

Abstract
We report design, synthesis, and evaluation of polyaminocarboxylate ligand-based antibody conjugate for potential application in targeted cancer therapy and near infrared fluorescent imaging. We synthesized a new polyaminocarboxylate chelate (CAB-NE3TA) as a potential anti-cancer agent. CAB-NE3TA displayed potent inhibitory activities against different cancer cell lines. We then designed a multifunctional theranostic platform (CAB-NE3TA-PAN-IR800) constructed on an EGFR-targeted antibody (panitumumab, PAN) labeled with a near IR fluorescent dye. We also built the first atomistic model of the EGFR-PAN complex and loaded it with the cytotoxic CAB-NE3TA and the near IR dye. The therapeutic (CAB-NE3TA-PAN) and theranostic (CAB-NE3TA-PAN-IR800) conjugates were evaluated using EGFR-positive A431 (human skin cancer) cell xenograft mouse model. Biodistribution studies using near IR fluorescence imaging demonstrated that the CAB-NE3TA-PAN labeled with the IR800 dye selectively targeted the A431 tumors in mice and resulted in prolonged retention in the tumor tissue and displayed excellent clearance in blood and normal organs. The therapeutic conjugate was capable of significantly inhibiting tumor growth, leading to nearly complete disappearance of tumors in the mice. The results of our pilot in vivo studies support further evaluation of the novel ligand-based therapeutic and theranostic conjugates for targeted iron chelation cancer therapy and imaging applications.
Keywords: theranostic agent, anti-cancer therapy, near IR fluorescent imaging, antibody drug conjugate
Table of Contents
We report therapeutic and theranostic antibody drug conjugates (ADCs) for targeted iron chelation cancer therapy and imaging. A new polyaminocarboxylate chelate (CAB-NE3TA) as a potential anticancer agent was conjugated to an EGFR-targeted antibody (panitumumab, PAN) for targeted delivery to skin carcinoma. The therapeutic ADC (CAB-NE3TA-PAN) significantly inhibited tumor growth, leading to nearly complete disappearance of tumors in the mice. Whole body fluorescence imaging demonstrated that the theranostic ADC (CAB-NE3TA-PAN- IR800) labeled with a near IR probe selectively targeted the skin carcinoma in
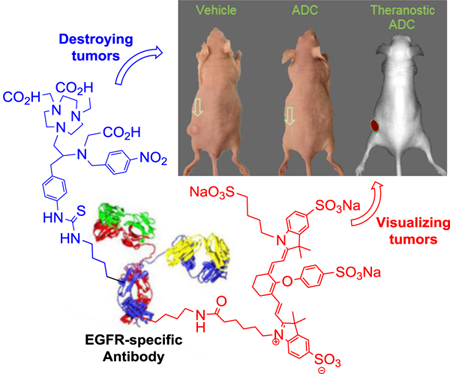
Introduction
Iron is a biologically important transition metal and involved in many cellular biochemical pathways through its versatile coordination chemistry and redox process between the two oxidation states, Fe(II) and Fe(III).[1–3] Rapidly dividing cancer cells require more iron than normal healthy cells and are particularly sensitive to depletion of cellular iron.[3–5] The enhanced requirement of iron in cancer cells is related to overexpression of iron-related proteins and enzymes including the iron storage protein ferritin, the transferrin receptor (TfR) involved in endocytosis of iron-bound transferrin (Tf), and ribonucleotide reductase (RR) required for DNA synthesis.[3–8]
Iron chelators that can target the iron-related proteins and enzymes and inhibit growth of cancer cells have been designed and explored as cancer therapeutics.[3,4,9] Among the iron chelators being evaluated in clinical settings are triapine (3-aminopyridine-2-carboxaldehyde thiosemi- carbazone, 3-AP) and DFO (deferoxamine).[9] Triapine and DFO (Figure 1) have been shown to inhibit the activity of the RR enzyme overexpressed in cancer cells, and iron depletion was proposed as a mechanism for the inhibitory action of the chelating agents on the cancer cells.[3–5]
Figure 1.
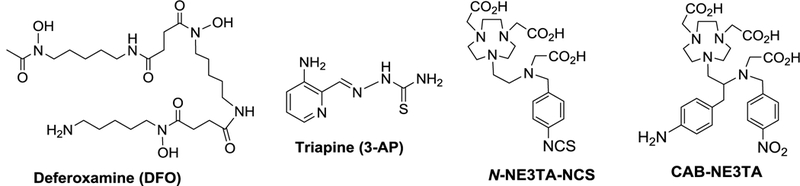
Iron-Chelating Antitumor Agents in clinical and preclinical evaluations
Triapine is a thiosemicarbazone-based RR inhibitor and was shown to sequester a tyrosyl radical in the R2 protein of the tetrameric enzyme, and anti-tumor therapeutic and iron depleting efficacy of the tridentate chelator was demonstrated in numerous preclinical and clinical trials.[10–12] DFO, a natural siderophore, is the first iron chelator that was approved by the US FDA for the treatment of iron overload in patients with β-thalassemia major.[9] The hexadentate DFO was shown to be highly selective in binding Fe(III) over Fe(II) or other biologically important metals.[9] DFO was also found to induce apoptotic cell death and display inhibitory and antiproliferative activity on tumor cells including leukemia, bladder carcinoma, and hepatocellular carcinoma.[13–16] We developed polyaminocarboxylate-based chelators in the series of NE3TA (7- [2-[carboxymethyl)amino]ethyl]-1,4,7-triazacyclononane-1,4-diacetic acid, Figure 1) and reported that the NE3TA chelators exhibited significantly higher anti-proliferative activity against HeLa and colon cancer cells than the clinically available DFO.[17–18] Within our continued effort towards development of metal chelation chemistry for cancer therapeutic and imaging applications.[17–22], we were interested in utilizing the NE3TA-based cytotoxic agent for construction of a multifunctional conjugate that can target, detect, and destroy cancer cells. Development of such tumor-specific theranostic agents that combine advantages of therapeutic and imaging components into a single platform remains an active area of cancer research.[23–24]
We herein report synthesis and evaluation of a new cytotoxic chelating agent (CAB-NE3TA, Figure 1) for use in antibody-targeted iron chelation therapy and near-infrared (NIR) optical imaging of cancers. We selected panitumumab (PAN) as a model targeting vector for the present study. PAN is a fully humanized and internalizing antibody with high binding affinity to epidermal growth factor receptor (EGFR) overexpressed in various cancer cells.[25–28] The new chelate CAB-NE3TA was evaluated for anti-proliferative activity against cancer cells and further conjugated to PAN for pilot in vivo therapeutic and theranostic studies using EGFR-positive tumor-bearing mice. A docking simulation was conducted for a complex of EGFR and panitumumab conjugate containing the theranostic components, CAB-NE3TA and near IR fluorescent dye.
Results and Discussion
Synthesis of the bifunctional chelating agent CAB-NE3TA.
We previously reported evaluation of N-NE3TA-based transferrin conjugate as a potential theranostic agent for cancer therapy and imaging.[22] The theranostic conjugate was built on transferrin (Tf), a glycoprotein targeting the extracellular domain of transferrin receptor. While the transferrin-based theranostic conjugate was well-tested against cancer cells, high uptake of the optical tracer in liver, spleen, and kidneys was observed in the subsequent in vivo biodistribution and near IR fluorescent imaging studies using tumor-bearing mice. It is speculated that the labile N-benzylic moiety (p- NCS-Bn) in N-NE3TA-NCS (Figure 1) used for conjugation to transferrin could not be tolerated and be cleaved in vivo. In an effort to synthesize a functional analogue of the anti-tumor agent N- NE3TA with a more robust C-substituted linker, we designed the new chelator CAB-NE3TA (Scheme 1) where the amino group can be converted to the isothiocyanate group for conjugation to an antibody. The synthetic route to CAB-NE3TA is centered on preparation of the key precursor molecule 11 via the regiospecific ring opening of aziridinium ion 9 with NO2A (di- tert-butyl-1,4,7-triazacyclononane-1,4-diacetate, 10). The starting material, N-BOC (tert- butyloxycarbonyl) protected p-NO2-phenylalaninol 1 was subjected to hydrogenation for conversion of the nitro group to the amino group in 2, which was further reacted with benzyl chloroformate to afford 3. The BOC group in 3 was removed by treatment of 3 with TFA to provide compound 4. The amino group in 4 was selectively substituted with tert- butylbromoacetate to afford 5 in 72% yield, and subsequent alkylation of 5 with p-nitrobenzyl bromide produced N,N’-bisubstituted β-amino alcohol 6. Bromination of 6 using N-bromosuccinimide (NBS) and triphenylphospine (PPh3) provided secondary β-amino bromide 8a in 44% isolated yield. The aziridinium ion 7 was initially formed from intramolecular reaction of 6 and ring opening of 7 at the more substituted carbon with the counter ion bromide led to the formation of 8a. Iodination of 6 using I2 and imidazole provided secondary β-amino iodide 8b in an improved isolated yield (86%).
Scheme 1.
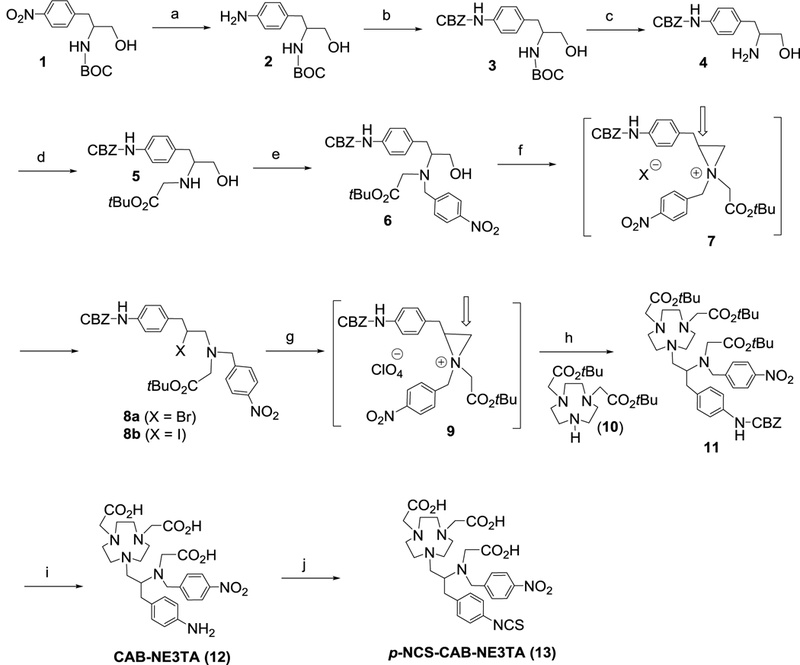
Synthesis of CAB-NE3TA analogues. Reagents and Conditions: a) H2 (15 psi), 10% Pd/C, EtOH, RT, 5.5 h, 97%; b) CBZ-Cl, K2CO3, CH3CN, RT, 24 h, 56%; c) TFA, CHCl3, RT, 3 h, 87%; d) BrCH2CO2tBu, K2CO3, CH3CN, RT, 18 h, 72%; e) p-NO2-BnBr, K2CO3, CH3CN, RT, 18 h, 53%; f) NBS or I2, Imidazole, CH2Cl2, 0 °C 4 h, RT, 1 h, 44% (8a), 86% (8b); g) AgClO4, CH3CN, - 5 °C, 15 min; h) 10, DIPEA, CH3CN, 0 °C to RT, 89 h, 70% (from 8a), 48 h, 76% (from 8b), 70–76%; i) 4M HCl/1,4-dioxane, RT, 18 h, 95%; j) CSCl2, CHCl3/H2 O, RT, 4 h, 93%.
Compound 8a was converted to aziridinium ion 9 using AgClO4 as a halide sequestering agent. Aziridinium ion 9 formed in situ was reacted with 10 for 89 h to provide the desired substitution product 11 (70% isolated yield). The substitution reaction of 8b with 10 was faster in producing 11 (48 h, 76% isolated yield). Removal of the CBZ group and tert-butyl groups in 11 via acidic hydrolysis furnished the desired product CAB-NE3TA (12). The amino group in 12 was converted to the isothiocyanate (NCS) group in p-NCS-CAB-NE3TA (13).
In vitro anti-tumor activity of CAB-NE3TA.
HeLa (cervical cancer), MDA-MB-231 (breast cancer), A375 (melanoma), PC3 (prostate), HT29 (colon cancer), and A431 (human skin cancer), LS174T (colorectal cancer) cells are reported to overexpress EGFR receptors.[29–33] CAB-NE3TA and the clinically available iron chelator DFO (10 μM) as a control agent were comparatively evaluated for cytotoxicity against the EGFR-positive cancer cell lines (Figure 2). CAB-NE3TA was significantly more effective in inhibiting all cancer cell lines tested than the known iron chelator DFO. Among the cancer cells tested, HT29 cells were the most sensitive to the cytotoxic effect of CAB-NE3TA (1.2% viability). CAB-NE3TA displayed the lowest inhibitory activity against LS74T cells (29.4% viability). Viability values of the cancer cells treated with DFO at 10 μM were in the range between 20.4% (A375) and 89.6% (HeLa).
Figure 2.
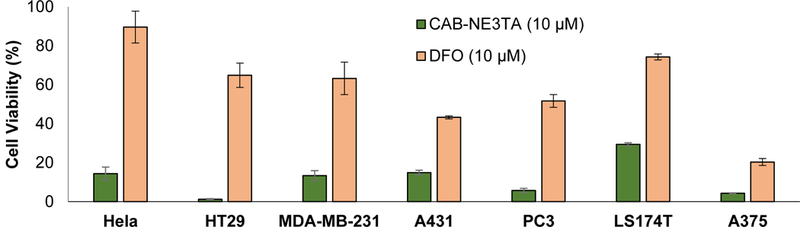
Anti-proliferative activity of CAB-NE3TA (10 μM) and DFO (10 μM) against cancer cells. The compound was incubated with the cells for 72 h, and cell viability was determined using MTS assay. The reported data on the anti-proliferative activity of the known compound DFO against HeLa, HT29, and PC3 are included for comparison [17,22].
CAB-NE3TA at a range of concentrations was further tested for cytotoxicity against the cancer cells (Figure 3). CAB-NE3TA exhibited potent anti-proliferative activity on all cancer cell lines. HeLa (IC50 = 9.0 ± 0.6 μM), HT29 (IC50 = 4.8 ± 0.5 μM), PC3 (IC50 = 4.8 ± 0.5 μM), A375 (IC50 = 3.4 ± 0.3 μM), A431 (IC50 = 3.2 ± 0.2 μM), and MDA-MB-231 (IC50 = 7.1 ± 0.8 μM) cancer cells remained sensitive to the inhibitory activities of CAB-NE3TA. When treated with CAB- NE3TA at 20 μM, nearly all HT29, PC3, and A375 cells were destroyed (<4% viability). A slightly enhanced viability (<14% viability) was observed with HeLa, MDA-MB-231, and A431 cells relative to HT29, PC3, and A375 cells treated with CAB-NE3TA. The LS174T colorectal cancer cells was less responsive to cytotoxicity of CAB-NE3TA at high concentration (~30% viability at 10~50 μM) than other cancer cell lines. Viability of LS174T when treated with CAB- NE3TA was not shown to be dose-dependent (~30% viability, 10~50 μM).
Figure 3.
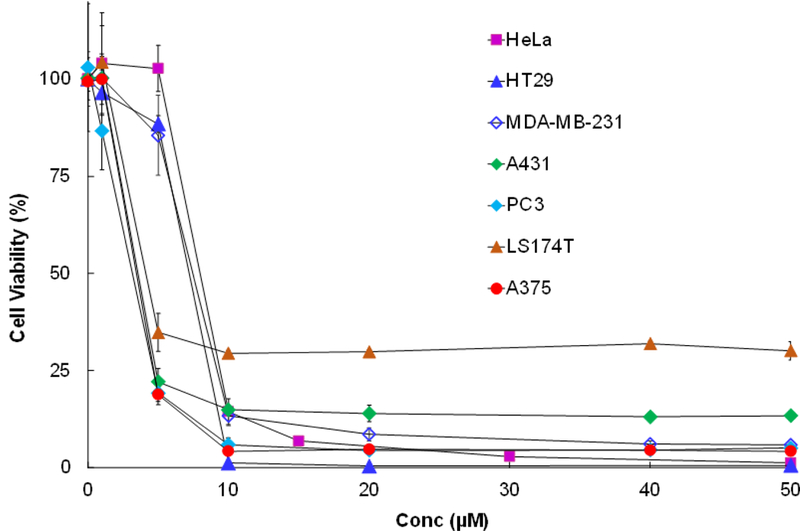
Anti-proliferative activity of CAB-NE3TA on cancer cells. CAB-NE3TA was incubated with the cells for 72 h, and cell viability was determined using MTS assay.
Preparation and Characterization of therapeutic and theranostic conjugates (CAB- NE3TA-PAN and CAB-NE3TA-PAN-IR800).
With the encouraging cytotoxicity data of the anti-tumor chelating agent CAB-NE3TA, we aim to develop a receptor-specific antibody conjugates of CAB-NE3TA (Scheme 2). A humanized antibody panitumumab (PAN) and a NIR fluorescent dye (IR800) were selected for construction of CAB-NE3TA-based therapeutic and theranostic conjugates. Epidermal growth factor receptor (EGFR) is overexpressed in many cancers including gliomas, colorectal, skin, breast, head and neck cancers.[29–33] Panitumumab binds to EGFR with high specificity and affinity and inhibits ligand binding to EGFR inducing receptor internalization and blocking receptor-mediated signaling.[34] Panitumumab (Vectibix®) was approved by US FDA for treatment of metastatic colorectal cancer.[35,36]
Scheme 2.
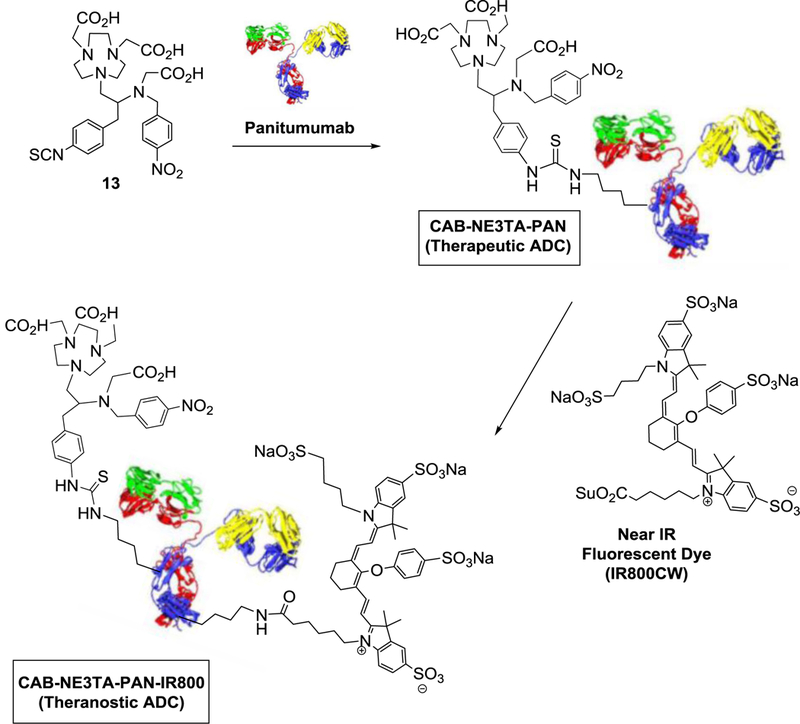
Construction of CAB-NE3TA-Based Therapeutic and Theranostic ADCs.
The NIR fluorescent cyanine dye IR800 is cGMP produced and has undergone preliminary toxicology studies and has a fluorescence emission spectrum in the range of 800–850 nm where mouse auto-fluorescence (background fluorescence) is very low.[37]
The chelate CAB-NE3TA in the NCS form (13, Scheme 2) was conjugated to panitumumab (PAN), and the corresponding conjugate CAB-NE3TA-PAN was purified. The number of ligands bound to protein (L/P ratio = 2 or 5) in the purified PAN conjugate was controlled and determined by the spectroscopic method using Cu(II)-AAIII as previously reported.[22] The theranostic antibody conjugate (CAB-NE3TA-PAN-IR800, Scheme 2) was prepared from reaction of CAB-NE3TA-PAN with IR800CW NHS ester and purified via dialysis and characterized by HPLC, UV-Vis, and fluorescence spectrometric analysis (Figure 4).
Figure 4.
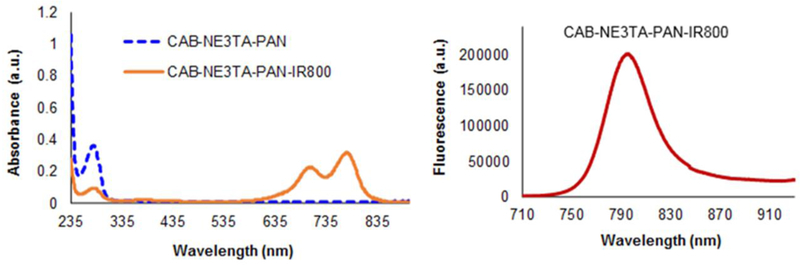
UV-Vis and fluorescence emission spectra of CAB-NE3TA-PAN and CAB-NE3TA- PAN-IR800 conjugates.
Incorporation of IR800 to the CAB-NE3TA-PAN conjugate was evidenced by HPLC analysis (Supporting Information). The CAB-NE3TA-PAN-IR800 conjugate was eluted at around 6 min (λ = 280 nm, 778 nm), while the unbound dye IR800 gave the major HPLC traces at around 13 and 17 min (λ = 778 nm). The CAB-NE3TA-PAN-IR800 conjugate gave the two major near IR absorption peaks at 703 nm and 778 nm along with the peak at 280 nm (Figure 4). The near IR fluorescence emission of CAB-NE3TA-PAN-IR800 conjugate peaked at 797 nm (Figure 4). The concentration of IR800 and PAN in the conjugate was determined by UV-Vis spectroscopic method.[22] The ratio of dye to protein (D/P = 3.9 or 0.06) in the conjugate was controlled and determined by spectroscopic method based on absorbance of dye and protein at 710 nm and 280 nm.[22]
Pilot in vivo anti-tumor activity of CAB-NE3TA-PAN.
We conducted preliminary therapeutic studies to evaluate antitumor effect of therapeutic conjugate (CAB-NE3TA-PAN) using the A431 cell xenograft mouse model (Figure 5 and Supporting Information). We selected the A431 (human skin cancer) cell xenograft mouse model for our in vivo studies as elevated expression of EGFR on A431 cells is well documented.[30–31] Mice received a subcutaneous frank injection of the A431 cells. When tumors in the mice were tumors were measurable using digital caliper, treatment was initiated. The mice were treated four times over a period of 8 days by IP injections of the therapeutic conjugate (CAB-NE3TA-PAN, 0.5 mg) and sacrificed at 21 days posttreatment (Figures 5A and5B). The mice in the control group were left untreated and received IP injections of PBS solution during the same 8-day treatment period. The photographic images of representative mice and tumors removed from the mice are shown in Figure 5C. Significant tumor growth suppression was observed with the mice treated with the CAB-NE3TA-PAN, and the treatment resulted in nearly complete disappearance of tumors in the mice by the end of the study period. The treated mice remained well tolerated and active until the end of the study period (21 days, Figure 5B) and had no significant change in body weight of the mice during the study period. The untreated mice were faster in tumor growth and developed significantly larger tumors than the treated mice (Figure 5C) and had a median survival of 16 days (Figure 5B). The median tumor volume measured after dissection of the treated mice and untreated mice was 4.5 ± 2.9 mm3 and 325.7 ± 64.5 mm3, respectively (P < 0.0010, Figure 5D). As shown in Figure 5E, tumor weight was significantly higher in the untreated mice (160.1 ± 79.6 mg) than in the treated mice (0.6 ± 0.3 mg). Overall, the results of our pilot in vivo therapy studies indicate that the CAB-NE3TA-PAN conjugate was highly effective in tumor suppression and delayed tumor growth in the mice with subsequent increase in median survival time over the control group.
Figure 5.
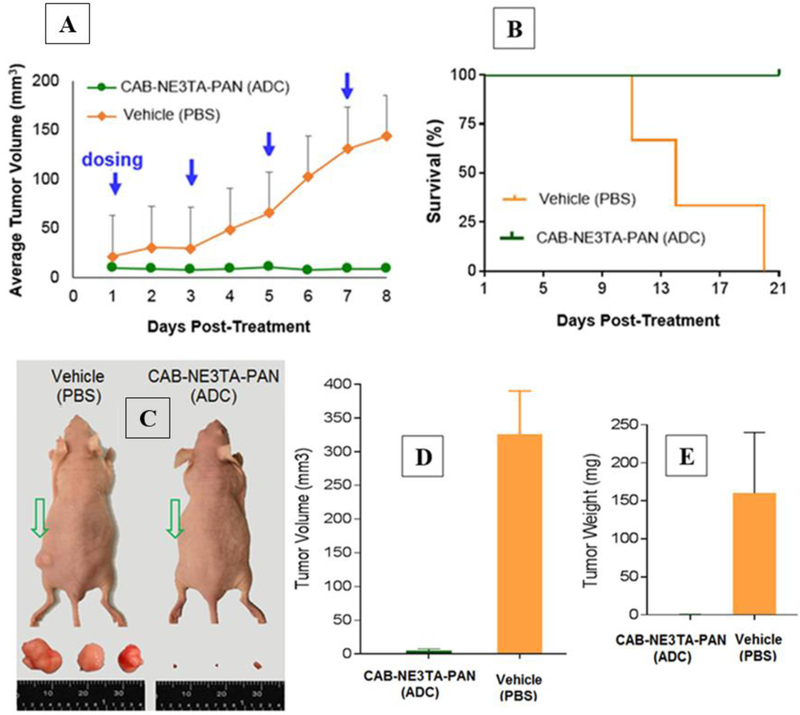
Pilot in vivo anti-cancer therapy study. A431 (human skin cancer)-bearing mice received IP injections of the therapeutic conjugate (CAB-NE3TA-PAN, L/P = 5) or vehicle (PBS only) four times for a period of 8 days and were sacrificed at 21 days post-treatment. (A) Median volumes of tumors in the treated and untreated mice measured during the treatment period. (B) Kaplan-Meier survival curves of the treated and untreated mice bearing A431 tumors. (C) Images of representative A431-bearing mice and tumors removed from the mice sacrificed at 11–21 days post-treatment. (D) Median volumes of tumors removed from the treated and untreated mice. (E) Median weight of tumors removed from the treated and untreated mice.
In vivo NIR fluorescence imaging of theranostic CAB-NE3TA-PAN-IR800 conjugate.
The therapeutic conjugate (CAB-NE3TA-PAN) labeled with the near IR fluorescent dye was evaluated for biodistribution in tumor-bearing mice using near IR fluorescent imaging (Figure 6 and Supporting Information). The CAB-NE3TA-PAN-IR800 conjugate was injected intraperitoneally to the mice bearing A431 xenograft tumors, and fluorescence images were sequentially captured using a PEARL Imager. Tumor uptake and whole body clearance of the theranostic agent were monitored, and the fluorescence images collected for 10 days (Figure 6).
Figure 6.
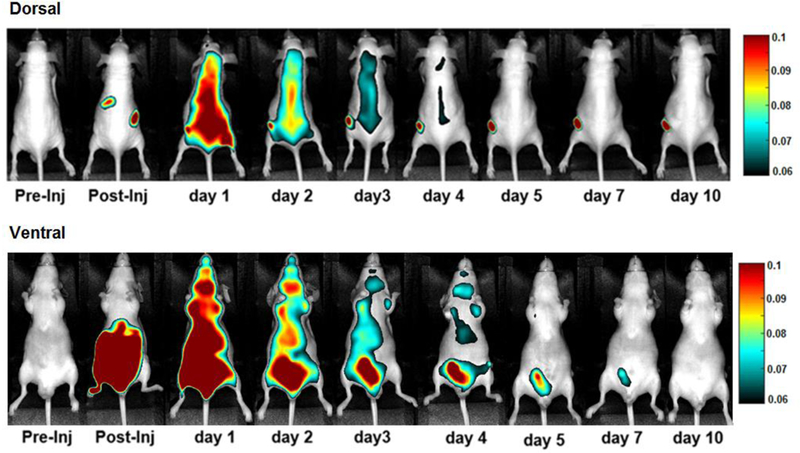
In vivo NIR fluorescence images of A431-bearing male nude mice intraperitoneally (IP) injected with the theranostic conjugate (CAB-NE3TA-PAN-IR800, 5 μM).
Tumor uptake of the fluorescent antibody conjugate was clearly visualized in mice over the time period of evaluation. Liver uptake of the conjugate was high at earlier time points. This was expected from increased lipophilicity of the conjugate by addition of IR800. Retention of the conjugate in the kidney, liver, and bladder was shown to be substantial in early time points but cleared well over the time period of evaluation. Visual comparison of the whole body fluorescence images of the A431-bearing mice acquired for 10 days (Figure 6) indicates that the theranostic ADC displayed rapid blood clearance. The concentration of the dye in the blood samples removed from the mice dissected at 10 days post-treatment was considered negligible (Supporting Information). The conjugate was highly localized in the A431 tumor with prolonged retention and had excellent clearance in blood and normal organs.
Homology modeling of panitumumab (PAN) and docking of CAB-NE3TA-PAN-IR800 to EGFR.
Our forthcoming investigation on optimization of the theranostic conjugate for effective cancer therapy and imaging involves preparation of the conjugate with different drug to antibody (L/P) and dye to antibody (D/P) ratios and their biological evaluation for anti-cancer activity and fluorescence imaging using EGFR-positive tumor model. We expect loading of the anti-cancer agent (CAN-NE3TA) and the fluorescent dye to the antibody can be controlled for highly specific, sensitive, and potent NIR fluorescent image-guided therapy, while preserving the biological activity of the sensitive antibody. We sought to construct a complex of the CAB- NE3TA-PAN-IR800 conjugate bound to EGFR for better understanding of the structural features in the antibody panitumumab and the antibody-EGFR complex in line with our future studies for production of optimal theranostic panitumumab drug conjugates. While X-ray crystal structures of the extracellular domain of EGFR are publicly available, no structure of panitumumab has been reported. We built a homology model for panitumumab and docked it to EGFR using epitope-derived constraints. In our modeled complex, a total of 10 lysine residues in the Fc region of panitumumab were exposed to the solvent. The number of exposed lysines is consistent with our measured ligand to protein ratios. Six of the lysine residues of the antibody were randomly selected for conjugation to two CAB-NE3TA and four IR800 moieties (Figure 7). Our modeled complex is consistent with Voigt et al.[38] and suggests that EGFR binds to panitumumab in a manner similar to cetuximab. Residues critical for recognizing panitumumab[38] are situated along the EGFR-antibody interface, in contact with the CDR loops of the antibody (Figure 8a). The modeled EGFR-panitumumab interface overlaps with but is larger than the crystallographic interface of EGFR-cetuximab.[39] (Figure 8b).
Figure 7.
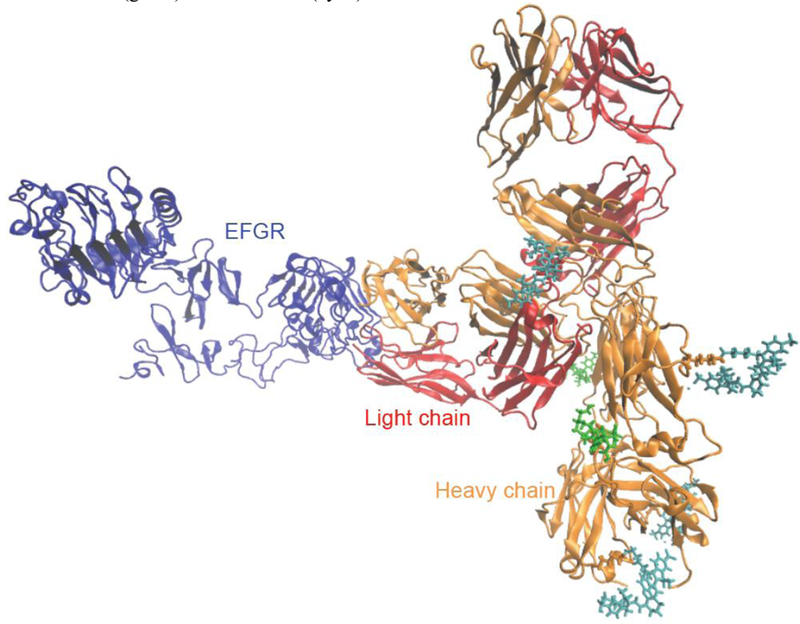
A proposed docking model for a complex of EGFR and Panitumumab conjugated with CAB-NE3TA (green) and IR800CW (cyan).
Figure 8.
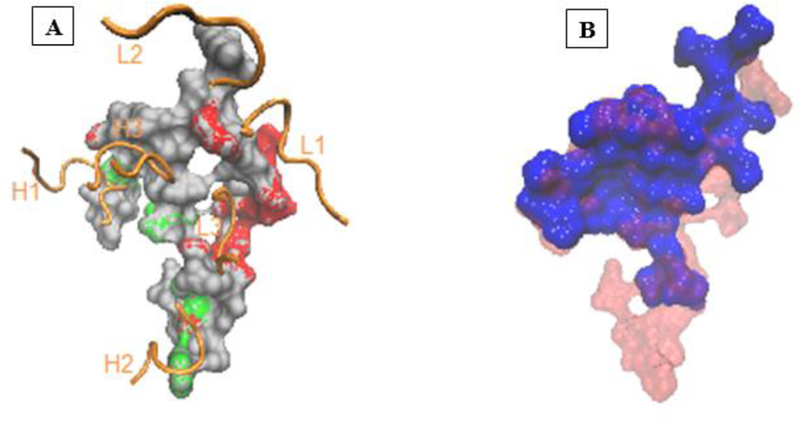
(A) The panitumumab binding surface of EGFR obtained by docking. Amino acid residues critical for panitumumab binding (P349, F352, D355, W386, P387, E388, N389, R390, T391)[38] are shown in red. The EGF binding residues (V350, D355, F357, L382, Q384, F412, I438)[40] are shown in green. The CDR loops of panitumumab are shown in orange ribbons. (B) Superposition of the panitumumab binding surface obtained by docking (red) with the cetuximab binding surface (blue) obtained from the X-ray crystal structure[39] of cetuximab-EGFR complex.
As a caveat, the larger interface may be an artifact of docking in the gas phase, which can over-stabilize the binding interface. The model also suggests that panitumumab inhibits the interaction between EGFR and EGF in a similar way to cetuximab; EGF binds to domains I and III of EGFR.[40] The interface between EGFR domain I and EGF overlaps the EGFR-antibody interface in the panitumumab model and cetuximab crystal structure. The overlapping interfaces are consistent with the suggestion by Li et al. that panitumumab binding may deactivate the receptor by preventing it from binding to the activating ligand EGF.[39] Furthermore, our model suggests that antibody binding also prevents EGFR from adopting the active extended (untethered) conformation in which domains I and III are close enough to both bind EGF. Superposing EGFR domain III from our model to the same domain in the crystal structure of the extended EGFR (PDB ID: 1IVO) causes a steric clash between the receptor and antibody.[39] These structural interpretations of our docking model are consistent with the known mechanism of action of panitumumab.[41,42]
Conclusion
A new cytotoxic polyaminocarboxylate-based ligand CAB-NE3TA was synthesized and evaluated for potential targeted iron chelation cancer therapy and imaging applications. CAB- NE3TA exhibited significant cytotoxic effect on EGFR-positive cancer cells. CAB-NE3TA conjugated to a humanized antibody (Panitumumab, PAN) was shown to be effective in inhibiting tumor growth in the A431 cell xenograft mouse model. The biodistribution studies using near IR fluorescence imaging were conducted on the CAB-NE3TA-PAN conjugate labeled with the IR800 dye. The CAB-NE3TA-PAN conjugate displayed highly selective tumor localization and excellent clearance in blood and normal organs and prolonged retention in the tumors. The structure of the theranostic panitumumab conjugate bound to EGFR was predicted through the computational modeling and docking and will be applied for optimization of the therapeutic and theranostic conjugates for high specificity, potency, and sensitivity. The results of our pilot in vivo studies suggest that the new chelate-based therapeutic and theranostic conjugates deserves further evaluation for targeted iron chelation therapy and near IR fluorescent imaging of cancers.
Experimental Section
Instruments and methods.
1H and 13C NMR spectra were obtained using a Bruker 300 instrument, and chemical shifts are reported in ppm on the δ scale relative to TMS, TSP, or solvent. Electrospray iodization (ESI) high resolution mass spectra (HRMS) were obtained on JEOL double sector JMS-AX505HA mass spectrometer (University of Notre Dame, IN). Analytical SE-HPLC was performed on an Agilent 1200 (Agilent, Santa Clara, CA) equipped with a diode array detector (λ = 254 and 280 nm), with the themostat set at 35°C. All absorbance measurements for the protein concentration and ligand protein ratio were obtained on an Agilent 8453 diode array UV-Vis spectrometer equipped with an 8-cell transport system (designed for 1 cm cells).
tert-butyl N-[1-(4-aminophenyl)-3-hydroxypropan-2-yl]carbamate (2).
10% Pd/C (1.70 g) was added to a solution of compound 1 (8.70 g, 29.4 mmol) in ethanol (200 mL) under Ar (g). The reaction mixture was placed under a hydrogenation apparatus (15 Psi) for 5.5 h. The resulting mixture was filtered via celite bed and washed thoroughly with methanol. The filtrate was concentrated to provide 5 (7.6 g, 97%). 1H NMR (CDCl3, 300 MHz) δ 1.42 (s, 9H), 2.71 (d, J = 6.9 Hz, 3H), 3.42–3.56 (m, 1H), 3.62 (s, 3H), 3.78 (s, 1H), 4.78 (d, J = 7.5 Hz, 1H), 6.63 (d, J = 8.1 Hz, 2H), 6.98 (d, J = 8.1 Hz, 2H); 13C NMR (CDCl3, 75 MHz) δ 28.1 (q), 36.6 (t), 53.9 (d), 64.4 (t), 79.7 (s), 115.4 (d), 127.6 (s), 130.1 (d), 144.9 (s), 156.3 (s). HRMS (Positive ion ESI) Calcd for C14H23N2O3 [M + H]+ m/z 267.1703. Found: [M + H]+ m/z 267.1719.
benzyl N- [4-(2- {[(tert-butoxy)carbonyl]amino}−3-hydroxypropyl)phenyl]car-bamate (3).
K2CO3 (4.20 g, 30.2 mmol) was added to a stirred solution of 2 (6.70 g, 25.2 mmol) in CH3CN (60 mL) at 0 °C. A solution of benzyl chloroformate (4.70 g, 27.7 mmol) in CH3CN (40 mL) was added dropwise to the resulting mixture over 15 min. The resulting mixture was warmed to room temperature and stirred for 24 h. The resulting mixture was evaporated to dryness. The residue was purified via column chromatography on silica gel (60–230 mesh) eluting with 25% ethyl acetate in hexanes to provide the pure product 3 (5.7 g, 56%). 1H NMR (CDCl3, 300 MHz) δ 1.41 (s, 9H), 2.77 (d, J = 6.6 Hz, 3H), 3.42–3.54 (m, 1H), 3.54–3.67 (m, 1H), 3.80 (s, 1H), 4.86 (d, J = 7.2 Hz, 1H), 5.18 (s, 2H), 7.01 (s, 1H), 7.12 (d, J = 8.1 Hz, 2H), 7.26–7.45 (m, 6H); 13C NMR (CDCl3, 75 MHz) δ 28.4 (q), 36.7 (t), 53.7 (d), 64.0 (t), 67.0 (t), 79.8 (s), 119.0 (d), 128.3 (d), 128.4 (d), 128.6 (d), 129.9 (d), 133.0 (s), 136.1 (s), 136.3 (s), 153.6 (s), 156.2 (s). HRMS (Positive ion ESI) Calcd for C22H28N2NaO5 [M + Na]+ m/z 423.1890. Found: [M + Na]+ m/z 423.1873.
benzyl N-[4-(2-amino-3-hydroxypropyl)phenyl]carbamate (4).
To a solution of 3 (5.20 g, mmol) in CDCl3 (25 mL) at 0–5°C, TFA (25 mL) was added dropwise over 10 min. The resulting mixture was warmed to room temperature. After 3 h, the reaction mixture was concentrated, and deionized water (30 mL) was added to the residue. The aqueous layer was first extracted with CDCl3 (3 × 30 mL). The aqueous layer was further adjusted to pH 13 using 1M NaOH (aq) and extracted with CDCl3 (3 × 30 mL). The organic layers obtained from extraction of the aqueous solution at pH 13 were combined and dried over MgSO4, filtered, and concentrated in vacuo to provide product 4 (3.4 g, 87%). 1H NMR (MeOD, 300 MHz) δ 2.50 (dd, J = 13.5, 7.5 Hz, 1H), 2.71 (dd, J = 13.5, 6.0 Hz, 1H), 2.92–3.06 (m, 1H), 3.33 (dd, J = 10.8, 3.6 Hz, 1H), 3.51 (dd, J = 10.8, 4.5 Hz, 1H), 5.15 (s, 2H), 7.12 (d, J = 8.1 Hz, 2H), 7.25–7.45 (m, 6H); 13C NMR (MeOD, 75 MHz) δ 38.7 (t), 54.1 (d), 65.3 (t), 66.1 (t), 118.8 (d), 127.6 (d), 127.7 (d), 128.1 (d), 129.3 (d), 133.3 (s), 136.7 (s), 137.1 (s), 154.5 (s). HRMS (Positive ion ESI) Calcd for C17H21N2O3 [M + H]+ m/z 301.1547. Found: [M + H]+ m/z 301.1562.
tert-butyl 2-{[1-(4-{ [(benzyloxy)carbonyl]amino}phenyl)-3-hydroxypropan-2-yl]amino}acetate (5).
To a solution of 4 (2.0 g, 6.66 mmol) in CH3CN (230 mL) at 0 °C, K2CO3 (0.97 g, 6.99 mmol) was added. A solution of tert-butyl bromoacetate (1.30 g, 6.66 mmol) in CH3CN (20 mL) was added dropwise to the resulting mixture over 30 min. The resulting mixture was stirred for 18 h at room temperature, while monitoring the reaction progress using TLC. The resulting mixture was evaporated to dryness. The residue was purified via column chromatography on silica gel (60–230 mesh), eluting with 50% ethyl acetate in hexanes to provide the pure product 5 (2.0 g, 72%). 1H NMR (CDCl3, 300 MHz) δ 1.45 (s, 9H), 2.62–2.74 (m, 3H), 2.74–2.86 (m, 1H), 3.18–3.37 (m, 3H), 3.45–3.58 (m, 1H), 5.19 (s, 2H), 6.83 (s, 1H), 7.12 (d, J = 8.4 Hz, 2H), 7.26–7.45 (m, 6H); 13C NMR (CDCl3, 75 MHz) δ 28.1 (q), 37.6 (t), 49.3 (t), 60.7 (d), 62.2 (t), 67.0 (t), 81.7 (s), 118.9 (d), 128.3 (d), 128.4 (d), 128.6 (d), 129.8 (d), 133.6 (s), 136.1 (s), 136.2 (s), 153.4 (s), 172.4 (s). HRMS (Positive ion ESI) Calcd for C23H31N2O5 [M + H]+ m/z 415.2227. Found: [M + H]+ m/z 415.2211.
tert-butyl 2-{[1-(4-{[(benzyloxy)carbonyl]amino}phenyl)-3-hydroxypropan-2-yl][(4- nitrophenyl)methyl]amino}acetate (6).
To a solution of 5 (1.0 g, 2.413 mmol) in CH3CN (15 mL) at 0 °C K2CO3 (366.8 mg, 2.654 mmol) was added. A solution of 4-nitrobenzyl bromide (547.2 mg, 2.533 mmol) in CH3CN (5 mL) was added dropwise to the resulting mixture over 30 min. The resulting mixture was stirred for 18 h at room temperature, while monitoring the reaction progress using TLC. The resulting mixture was evaporated to dryness. The residue was purified via column chromatography on silica gel (60–230 mesh), eluting with 50% ethyl acetate in hexanes to provide the pure product 6 (700 mg, 52.8%). 1H NMR (CDCl3, 300 MHz) δ 1.39 (s, 9H), 2.47 (dd, J = 13.8, 7.5 Hz, 1H), 2.76 (dd, J = 13.5, 6.9 Hz, 1H), 3.04–3.22 (m, 2H), 3.31–3.45 (m, 2H), 3.46–3.57 (m, 1H), 3.86 (dd, J = 28.2, 18.4 Hz, 2H), 5.20 (s, 2H), 6.93 (s, 1H), 7.06 (d, J = 8.1 Hz, 2H), 7.28–7.45 (m, 8H), 8.10 (d, J = 8.4, 2H); 13C NMR (CDCl3, 75 MHz) δ 28.0 (q), 33.3 (t), 51.3 (t), 56.1 (t), 62.3 (t), 65.7 (d), 67.0 (t), 82.1 (s), 119.0 (d), 123.6 (d), 128.3 (d), 128.4 (d), 128.6 (d), 129.5 (d), 129.5 (d), 133.8 (s), 136.1 (s), 136.4 (s), 146.8 (s), 147.2 (s) 153.4 (s), 172.4 (s). HRMS (Positive ion ESI) Calcd for C22H29N3O5 [M + H]+ m/z 415.2102. Found: [M + H]+ m/z 415.2117.
tert-butyl 2-{[3-(4-{[(benzyloxy)carbonyl]amino}phenyl)-2-bromopropyl][(4-nitrophenyl)methyl]amino}acetate (8a).
To a solution of 6 (630 mg, 1.146 mmol) and PPh3 (451 mg, 1.719 mmol) in CDCl3 (8 mL) at 0 C, NBS (630 mg, 1.719 mmol) was added portion- wise over 5 min. The resulting mixture was stirred for 4.5 h, while the reaction mixture was gradually warmed to room temperature. The solvent was evaporated, and the residue was purified by silica gel column chromatography and eluted with 15–20% ethyl acetate in hexanes to afford 8a (310 mg, 44.2%) as a yellow oil. 1H NMR (CDCl3, 75 MHz) 1.46 (s, 9H), 1.62–2.17 (m, 4H), 2.60–2.81 (m, 2H), 2.96 (dd, J = 13.8, 8.7 Hz, 1H), 3.17 (dd, J = 13.8, 5.4 Hz, 1H), 3.24 (s, 2H), 3.66–3.82 (m, 7H), 3.87 (d, J = 7.4 Hz, 1H), 3.94–4.12 (m, 1H), 6.44–6.49 (m, 2H), 7.20 (d, J = 8.7 Hz, 1H), 7.33 (d, J = 8.7 Hz, 2H), 8.13 (d, J = 8.7 Hz, 2H); 13C NMR (CDCl3, 75 MHz) δ 28.2 (q), 28.5 (t), 35.1 (t), 35.2 (t), 51.9 (t), 55.1 (d), 55.3 (q), 56.1 (t), 61.8 (t), 80.9 (s), 98.4 (d), 103.9 (d), 119.1 (s), 123.6 (d), 129.2 (d), 131.2 (d), 146.3 (s), 150.0 (s), 158.8 (s), 160.2 (s), 170.9 (s). HRMS (Positive ion ESI) Calcd for C30H35BrN3O6 [M + H]+ m/z 612.1704. Found: [M + H]+ m/z 612.1690.
tert-butyl 2-{[3-(4-{[(benzyloxy)carbonyl]amino}phenyl)-2-iodopropyl][(4-nitrophenyl)methyl]amino}acetate (8b).
To a solution of 6 (150 mg, 0.27 mmol) and PPh3 (104.9 mg, 0.40 mmol) and imidazole (27.2mg, 0.40 mmol) in CHCl3 (3 mL) at 0 °C was added portionwise Iodine (101.52 mg, 0.40 mmol) over 10 min. The reaction was stirred in 0 °C for 4 h and room temperature for 1 h. The solvent was evaporated, and the residue was purified by silica gel column chromatography eluted with 15–20% ethyl acetate in hexanes to afford 8b (151 mg, 86%) as a yellow oil. 1H NMR (CDCl3, 300 MHz δ 1.45 (s, 9H), 3.0–3.18 (m, 3H), 3.29–3.33 (m, 3H), 3.98 (s, 2H), 4.14–4.20 (m, 1H), 5.20 (s, 2H) 6.79 (s, 1H), 7.09 (d, J = 8.4 Hz, 2H), 7.31–7.40 (m, 7H), 7.58 (d, J = 8.7 Hz, 2H), 8.18 (d, J = 8.7, 2H); 13C NMR (CDC13, 75 MHz) δ 28.1 (q), 28.2 (t), 36.2 (d), 43.6 (t), 55.1 (t), 57.8 (t), 62.5 (t), 67.1 (t), 81.6 (s), 118.7(s), 123.4 (d), 124.7 (d), 128.4 (d), 129.6 (d), 134.5 (s), 136.1 (s),136.6 (s), 146.8 (s), 147.3 (s), 153.3(s), 170.2(s).
tert-butyl 2-{[1-(4-{[(benzyloxy)carbonyl]amino}phenyl)-3-{4,7-bis[2-(tert-butoxy)-2- oxoethyl]-1,4,7-triazonan-1-yl}propan-2-yl][(4-nitrophenyl)methyl]amino}acetate (11).
To a solution of 8a (150 mg, 0.245 mmol) in CH3CN (3 mL) at - 5 °C, AgClO4 (50.8 mg, 0.245 mmol) was added. The resulting mixture was stirred for 15 min at the same temperature. Compound 1018 (87.6 mg, 0.245 mmol) and DIPEA (95 mg, 0.735 mmol) were sequentially added to the reaction mixture at −5°C. The resulting mixture was gradually warmed to room temperature and stirred for 89 h. The reaction mixture was filtered and concentrated to the dryness in vacuo. 0.1 M HCl solution (10 mL) was added to the residue, and the resulting mixture was extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified via column chromatography on silica gel (60–220 mesh) and sequentially eluted with 50% ethyl acetate in hexanes and then with 3% CH3OH in CH2Cl2 to provide pure product 11 (153 mg, 70.2%) as a yellowish oil. 1H NMR (CDCl3, 300 MHz) δ 1.36–1.43 (m, 27H), 2.71–4.13 (m, 25H), 5.19 (s, 2H), 6.92 (s, 2H), 7.21 (d, J = 8.4 Hz, 2H), 7.31–7.39 (m, 7H), 7.50 (d, J = 8.4 Hz, 2H), 8.17 (d, J = 8.4 Hz, 2H). 13C NMR (CDCl3, 75 MHz) δ 27.9 (q), 28.0 (q), 28.1 (q), 33.1 (t), 49.3 (t), 51.7 (t), 52.3 (t), 52.8 (t), 53.1 (t), 53.4 (t), 55.2(t), 56.0 (t), 58.0 (t), 60.5 (d), 67.0 (t), 81.7 (s), 81.9 (s), 82.5 (s), 119.4 (s), 123.7 (d), 128.2 (d), 128.3 (d), 128.6 (d), 129.9 (d), 132.5 (s), 136.1 (s), 136.8 (s), 145.5 (s), 147.5 (s), 153.4 (s), 170.3 (s), 172.0 (s). HRMS (Positive ion ESI) Calcd for C48H68N6O10 [M + H]+ m/z 889.0877. Found: [M + H]+ m/z 889.5048.
tert-butyl 2-{[1-(4-{[(benzyloxy)carbonyl]amino}phenyl)-3-{4,7-bis[2-(tert-butoxy)-2- oxoethyl]-1,4,7-triazonan-1-yl}propan-2-yl][(4-nitrophenyl)methyl]amino}acetate (11).
To a solution of 8b (100.0 mg, 0.15 mmol) in CH3CN (2 mL) at - 5 °C was added AgClO4 (39.9 mg, 0.15 mmol). The resulting mixture was stirred for 15 min at the same temperature. Compound 1018 (53.6 mg, 0.15 mmol) and DIPEA (58.2 mg, 0.45 mmol) was sequentially added to the reaction mixture at - 5 °C. The resulting mixture was gradually warmed to room temperature and stirred for 48 h. The reaction mixture concentrated in vacuo. The residue was purified via column chromatography on silica gel (60–220 mesh) sequentially eluted with 50% ethyl acetate in hexanes and then with 2% CH3OH in CH2Cl2 to provide pure product 11 (102 mg, 76.1%) as a yellowish oil. The product was characterized by 1H and 13C NMR as described above.
2-{[1-(4-aminophenyl)-3-[4,7-bis(carboxymethyl)-1,4,7-triazonan-1-yl]propan-2-yl][(4- nitrophenyl)methyl]amino}acetic acid (12).
Compound 11 (55 mg, 0.0619 mmol) was treaeted with 6M HCl (aq) (6 mL). The resulting mixture was refluxed for 1.5 h. The aqueous solution was washed with CDCl3 (10 mL) and then concentrated in vacuo to provide 12 (45 mg, 94.6%). 1H NMR (D2O, 300 MHz) δ 2.95–3.46 (m, 17H), 3.59–3.75 (m, 5H), 3.92 (s, 3H), 7.16–7.39 (m, 6H), 7.99 (d, J = 6.0 Hz, 2H); 13C NMR (D2O, 75 MHz) δ 32.1 (t), 50.4 (t), 50.5 (t), 56.3 (t), 58.4 (t), 60.3 (d), 123.6 (d), 124.1 (d), 128.7 (s), 130.3 (d), 130.9 (d), 137.9 (s), 143.7 (s), 147.3 (s), 171.5 (s). HRMS (Positive ion ESI) Calcd for C28H39N6O8 [M + H]+ m/z 587.2824. Found: [M + + m/z 587.2844.
2-({1-[4,7-bis(carboxymethyl)-1,4,7-triazonan-1-yl]-3-(4-isothiocyanatophenyl)propan-2- yl}[(4-nitrophenyl)methyl]amino)acetic acid (13).
To a solution of 12 (11.6 mg, 0.0151 mmol) in water (100 μL), 1 M thiophosgene in CHCl3 (18.1 μL, 0.0181 mmol) was added dropwise. The resulting mixture was stirred at room temperature for 3h. The aqueous layer was concentrated in vacuo to provide pure 13 (10.9 mg, 93.3%). 1H NMR (D2O, 300 MHz) δ 2.65–4.40 (m, 25H), 7.15–7.82 (m, 6H), 8.26 (d, J = 5.7 Hz, 2H). HRMS (Positive ion ESI) Calcd for C29H37N6O8S [M + H]+ m/z 629.2388. Found: [M + H]+ m/z 629.2378.
Conjugation of CAB-NE3TA to panitumumab (PAN).
CAB-NE3TA-NCS was conjugated to the antibody, and the corresponding chelate-antibody conjugates were purified and characterized as previously reported. PAN (5.0 mg) was purified using a PD-10 column charged with HEPES buffer prior to conjugation. To a sterile test tube containing the recovered panitumumab (4.8 mg, 640 μL, 0.050 mM), a 40-fold excess of CAB-NE3TA (130 μL, 10 mM) was added. The resulting solution was gently agitated for 16 h at room temperature and placed on a Centricon C-50 membrane and spun down to reduce volume. PBS (3 × 2 mL) was added to the remaining solution of the panitumumab conjugates, followed by centrifugation to remove unreacted ligand. Concentration of panitumumab in each conjugate (4.8 × 10−5 M) was measured by UV spectral analysis (λ = 280 nm). A L/P ratio of antibody conjugates was determined by Arsenazo III (AAIII)-based spectroscopic assay as previously reported.[19,20] Concentration of CAB-NE3TA and PAN in CAB-NE3TA-PAN was 4.8 × 10−5 M and 2.53 × 10−4 M, respectively. The L/P ratio of the CAB-NE3TA-PAN conjugate was measured to be 5.3.
Preparation of CAB-NE3TA-PAN-IR800 conjugate.
IR Dye 800CW was purchased from LI-COR Biosciences (Lincoln, NE). CAB-NE3TA-PAN (1.0 mg, 6.8 nmol) in PBS was reacted with IR Dye 800CW (540 ng, 0.465 nmol) at 1000 rpm for 35 min at room temperature. The resulting solution was placed on a Centricon C-50 membrane (Millipore) and spun down to reduce volume. PBS (2 × 2 mL) was added to the remaining solution of the conjugate, followed by centrifugation to remove unreacted IR800. The solution of the purified conjugate in PBS was diluted to 300 μL. The purified conjugate was characterized by analytical SE-HPLC (TSKgel G3000PW column, Tosoh Biosep) using the isocratic mobile phase (PBS, pH 7.4, 45 min, flow rate of 1 mL/min, λ = 280 and 778 nm). CAB-NE3TA-PAN-IR800 conjugate (tR = ~ 6 min) was eluted earlier than unbound IR800 (tR = ~13, 17 min) at 778 nm.
Spectroscopic Determination of Dye to Protein (D/P) Ratio for CAB-NE3TA-PAN- IR800CW conjugate.
To measure concentrations of protein and IR800CW in the conjugate, a UV/Vis spectrometer was zeroed against a cuvette filled with 2.0 mL of PBS with a window open from 190 nm to 1100 nm. A 3-μL portion of PBS was removed and discarded, 3 μl of the conjugate solution was added, and absorbance at 280 nm and 778 nm was noted. Beer’s Law was used to calculate concentration of antibody in the conjugate using equation A = εbc, where A is absorbance at 280 nm, ε is extinction coefficient of panitumumab (225,000 M−1cm−1), b is path length (1 cm), and c is concentration of the protein with dilution factor of 2000/3. Concentration of IR800 in the conjugate was also obtained using Beer’s Law with molar absorptivity of 24,000 M−1cm−1 and dilution factor of 2000/3.[22] Absorbance at 280 nm for the protein concentration was corrected (A280,actual = A280,observed - (A778 × 0.03)) due to the contribution of IR800 to the absorbance at 280 nm according to the method provided by the manufacturer (LI-COR Biosciences). Concentration of IR800 and PAN in CAB-NE3TA-PAN-IR800CW was determined to be 4.44 × 10−5 M, and 2.42 × 10−6 M, respectively. The resulting dye to protein ratio (D/P) of 0.055 was obtained.
UV-Vis and Fluorescence spectral characterization of CAB-NE3TA-PAN and CAB- NE3TA-PAN-IR800CW conjugates.
A stock solution of CAB-NE3TA-PAN (1.60 μM) and CAB-NE3TA-PAN-IR800 (0.4 μM) in PBS (pH 7.4) was prepared. UV-Vis spectra were recorded on an Agilent 8453 diode array spectrophotometer. UV-Vis measurements were carried out by adding an aliquot of the stock solution of CAB-NE3TA-PAN or CAB-NE3TA-PAN- IR800 into a quartz cuvette (2 mL). Fluorescence spectra were recorded on a Horiba Flurolog-3 spectrofluorometer (Horiba Jobn Yvon, Inc., NJ). The measurement of fluorescence was carried out by adding 1 mL aliquot of the stock solution of CAB-NE3TA-PAN-IR800 (0.2 μM) into a quartz cuvette. Emission fluorescence spectra of CAB-NE3TA-PAN-IR800 (0.2 μM) was recorded at excitation wavelength 700 nm and bandwidth 5 nm, and the data were collected at every 1 nm at 20°C. The emission data were collected from 655 nm to 800 nm. Emission fluorescence spectra were recorded at excitation wavelength 830 nm and bandwidth 2 nm, and the data were collected at every 1 nm at 20°C.
Cell culture.
Human cervical cancer (HeLa), human colon cancer (HT29) were kindly provided by Dr. Jialing Xiang (Illinois Institute of Technology, Chicago, IL) and Dr. Rajendra Metha (Illinois Institute of Technology Research Institute, Chicago, IL), respectively. The human prostate cancer (PC3), human epidermoid cancer (A431), human breast cancer (MDA-MB-231), human colon cancer (LS174T), human malignant melanoma (A375) cell lines was obtained from ATCC. The cancer cells were cultured in RPMI-1640 medium containing 10% fetal bovine serum (FBS) in a humidified atmosphere with 5% CO2 at 37 °C.
Anti-proliferative activity.
Cells were seeded onto a 96-well plate at a density of 2000 (HeLa, PC3, A375, A431, LS174T) and 5000 (HT29, MDA-MB-231) cells per well in complete medium (100 μL) and allowed to attach for 24 h. The medium was replaced with fresh medium (100 μL) containing a series of concentrations (1, 5, 10, 20, 40, 50 and 100 μM) of an antitumor agent. The cells were incubated for 72 h after which the medium was replaced with fresh medium (100 μL) prior to MTS assay.[17] To measure cell proliferation, the Cell Titer 96 aqueous nonreactive cell proliferation assay (Promega Life Sciences, Madison, WI, USA) was used according to the manufacturer’s instructions. Briefly, MTS (2 mg mL-1) and PMS (0.92 mg mL- 1) were mixed at a ratio of 20:1. The MTS/PMS mixture (20 μL) was added into the complete medium (0.1 mL) in each well. The plate was incubated for 3 h at 37°C. Optical absorbance at 490 nm was then recorded with an enzyme-linked immunosorbent assay (ELISA) microtiter plate reader (Biotek). Each experiment was performed in triplicate. Anti-proliferative activity of the compounds tested was expressed as the fraction of optical absorbance of the treated cells relative to the untreated control cells. The data were plotted in GraphPad Prism 6.0. Nonlinear regression analysis was used to determine IC50 values, which are defined as the drug concentration required to inhibit cell growth by 50 %.
In vivo anti-tumor activity of therapeutic conjugate (CAB-NE3TA-PAN).
The animal study protocol was approved by the Animal Use and Care Committee at the Illinois Institute of Technology. The antibody drug conjugate (CAB-NE3TA-PAN, L/P = 5, PBS, pH 7.4) was freshly prepared for the therapy studies. 1.5 million cells of A431 human epidermoid cancer cells were implanted into left thighs of three male nude mice to establish the animal model. Mice were daily monitored for tumor development by palpation, and treatment was initiated when tumors were measurable (20–40 mm3) using digital caliper. A group of mice (N = 3) were intraperitoneally injected with the CAB-NE3TA-PAN conjugate (pantitumumb at 17 μM and CAB-NE3A at 85 μM, 0.2 mL PBS, L/P = 5.3) four times over a period of 8 days. A control group of mice (N = 3) was intraperitoneally injected with PBS (0.45 mL) four times over a period of 8 days. Mice were monitored daily, and their weight and tumors were measured every weekday for 21 days. Mice were euthanized if their tumor diameter exceeded 1 cm or showed skin ulcers. The mice were sacrificed at 21 days post-treatment, and tumors and organs of the mice were resected. The tumors removed from the mice were frozen and dehydrated and dried under vacuum and weighed. The tumors were measured with the digital caliper, and the volumes of tumors were calculated based on the formula (V = 0.5 x a x b2) and expressed as mean ± standard deviation mm3 (SD). Kaplan-Meier curves were constructed using GraphPad Prism 7 (GraphPad Software Inc.). The data of tumor volume and weight were analyzed by Student’s t test. Difference was considered significant if p values are less than 0.05.
In vivo fluorescence imaging of IP injected CAB-NE3TA-PAN-IR800.
1.5 million cells of A431 human epidermoid cancer were implanted into left thighs of three male nude mice to establish the animal model. Mice were daily monitored for tumor development by palpation, and treatment was initiated when tumors were measurable (20–40mm3) using digital caliper. Prior to imaging, the mice were anesthetized with 2% isoflurane and background images for each animal were taken before imaging agent injection. After initial imaging, mice were IP injected with 3.4 nmol (0.85.μM IR800 in 200 μL PBS) of the theranostic conjugate CAB-NE3TA-PAN-IR800 and imaged at 30 min post-injection and then daily for 10 days in both dorsal and ventral orientations. At 10 days post-injection, animals were sacrificed to perform bio-distribution tests. All imaging was carried out on a wide-field dual-wavelength commercial small animal fluorescence imaging system (Pearl® Impulse, LI-COR Biosciences). In brief, the system employs wide-field illumination at two wavelengths, 685 nm and 785 nm, and respectively accepts emitted fluorescence at 700–745 nm and 800–845 nm in “reflectance” mode. Only the 800 nm channel imaging was used in the current study.
Homology modeling and docking.
The sequence of panitumumab (PAN) was obtained from http://www.commonchemistry.org/ (CAS ID: 339177–26-3). The Abnum web server[43] (http://www.bioinf.org.uk/abs/abnum/) was used to identify sequences of the variable domains in the light and heavy chains (VL and VH domains). The sequences of VL and VH were used as an input to the Rosetta Antibody program.[44] to obtain homology models for the Fv region of the antibody. The HADDOCK2.2 web server.[45] was used to dock the best homology model for Fv to the crystal structure of the extracellular domain of EGFR downloaded from PDB (PDB ID: 1YY9). Residues in EGFR belonging to the panitumumab epitope were taken from Voigt et al. and used as constraints in the docking calculation.[38] To model the constant region of panitumumab, we used Modeller 9.17.[46] to build a homology model for the whole dimeric antibody using the crystal structure of pembrolizumab (PDB ID: 5DK3) as a template. Then the structure of EGFR-Fv complex obtained above by Rosetta and HADDOCK calculations was superimposed onto the homology model of the full-length dimeric antibody. The atom coordinates in the Fv region modeled by Modeller 9.17 were removed. The complex was minimized with AMBER ff14 force field.[47] using NAMD 2.9.[48] The backbone atoms of EGFR and Fv region were fixed during the energy minimization. Six solvent-exposed LYS residues were randomly selected from the constant region to attach four dyes and two chelators. The LYS-chelator and LYS-dye moieties were parameterized with the generalized AMBER force field.[49] and AM1BCC partial charges.[50–51] using antechamber.[52] The chelator and dye molecules were placed near the corresponding LYS residues using VMD.[53] The structure of the whole chelated EGFR-panitumumab complex was minimized using NAMD 2.9. During the minimization, backbone atoms of EGFR and panitumumab were fixed.
Supplementary Material
Acknowledgement
This research was partly supported by National Institutes of Health (5RG1CA1125G3 to H-S. Chong and R15GM114781 to D. D. L. Minh). We thank Dr. Martin Brechbiel (Radioimmune and Inorganic Chemistry Section, Radiation Oncology Branch, NIH) for generous supply of panitumumab.
References
- [1].Lieu PT, Heiskala M, Peterson PA, Yang Y, Mol. Aspects Med. 2001, 22, 1–87. [DOI] [PubMed] [Google Scholar]
- [2].Papanikolaou G, Pantopoulos K, Toxicol. Appl. Pharmacol. 2005, 202, 199–211. [DOI] [PubMed] [Google Scholar]
- [3].Torti SV, Torti FM, Nat. Rev. Cancer 2013, 13, 342–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kalinowski DS, Richardson DR, Pharmacol. Rev. 2005, 57, 547–583. [DOI] [PubMed] [Google Scholar]
- [5].Buss JL, Torti FM, Torti SV, Curr. Med. Chem. 2003, 10, 1021–1034. [DOI] [PubMed] [Google Scholar]
- [6].Chaston TB, Lovejoy DB, Watts RN, Richardson DR, Clin. Cancer Res 2003, 9, 402–414. [PubMed] [Google Scholar]
- [7].Qian ZM, Li H, Sun H, Ho K, Pharmacol. Rev. 2002, 54, 561–587. [DOI] [PubMed] [Google Scholar]
- [8].Thorstensen K, Romslo I, Biochem. J. 1990, 271, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Birch N, Wang X, Chong HS, Expert Opin. Ther. Pat. 2006, 16, 1533–1557. [Google Scholar]
- [10].Alvero AB, Chen W, Sartorelli AC, Schwartz P, Rutherford T, Mor G, J. Soc. Gynecol. Investig. 2006, 13, 145–152. [DOI] [PubMed] [Google Scholar]
- [11].Krishna G, Mao J, Almassian B, Lang W, Pharm. Dev. Technol. 1999, 4, 461. [DOI] [PubMed] [Google Scholar]
- [12].Murren J, Modiano M, Clairmont C, Lambert P, Savaraj N, Doyle T, Sznol M, Clin. Cancer Res. 2003, 9, 4092–4100. [PubMed] [Google Scholar]
- [13].Estrov Z, Tawa A, Wang XH, Dube ID, Sulh H, Cohen A, Gelfand EW, Freedman MH, Blood 1987, 69, 757–761. [PubMed] [Google Scholar]
- [14].Donfrancesco A, Deb G, DeSio L, Cozza R, Castellano A, Acta Haematol. 1996, 95, 66–69. [DOI] [PubMed] [Google Scholar]
- [15].Blatt J, Stitely S. Cancer Res. 1987, 47, 1749–1750. [PubMed] [Google Scholar]
- [16].Lederman HM, Cohen A, Lee JW, Freedman MH, Gelfand EW, Blood 1984, 64, 748–753. [PubMed] [Google Scholar]
- [17].Chong H-S, Ma X, Lee H, Bui P, Song HA, Birch N, J. Med. Chem. 2008, 51, 2208–2215. [DOI] [PubMed] [Google Scholar]
- [18].Chong H-S, Song HA, Ma X, Lim SY, Sun X, Mhaske S, Chem. Commun. 2009, 21, 3011–3013. [DOI] [PubMed] [Google Scholar]
- [19].Song HA, Kang CS, Baidoo KE, Milenic DE, Chen Y, Dai A, Brechbiel MW, Chong HS, Bioconjugate Chem. 2011, 22, 1128–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kang CS, Sun X, Fang J, Song HA, Chen Y, Lewis M, Chong HS, Bioconjugate Chem. 2012, 23, 1775–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Sun X, Chen Y, Wu N, Kang CS, Song HA, Jin S, Fu Y, Bryant JH, Frank JA, Chong H-S, RSC Adv. 2015, 5, 94571–94581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kang CS, Ren S, Sun X, and Chong H-S, ChemMedChem 2016, 11, 1 – 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Lee DE, Koo H, Sun IC, Ryu JH, Kim K, Kwon IC, Chem. Soc. Rev. 2012, 41, 2656–2672. [DOI] [PubMed] [Google Scholar]
- [24].Kelkar SS, Reineke TM, Bioconjugate Chem. 2011, 22, 1879–1903. [DOI] [PubMed] [Google Scholar]
- [25].Scott AM, Allison JP, Wolchok JD, Cancer Immun. 2012, 12, 14–21. [PMC free article] [PubMed] [Google Scholar]
- [26].Bareschino MA, Schettino C, Maione P, Rossi A, Nicolella D, Ciardiello F, Gridelli C, Cancer Ther. 2008, 6, 477–490. [Google Scholar]
- [27].Yang X-D, Jia X-C, Corvalan JRF, Wang P, Davis CG, Development of ABX- EGF, Crit. Rev. Oncol. Hematol. 2001, 38, 17–23. [DOI] [PubMed] [Google Scholar]
- [28].Power DG, Shah MA, Asmis TR, Garcia JJ, Kemeny NE, Safety and efficacy of panitumumab following cetuximab: retrospective review of the Memorial Sloan-Kettering experience. Invest New Drugs. 2010, 28, 353–360. [DOI] [PubMed] [Google Scholar]
- [29].Baselga J J, Arteaga CL. J. Clin. Oncol. 2005, 23, 2445–2459. [DOI] [PubMed] [Google Scholar]
- [30].Masui H, Kawamoto T, Sato JD, Wolf B, Sato G, J. Mendelsohn. Cancer Res. 1984, 44, 1002–7. [PubMed] [Google Scholar]
- [31].Ray GL, Baidoo KE, Wong KJ, Williams M, Garmestanil K, Brechbiel MW, Milenic DE. British J. Pharmacol. 2009, 157, 1541–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Salomon DS, Brandt R R, Ciardiello F, Normanno N. Crit Rev OncolHematol. 1995, 19, 183–232. [DOI] [PubMed] [Google Scholar]
- [33].Yang X-D, Jia X-C, Corvalan JRF, Wang P, Davis CG, Crit. Rev. Oncol. Hematol. 2001, 38, 17–23. [DOI] [PubMed] [Google Scholar]
- [34].Bareschino MA, Schettino C, Maione P, Rossi A, Nicolella D, Ciardiello F, Gridelli C, Cancer Ther. 2008, 6, 477–490. [Google Scholar]
- [35].Cohenuram M M, Saif MW. Anticancer Drugs. 2007, 18, 7–15. [DOI] [PubMed] [Google Scholar]
- [36].Scott AM, Allison JP, Wolchok JD, Cancer Immun. 2012, 12, 14–21. [PMC free article] [PubMed] [Google Scholar]
- [37].Marshall MV, Draney D, Sevick-Muraca EM, Olive DM, Mol. Imaging Biol. 2010, 12, 583–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Voigt M, Braiq F, Göthel M, Schulte A, Lamszus K, Bokemeyer C, Binder M, Neoplasia 2012, 14, 1023–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Li S, Schmitz KR, Jeffrey PD, Wiltzius JJ, Kussie P, Ferquson KM, Cancer Cell 2005, 7, 301–311. [DOI] [PubMed] [Google Scholar]
- [40].Ogiso H, Ishitani R, Nureki O, Fukai S, Yamanaka M, Kim JH, Saito K, Inoue M, Shirouzu M, Yokoyama S, Cell 2002, 110, 775–787. [DOI] [PubMed] [Google Scholar]
- [41].Messersmith WA, Hidalgo M, Clin. Cancer. Res. 2007, 13, 4664–4666. [DOI] [PubMed] [Google Scholar]
- [42].Dubois EA, Cohen AF, Br. J. Clin. Pharmacol. 2009, 68, 482–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Abhinandan KR, Martin ACR, Mol. Immunol. 2008, 45, 3832–3839. [DOI] [PubMed] [Google Scholar]
- [44].Sivasubramanian A, Sircar A, Chaudhury S, Gray JJ, Proteins 2009, 74, 497–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Van Zundert GC, Rodrigues JP, Trellet M, Schmitz C, Kastritis PL, Karaca E, Melquiond AS, Van Dijk M, de Vries SJ, Bonvin AM, J. Mol. Biol. 2016, 428, 720–725. [DOI] [PubMed] [Google Scholar]
- [46].Sali A, Blundell TL, J. Mol. Biol. 1993, 234, 779–815. [DOI] [PubMed] [Google Scholar]
- [47].Ponder JW, Case DA, Adv. Protein Chem. 2003, 66, 27–85. [DOI] [PubMed] [Google Scholar]
- [48].Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, J. Comput. Chem. 2005, 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA, J. Comput. Chem. 2004, 25, 1157–1174. [DOI] [PubMed] [Google Scholar]
- [50].Jakalian A, Bush BL, Jack DB, Bayly CI, J. Comput. Chem. 2000, 21, 132–146. [Google Scholar]
- [51].Jakalian A, Jack DB, Bayly CI, J. Comput. Chem. 2002, 23, 1623–1641. [DOI] [PubMed] [Google Scholar]
- [52].Wang J, Wang W, Kollman PA, Case DA, J. Mol. Graph. Model. 2006, 25, 247–26G. [DOI] [PubMed] [Google Scholar]
- [53].Humphrey W, Dalke A, Schulten K, J. Mol. Graph. 1996, 14, 33–38. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.