

Abstract
Chemoproteomic approaches to identify ligand-receptor interactions have gained popularity. However, identifying transmembrane receptors remains challenging. A new trifunctional probe to aid the nonbiased identification of such receptors was developed and synthesized using a convenient seven-step synthesis. This probe contained three functional groups: 1) an N-hydroxysuccinimide ester for ligand-coupling through free amines, 2) a diazirine moiety to capture the receptor of interest upon irradiation with UV light, and 3) a biotin group which allowed affinity purification of the final adduct using streptavidin. The interaction between the G protein–coupled tachykinin neurokinin 1 (NK1) receptor, expressed in an inducible manner, and the peptidic ligand substance P was used as a test system. Liquid chromatography–mass spectrometry analysis confirmed successful coupling of the probe to substance P, while inositol monophosphate accumulation assays demonstrated that coupling of the probe did not interfere substantially with the substance P–NK1 receptor interaction. Confocal microscopy and western blotting provided evidence of the formation of a covalent bond between the probe and the NK1 receptor upon UV activation. As proof of concept, the probe was used in full ligand-based receptor-capture experiments to identify the substance P–binding receptor via liquid chromatography–tandem mass spectrometry, resulting in the successful identification of only the NK1 receptor. This provides proof of concept toward general utilization of this probe to define interactions between ligands and previously unidentified plasma-membrane receptors.
Introduction
Membrane proteins are vital components of drug discovery, being targeted by some 60% of the currently marketed therapeutic medicines, with roughly half of these interacting with one or more members of the G protein–coupled receptor (GPCR) superfamily (Overington et al., 2006; Hauser et al., 2017; Santos et al., 2017). Identification of transmembrane receptor targets of poorly characterized ligands can provide new starting points for drug innovation, provide valuable information about off-target effects, and enhance mechanistic understanding of molecular pathways (Hopkins and Groom, 2002; Schirle and Jenkins, 2016).
Combining affinity chromatography with mass spectrometry (MS) is a popular chemoproteomic method that has been used to identify intracellular ligand-receptor interactions (Lenz et al., 2011); however, transmembrane receptors possess some unfavorable characteristics which hamper this approach. For example, their hydrophobicity and high number of post-translational modifications can result in aggregation and loss of ligand-binding activity upon solubilization. Moreover, these receptors are regularly of low abundance, and their interactions with ligands are often transient in nature, making it challenging to maintain ligand-receptor interactions throughout experiments (Wright, 2009; Helbig et al., 2010; Savas et al., 2011; Vuckovic et al., 2013). Various groups have endeavored to overcome these issues by developing trifunctional probes either structurally based on a ligand of interest or capable of coupling to such a ligand (Frei et al., 2012; Blex et al., 2017; Thomas et al., 2017; Tremblay and Hill, 2017; Sobotzki et al., 2018). Apart from a ligand or ligand-binding moiety, such probes must contain a receptor-capture moiety, which enables formation of a covalent bond between the probe and target receptor, and an affinity tag to assist with receptor purification. The covalent bond formed between the probe and target receptor prevents dissociation during such studies. Moreover, as such a bond will stay intact during cell lysis, it allows for ligand-receptor binding and receptor capture to be performed on living cells, thus circumventing binding issues faced following receptor solubilization.
One of the first trifunctional probes specifically designed to identify membrane receptors on living cells is TRICEPS, a potentially universal probe that is coupled to ligands of interest through an amine-reactive N-hydroxysuccinimide (NHS) ester (Frei et al., 2012). The ligand-probe adduct is then used to capture and purify glycosylated target receptors, as transplasma membrane proteins routinely become N-glycosylated during transport from the endoplasmic reticulum. Conceptually, the target can then be identified through MS analysis. Although this elegant ligand-based receptor-capture (LRC) strategy has resulted in successful identification of certain membrane receptors (Li et al., 2016, 2018; Ponda and Breslow, 2016), identification can only occur for receptors carrying an N-linked glycan in close proximity to the ligand-binding site. To overcome this glycan dependency, others have recently developed ligand-derived probes harboring photoaffinity labels to capture target receptors (Blex et al., 2017; Thomas et al., 2017). Nowadays such labels, which are activated upon irradiation with UV light, are commonly used to enhance capture of cytosolic receptors (Hatanaka, 2015).
Described herein is the design, synthesis, and evaluation of a new trifunctional probe for LRC that combines the universality of a ligand-binding NHS ester with receptor capture through a photoaffinity label. The interaction between the tachykinin neurokinin 1 (NK1) receptor and substance P (SP) was used as a test system. The NK1 receptor is a GPCR belonging to the tachykinin family, and the undecapeptide SP is its major and most potent endogenous ligand. Interaction between the peptide and the NK1 receptor is associated with various physiologic processes, including mitogenesis; wound healing; and neuronal transmission related to pain, depression, stress, anxiety, and emesis (Garcia-Recio and Gascón, 2015). Initially, this system was used to investigate individual steps in the LRC protocol, including ligand coupling, potential interference of the probe on the ligand-receptor interaction, and ability of the probe to covalently couple to the receptor. Finally, it was also used in a full LRC experiment to provide a proof of concept, demonstrating that this novel probe can be used as a general tool to help identify target receptors for a variety of ligands.
Materials and Methods
Unless otherwise stated, all (bio)chemicals and reagents were from Sigma-Aldrich (Dorset, UK). Tissue culture reagents and buffers were from Thermo Fisher Scientific (Paisley, UK). Molecular biology enzymes and reagents were from New England BioLabs (Hitchin, UK). Primers were ordered at Eurofins Genomics (Ebersberg, Germany). For western blotting, the following primary antibodies were used: mouse antitubulin (ab7291; 1:4000; Abcam, Cambridge, UK); rabbit anti–hexa-histidine tag (6xHis, ab9108; 1:4000; Abcam); and sheep anti–green fluorescent protein (produced in house). Secondary antibodies (800CW goat anti-mouse, 680RD goat anti-mouse, 800CW donkey anti-rabbit, and 800CW donkey anti-goat) and 680LT streptavidin were purchased from LI-COR (Cambridge, UK) and were all used at 1:10,000. Acetyl–norleucine–substance P (Ac-Nle-SP) and probe 1 were synthesized as described here.
Chemical Synthesis
All chemicals were commercially obtained and used without further purification unless stated otherwise. Reactions involving air-sensitive reagents and dry solvents were performed in flame-dried glassware under nitrogen atmosphere. Reactions were monitored by thin layer chromatography using plates with a UV fluorescent indicator (60 F254, 0.25 mm; Merck, Feltham, UK). Compounds were visualized either by UV absorption or by dipping in molybdenum or a ninhydrin-based stain, followed by heating. Column chromatography was performed using silica gel 60 (35–70 μm; Merck) or with an Isolera One purification system (Biotage, Ystrad Mynach, UK) and use of prepacked silica ULTRA SNAP cartridges (Biotage). An Alpha-2-4 lyophilizer (Christ, Osterode am Harz, Germany) equipped with a high vacuum pump was used for lyophilization. All NMR spectra were recorded using 400/500 NMR spectrometers (Bruker, Glasgow, UK). Values of δH are given in parts per million relative to an internal signal of Me4Si (0.00 ppm) or the signal of CDCl3 (7.26 ppm). Values of δc are given in parts per million relative to the signal of Me4Si (0.00 ppm), the signal of CDCl3 (77.16 ppm), or the signal of (CD3)2SO (39.52 ppm). For 1H NMR spectra integration, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, sept = septet, m = multiplet, br = broad, or a combination of these), coupling constants [J (Hz)], and assignments are also reported. For 13C NMR spectra, multiplicity and coupling constants are reported where appropriate. High-resolution mass spectrometry (HRMS) was performed by the analytical service of the University of Glasgow, either on a MStation JMS-700 instrument (Jeol, Welwyn Garden City, UK) using positive chemical ionization or a positive ion impact, or on a micro TOFq High Resolution instrument (Bruker) using positive ion electrospray. Analytical high-performance liquid chromatography (HPLC) was performed on the Prominence HPLC system (Shimadzu, Wolverton, UK) with a UV detector operating at 214 and 254 nm using either a Phenomenex (Torrance, CA) Gemini C18 column (110 Å, 5 μm, 250 × 4.60 mm) or a Dr. Maisch (Ammerbuch, Germany) Reprosil Gold C18 column (200 Å, 5 μm, 250 × 4.6 mm) at a flow rate of 1 ml/min using a 40-minute protocol (buffer A for 2 minutes, a linear gradient of 0%–100% buffer B in buffer A for 28 minutes, buffer B for 2 minutes, a linear gradient of 100%–0% buffer B in buffer A for 3 minutes, and buffer A for 5 minutes). Buffer A consisted of H2O/MeCN 95:5 with 0.1% trifluoroacetic acid (TFA), and buffer B consisted of MeCN/H2O 95:5 with 0.1% TFA. Liquid chromatography–mass spectrometry (LC-MS) was carried out with an LCQ Fleet quadrupole mass spectrometer (Thermo Fisher Scientific) and an Ultimate 3000 LC (Dionex, Camberley, UK) with use of a Dr. Maisch Reprosil Gold 120 C18 column (150 Å, 3 μm, 150 × 4 mm). Buffers and protocols used were identical to those of analytical HPLC.
Synthesis Probe 1
For long-term storage, diazirine-containing products were protected from light. During synthesis, however, no extra care was taken to avoid light, and no activation of the diazirine moiety was observed.
Compound 2: (9H-fluoren-9-yl)methyl tert-butyl (6-oxo-6-(prop-2-yn-1-ylamino)hexane-1,5-diyl)(S)-dicarbamate
Fmoc-Lys(Boc)-OH (23.61 g, 50 mmol), Oxyma Pure Novabiochem (10.48 g, 74 mmol) (Merck, Darmstadt, UK), and propargylamine (3.4 ml, 53 mmol) were dissolved in dichloromethane (DCM; 1 l) and cooled on ice. 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC; 18.87 g, 98 mmol) was added, after which the (now turbid) mixture was stirred at room temperature (RT) for 40 minutes. The reaction mixture was subsequently washed with H2O (500 ml, 3×), citric acid (1 M, 500 ml, 2×), H2O (1 l), NaHCO3 (saturated, 500 ml, 2×), and H2O (1 l). The organic layer was dried over MgSO4 and concentrated under vacuum to yield a yellow solid (26.52 g, 95%). Analytical data corresponded to previously reported data (Hartwig and Hecht, 2010).
Compound 3: Tert-butyl (S)-(6-oxo-6-(prop-2-yn-1-ylamino)-5-(3-(4-(3-(trifluoromethyl)-3H-diazirin-3-yl)phenyl)propanamido)hexyl)carbamate
Compound 2 (748.4 mg, 1.5 mmol) was dissolved in piperidine/dimethylformamide [DMF; 1:15 (v/v) 25 ml], and the mixture was stirred at RT. After 10 minutes, the mixture was concentrated under vacuum, and the piperidine was removed by coevaporations with toluene. This crude mixture was dissolved in 10 ml of DCM, and 3-(4-(3-(trifluoromethyl)-3H-diazirin-3-yl)phenyl)propanoic acid [304.0 mg, 1.2 mmol, synthesized as described before by Geurink et al. (2010)], hexafluorophosphate benzotriazole tetramethyl uronium (448.1 mg, 1.2 mmol), and N,N-diisopropylethylamine (DIPEA; 790 μl, 4.6 mmol) were added. The total mixture was stirred for 2 hours at RT before it was washed with H2O (50 ml), KHSO4 (1 M, 50 ml), and H2O (50 ml). The organic layer was dried over MgSO4, concentrated under vacuum, and purified by flash chromatography (DCM/MeOH 20:0→19:1), yielding a white solid (556 mg, 91%).
Analytical Data.
The analytical data for compound 3 were as follows: 1H NMR (400 MHz, chloroform-d) δ 7.26 (br s, 1H, NH), 7.20 (d, J = 8.4 Hz, 2H, 2× CAR-H), 7.09 (d, J = 8.0 Hz, 2H, 2× CAR-H), 6.73 (d, J = 8.0 Hz, 1H, NH), 4.79 (t, J = 6.0 Hz, 1H, NH), 4.52 (td, J = 7.8, s5.8 Hz, 1H, Lys-αCH), 4.06–3.85 (m, 2H, CH2C-CH), 3.08 (dd, J = 11.9, 6.2 Hz, 2H, Lys-εCH2), 2.94 (td, J = 7.6, 4.4 Hz, 2H, CH2CH2CAR), 2.52 (t, J = 7.7 Hz, 2H, CH2CH2CAR), 2.20 (t, J = 2.5 Hz, 1H, C-CH), 1.92–1.19 (m, 15H, Lys-β,γ,δCH2 and Boc-CH3); 13C NMR (101 MHz, CDCl3) δ 172.0, 171.6, 156.2, 142.5, 128.8, 127.1, 126.7, 122.16 (q, J = 274.7 Hz, CF3), 79.3, 77.3, 71.6, 52.7, 40.0, 37.4, 32.1, 31.1, 29.6, 29.1, 28.5 28.33 (q, J = 40.4 Hz, CN2-CF3), 22.5. Heteronuclear single quantum coherence spectroscopy (HSQC) and correlation spectroscopy (COSY) spectra were in accordance with assignments given earlier. C25H32F3N5O4 [M+Na]+: 546.2299, found: 546.2284.
Compound 4: (S)-4-oxo-4-((6-oxo-6-(prop-2-yn-1-ylamino)-5-(2-(4-(3-(trifluoromethyl)-3H-diazirin-3-yl)phenyl)acetamido)hexyl)amino)butanoic Acid
Compound 3 (554.2 mg, 1.1 mmol) was dissolved in DCM/TFA [1:1 (v/v) 60 ml] and stirred for 10 minutes before it was concentrated under vacuum. The last traces of TFA were removed by three coevaporations with toluene. The yellow solid that yielded was dissolved in DMF (50 ml), and succinic anhydride (128.4 mg, 1.3 mmol) and DIPEA (880 μl, 5.1 mmol) were added. The mixture was stirred for 45 minutes at RT, after which it was concentrated under vacuum. The remaining solid was dissolved in DCM (100 ml) and extracted with NaHCO3 (saturated, 150 ml, 2×). The aqueous layers were acidified with HCl (37%) and extracted with EtOAc (100 ml, 3×). The organic layers were combined, dried over MgSO4, concentrated under vacuum, and purified by flash chromatography (DCM/MeOH/AcOH, 19:1:0.2) to yield the acid as a white solid (418 mg, 75% over two steps).
Analytical Data.
The analytical data for compound 4 were as follows: 1H NMR (400 MHz, methanol-d4) δ 7.32 (d, J = 8.4 Hz, 2H, 2× CAR-H), 7.16 (d, J = 8.0 Hz, 2H, 2× CAR-H), 4.25 (dd, J = 8.7, 5.4 Hz, 1H, Lys-αCH), 4.05–3.79 (m, 2H, CH2C-CH), 3.14 (t, J = 7.0 Hz, 2H, Lys-εCH2), 2.94 (t, J = 7.7 Hz, 2H, CH2CH2CAR), 2.66–2.50 (m, 5H, CH2CH2CAR and CH2CH2COOH), 2.45 (t, J = 6.9 Hz, 2H, CH2CH2COOH), 1.83–1.20 (m, 6H, Lys-β,γ,δCH2); 13C NMR (101 MHz, MeOD) δ 176.4, 174.8, 174.5, 174.0, 144.7, 130.3, 127.8, 127.7, 126.4 (q, J = 274.0 Hz, CF3), 80.5, 72.3, 54.5, 40.0, 38.0, 32.7, 32.3, 31.7, 30.5, 29.9, 29.6 (q, J = 39.7 Hz, CN2-CF3), 29.5, 24.0. HSQC and COSY spectra were in accordance with assignments given earlier. HRMS calculated for C24H28F3N5O5 [M+Na]+: 546.1935, found: 546.1926.
Compound 5: 4-Oxo-4-(((S)-6-oxo-6-(((1-(15-oxo-19-((3aS,4S,6aR)-2-oxohexahydro-1H-thieno[3,4-d]imidazol-4-yl)-4,7,10-trioxa-14-azanonadecyl)-1H-1,2,3-triazol-4-yl)methyl)amino)-5-(3-(4-(3-(trifluoromethyl)-3H-diazirin-3-yl)phenyl)propanamido)hexyl)amino)butanoic Acid
Compound 4 (58.1 mg, 111 μmol) and biotin–polyethylene glycol (PEG)–azide (65.1 mg, 138 μmol) [synthesized as described before by Chambers et al. (2013)] were dissolved in MeOH (3 ml). Next, aqueous CuSO4 (10 mM, 2.8 ml) and aqueous sodium ascorbate (30 mg/ml, 335 μl) were added. The mixture was stirred under N2 atmosphere for 2 hours before it was concentrated under vacuum. The crude product was purified by flash chromatography (DCM/MeOH/AcOH, 19:1:0.2→16:4:0.2) to yield the biotinylated product as a white solid (71 mg, 65%).
Analytical Data.
The analytical data for compound 5 were as follows: 1H NMR [500 MHz, dimethylsulfoxide (DMSO)-d6] δ 8.44 (br s, 1H, NH), 8.16 (br s, 1H, NH), 7.90–7.83 (m, 2H, NH, and triazole-CH), 7.76 (t, J = 5.7 Hz, 1H, NH), 7.35 (d, J = 8.0 Hz, 2H, 2× CAR-H), 7.17 (d, J = 7.9 Hz, 2H, 2× CAR-H), 6.44 (s, 1H, NH), 6.37 (s, 1H, NH), 4.37 (t, J = 7.0 Hz, 2H, PEG-CH2Ntriazole), 4.33–4.24 (m, 3H, NHCH2Ctriazole, and biotin-SCH2CH), 4.24–4.14 (m, 1H, Lys- αCH), 4.13 (ddd, J = 7.3, 4.5, 1.8 Hz, 1H, biotin-SCHCH), 3.54–3.34 (m, 12H, 2× PEG-OCH2CH2O, and 2× PEG-CH2CH2CH2O, overlapping with water signal), 3.13–3.02 (m, 3H, biotin-SCH, and PEG-CH2NH), 2.96 (q, J = 6.5 Hz, 2H, Lys-εCH2), 2.88–2.78 [m, 3H, biotin-SCH2 (1H), and CH2CH2CAR], 2.58 [d, J = 12.4 Hz, 1H, biotin-SCH2 (1H)], 2.50–2.40 (m, 2H, CH2CH2CAR), 2.26 (s, 4H, CH2CH2COOH), 2.09–1.96 (m, 4H, PEG-CH2CH2Ntriazole, and biotin-αCH), 1.81–1.09 (m, 14H, Lys-β,γ,δCH2, biotin- β,γ,δCH2, and PEG-CH2CH2NH); 13C NMR (126 MHz, DMSO) δ 171.9, 171.2, 162.7, 144.8, 144.0, 129.3, 126.3, 125.1, 122.8, 122.0 (d, J = 275.5 Hz, CF3), 69.8, 69.7, 69.6, 69.5, 68.1, 66.9, 61.1, 59.2, 55.4, 46.5, 40.0, 38.2, 36.2, 35.7, 35.2, 34.3, 31.6, 30.6, 30.0, 29.4, 28.7, 28.2, 28.0, 28.0 (d, J = 39.8 Hz, CN2-CF3), 25.3, 22.7. HSQC, heteronuclear multiple bond correlation, COSY and total correlated spectroscopy spectra were in accordance with assignments given earlier. HRMS calculated for C44H64F3N11O10S [M+Na]+: 1018.4403, found: 1018.4394.
Probe 1: 2,5-Dioxopyrrolidin-1-yl 4-oxo-4-(((S)-6-oxo-6-(((1-(15-oxo-19-((3aS,4S,6aR)-2-oxohexahydro-1H-thieno[3,4-d]imidazol-4-yl)-4,7,10-trioxa-14-azanonadecyl)-1H-1,2,3-triazol-4-yl)methyl)amino)-5-(3-(4-(3-(trifluoromethyl)-3H-diazirin-3-yl)phenyl)propanamido)hexyl)amino)butanoate
Compound 5 (27.5 mg, 27.6 μmol) was dissolved in 3 ml of dry DMF, and NHS (33.5 mg, 291 μmol) and EDC (55.3 mg, 288 μmol) were added. The mixture was stirred under N2 atmosphere for 2.5 hours, after which it was concentrated under vacuum. The crude yellow oil was triturated with ice-cold H2O (3× 1 ml) to yield a white solid (13.5 mg, 45%).
Analytical Data.
LC-MS data are shown in Supplemental Fig. S1. HRMS calculated for C48H67F3N12O12S [M+Na]+: 1115.4566, found: 1115.4607.
Synthesis Ac-Nle-SP
Ac-Nle-SP was assembled by solid phase peptide synthesis on an automatic Tribute-UV peptide synthesizer (Protein Technologies, Manchester, UK) on a 0.25-mmol scale using Rink Amide resin (0.24 mmol/g; Rapp Polymere, Tübingen, Germany). First, the resin was allowed to swell in DMF (3× 10 minutes). The Fmoc group was then removed with a mix of piperidine/DMF [8 ml, 1:4 (v/v)] using the Tribute-UV peptide synthesizer RV_top_UV_Xtend protocol, followed by washes with DMF (5× 30 seconds). Next, the correct Fmoc-protected amino acid was pre-activated for 2 min with HCTU (4 eq) and DIPEA (8 eq) in DMF, before it was added to the resin and shaken (20 min). The resin was washed with DMF (6× 30 seconds), after which Fmoc deprotection and coupling of the next amino acid took place as before. After the coupling of the last amino acid, one final Fmoc deprotection step took place. The resin was then manually treated with Ac2O (250 μl, 2.3 mmol) and DIPEA (185 μl, 1 mmol) in DMF (10 ml) for 30 minutes. The resin was washed with DMF (5× 30 seconds) and DCM (5× 30 seconds) and dried under high vacuum, and peptides were then cleaved and deprotected in TFA/H2O/triisopropylsilane [10 ml, 95/2.5/2.5 (v/v/v), 1 hour]. To precipitate the peptide, the TFA mixture was added dropwise to an ice-cold solution of methyl tert-butyl ether/hexanes [90 ml, 1:1 (v/v)]. After centrifugation (2000g, 5 minutes), the supernatant was discarded and the peptide was washed twice more with methyl tert-butyl ether/hexanes. The peptide was then dissolved in tBuOH/H2O [15 ml, 1:1 (v/v)] and lyophilized to yield a crude peptide. This crude was further purified using the Agilent Technologies (Santa Clara, CA) 1260 Infinity LC System equipped with a Gemini C18 column (110 Å,10 μm, 250 × 20 mm) using a 60-minute linear gradient of 0%–100% buffer B in buffer A at 12.5 ml/min. Fractions were autocollected based on the UV measurements at 214 nm. Fractions containing pure peptide, as assessed by analytical HPLC, were combined and lyophilized to give Ac-Nle-SP with a 13% yield.
Analytical Data.
For HPLC trace, see Supplemental Fig. S2. LC-MS calculated for C66H102N18O14 [M+2H]2+: 686.40, found: 686.50.
UV Activation of Probe 1
The free acid derivative of the probe (compound 5) was dissolved in MeOH (1 mg/ml) and either kept in the dark or exposed to light of 365 nm for 15 minutes with a high-intensity UV lamp (SB-100p/FB; Spectroline, Westbury, NY). Samples were analyzed using LC-MS.
Probe 1 Coupling to Ac-Nle-SP and Glycine
Probe 1 in DMSO (50 mM, 25 µl) was added to a solution of Ac-Nle-SP in phosphate-buffered saline (PBS) (pH 8.2, 1.05 mM, 475 µl) to form Ac-Nle-SP-probe 8. The mixture was incubated for 1 hour at RT, during which time a precipitate formed. This was redissolved by addition of 500 µl of DMSO, yielding a 0.5-mM solution of 8 (Supplemental Fig. S3A). Glycine (Gly)-probe 9 was prepared in a similar fashion using a solution of glycine in PBS (pH 8.2, 1 mg/ml, 475 µl) (Supplemental Fig. S3B). Although no precipitate formed, this mixture was also diluted with 500 µl of DMSO. Moreover, to keep the amount of probe, probe by-products, and DMSO between Ac-Nle-SP-probe and Gly-probe equal, the volume of Gly-probe used in all experiments was always identical to the volume of Ac-Nle-SP-probe used. Both mixtures were stored at −20°C and went through numerous freeze-thaw cycles over the span of a year without any obvious loss of activity.
Generation of Flp-In T-REx 293 Cell Lines
Hemagglutinin-NK1-eGFP.
A human NK1 construct in pcDNA3.1+ (TACR100000; cDNA Resource Center, Bloomsberg, CA) was used as a polymerase chain reaction template. Primers were designed to introduce a hemagglutinin (HA) epitope (YPYDVPDYA) on the N terminus of the NK1 construct. Moreover, XhoI and BamHI restriction sites were introduced at the 3′ and 5′ termini, respectively. The primers used were 5′ ACGTACGTACGTCTCGAGGCCACCATGTACCCCTATGACGTGCCCGATTACGCGGATAAC GTCCTCCCGGTGGACTCAGACCTCTCC 3′ and 3′ ACGTACGTACGTTGGATCCCGGGAGAGCACATTGGAGGAGAAGCTGAAGC 5′. The resulting DNA fragment was subcloned between the XhoI and BamHI sites of the pEGFP-N1 plasmid–harboring monomeric Ala206Lys enhanced green fluorescent protein (eGFP), a variant of eGFP that is less prone to dimerization (von Stetten et al., 2012), resulting in an HA-NK1-eGFP construct. HA-NK1-eGFP was excised from pEGFP-N1 with NheI and NotI. The NheI end was blunted, and the fragment was subcloned into pcDNA5/FRT/TO (Thermo Fisher Scientific) between EcoRV and NotI. This plasmid was used in the Flp-In T-REx system to create cells inducibly able to express HA-NK1-eGFP as described previously (Ward et al., 2011).
HA-NK1-6xHis.
Using NotI and AgeI, eGFP was excised from the HA-NK1-eGFP construct in pcDNA5/FRT/TO. DNA encoding a hexa-His tag, created by annealing the two primers 5′ CCGGTCCACCATCACCATCACCATTAGGGCGCGCCGC 3′ and 3′ AGGTGGTAGTGGTAGTGGTAATCCCGCGCGGCGCCGG 5′, was subcloned into the same site, thereby creating an HA-NK1-6xHis construct. This was used in the Flp-In T-REx system, as done earlier, to create cells able to inducibly express HA-NK1-6xHis.
Cell Maintenance
Flp-In T-REx 293 cells able to express either HA-NK1-eGFP or HA-NK1-6xHis upon addition of doxycycline (Dox) were maintained in complete Dulbecco’s modification of Eagle’s medium with glucose (4500 mg/l) and sodium bicarbonate (without sodium pyruvate); supplemented with 10% (v/v) fetal bovine serum, penicillin (100 U/ml), streptomycin (100 μg/ml), hygromycin (200 μg/ml), and blasticidin (5 μg/ml) in a humidified 5% CO2 atmosphere at 37°C; and subcultured every 3 days.
Dox Titration
Western Blotting.
Flp-In T-REx 293 cells harboring HA-NK1-eGFP or HA-NK1-6xHis were grown in poly-d-lysine–coated six-well plates and incubated with various concentrations of Dox overnight. Cells were harvested in radioimmunoprecipitation assay buffer [50 mM HEPES (pH 7.4), 150 mM NaCl, 1% (v/v) Triton X-100, 0.5% (w/v) sodium deoxycholate, 0.1% (w/v) SDS, 10 mM NaF, 5 mM EDTA, 10 mM Na2HPO4, 5% (v/v) ethylene glycol, pH 7.3], passed through a 25-gauge needle (6×), rotated for 45 minutes at 4°C, and centrifuged for 10 minutes at 21,000g at 4°C to obtain cleared lysates. Lysates were analyzed by western blotting.
Epifluorescence Quantification.
Flp-In T-REx cells harboring HA-NK1-eGFP were cultured on black clear-bottom 96-well plates coated with poly-d-lysine. After induction with various concentrations of Dox, the plates were incubated overnight. Cells were washed twice with Hanks’ balanced salt solution (HBSS) before they were incubated for 20 minutes at 37°C with 10 μg/ml Hoechst nuclear stain to enable cell number determination. After two more washes with HBSS, fluorescence was measured with a CLARIOstar plate reader (BMG Labtech, Ortenberg, Germany). To quantify the Hoechst-stained nuclei, an excitation wavelength of 355/20 nm and an emission wavelength of 455/30 nm were used for eGFP 470/15 nm and 515/20 nm were used.
Inositol Phosphate Accumulation Assays
Flp-In T-REx 293 cells harboring HA-NK1-eGFP or HA-NK1-6xHis were grown in T75 tissue culture flasks and induced by addition of 100 ng/ml Dox overnight. Uninduced cells, grown in a similar fashion, were used as negative controls. Inositol monophosphate (IP1) experiments were performed using a homogeneous time-resolved fluorescence-based detection kit (CisBio Bioassays, Codolet, France) according to the manufacturer’s protocol. Cells were plated at 7500 cells/well into white, solid-bottom, 384-well plates with appropriate ligand concentrations present in the wells and incubated at 37°C for 2 hours. The reactions were stopped by the addition of the supplied lysis buffer and supplemented with the homogeneous time-resolved fluorescence reagents. Plates were incubated for 1–24 hours at RT before the output was measured with a Pherastar FS plate reader (BMG Labtech).
Live-Cell Imaging Assays
Flp-In T-REx 293 cells harboring HA-NK1-eGFP were grown on poly-d-lysine–coated coverslips and induced with 100 ng/ml Dox in serum-free medium overnight. Uninduced cells, grown in a similar fashion, were used as a negative control. All subsequent steps, apart from the actual imaging, were performed on ice or at 4°C. HBSS was used for all washes and dilutions. Cells were washed three times, incubated with Ac-Nle-SP-probe 8 (1 μM) or Gly-probe 9 for 1 hour in the dark, and washed an additional three times. For competition experiments, Ac-Nle-SP (compound 7) (10 μM) was added, and cells were incubated for 3 hours at 4°C in the dark, followed by three more washes. The cells were exposed to light of 365 nm for 15 minutes with a high-intensity UV lamp (SB-100p/FB; Spectroline). Cells were washed three times and incubated with DyLight 594–conjugated streptavidin (15 μg/ml; Vectorlabs, Peterborough, UK) for 30 minutes at RT in the dark. After three final washes, cells were imaged using an 880 laser-scanning confocal microscope (Zeiss, Cambridge, UK).
Simultaneous Dual-Color eGFP/DyLight 594 Live-Cell Microscopy Imaging
A Zeiss 880 laser-scanning confocal microscope (invert configuration), equipped with a 63× oil immersion Plan Apochromat objective lens (numerical aperture = 1.4), was used to acquire high-resolution, 12-bit-depth channel images (image size = 512 × 512). Using the single-track multiple-channel capability of the Zeiss Zen Black software (version 2.3), HA-NK1-eGFP and DyLight 594–labeled ligand were excited simultaneously at 488 and 561 nm, respectively. Well separated spectral emission detection windows (495–555 nm for eGFP and 600–700 nm for DyLight 594) were set up to ensure that the emission signals elicited from each fluorophore were recorded without any bleed through or time delay issues. The pinhole diameter was set to 1 Airy unit, and frame averaging was set to 4 to minimize noise and optimize signal collection. Bright field transmission images were simultaneously detected along with the fluorescence images using the dedicated transmitted light detector.
eGFP/DyLight 594 Colocalization Analysis
Each recorded channel image was exported into Metamorph (version 7.8.13; Molecular Devices, San Jose, CA), and a green or red look-up table was assigned to the eGFP and DyLight 594 channel images, respectively. The total background level of autofluorescence was subtracted (using a black-colored region adjacent to the fluorescing cell) from every matrix pixel used to form each 512 × 512 channel image. To quantify the colocalization between HA-NK1-eGFP and DyLight 594, green-red pixel intensity scatterplots were generated for each image. Pearson correlation coefficient values were generated from the generated scatter plots that described the degree to which eGFP and DyLight 594 fluorescence at each homologous pixel varied from a perfect positive correlation slope value of 1.0. This was done for four representative images for each experimental group. Significant statistical difference between the experimental groups was determined using analysis of variance with Tukey’s multiple comparison test.
Receptor Capture by Ac-Nle-SP-Probe and Gly-Probe
Flp-In T-REx 293 cells harboring HA-NK1-6xHis were grown in a 10-cm dish and induced with 100 ng/ml Dox overnight. If needed for a negative control, uninduced cells were grown in a similar fashion. Cells were harvested using Versene. All subsequent steps were performed on ice or at 4°C. Cells were resuspended in HBSS (600 µl) supplemented with Ac-Nle-SP-probe 8 (1 µM) or Gly-probe 9. Cells were incubated for 1 hour in the dark and under constant agitation. Cells were spun down at 300g for 3 minutes and washed twice with HBSS before being resuspended in HBSS (600 µl), transferred to a 12-well plate, and exposed to light of 365 nm for 15 minutes (SB-100p/FB; Spectroline). Cells were transferred back to a 15-ml Falcon tube, centrifuged at 3200g for 5 minutes, and washed twice with PBS before they were frozen at −20°C for at least an hour. From there, all buffers were supplemented with cOmplete EDTA-free Protease Inhibitor Cocktail (Roche, Burgess Hill, UK). Frozen cell pellets were resuspended in PBS buffer (500 µl), passed through a 25-gauge needle (10×), homogenized using a glass on a Teflon homogenizer (50×), and again passed through a needle (10×). To remove cell debris, the mixture was centrifuged at 200g for 5 minutes, after which the supernatant was centrifuged at 50,000g for 30 minutes. The pellets were resuspended in PBS supplemented with 1% (v/v) NP40 (150 µl), after which the samples were rotated for 1 hour. The lysates were then centrifuged at 21,000g for 10 minutes to get rid of any nonsolubilized material. Lysates were analyzed by western blotting or used for the full LRC experiment.
Western Blotting
Lysates of the Dox titration and receptor-capture experiments were analyzed through western blotting. A bicinchoninic assay (Expedeon, Cambridge, UK) was used according to the manufacturer’s protocol to determine and equalize the protein concentrations of the samples. SDS-PAGE sample buffer was added to the samples, and they were heated to 65°C for 5 minutes. Ten to 20 μg of protein per sample was loaded into wells of 4%–12% BisTris precast NuPAGE or BOLT gels (Thermo Fisher Scientific) and subjected to SDS-PAGE in NuPAGE or BOLT MOPS SDS running buffer (Thermo Fisher Scientific). The proteins were then electrophoretically transferred to a nitrocellulose membrane, which was blocked in PBS blocking buffer (LI-COR) and subsequently probed for HA-NK1-eGFP, HA-NK1-6xHis, tubulin, and/or biotin using the appropriate primary and secondary antibodies, or streptavidin diluted in PBS blocking buffer (LI-COR) supplemented with 0.2% Tween 20. An Odyssey Scanner (LI-COR) was used to image the membranes.
Full Ligand-Based Receptor-Capture Experiment
For the full LRC experiment, receptor capture took place as described earlier, with the exception that five confluent T150 flasks were used, buffer quantities were multiplied by 10, and UV activation was performed in a 10-cm dish. All subsequent steps were performed on ice or at 4°C, and all buffers were supplemented with cOmplete EDTA-free Protease Inhibitor Cocktail (Roche). Lysates were added to Pierce Streptavidin Agarose beads (250 µl; Thermo Fisher Scientific), and the total volume was up to 3 ml using PBS supplemented with 1% (v/v) NP40. The beads were incubated overnight before being washed four times with radioimmunoprecipitation assay buffer, four times with PBS supplemented with 880 mM NaCl, and four times with PBS. Beads were frozen at −80°C until they were analyzed by liquid chromatography–tandem mass spectrometry (LC-MS/MS). For each condition, three biologic repeats were performed.
LC-MS/MS Analysis
Proteomics was carried out by the University of Leicester Proteomics Facility (Protein Nucleic Acid Chemistry Laboratory, University of Leicester, Leicester, UK). The streptavidin-conjugated beads were resuspended by gentle mixing and transferred to a 30,000–molecular weight cutoff filtration unit (Vivacon 500; Satorius, Göttingen, Germany). Excess liquid was removed by spinning at 14,000g for 15 minutes. The beads were then resuspended a total of two times in 200 µl of freshly prepared 8 M urea in 0.1 M Tris/HCl (pH 8.5) and spun at 14,000g for 15 minutes, discarding the flow through solution. Reduction was carried out by addition of 100 µl of 5 mM dithiothreitol solution for 30 minutes at 60°C. After cooling, samples were incubated with addition of 100 µl of 50 mM iodoacetamide solution for 30 minutes at RT in the dark. After incubation, solutions were removed by spinning at 14,000g for 15 minutes. Beads were then resuspended a total of two times in 100 µl of 8 M urea in 0.1 M Tris/HCl (pH 8.5) and spun at 14,000g for 15 minutes, discarding the flow through solution. Beads were then resuspended twice in 100 µl of 50 mM triethylammonium bicarbonate and spun at 14,000g for 15 minutes, discarding the flow through solution. Just prior to use, lyophilized trypsin was resuspended in 50 mM triethylammonium bicarbonate, and 5 µg was added to each sample. Digestion took place overnight at 37°C in a humidified chamber. Filter units were transferred to a clean collection tube, and samples were spun down for 10 minutes at 14,000g. A further 40 µl of 50 mM triethylammonium bicarbonate was added to beads, and samples were spun again for 10 minutes at 14,000g. The samples were concentrated to approximately 20-µl volume in a SpeedVac centrifuge (Thermofisher Scientific, Paisley, UK), and digests were then acidified with formic acid (final concentration 0.1%). LC-MS/MS was carried out using an RSLCnano HPLC system (Dionex) and an LTQ-Orbitrap-Velos mass spectrometer (Thermo Fisher Scientific). Samples were loaded at a high flow rate onto a reverse-phase trap column (0.3 mm i.d. × 1 mm) containing 5 μm C18 300 Å Acclaim PepMap medium (Dionex) maintained at a temperature of 37°C. The loading buffer was 0.1% formic acid/0.05% trifluoroacetic acid/2% acetonitrile. Peptides were eluted from the trap column at a flow rate of 0.3 µl/min and through a reverse-phase capillary column (75 μm i.d. × 250 mm) containing Symmetry C18 100 Å medium (Waters, Elstree, UK) that was manufactured in house using a high-pressure packing device (Proxeon Biosystems, Odense, Denmark). The output from the column was sprayed directly into the nanospray ion source of the LTQ-Orbitrap-Velos mass spectrometer. The LTQ-Orbitrap-Velos mass spectrometer was set to acquire a 1 microscan Fourier transform (FT) MS scan event at 60000 resolution over the m/z range 300-2000 Da in positive ion mode. The maximum injection time for MS was 500 ms, and the AGC target setting was 1e6. Accurate calibration of the FTMS scan was achieved using a background ion lock mass for C6H10O14S3 (401.922718 Da). Subsequently, up to 10 data-dependent higher energy collisional dissociation MS/MS were triggered from the FTMS scan. The isolation width was 2.0 Da, and normalized collision energy was 42.5. Dynamic exclusion was enabled. The maximum injection time for MS/MS was 250 ms, and the AGC target setting was 5e4. The .raw data file obtained from each LC-MS/MS acquisition was processed using Proteome Discoverer (version 1.4; Thermo Fisher Scientific), searching each file in turn using Mascot (version 2.2.04; Matrix Science Ltd., London, UK) (Perkins et al., 1999) against the human reference proteome downloaded from UniProtKB (proteome identifier: UP000005640; UniProt Consortium, 2010). The peptide tolerance was set to 5 ppm, and the MS/MS tolerance was set to 0.05 Da. Fixed modifications were set as carbamidomethyl (C) with variable modification of oxidation (M) and phosphorylation (S, T, and Y). Trypsin was selected as the enzyme, and up to three missed cleavages were allowed. A decoy database search was performed. The output from Proteome Discoverer was further processed using Scaffold Q+S (version 4.0.5; Proteome Software, Portland, OR) (Searle, 2010). Upon import, the data were searched using X!Tandem (The Global Proteome Machine Organization) (Craig and Beavis, 2004). PeptideProphet (Keller et al., 2002) and ProteinProphet (Institute for Systems Biology, Seattle, WA) (Nesvizhskii et al., 2003) probability thresholds of 95% were calculated from the decoy searches, and Scaffold was used to calculate an improved 95% peptide and protein probability threshold on the data from the two different search algorithms. Protein identifications were accepted if they contained at least three identified peptides.
Results
Design and Synthesis of the Probe.
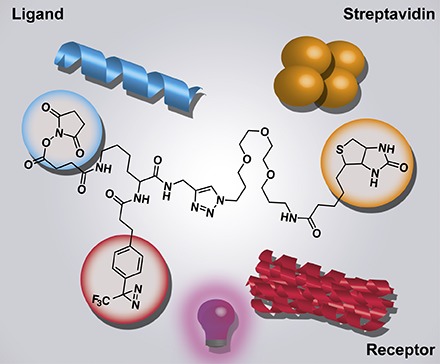
A probe for ligand-based receptor capture should permit the attachment of a ligand, enable receptor capture, and allow enrichment for identification via, for example, MS analysis (Fig. 1A). Similar to other probes, an NHS ester was chosen to attach the ligand to the probe. Under physiologic conditions, the NHS ester reacts with primary amines, present in peptide and protein ligands, to yield amide bonds (Anderson et al., 1963; Mädler et al., 2009). To enable receptor capture, a photoaffinity label was introduced. Irradiation of such labels results in covalent coupling to nearby molecules (Sumranjit and Chung, 2013). A diazirine moiety was chosen over other photoaffinity labels due to its reported short activation time, high stability, and low background photo cross-linking (Kleiner et al., 2017; Hill and Robertson, 2018). For enrichment of the captured receptor(s) after cell lysis, a biotin moiety was incorporated into the probe. The required functional groups were introduced into a trifunctional molecular scaffold based on lysine, creating probe 1, which can be used for ligand-based receptor capture (Fig. 1). Probe 1 was conveniently synthesized in seven steps from Fmoc-N-ε-Boc-lysine with an overall yield of 19% (Fig. 1B). The synthesis started with the coupling of propargylamine to the free carboxylic acid, creating an alkyne handle in amide 2. This was followed by Fmoc deprotection and coupling of the photoaffinity label 3-{4-[3-(trifluoromethyl)-3H-diazirin-3-yl]phenyl}propanoic acid, leading to compound 3 (Geurink et al., 2010). After Boc-deprotection, succinic anhydride was used to incorporate a carboxylic acid moiety, thereby affording compound 4. Addition of biotin-PEG-azide via copper(I)-catalyzed azide alkyne cycloaddition (CuAAC) resulted in the formation of compound 5, which was activated with an NHS ester to yield the final product, probe 1. Before proceeding to the biologic evaluation, it was confirmed that UV irradiation of the probe did indeed activate the diazirine moiety followed by insertion of the resulting carbene into nearby molecules (Supplemental Fig. S4).
Fig. 1.

A flowchart of ligand-based receptor capture and the synthesis of probe 1. (A) To initiate ligand-based receptor capture, the probe is first coupled to the ligand of interest (top of scheme) or a control ligand (glycine, bottom of scheme), after which the adduct is added to cells potentially expressing a target receptor. After an appropriate incubation time, during which the ligand binds the receptor, cells are washed to rinse away unbound probe. Cells are then exposed to UV light to activate the diazirine moiety which captures the receptor. Cells are lysed, and lysates are added to streptavidin beads. Captured proteins are digested on the beads with trypsin, and released peptides are analyzed using mass spectrometry. Comparison between the proteins identified for ligand-treated and glycine-treated cells should reveal the ligand-binding receptor. (B) Synthesis of probe 1. HBTU, hexafluorophosphate benzotriazole tetramethyl uronium.
Synthesis of Substance P and Coupling to the Probe.
The NK1 receptor and its ligand SP were chosen as a test system. A slightly modified version of SP (Ac-Nle-SP), in which the C-terminal methionine residue was replaced with a norleucine (Nle) residue and the N terminus was capped with an acetyl (Ac) group to leave only one free amine available, was synthesized using solid phase peptide synthesis (Fig. 2A; Supplemental Fig. S2). Addition of probe 1 (2.5 eq) to Ac-Nle-SP in PBS (pH 8.2) resulted in coupling of Ac-Nle-SP to probe 1, yielding Ac-Nle-SP-probe 8, while no free Ac-Nle-SP remained (Fig. 2B; Supplemental Fig. S3A). The remainder of the probe either hydrolyzed back to the free acid 5 or formed cyclization product 6 (Supplemental Fig. S3A). To function as a negative control, probe 1 was also coupled to glycine, resulting in a similar mixture of Gly-probe 9 (Fig. 2C), and hydrolyzed probe 5 and cyclization product 6 (Supplemental Fig. S3B).
Fig. 2.

Structures of Ac-Nle-SP (A), Ac-Nle-SP-probe (B), and Gly-probe (C).
Generation of NK1-Expressing Cell Lines.
The Flp-In T-REx 293 system was used to create cell lines designed to allow inducible expression of a receptor of interest upon addition of the antibiotic Dox (Fig. 3). In the first of these, an NK1 receptor construct was generated in which the HA epitope tag (amino acid sequence YPYDVPDYA) was engineered into the extracellular N-terminal domain, and eGFP was added in frame with the intracellular C-terminal tail of the receptor (HA-NK1-eGFP). In a second iteration, eGFP was replaced with a hexa-histidine tag (HA-NK1-6xHis) to facilitate detection using an anti-6xHis antiserum.
Fig. 3.

Doxycycline-dependent expression of HA-NK1-eGFP and HA-NK1-6xHis. Flp-In T-REx 293 cells expressed HA-NK1-eGFP (A–C) or HA-NK1-6xHis (D) following induction by treatment with Dox. (A) Expression of HA-NK1-eGFP following overnight treatment with various concentrations of Dox was visualized by immunoblotting with anti–green fluorescent protein after cell lysates were resolved with SDS-PAGE and transferred to blotting membrane. An antitubulin antiserum provided a loading control. (B) Expression of HA-NK1-eGFP following overnight treatment of cells with various concentrations of Dox and treatment with Hoechst nuclear stain (10 μg/ml, 30 minutes) was quantified with a microplate reader by measuring fluorescence of eGFP and the Hoechst-stained nuclei. (C) Cells as aforementioned were treated in the absence (left panel) or presence of Dox (100 ng/ml; right panel) overnight and imaged using a confocal microscope while HA-NK1-eGFP was excited. (D) Expression of HA-NK1-6xHis following overnight treatment with various concentrations of Dox was visualized by immunoblotting with anti-6xHis after cell lysates were resolved with SDS-PAGE and transferred to blotting membrane. As in (A), an antitubulin antiserum provided a loading control.
Binding of Ac-Nle-SP-Probe to the NK1 Receptor.
The NK1 receptor interacts with G proteins of the Gq class, and this results in the hydrolysis of phosphatidylinositol 4,5-bisphosphate. To confirm that Ac-Nle-SP-probe retained affinity for the receptor, accumulation of the IP1 end products of this reaction was assessed in Flp-In T-REx 293 cells that had been treated with Dox to induce expression of either HA-NK1-eGFP or HA-NK1-6xHis. These studies showed that although the potency of Ac-Nle-SP-probe was reduced by a factor of between 4- and 8-fold compared with free Ac-Nle-SP, it retained binding and agonism at the receptor (Fig. 4). Control experiments confirmed that no response was obtained in cells not induced to express the NK1 receptor constructs or in cells treated with the Gly-probe.
Fig. 4.

Ac-Nle-SP-probe activates the NK1 receptor constructs. IP1 accumulation was assessed in both Dox-induced and uninduced Flp-In T-REx 293 cells harboring either the HA-NK1-eGFP (A) or the HA-NK1-6xHis (B) construct. The Ac-Nle-SP-probe was clearly active, although less potent that native Ac-Nle-SP. Data are means ± S.D. pooled from n = 3 independent experiments, each performed in triplicate.
To study the binding of Ac-Nle-SP-probe to the NK1 receptor directly, confocal microscopy was performed on HA-NK1-eGFP–expressing cells. The eGFP component of this receptor construct can be visualized directly when the receptor is induced, and indeed, cell surface localization of the expressed receptor was clearly observed in Dox-induced cells but not in uninduced cells (Fig. 5, upper panels). Following treatment of cells with 1 μM Ac-Nle-SP-probe for 1 hour at 4°C, after which cells were washed and exposed to UV light for 15 minutes, DyLight 594–conjugated streptavidin, which interacts with the biotin component of the probe, was used to visualize binding of Ac-Nle-SP-probe to HA-NK1-eGFP (Fig. 5, middle panels). This was obvious only when the receptor was expressed and was not replicated when Gly-probe was used (Fig. 5, middle panels). Merging of the images showed colocalization at the level of light microscopy of the eGFP-containing receptor and the DyLight 594–labeled probe (Fig. 5, lower panels).
Fig. 5.

Ac-Nle-SP-probe binds to the NK1 receptor constructs at the cell surface. Dox-induced or uninduced Flp-In T-REx 293 cells harboring HA-NK1-eGFP were treated with Ac-Nle-SP-probe or Gly-probe (1 μM at 4°C for 1 hour) before exposure to UV light (15 minutes) and were then treated with DyLight 594-conjugated streptavidin before they were imaged using a confocal microscope. HA-NK1-eGFP and the DyLight 594–labeled probe were excited simultaneously at 488 nm (upper panels) and 561 nm (middle panels), respectively. Colocalization between the receptor and the probe was visualized (yellow color; lower panels). The bright field image demonstrates that cells were present in the uninduced −Dox panels.
NK1 Receptor Capture by Ac-Nle-SP-Probe.
To confirm that the observed interaction between probe 1 and HA-NK1-eGFP was indeed covalent and not simply a reflection of a slow off-rate of Ac-Nle-SP-probe from the receptor following initial binding, competition experiments were performed. As before, cells expressing HA-NK1-eGFP were treated with Ac-Nle-SP-probe followed by irradiation with UV light. Now, however, before adding the DyLight 594–conjugated streptavidin to visualize probe binding to the receptor, cells were incubated with excess free Ac-Nle-SP. Treatment with free ligand did not result in reduction of colocalization between the probe and the receptor for cells exposed to UV light (Fig. 6, samples 1 vs. 3). By contrast, when cells were maintained in the dark, there was a significant (P ≤ 0.001) reduction in colocalization (Fig. 6, samples 2 vs. 4), indicating partial dissociation of Ac-Nle-SP-probe from the receptor over this time period. This suggests that UV activation did indeed induce covalent linkage between the probe and the NK1 receptor, and although dissociation of Ac-Nle-SP-probe from the receptor in the absence of cross-linking was slow, it did occur.
Fig. 6.

Ac-Nle-SP-probe covalently captures NK1 receptor constructs upon UV activation. (A) Dox-induced Flp-In T-REx 293 cells expressing HA-NK1-eGFP were treated with Ac-Nle-SP-probe (1 μM at 4°C for 1 hour) before they were activated with UV light for 15 minutes (samples 1 and 3) or kept in the dark (samples 2 and 4). Cells were then either treated immediately with DyLight 594–conjugated streptavidin (samples 1 to 2) or were first exposed to Ac-Nle-SP (10 μM at 4°C for 3 hours) (samples 3 to 4). They were then imaged using a confocal microscope. HA-NK1-eGFP and the DyLight 594–labeled probe were excited simultaneously at 488 nm (upper panels) and 561 nm (middle panels), respectively. Colocalization between the receptor and the probe was visualized (yellow color; lower panels). (B) Colocalization was quantified by generating green-red pixel intensity scatterplots for each pixel and determining the Pearson correlation coefficient for four representative images for each condition. Data are means + S.D. Significant statistical difference was determined using analysis of variance with Tukey’s multiple comparison test (***P ≤ 0.001).
To further confirm UV-induced formation of a covalent bond between the photoaffinity probe and HA-NK1-eGFP, western blotting was performed. Since Flp-In T-REx 293 cells constitutively express a number of endogenously biotinylated proteins, which unfortunately migrated at the same molecular mass on SDS-PAGE gels as the HA-NK1-eGFP receptor, these experiments were performed on cells expressing the HA-NK1-6xHis receptor construct, which is predicted to have a substantially lower molecular mass (by some 27 kDa). Following Dox-induced expression of HA-NK1-6xHis, cells were treated with Ac-Nle-SP-probe, washed, and exposed to UV light. Cells were then lysed and membrane preparations collected. Streptavidin detection confirmed the presence of a biotinylated protein of the same mobility as the HA-NK1-6xHis receptor in UV-irradiated samples treated with Ac-Nle-SP-probe (Fig. 7, sample 2) but not in samples treated with Gly-probe (Fig. 7, sample 3), nor in samples that were not exposed to UV light (Fig. 7, sample 4) or in samples not expressing the NK1 receptor (Fig. 7, sample 5). This confirmed that a covalent bond was indeed formed between Ac-Nle-SP-probe and the NK1 receptor but not between Gly-probe and the receptor.
Fig. 7.

Detection of Ac-Nle-SP-probe NK1 receptor interactions. Dox-induced (lanes 1–4) or uninduced (lane 5) Flp-In T-REx 293 cells harboring HA-NK1-6xHis were treated with either Ac-Nle-SP-probe (lanes 2, 4, and 5) or Gly-probe (lane 3) and then exposed to UV light (lanes 1–3 and 5) or maintained in the dark (lane 4). Following cell lysis, membrane preparations were generated and resolved by SDS-PAGE. These were immunoblotted with an anti-6xHis antiserum (left panel) or streptavidin (center panel). Merging of these two images and pseudocolor labeling identifies the receptor-probe complex as yellow (right panel).
Ligand-Based Receptor Capture Using Ac-Nle-SP-Probe.
Confidence in the ability to selectively label the NK1 receptor now allowed attempts to identify the covalently captured receptor in a nonbiased manner via ligand-based receptor capture and MS according to the procedure depicted in Fig. 1. After treatment of cells expressing HA-NK1-6xHis with either Ac-Nle-SP-probe or Gly-probe followed by exposure to UV, membrane preparations were obtained, detergent solubilized, and added to streptavidin-coated beads. An “on-bead” trypsin digest was performed, and resulting peptides were analyzed using LC-MS/MS. This resulted in a total list of 49 identified human proteins, 48 of which were identified in both Gly-probe- and Ac-Nle-SP-probe–treated samples (Fig. 8A; Supplemental Material). This group mostly consisted of endogenously biotinylated proteins, including carboxylases and common contaminants such as keratin. The NK1 receptor (UniProt identifier P25103), which was recovered as five distinct peptide fragments (Fig. 8B; Table 1), was the only protein identified in Ac-Nle-SP-probe- but not in Gly-probe–treated samples. Moreover, apart from NK1, none of the other protein hits showed significant differences (P ≤ 0.05) in either total spectrum count or total ion current between the samples, illustrating the specificity of the system and probe developed (Supplemental Material).
Fig. 8.

Identification of proteins by mass spectrometry after ligand-based receptor capture. (A) Schematic view of human proteins identified by LC-MS/MS after LRC experiments with either Gly-probe or Ac-Nle-SP-probe. (B) The amino acid sequence of the human NK1 receptor is displayed. Peptides that were identified using LC-MS/MS after LRC with Ac-Nle-SP-probe are colored. See Table 1 for further details (1 = blue, 2 = green, 3 = yellow, 4 = purple. 5 = green).
TABLE 1.
Peptide sequences from NK1 receptor identified by mass spectrometry
| Peptide sequence | Total Number of Spectra | Found in # Samples | Modifications | |
|---|---|---|---|---|
| 1 | (R)LETTISTVVGAHEEEPEDGPK(A) | 2 | 2 | |
| 2 | (R)YLQTQGSVYKVSR(L) | 1 | 1 | |
| 3 | (K)ATPSSLDLTSNcSSR(S) | 2 | 2 | Carbamidomethyl |
| 4 | (R)YMAIIHPLQPR(L) | 1 | 1 | |
| 5 | (R)YLQTQGSVYK(V) | 1 | 1 |
Discussion
Herein, the development, synthesis, and evaluation of a new trifunctional probe for ligand-based receptor capture have been documented. This probe uses a diazirine photoaffinity label to capture nearby target receptors. This has several advantages over other capture techniques. First, all receptors with an extracellular domain can be captured in intact cells by such a photo-reactive group. This is in contrast to, for example, the protected hydrazine group used in TRICEPS, which can only capture receptors that are N-glycosylated in proximity to the ligand-binding site (Frei et al., 2012). Second, a washing step after ligand-receptor binding but before receptor capture can be introduced, thus reducing nonspecific binding and background labeling by the probe. Third, as these groups are very stable in the absence of light, the ligand can be coupled to the probe and the complex can be stored, limiting the need to synthesize new batches of the probe for each experimental run. For affinity purification, a biotin moiety was incorporated into the probe. Biotin is the most widely used affinity tag due to its high-affinity binding to streptavidin (Hermanson, 2013); however, a new trend in the development of cytosolic probes has emerged recently. This involves the incorporation of an alkyne or azide handle into the probe instead of the bulky biotin group, which would hinder cell entrance of the probe. CuAAC can then be used to couple biotin, or any other reporter tag, to the probe after cell lysis, or even couple the probe directly to azide-conjugated beads (Smith and Collins, 2015). Originally such a strategy was explored, and a variant of probe 1, bearing an alkyne handle instead of a biotin moiety, was created by directly activating compound 4 with an NHS ester. This probe was coupled to Ac-Nle-SP and added to cells expressing the NK1 receptor. Then, CuAAC was attempted on lysates of these cells to couple this probe to either biotin- or azide-conjugated agarose beads. In our hands, however, this strategy was not successful. For the purpose of identifying transmembrane receptors on living cells, no cell entrance is required, hence it was decided to attach biotin to the probe from the outset, resulting in probe 1.
It should be noted that probe 1 was not fully purified. LC-MS analysis revealed the presence of some hydrolyzed probe 5, as well as a probe product which had undergone cyclic imide formation (6) (Supplemental Fig. S1). Whether these by-products were formed during the analysis or whether they were actually present in the product was not investigated, since they were not expected to interfere with coupling of a ligand. For ligand coupling, probe 1 was dissolved in PBS (pH 8.2) and partial hydrolysis as well as intramolecular cyclization of the active NHS ester were inevitable under these conditions (Supplemental Fig. S3) (Lim et al., 2014; Klykov and Weller, 2015). These probe by-products were easily eliminated when the cells were washed before irradiation. Indeed, when cells lacking expression of NK1 receptor were treated with the Ac-Nle-SP-probe, no substantial background labeling was observed (Fig. 5, right panels).
For ligand-based receptor-capture experiments to be successful, it is important that the ligand-probe adduct binds to the receptor(s) of interest. In this case, this was tested by using accumulation of IP1, which is a consequence of binding of SP and related peptides to the NK1 receptor. After binding of the ligand-probe adduct to the receptor, it is vital that the ligand remains associated with the receptor until the probe is activated by UV light. Incubation of cells with Ac-Nle-SP-probe was first performed at 37°C; however, it was soon appreciated that the NK1 receptor rapidly (3 minutes) internalizes upon activation by its agonist SP (Garland et al., 1994). Internalization of the NK1-SP complex is known to be linked with dissociation and degradation of SP; therefore, it was important to perform receptor-capture experiments at lower temperatures, preventing internalization of the receptor (Garland et al., 1994). That the Ac-Nle-SP-probe still bound to the NK1 receptor at 4°C but did not result in substantial internalization of the receptor was apparent from the microscopy studies. These low temperatures also slowed down the dissociation of the ligand from the receptor. Potentially, this may be beneficial for receptor capture, as it should keep the probe in close proximity to the receptor. It did, however, limit dissociation and competition of the non-crosslinked Ac-Nle-SP-probe from the NK1 receptor in the presence of an excess of free Ac-Nle-SP.
After initial characterization of the probe, a proof of concept for probe 1 was provided in which Ac-Nle-SP was coupled to this probe and used to identify the GPCR NK1 through LC-MS/MS. This is one of the few successful examples in which a GPCR has been identified using a universal probe and proteomics. TRICEPS was used previously to identify the apelin receptor; however, this probe clearly has shortcomings with respect to the requirement for glycosylation of the receptor (Frei et al., 2012). While we were working on the development of these photoreactive universal probes, others also realized the benefit of incorporating photoreactive groups into probes targeting transmembrane receptors. Two reports describing such probes specifically targeting GPCRs have been published recently. In both cases, the probes were designed on known pharmacological ligands rather than being potentially universal. Blex et al. (2017) generated the probe CPT-00031 based on the structure of sertindole, a well known dopamine D2 receptor antagonist, whereas Thomas et al. (2017) created a probe based on the structure of an active parent compound known to bind G protein–coupled receptor 39. Akin to the probe described herein, both of these probes also contained a 3-aryl-3-(trifluoromethyl)diazirine moiety and a biotin group and were tested on systems overexpressing the receptor of interest.
A major challenge in progressing studies of this nature to true unbiased detection of genuinely novel ligand-receptor interactions is the number of “off-target” interactions that are identified, and including appropriate negative controls is an important aspect of LRC experiments. In the studies using CPT-00031 on dopamine D2 receptor–expressing HEK293 cells, a competition experiment with an excess of sertindole was performed as a potential negative control. However, there were over 150 proteins enriched in the noncompetition over the competition samples (Blex et al., 2017). Although this study did attempt to assess sensitivity by diluting transfected cells with nontransfected control cells, there were always other proteins identified as well as the dopamine D2 receptor. Moreover, when diluting the transfected cells to a level in which the target receptor was not enriched, 18 other membrane proteins were enriched. Whether any of these 18 proteins truly bound to sertindole was not further investigated. Unfortunately, Thomas et al. (2017), who used a similar competition experiment as a negative control, did not include the full data set obtained with their LC-MS/MS experiments, and therefore, it is hard to ascertain how many proteins were identified in the LRC studies. It is clear, however, that there were at least seven transmembrane receptors, including G protein–coupled receptor 39, enriched in the noncompetition samples (Thomas et al., 2017). In the studies reported herein, instead of competition experiments, an LRC experiment using Gly-probe was used as a negative control. In contrast to the two probes discussed earlier, comparison between the Gly-probe- and the Ac-Nle-SP-probe–treated samples resulted in the identification of only one protein: the NK1 receptor.
Establishing that probe 1 can indeed be used to identify the NK1 receptor as the Ac-Nle-SP–binding receptor in a test system is an important proof of concept toward general utilization of this probe to help identify unknown plasma-membrane receptors for ligands of interest. Nevertheless, the goal of developing a truly high-specificity and universal probe for ligand-based receptor capture that can be used with confidence in natively expressing cells and tissue remains a substantial challenge.
Acknowledgments
We thank John Pediani for help with the confocal microscopy experiments and Andrew Bottrill and Sharad Mistry for the performance of the LC-MS/MS analysis.
Abbreviations
- Ac
acetyl
- COSY
correlation spectroscopy
- CuAAC
copper(I)-catalyzed azide alkyne cycloaddition
- DCM
dichloromethane
- DIPEA
N,N-diisopropylethylamine
- DMF
dimethylformamide
- DMSO
dimethylsulfoxide
- Dox
doxycycline
- EDC
1-ethyl-3-(3-dimethylaminopropyl)carbodiimide
- eGFP
enhanced green fluorescent protein
- eq
equivalent
- FT
Fourier transform
- Gly
glycine
- GPCR
G protein–coupled receptor
- HA
hemagglutinin
- HBSS
Hanks’ balanced salt solution
- 6xHis
hexa-histidine tag
- HPLC
high-performance liquid chromatography
- HRMS
high-resolution mass spectrometry
- HSQC
heteronuclear single quantum coherence spectroscopy
- IP1
inositol monophosphate
- LC-MS
liquid chromatography–mass spectrometry
- LC-MS/MS
liquid chromatography–tandem mass spectrometry
- LRC
ligand-based receptor capture
- MS
mass spectrometry
- NHS
N-hydroxysuccinimide
- NK1
neurokinin 1
- Nle
norleucine
- PBS
phosphate-buffered saline
- PEG
polyethylene glycol
- RT
room temperature
- SP
substance P
- TFA
trifluoroacetic acid
Authorship Contributions
Participated in research design: Müskens, Ward, van de Langemheen, Tobin, Liskamp, Milligan.
Conducted experiments: Müskens.
Contributed new reagents or analytic tools: Müskens, Herkt.
Performed data analysis: Müskens.
Wrote or contributed to the writing of the manuscript: Müskens, Liskamp, Milligan.
Footnotes
F.M.M. thanks the University of Glasgow for a Lord Kelvin-Adam Smith studentship.
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Anderson GW, Zimmerman JE, Callahan FM. (1963) N-hydroxysuccinimide esters in peptide synthesis. J Am Chem Soc 85:3039. [DOI] [PubMed] [Google Scholar]
- Blex C, Michaelis S, Schrey AK, Furkert J, Eichhorst J, Bartho K, Gyapon Quast F, Marais A, Hakelberg M, Gruber U, et al. (2017) Targeting G Protein-coupled receptors by capture compound mass spectrometry: a case study with sertindole. ChemBioChem 18:1639–1649. [DOI] [PubMed] [Google Scholar]
- Chambers JM, Lindqvist LM, Webb A, Huang DCS, Savage GP, Rizzacasa MA. (2013) Synthesis of biotinylated episilvestrol: highly selective targeting of the translation factors eIF4AI/II. Org Lett 15:1406–1409. [DOI] [PubMed] [Google Scholar]
- Craig R, Beavis RC. (2004) TANDEM: matching proteins with tandem mass spectra. Bioinformatics 20:1466–1467. [DOI] [PubMed] [Google Scholar]
- Frei AP, Jeon O-Y, Kilcher S, Moest H, Henning LM, Jost C, Plückthun A, Mercer J, Aebersold R, Carreira EM, et al. (2012) Direct identification of ligand-receptor interactions on living cells and tissues. Nat Biotechnol 30:997–1001. [DOI] [PubMed] [Google Scholar]
- Garcia-Recio S, Gascón P. (2015) Biological and pharmacological aspects of the NK1-receptor. BioMed Res Int 2015:495704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garland AM, Grady EF, Payan DG, Vigna SR, Bunnett NW. (1994) Agonist-induced internalization of the substance P (NK1) receptor expressed in epithelial cells. Biochem J 303:177–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geurink PP, Klein T, Prely L, Paal K, Leeuwenburgh MA, Van Der Marel GA, Kauffman HF, Overkleeft HS, Bischoff R. (2010) Design of peptide hydroxamate-based photoreactive activity-based probes of zinc-dependent metalloproteases. Eur J Org Chem 11:2100–2112. [Google Scholar]
- Hartwig S, Hecht S. (2010) Polypseudopeptides with variable stereochemistry: synthesis via click-chemistry, postfunctionalization, and conformational behavior in solution. Macromolecules 43:242–248. [Google Scholar]
- Hatanaka Y. (2015) Development and leading-edge application of innovative photoaffinity labeling. Chem Pharm Bull (Tokyo) 63:1–12. [DOI] [PubMed] [Google Scholar]
- Hauser AS, Attwood MM, Rask-Andersen M, Schiöth HB, Gloriam DE. (2017) Trends in GPCR drug discovery: new agents, targets and indications. Nat Rev Drug Discov 16:829–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helbig AO, Heck AJR, Slijper M. (2010) Exploring the membrane proteome--challenges and analytical strategies. J Proteomics 73:868–878. [DOI] [PubMed] [Google Scholar]
- Hermanson GT. (2013) (Strept)avidin–biotin systems, in Bioconjugate Techniques pp 465–505, Elsevier, Boston. [Google Scholar]
- Hill JR, Robertson AAB. (2018) Fishing for drug targets: a focus on diazirine photoaffinity probe synthesis. J Med Chem 61:6945–6963. [DOI] [PubMed] [Google Scholar]
- Hopkins AL, Groom CR. (2002) The druggable genome. Nat Rev Drug Discov 1:727–730. [DOI] [PubMed] [Google Scholar]
- Keller A, Nesvizhskii AI, Kolker E, Aebersold R. (2002) Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem 74:5383–5392. [DOI] [PubMed] [Google Scholar]
- Kleiner P, Heydenreuter W, Stahl M, Korotkov VS, Sieber SA. (2017) A whole proteome inventory of background photocrosslinker binding. Angew Chem Int Ed Engl 56:1396–1401. [DOI] [PubMed] [Google Scholar]
- Klykov O, Weller MG. (2015) Quantification of N-hydroxysuccinimide and N-hydroxysulfosuccinimide by hydrophilic interaction chromatography (HILIC). Anal Methods 7:6443–6448. [Google Scholar]
- Lenz T, Fischer JJ, Dreger M. (2011) Probing small molecule-protein interactions: a new perspective for functional proteomics. J Proteomics 75:100–115. [DOI] [PubMed] [Google Scholar]
- Li Y, Ozment T, Wright GL, Peterson JM. (2016) Identification of putative receptors for the novel adipokine CTRP3 using ligand-receptor capture technology. PLoS One 11:e0164593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Zeppa JJ, Hancock MA, McCormick JK, Doherty TM, Hendy GN, Madrenas J. (2018) Staphylococcal superantigens use LAMA2 as a coreceptor to activate T cells. J Immunol 200:1471–1479. [DOI] [PubMed] [Google Scholar]
- Lim CY, Owens NA, Wampler RD, Ying Y, Granger JH, Porter MD, Takahashi M, Shimazu K. (2014) Succinimidyl ester surface chemistry: implications of the competition between aminolysis and hydrolysis on covalent protein immobilization. Langmuir 30:12868–12878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mädler S, Bich C, Touboul D, Zenobi R. (2009) Chemical cross-linking with NHS esters: a systematic study on amino acid reactivities. J Mass Spectrom 44:694–706. [DOI] [PubMed] [Google Scholar]
- Nesvizhskii AI, Keller A, Kolker E, Aebersold R. (2003) A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem 75:4646–4658. [DOI] [PubMed] [Google Scholar]
- Overington JP, Al-Lazikani B, Hopkins AL. (2006) How many drug targets are there? Nat Rev Drug Discov 5:993–996. [DOI] [PubMed] [Google Scholar]
- Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. (1999) Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20:3551–3567. [DOI] [PubMed] [Google Scholar]
- Ponda MP, Breslow JL. (2016) Serum stimulation of CCR7 chemotaxis due to coagulation factor XIIa-dependent production of high-molecular-weight kininogen domain 5. Proc Natl Acad Sci USA 113:E7059–E7068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos R, Ursu O, Gaulton A, Bento AP, Donadi RS, Bologa CG, Karlsson A, Al-Lazikani B, Hersey A, Oprea TI, et al. (2017) A comprehensive map of molecular drug targets. Nat Rev Drug Discov 16:19–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savas JN, Stein BD, Wu CC, Yates JR., III (2011) Mass spectrometry accelerates membrane protein analysis. Trends Biochem Sci 36:388–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirle M, Jenkins JL. (2016) Identifying compound efficacy targets in phenotypic drug discovery. Drug Discov Today 21:82–89. [DOI] [PubMed] [Google Scholar]
- Searle BC. (2010) Scaffold: a bioinformatic tool for validating MS/MS-based proteomic studies. Proteomics 10:1265–1269. [DOI] [PubMed] [Google Scholar]
- Smith E, Collins I. (2015) Photoaffinity labeling in target- and binding-site identification. Future Med Chem 7:159–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobotzki N, Schafroth MA, Rudnicka A, Koetemann A, Marty F, Goetze S, Yamauchi Y, Carreira EM, Wollscheid B. (2018) HATRIC-based identification of receptors for orphan ligands. Nat Commun 9:1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumranjit J, Chung SJ. (2013) Recent advances in target characterization and identification by photoaffinity probes. Molecules 18:10425–10451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JR, Brittain SM, Lipps J, Llamas L, Jain RK, Schirle M. (2017) A photoaffinity labeling-based chemoproteomics strategy for unbiased target deconvolution of small molecule drug candidates. Methods Mol Biol 1647:1–18. [DOI] [PubMed] [Google Scholar]
- Tremblay T-L, Hill JJ. (2017) Biotin-transfer from a trifunctional crosslinker for identification of cell surface receptors of soluble protein ligands. Sci Rep 7:46574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt Consortium (2010) The universal protein resource (UniProt) in 2010. Nucleic Acids Res 38:D142–D148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Stetten D, Noirclerc-Savoye M, Goedhart J, Gadella TWJ, Jr., Royant A. (2012) Structure of a fluorescent protein from Aequorea victoria bearing the obligate-monomer mutation A206K. Acta Crystallogr Sect F Struct Biol Cryst Commun 68:878–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuckovic D, Dagley LF, Purcell AW, Emili A. (2013) Membrane proteomics by high performance liquid chromatography-tandem mass spectrometry: analytical approaches and challenges. Proteomics 13:404–423. [DOI] [PubMed] [Google Scholar]
- Ward RJ, Pediani JD, Milligan G. (2011) Heteromultimerization of cannabinoid CB(1) receptor and orexin OX(1) receptor generates a unique complex in which both protomers are regulated by orexin A. J Biol Chem 286:37414–37428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright GJ. (2009) Signal initiation in biological systems: the properties and detection of transient extracellular protein interactions. Mol Biosyst 5:1405–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]