) The cytochrome P450 2B6 (CYP2B6) is the main catalyst of efavirenz primary and secondary metabolism: implication for HIV/AIDS therapy and utility of efavirenz as a substrate marker of CYP2B6 catalytic activity. J Pharmacol Exp Ther 306:287–300. [DOI] [PubMed] [Google Scholar]
- Yimer G, Amogne W, Habtewold A, Makonnen E, Ueda N, Suda A, Worku A, Haefeli WE, Burhenne J, Aderaye G, et al. (2012) High plasma efavirenz level and CYP2B6*6 are associated with efavirenz-based HAART-induced liver injury in the treatment of naïve HIV patients from Ethiopia: a prospective cohort study. Pharmacogenomics J 12:499–506. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. (2001) XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107:881–891. [DOI] [PubMed] [Google Scholar]
- Zhang K, Wang S, Malhotra J, Hassler JR, Back SH, Wang G, Chang L, Xu W, Miao H, Leonardi R, et al. (2011) The unfolded protein response transducer IRE1α prevents ER stress-induced hepatic steatosis. EMBO J 30:1357–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]