

Abstract
Aziridinium ions are useful reactive intermediates for synthesis of enantiomerically enriched building blocks. However, N,N-dialkyl aziridinium ions are relatively underutilized in synthesis of optically active molecules as compared to other three-membered ring cogeners, aziridines and epoxides. Characterization of both optically active aziridinium ions and secondary β-halo amines as precursor molecules of aziridinium ions has been scarcely reported and is often unclear. In this paper, we report for the first time preparation and experimental and theoretical characterization of optically active aziridinium ions and secondary β-halo amines. Optically active secondary N,N-substituted β-halo amines were efficiently synthesized from N,N-substituted alaniol via formation and ring opening at the more hindered carbon of aziridinium ions by halides. Optically active β-halo amines and aziridinium ions were characterized by NMR and computational analysis. The structure of an optically active β-chloro amine was confirmed via X-ray crystallographic analysis. The aziridinium ions derived from N,N-dibenzyl alaniol were attempted for X-ray crystallographic structural determination but remained stable only for several hours which was long enough for analyses of NMR and optical activity. The stereospecific ring opening of aziridinium ions by halides were computationally studied using DFT and highly-accurate DLPNO-CCSD(T) methods. Highly regioselective and stereoselective ring opening of aziridinium ions was applied to efficient one-pot conversion of β-alaniols to enantiomerically enriched β-amino alcohols, β-amino nitriles, and vicinal diamine derivatives.
Graphical Abstract
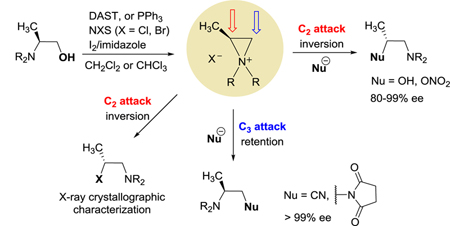
Introduction
Aziridinium ions are strained three-membered ring systems containing two electrophilic carbons and positively charged nitrogen.1 Optically active small and complex molecules such as vicinal diamines, 1,2-amino alcohols, 1,2-amino ethers, 3,4-diamino nitriles, and α,β-diamino esters have been prepared via nucleophilic ring opening reactions of aziridinium ions.1,2 In addition, aziridinium ions are involved in anti-cancer activity of nitrogen mustards such as chlorambucil, mechlorethamine, and phosphamide derivatives.3 The biological activity of the mustards results from reaction of the aziridinium ions derived from the mustards with guanine residues in DNA to form interstrand cross-links.3b Since the Leonard group reported various aziridinium ions as reactive intermediates in the 1960s,4 formation of aziridinium ions and their applications in the synthesis of optically active synthons have been explored.5 In contrast to relatively well known chemistry of other heterocyclic cogeners, aziridines and epoxides, ring opening chemistry of N,N-dialkyl aziridnium ions remain underutilized in synthesis of enantiomerically enriched small and complex molecules 6
We recently reported that aziridinium ions with various structural functionality were prepared in two steps starting from primary β-amino alcohols via formation of secondary β-halo amines, and ring opening of aziridinium ions was applied for efficient synthesis of nitrogen-containing optically active molecules with potential biomedical applications.7 Although the synthetic methods to secondary β-halo amines including mesylation and halogenation of β-amino alcohols and N-alkylation of aziridines are known, preparation of both optically active secondary β-halo amines and aziridinium ions and their synthetic applications have been limitedly reported in literature, and characterization of the labile species is often unclear.8
In the present paper, we describe preparation and experimental and theoretical aspects of optically active secondary β-halo amines and aziridinium ions and their applications for nucleophilic substitution reactions (Scheme 1). Optically active β-halo amines were prepared by one-pot halogenation of β-alaniols and ring opening of aziridinium ions by halides. Optically active β-halo amines and aziridinium ions were characterized by NMR and X-ray crystallographic and computational analysis. The stereospecific ring opening of aziridinium ions by halides were computationally studied using DFT and highly-accurate DLPNO-CCSD(T) methods. Highly regioselective and stereoselective ring opening of aziridinium ions was applied to efficient one-pot conversion of β-alaniols to enantiomerically enriched small molecules including β-amino alcohols, β-amino nitriles, and vicinal diamine derivatives.
Scheme 1.
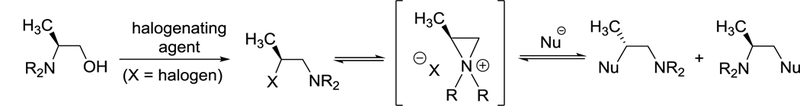
Formation and reactions of β-haioamines and aziridinium ions derived from β-alaniol
Results and Discussion
One-pot halogenation and ring opening of aziridinium ions by halides for preparation of optically active β-halo amines.
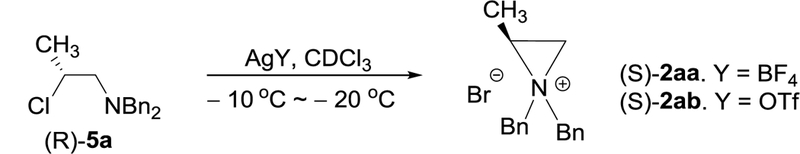
Using N,N-dibenzylated β-alaniol (S)-1a9 (R = Bn) as the model system, enantiomerically enriched β-halo amines 3-8 were prepared using different halogenating agents (Table 1). N,N-dibenzylated β-alaniol (S)-1a (1 equiv) in CH2Cl2 at 0 oC was treated with diethylaminosulfur trifluoride (DAST, 1.2 equiv), and the resulting mixture was reacted for 4 h at 0 oC and 20 h at room temperature. Reaction of (S)-1a with DAST provided secondary β-fluoro amine (R)-3a as the major product which is formed from nucleophilic attack of fluoride counter ion at the more hindered methine carbon (C2) in aziridinium ion 2a formed by intramolecular rearrangement (Table 1, entry 1). A small amount of primary β-fluoro amine (S)-4a as the minor product was formed from ring opening of aziridinium ion 2a at the less hindered carbon (C1) by fluoride.
Table 1.
Formation and ring opening of aziridinium ions: synthesis of optically active β-haloamines
 |
| entry | substrate | halogenating agent | product | #ratio of isomers | yield (%) |
|---|---|---|---|---|---|
| 1 | 1a | +DAST | 3a/4a | 5:1 | 71 |
| 2 | 1a | NCS/PPh3 | 5a | 70 | |
| 3 | 1a | NBS/PPh3 | 6a | 86 | |
| 4 | 1b | NBS/PPh3 | 6b | 63 | |
| 5 | 1c | NBS/PPh3 | 6c | 44 | |
| 6 | 1a | I2/PPh3/imidazole | 7a/8a | 6:1 | 73 |
Determined by 1H NMR;
diethylaminosulfur trifluoride (DAST)
(S)-1a was studied for chlorination and bromination. (S)-1a (1 equiv) in CHCl3 at 0 oC was sequentially reacted with PPh3 (1.2 equiv) and N-halosuccinimide (NXS, X = Cl or Br, 1.2 equiv), and the resulting mixture was reacted for 4 h at 0 oC and 20 h at room temperature. Halogenation of 1a with NCS/PPh3 and NBS/PPh3 followed by ring opening of aziridinium ions by halides afforded secondary β-chloro amine (R)-5a10 (Table 1, entry 2) and secondary β-bromo amine (R)-6a8 (Table 1, entry 3) as the regiospecific isomer, respectively. The structure of β-chloro amine (R)-5a was unambiguously confirmed via X-ray crystallography (Figure 1) and NMR analysis (Figure 2). The inverted stereochemistry in (R)-5a indicates that aziridinium ion (S)-2a was formed by intramolecular rearrangement of activated β-amino alcohol (S)-1a and subsequent stereospecific and regiospecific reaction of (S)-2a with the counter anion chloride at the methine carbon afforded β-chloro amine 5a. N,N-dibenzyl groups in 1a were replaced with an electron withdrawing group (1b) or a bulky piperazine ring (1c) for the study of N-substitution effect on formation of β-halo amines (Table 1, entries 4 and 5). Bromination of 1b and 1c provided the corresponding regiospecific isomers 6b and 6c in a slightly lower isolated yield (63% and 44%), respectively. β-Chloro amine (R)-5a and β-bromo amine (R)-6a were promptly isolated by column chromatography and remained stable at −20 oC for months.
Figure 1.
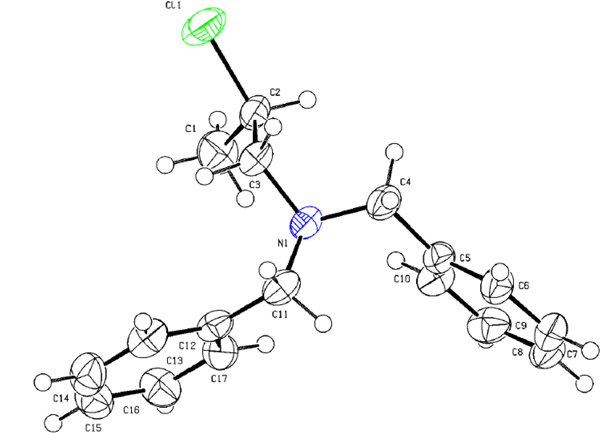
X-ray crystallographic structure of β-chloroamine (R)-5a.
Figure 2.
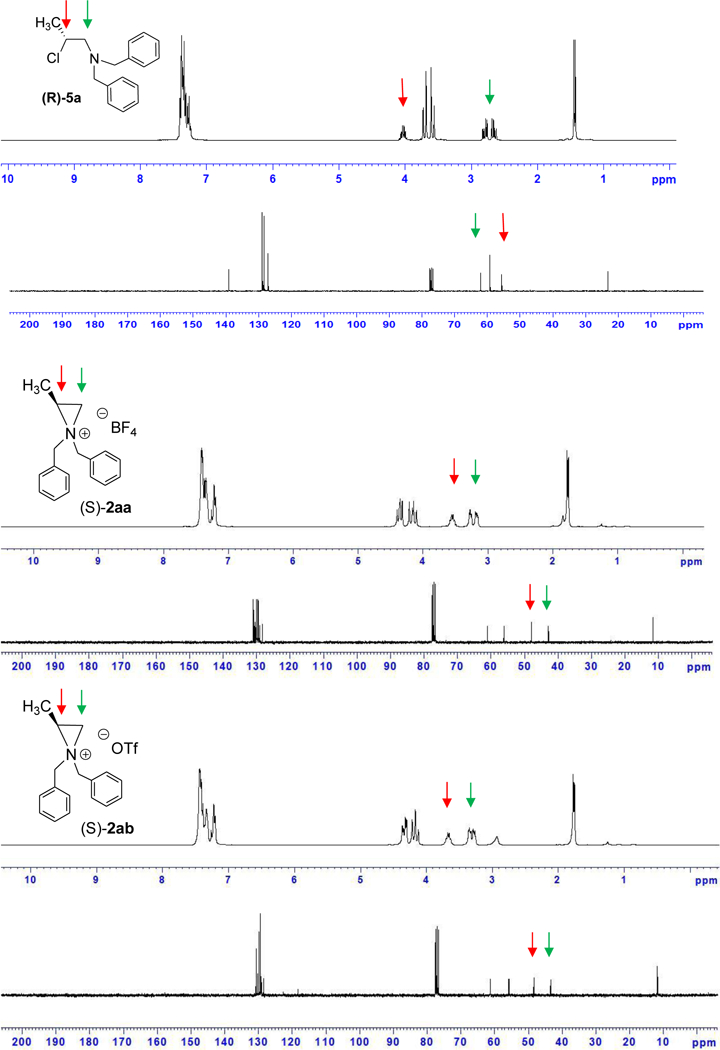
1H and 13C NMR spectra of β-chloroamine and aziridinium ions.
Lastly, iodination of (S)-1a was conducted to understand regioselectivity in ring opening of aziridinium ion. (S)-1a (1 equiv) in CHCl3 at 0 oC was sequentially reacted with PPh3 (1.2 equiv) and imidazole (1.2 equiv), and I2 (1.2 equiv), and the resulting mixture was reacted for 4 h at 0 oC and 20 h at room temperature. Reaction of (S)-1a with I2/PPh3 provided β-iodo amine (R)-7a as the major ring opening product along with a small amount of the regioisomer (S)-8a (Table 1, entry 6). Overall, halogenation of N,N-dibenzylated β-alaniol 1a was highly regioselective and provided (R)-halo amines as the major regioisomer from attack of halide at the more substituted carbon of aziridinium ions in an SN2 pathway.
Formation of isolable aziridinium ions.
Optically active aziridinium ions 2aa (X = BF4) and 2ab (X = OTf) were directly prepared by treatment of β-chloro amine (R)-5a with halo-sequestering silver salts containing weakly nucleophilic counteranions, AgBF4 and AgOTf, respectively (Scheme 2). β-chloro amine (R)-5a was rapidly converted to the corresponding aziridinium ion (S)-2ab containing triflate as a counter anion (25 min, −10 oC), while formation of aziridinium ion (S)-2aa containing tetrafluoroborate from (R)-6a took longer (1 h, −10 oC). Enantiomerically enriched aziridinium ions (S)-2aa and (S)-2ab remained stable at least for several hours at room temperature and were characterized by 1H and 13C and 2D HMQC (Heteronuclear Multiple-Quantum Correlation) NMR and optical rotation. 1H NMR spectra of aziridinium ions 2aa or 2ab were clearly distinguished from that of β-chloro amine 5a (Figure 2). As expected, the methine protons in aziridinium ions 2aa or 2ab (δ 3.5–3.7 ppm) is more shielded than those in β-chloro amine 5a (δ 4.1 ppm). The methylene protons in aziridinium ions 2aa and 2ab (δ3.2 and 3.3 ppm) are shifted downfield compared to those in β-chloro amine 5a (δ2.7 and 2.8 ppm) due to the positively charged nitrogen. The diastereotopic methylene protons in β-chloro amine 5a produce two sharp and distinctive doublets. The nonequivalent N-benzyl protons in aziridinium ions 2aa and 2ab give rise to four different resonance signals. Although a slightly different pattern in proton NMR was observed with aziridinium ions 2aa and 2ab, possibly due to counter ion effect, 2aa and 2ab produced the essentially identical 13C NMR signals. The methine carbon (C2) in β-chloro amine 5a (δ56 ppm) and aziridinium ions 2aa or 2ab (δ48 ppm) were clearly shown to resonate in different fields. The diastereotopic N-benzyl carbons in aziridinium ions 2aa and 2ab gave two different signals (δ56 and 61 ppm) with a large difference in chemical shift. The two magnetically nonequivalent phenyl rings in aziridinium ions 2aa and 2ab are clearly shown in 13C NMR.
Scheme 2.
Synthesis of enantiomerically enriched aziridinium ions
Nucleophilic ring opening reactions of aziridinium ions.
Nucleophilic reactions of aziridinium ions were initially studied using N-protected β-bromo amines (R)-6 and cyanide as a common carbon nucleophile (Table 2). The reaction of (R)-6a (R = Bn) with sodium cyanide in CH3CN provided the primary β-amino nitrile (S)-9aa as the major ring opening product along with a tiny amount of the secondary β-amino nitrile (R)-10aa (Table 2, entry 1, supporting information). Both of the regioisomeric substitution products were isolated and characterized by 1H and 13C NMR, optical rotation, and chiral HPLC. The formation of the two regioisomers (9 and 10) clearly indicates that the reaction involved formation and ring opening of aziridinium ion 2a. Ring opening of aziridinium ion 2a at the less hindered methylene carbon (C-3) provided (S)-9aa (82.2% isolated yield, >97.4% ee), while nucleophilic attack by the cyanide ion at the more substituted methine carbon (C-2) in 2a provided the minor regioisomer (R)-10aa (5.8% isolated yield, >99% ee). The nearly absolute stereoselectivity in (S)-9aa and (R)-10aa indicates that both formation and ring opening of aziridinium ions proceeded with inverted stereochemistry in a SN2 pathway. The ring opening reaction was significantly faster in CH3CN/H2O (10 min) or DMSO (30 min) as compared to CH3CN (4 days). The slow reaction observed in CH3CN is related to poor solubility of NaCN in CH3CN. Change of solvent to DMSO or a mixture of CH3CN and H2O (Table 2, entry 2) made a minimal impact on the preferential formation of the regioisomer (S)-9aa over (R)-10aa, although both regioisomers were produced in excellent stereoselectivity (>98% ee). The effect of N-substitution on the ring opening reaction was investigated. Interestingly, replacement of the N-protecting group from benzyl to p-NO2-Bn (6b, Table 2, entry 4) and piperazine ring (6c, Table 2, entry 5) as a bulky and/or electronwithdrawing group led to formation of the exclusive regiosiomer 9b and 9c in excellent isolated yield and ee (>93%, >99% ee). Other regioisomeric ring opening products (R)-10b or (R)-10c that are expected from nucleophilic attack of the cyanide ion at the more hindered methine carbon in aziridinium ion (S)-2 were not formed from the reaction. The exclusive regioselectivity observed in nucleophilic ring opening reactions of aziridinium ions 6b and 6c with cyanide can be explained by change in steric hindrance and/or electron withdrawal due to N-substitution. 6b and 6c were relatively slower in reaction with the cyanide ion than 6a. Overall, the nucleophilic attack at the less hindered carbon to provide (S)-9 was highly favored in ring opening of all aziridinium ions (S)-2 by cyanide nucleophile. No clear solvent effect on the preferred formation of the major ring opening products was observed. It should be noted that nucleophilic ring opening reactions of aziridinium ions 2 with cyanide provided the regioisomeric β-amino nitriles 9 and 10 in almost absolute stereoselectivity.
Table 2.
Ring opening of aziridiniums: Synthesis of optically active b-aminonitriles
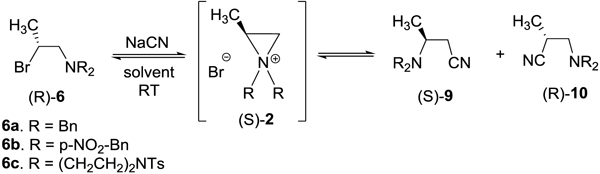 |
| entry | substrate | Nu | solvent | reaction time | product (yield%) | *ratio of (S)-9/(R)-10 | +ee (%) of (S)-9/(R)-10 |
|---|---|---|---|---|---|---|---|
| 1 | 6a | CN | CH3CN | 4 d | 9aa/10aa (82/6) | 14:1 | 97.4/ >99 |
| 2 | 6a | CN | DMSO | 30 min | 9aa/10aa (76/7) | 11:1 | 97.5/ >97.9 |
| 3 | 6a | CN | CH3CN/H2O | 10 min | 9aa/10aa (80/7) | 11:1 | 97.9/ 98.6 |
| 4 | 6b | CN | DMSO | 4 h | 9b (92.8) | >99 | |
| 5 | 6c | CN | CH3CN/H2O | 30 min | 9c (98.5) | >99 |
determined by isolated yield of the regioisomers by prep-TLC or flash LC.
determined by chiral HPLC.
With the high regioselectivity and sterespecificity observed in the ring opening of 2a with cyanide, we extended our investigation to other nucleophiles (Table 3). Reaction of 2a with sodium borohydride was prompt to provide a mixture of regioisomers 9ab and 10ab in a ratio of 2.4 to 1, with the preferred formation of the kinetic product (Table 3, entry 1). Relatively less nucleophilic azide as compared to cyanide and hydride led to the formation of 10ac as the major product from ring opening of 2a at the more hindered carbon (Table 3, entry 2). Both n-propyl amine and n-propane thiolate prefer to attack at the less substituted carbon (C3) of 2a to afford the kinetic product 9ad and 9ae, respectively (Table 3, entries 3 and 4). Particularly, ring opening of 2a at C3 by the strongly nucleophilic but bulky thiolate was highly favored over C2. Aziridinium ion 2a was sluggish in reaction with the sterically hindered nucleophiles n-PrNH2 or n-PrSNa. The formation of the major regioisomer 9ab, 9ad, and 9ae is expected from the kinetically controlled reaction of 2a at the less substituted carbon with a strong nucleophile (hydride, amine, or thiolate). Reaction of 2a with a relatively weak oxygen nucleophile H2O, CH3OH, or n-PrOH provided the thermodynamic product 10af, 10ag, and 10ah (Table 3, entries 5–7). The attack of the oxygen nucleophiles at the more hindered C2 of 2a was highly favored over C3 to produce more stable product 10 from the reaction wherein formation and ring opening of aziridinium ion are predicted to be in equilibrium. To understand the nature of the oxygen nucleophiles better, reaction of 2a with H2O and CH3OH in the presence of base (aq. NaOH) was conducted as control runs. Under the basic reaction condition, the back formation of aziridinium ions 2a from the ring opening product 9af and 9ag is nearly not feasible due to the disfavorable departure of the hydroxyl and methoxy group in 9af and 9ag and thus regiochemistry of azridinium opening is likely controlled by sterics. As predicted, reaction of 2a with H2O or CH3OH produced the kinetic products 9af and 9ag as the major isomer (Table 3, entris 8 and 9).
Table 3.
Ring opening of aziridinium ions: Synthesis of optically active molecules
 |
| entry | Reagent (Nu) | solvent | Additive* | time | temp | +product (yield%) | *ratio of (S)-9/(R)-10 |
|---|---|---|---|---|---|---|---|
| 1 | NaBH4 | CH3CN | 15 min | RT | 9ab/10ab (93) | 2.4/1 | |
| 2 | NaN3 | CH3CN/H2O | 10 min | RT | 9ac/10ac (96) | 1/2 | |
| 3 | n-PrNH2 | CH3CN | 4 h | RT | 9ad/10ad (96) | 2/1 | |
| 4 | n-PrSH | CH3CN | 1M NaOH | 4 h | RT | 9ae/10ae (90) | 5.7/1 |
| 5 | H2O | CH3CN/H2O | 5 h | RT | 9af/10af (91) | 1/8.6 | |
| 6 | CH3OH | CH3OH | 1 h | reflux | 9ag/10ag (96) | 1/6.4 | |
| 7 | n-PrOH | CH3CN/n-PrOH | 3 h | reflux | 9ah/10h (90) | 1/5.9 | |
| 8 | H2O | CH3CN/H2O | 1M NaOH | 3 h | RT | 9af/10af (92) | 1.2/1 |
| 9 | CH3OH | CH3CN/CH3OH | 1M NaOH | 1.5 h | RT | 9ag/10ag (97) | 1.6/1 |
a mixture of the regioisomeric products was isolated via flash column chromatography;
determined by 1H NMR.
One pot bromination and nucleophilic ring opening of aziridinium ions.
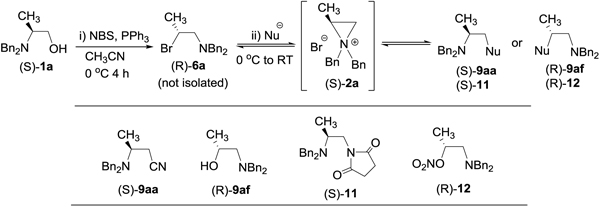
Regioselective and stereoselective ring opening of azirinidinum ions with a nucleophile was applied for a convenient one-pot synthesis of enantiomerically enriched building blocks from β-amino alcohol (S)-1a (Table 4). (S)-1a was subjected to bromination followed by nucleophilic substitution reaction in the presence or the absence of a halo-sequestering agent (Ag2CO3, AgNO3, or AgCN). When bromination of β-alaniol (S)-1a to provide (R)-6a was complete as monitored by TLC analysis, and a nucleophilic reagent (Table 4) was added to the reaction mixture. Reaction of aziridinium ion (S)-2a with NaCN in the presence of water and CH3CN as a co-solvent gave β-amino nitrile (S)-9aa as the major product in excellent stereoselectivity (Table 4, entry 1, 74%, >99% ee, 1 h). (S)-2a was reacted with AgCN in a mixture of CH3CN and H2O to provide secondary β-amino alcohol (R)-9af11 (Table 4, entry 2, 70%, 80% ee) from nucleophilic attack of aziridinium ion at the more hindered methine carbon. β-amino nitrile (S)-9aa was not formed from the substitution reaction, and this seems to reflect poor solubility of silver cyanide in CH3CN, and a sluggish reaction (24 h) of 2a with water instead of cyanide provided hydrolysis product (R)-9af. A significant loss of stereoselectivity observed in (R)-9af (80% ee) can be explained by possible formation of the secondary carbenium ion which is in equilibrium with aziridinium ion (S)-2a. Interestingly, treatment of aziridinium ion 2a with succinimide/1M NaOH or succinimide/Ag2CO3 led to the formation of β-amino succinimide (S)-11 in excellent stereoseletivity (>99% ee) (Table 4, entries 3 and 4). This unexpected formation of 11 resulted from direct reaction of 2a with succinimide (Su) counteranion present in the reaction mixture under basic condition. Steric hindrance seems to control the regioselectivity of the reaction which produced (S)-11 from ring opening of 2a at the less hindered methylene carbon. When 2a was treated with AgNO3 in the presence of water, β-amino nitrate (R)-12 was obtained from ring opening of 2a at the methine carbon by the nitrate ion (Table 4, entry 5, 67% isolated yield, >99% ee). The stereochemistry in (R)-12 was confirmed by conversion of (R)-12 to the known compound (R)-9af. The favored formation of more stable regioisomer (R)-12 is expected form the heat promoted ring opening of aziridinium ion 2a in equilibrium with (R)-12 containing nitrate ion as a leaving group. Unlike (R)-9af formed from the reaction of 2a with AgCN, an almost absolute stereoselectivity was observed with (R)-12, probably due to significantly shorter reaction time (1 h vs 24 h). It is noteworthy that the one-pot bromination and nucleophilic ring opening reactions of aziridinium ions provided the nucleophilic substitution products in excellent enantioselectivity (>99% ee, Table 4, entries 1, 3–5). The one-pot bromination and nucleophilic substitution reactions of aziridinium ions with carbon and nitrogen nucleophiles provided the kinetic products from ring opening of aziridinium ions at the less hindered carbon, while the aziridinium ions reacted with oxygen nucleophiles at the more hindered carbon as expected from the reactions controlled by thermodynamics.
Table 4.
One-pot synthesis of N-protected β-amino nitriie, β-amino alcohol, and vicinal diaminefrom primary β-amino alcohol (S)-1a
 |
| entry | Nu/reagent | time | product | ee (%) |
|---|---|---|---|---|
| 1 | NaCN/H2O | 1 h | (S)-9aa (74%) | >99 |
| 2 | AgCN/H2O | 24 h | (R)-9af (70%) | 80 |
| 3 | 1M NaOH | 2 h | (S)-11 (58%) | >99 |
| 4 | Ag2CO3 | 24 h | (S)-11 (52%) | >99 |
| 5 | AgNO3/H2O | 1 h | (R)-12 (67%) | >99 |
To confirm regiochemistry and stereochemistry in (S)-9aa and (S)-11 obtained from ring opening of aziridinium ions, syntheses of (S)-9aa and (S)-11 were independently carried out as outlined in Scheme 3. Activation of N-Boc protected β-amino alcohol 1312 afforded β-amino mesylate 14, which was reacted with sodium cyanide to provide β-amino nitrile 1513. Removal of N-Boc group in 15 using with TFA followed by N-alkylation with benzyl bromide provided the authentic product (S)-9aa. The structure and stereochemistry of the N-protected vicinal diamine (S)-11 prepared from the ring opening reaction of (S)-2a (Table 4) was confirmed by preparing (S)-11 starting from 13 by a modification of the literature procedure (Scheme 3).14 Reaction of β-amino mesylate 14 with succinimide and subsequent removal of N-Boc group in 16 provided 17 which was further alkylated to afford (S)-11. 1H and 13C NMR spectra and optical rotation data of (S)-9aa and (S)-11 prepared starting from 13 (Scheme 3) were essentially identical with (S)-9aa and (S)-13 prepared from ring opening of aziridinium ions (S)-2a, respectively (Tables 2 and 3). The optical rotation data of (S)-9aa and (S)-11 suggest that stereospecific rearrangement of β–bromoamine (R)-6a to aziridinium ions proceeds through a SN2 pathway with inversion of the chiral center and subsequent reaction of aziridinium ions at the less hindered methylene carbon with cyanide or succinimide provided the nucleophilic substitution products.
Scheme 3.
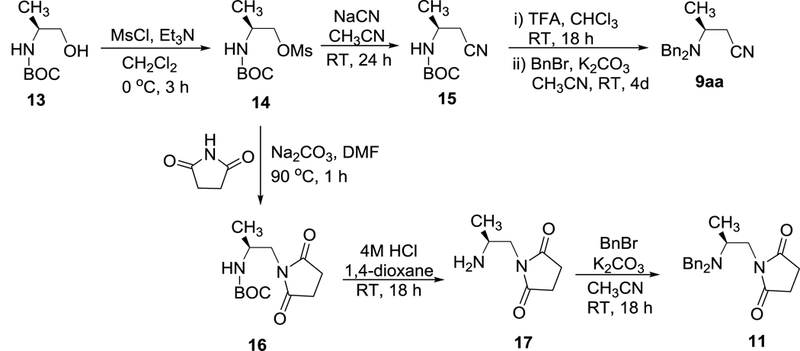
Structural determination of 9aa and 11
Computational studies.
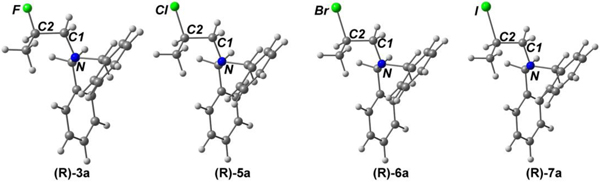
Formation and ring opening of aziridinium ions were theoretically studied by DFT (PBE0/cc-pVTZ) and DLPNO-CCSD(T) calculations. To understand the thermodynamic parameters, we initially optimized equilibrium geometries of the major ring opening products, (R)-β-halo amines (3a, 5a, 6a, and 7a) (Table 5). Based on the X-ray crystallographic parameters of β-chloroamine 5a, the geometrical optimization of 5a was achieved. The bond length between the methine carbon (C2) and halide (X = Cl) in 5a is in excellent agreement with the bond distance shown in the X-ray crystal structure of 5a (calc: 1.814Å vs exp: 1.815Å). A small deviation in the C1-C2 distance for X-ray and computational structure 5a was observed (calc: 1.527Å vs exp: 1.522Å). The length of the N-C1 bond in X-ray crystal structure of 5a was slightly longer than that of DFT-optimized 5a (calc: 1.457Å vs exp: 1.467Å). Structural optimization of other halo amines 3a, 6a, and 7a was conducted by replacing the chlorine atom in the optimized structure of 5a with other halide. As expected from electrostatic interaction and orbital overlap considerations, β-fluoro amine 3a containing fluorine (X = F) has the shortest X-C2 bond (1.395Å), while β-iodo amine 7a (X = I) is calculated to have the longest bond length of X-C2 (2.194Å). All (R)-β-halo amines are calculated to have a similar bond length of N-C1 (1.455~1.458Å) and C1-C2 (1.523~1.527Å). β-fluoro amine 3a has the longest N-C2 bond (2.489Å), while no significant difference in the bond was observed with other halo amines (~2.47Å). The atomic charges were calculated within a framework of natural population analysis (NPA). In all halo amines, nitrogen (−0.57) is the most negatively charged atom in β-halo amines, and the methine carbon (C2) in β-fluoro amine 3a (0.24) is positively charged, while C2 in other β-halo amines 5a (−0.20), 6a (−0.27), and 7a (−0.35) bears a negative charge. As expected, β-fluoro amine 3a has the largest difference in charge bewteen C2 and X (0.64) which is reduced for 5a (0.10), 6a (0.23), and 7a (0.39). The methine carbon (C2) attached to the most electronegative halogen in 3a (X = F) is significantly more positive than C1. Similar partial charge is calculated for the less hindered carbon (C1) in all β-halo amines (−0.28 for 3a and −0.26 for 5a, 6a, and 7a). The calculations show that the optimized structures of β-halo amines have similar parameters, except high positive charge on C2 in 3a (X = F).
Table 5.
Selected geometrical parameters of (R)-β-halo amines 3a, 5a, 6a, and 7a. (PBE0/cc-pVTZ).
 |
| Parameter | (R)-3a (X = F) |
(R)-5a (X = Cl) |
(R)-5a (X = Cl, X-ray) |
(R)-6a (X = Br) |
(R)-7a (X = I) |
|---|---|---|---|---|---|
| X-C1* | 2.338 | 2.702 | 2.837 | 3.031 | |
| X-C2* | 1.395 | 1.814 | 1.815 | 1.974 | 2.194 |
| N-C1* | 1.455 | 1.457 | 1.467 | 1.458 | 1.458 |
| N-C2* | 2.489 | 2.474 | 2.470 | 2.467 | |
| C1-C2* | 1.523 | 1.527 | 1.522 | 1.526 | 1.526 |
| X# | −0.40 | −0.10 | − | −0.04 | 0.04 |
| N# | −0.57 | −0.57 | − | −0.57 | −0.57 |
| C1# | −0.28 | −0.26 | − | −0.26 | −0.26 |
| C2# | 0.24 | −0.20 | − | −0.27 | −0.35 |
All bond lengths are in Angstrom.
Atomic charges are calculated within the NPA framework.
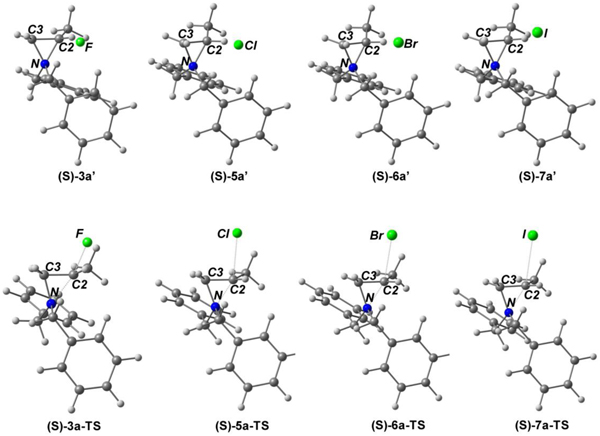
Next, we optimized the geometries of aziridinium ions 3a′, 5a′, 6a′, and 7a′ (Table 6). In all aziridinium ions, the more hindered carbon (C2) is more positive than the less hindered carbon (C3) regardless of the counter anions, and the N-C2 bond (average 1.53Å) is longer than the N-C3 bond (average 1.49Å) and the C2-C3 bond (average 1.47Å). As compared to β-halo amines, the aziridinium ions have shorter C2-C3 and N-C3 bonds by average 0.06Å and 0.03Å, respectively. The counter anion halides are more closely located to C2 than C3 in the aziridinium rings (X-C2: 2.18~3.07Å vs X-C3: 2.93~3.44Å). It is noteworthy that the counter anion halide is shown to form hydrogen bonding with the proton in the more substituted carbon (C2) and one of the benzylic protons leading to construction of a six-membered ring. The hydrogen bonding interaction is predicted to further weaken N-C2 bond and build up more positive charge in C2 that can facilitate ring opening by the halide. The calculated charge on the ring carbons and length of ring bonds explains the high regioselectivity observed with the ring opening reactions of aziridinium ions by halide (Table 1).
Table 6.
Optimized geometries and selected parameters of (S)-aziridinium ions 3a′, 5a′, 6a′, and 7a′ and transition states (TS) for formation of (R)-β-halo amines 3a, 5a, 6a, and 7a from ring opening reaction of aziridinium ion (Az+) with halide via a SN2 pathway (PBE0/cc-pVTZ).
 |
| parameter | (S)-3a′ (X =F) |
(S)-3a-TS (X = F) |
(S)-5a′ (X = Cl) |
(S)-5a-TS (X = Cl) |
(S)-6a′ (X = Br) |
(S)-6a-TS (X = Br) |
(S)-7a′ (X = I) |
(S)-7a-TS (X = I) |
|---|---|---|---|---|---|---|---|---|
| X-C2* | 2.610 | 2.184 | 3.184 | 2.705 | 3.351 | 2.846 | 3.591 | 3.068 |
| X-C3* | 3.228 | 2.931 | 3.434 | 3.086 | 3.564 | 3.217 | 3.779 | 3.445 |
| N-C2* | 1.527 | 1.813 | 1.517 | 1.693 | 1.516 | 1.715 | 1.515 | 1.749 |
| N-C3* | 1.487 | 1.474 | 1.494 | 1.485 | 1.494 | 1.482 | 1.494 | 1.477 |
| C2-C3* | 1.473 | 1.460 | 1.469 | 1.453 | 1.469 | 1.454 | 1.469 | 1.457 |
| X# | −0.75 | −0.68 | −0.82 | −0.82 | −0.82 | −0.80 | −0.84 | −0.79 |
| N# | −0.38 | −0.46 | −0.38 | −0.43 | −0.38 | −0.43 | −0.38 | −0.43 |
| C2# | −0.06 | −0.05 | −0.04 | 0.00 | −0.04 | 0.00 | −0.03 | 0.00 |
| C3# | −0.21 | −0.23 | −0.20 | −0.21 | −0.20 | −0.22 | −0.20 | −0.22 |
All bond lengths are in Angstrom.
Atomic charges are calculated within the NPA framework.
We calculated structures of transition states for the stereospecific ring opening of aziridinium ions by the halide ion leading to the formation of (R)-β-halo amines with inverted stereochemistry at C2 (Table 6). Intrinsic reaction coordinates (IRC) calculations confirm connection of aziridinium ions and (R)-β-halo amines through transition structures. Transition states of the ring opening reactions are shown to have reactant-like structures as supported by the calculated parameters (Tables 5 and 6) and energy profiles (Table 7). The X-C2 bond (average 3.18Å) in aziridinium ions is shown to be elongated in the transition structures (average bond length 2.70Å) as expected from the partial bond between the approaching nucleophilic halide and electrophilic C2. The X-C2 bond (average 1.84Å) in β-halo amines is significantly shorter than the X-C2 bond in the transition structures. The N-C2 bond in aziridinium ions is partially dissociated in transition state (average bond length from 1.52Å to 1.74Å), while the distance of the N-C3 bond remains almost unchanged along the pathway (average distance: 1.49 Å vs 1.48Å). In all transition structures, the methine carbon (C2) is more positively charged than C3, and the N-C2 bond (average 1.74Å) is much longer than the N-C3 bond (1.48Å). The change in bond length between atoms along the intrinsic reaction coordinates (IRC) is well reflected on progressive development of charges on the atoms. More positive charge is built on C2 in transition states relative to aziridinium ion, and no substantial change in charge on C3 and nitrogen is observed with conversion of aziridinium ions to transition structures.
Table 7.
Relative energies of aziridinium ions, transition states (TS), and (R)-β-halo amines involved in ring opening reactions of aziridinium ions with halides in a SN2 pathway (DLPNO-CCSD(T)/cc-pVTZ).
| energies (kcal/mol) |
|||
|---|---|---|---|
| X | aziridinium ion | transition state | (R)-β-halo amine |
| F | (S)-3a’ | 3a-TS | (R)-3a |
| 0.0 | 24.0 | −49.7 | |
| Cl | (S)-5a’ | 5a-TS | (R)-5a |
| 0.0 | 13.3 | −33.3 | |
| Br | (S)-6a’ | 6a-TS | (R)-6a |
| 0.0 | 12.1 | −29.0 | |
| I | (S)-7a’ | 7a-TS | (R)-7a |
| 0.0 | 11.1 | −24.9 | |
Relative energies of aziridinium ions, transition states (TS), and (R)-β-halo amines involved in the ring opening reaction of aziridinium ions (Az+) with halides via a SN2 pathway were calculated (Table 7). While using PBE0 functional for all geometric manipulations and electronic structure analysis, the energetics were evaluated at with help of DLPNO-CCSD(T) method15 (using DFT-optimized geometries) as providing more reliable estimations. The ring opening of aziridinium ions at the more substituted carbon (C2) for the formation of the major β-halo amines is calculated to be thermodynamically favored (Table 8). The activation barrier (24 kcal/mol) was highest for the ring opening of aziridinium ion 3a′ by fluoride being the least efficient nucleophile. The formation of β-fluoro amine 3a (−49.7 kcal/mol) was significantly more exothermic than that of other halo amines 5a (−33.3 kcal/mol), 6a (−29.0 kcal/mol), and 7a (−24.9 kcal/mol). Given the extremely high activation barrier (74 kcal/mol), the back reaction of fluoro amine 3a to aziridinium ion 3a′ traversing the highly labile transition state is shown to be nearly impossible. The calculation predicts that the reaction is kinetically controlled to produce the major regioisomer 3a which is estimated to be more stable than the minor regioisomer 4a (Table 8). The ring opening reactions of other aziridinium ions 5a′, 6a′, and 7a′ has smaller activation barrier (11~13 kcal/mol) as compared to that of 3a′ (X = F). The barrier in the back reaction of the ring opening products 5a, 6a, and 7a to aziridinium ions 5a′, 6a′, and 7a′ is not too large to overcome. The energy released in the formation of the ring opening products might be also used to surmount the kinetic barrier required for the back reaction. The ring opening reaction of 5a′, 6a′, and 7a′ is predicted to be dominantly controlled by stability of the products to favor formation of the thermodynamic products 5a, 6a, and 7a.
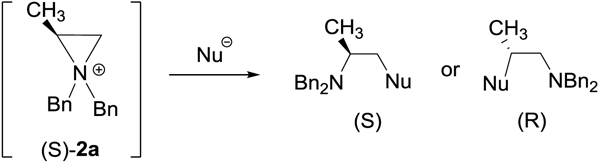
Table 8.
Difference in electronic energies (kcal/mol) between regioisomeric ring opening products (R or S isomer) from reaction of (S)-2a with nucleophile (DLPNO-CCSD(T)/cc-pVTZ).
 |
| Nu | Major isomer+ | Minor isomer+ | ΔE = E(S)-E(R) (kcal/mol) |
|---|---|---|---|
| F | (R)-3a | (S)-4a | 6.31 |
| Cl | (R)-5a | 4.94 | |
| Br | (R)-6a | 4.50 | |
| I | (R)-7a | (S)-8a | 3.81 |
| CN | (S)-9aa | (R)-10aa | 2.03 |
| H | (S)-9ab | (R)-10ab | 1.52 |
| N3 | (R)-9ac | (S)-10ac | 5.04 |
| n-PrNH | (S)-9ad | (R)-10ad | 5.28 |
| n-PrS | (S)-9ae | (S)-10ae | 3.41 |
| OH | (R)-9af | (S)-10af | 5.65 |
| CH3O | (R)-9ag | (S)-10ag | 3.96 |
| n-PrO | (R)-9ah | (S)-10ah | 2.56 |
| Su* | (S)-11 | 1.67 | |
| ONO2 | (R)-12 | 4.70 |
succinimidyl;
isolated
The major ring opening products (R)-β-halo amines 3a, 5a, 6a, and 7a are more stable than the regioisomeric minor products by 6.31, 4.94, 4.50, 3.81 kcal/mol, respectively (Table 8). Difference in relative energies between the fluorine-containing isomers was largest (ΔE = 6.31 kcal/mol). It seems that the difference in energies between the regisomeric β-halo amine parallels with efficiency of leaving group. While the reaction of chloride and bromide with the aziridinium ion provided more stable (R)-5a and (R)-6a as the exclusive regioisomer, a small amount of less stable regioisomers (S)-4a (X = F) and (S)-8a (X = I) were formed from attack of fluoride and iodide at the less hindered carbon (C3) of the aziridinium ions. The minor ring opening product (S)-8a (X = I) is less stable than the major regioisomer (R)-7a by 3.81 kcal/mol. This relatively small difference in energy between the regioisomers containing iodide as an efficient and bulky leaving group is expected to dilute the regiocontrol dominated by stability of product, leading to formation of less stable regioisomer (S)-8a. The formation of the minor product 4a (X = F) may be ascribed to the unusually large positive charge on C2 in 3a (Table 5) that can facilitate direct isomerization of (R)-3a to (S)-4a through back formation of aziridinium ion.
The regioisomeric products that can be obtained in nucleophilic reactions of aziridinium ions as outlined in Table 3 and 4 were compared for relative energies (Table 8). For all ring opening products, the (S)-isomers produced from nucleophilic attack at C2 of aziridinum ions are estimated to be more stable than the (R)-isomers by (ΔE = 1.5~5.7 kcal/mol). A smaller difference in energies between two isomers was calculated for ring opening products containing hydride (ΔE = 1.52 kcal/mol), succinimide (ΔE = 1.67 kcal/mol), and cyanide (ΔE = 2.03 kcal/mol). Given the small difference in stability of the isomers, sterics involving interaction of azirdinium ion and the relatively stronger nucleophiles in the presence of bromide counter anion is predicted to be a major factor in controlling regiochemistry in ring opening of aziridinum ion leading to the preferred formation of (S)-9aa, (S)-9ab, and (S)-11 from the respective nucleophilic attack at the less hindered carbon of cyanide, hydride, and succinimide. (R)-isomers formed from reaction of 2a with the nitrogen nucelophiles azide and n-propyl amine were calculated to be more stable than (S)-isomers by ~ 5 kcal/mol. A smaller difference in relative energies between the regioisomers was calculated with the ring opening product containing a thio propyl group ((S)-9ae/(R)-10ae, ΔE = 3.41 kcal/mol) as compared to an amino propyl group ((S)-9ad/ (R)-10ad, ΔE = 5.28 kcal/mol). The highly preferred formation of (S)-9ae as experimentally observed appeared to be outcome of a kinetically controlled reaction due to a negatively charged but sterically hindered thiolate. The formation of (R)-9af, (R)-9ag, (R)-9ah and (R)-12 over the regiosomeric counterparts from ring opening at the more hindered carbon by oxygen nucleophiles is thermodynamically favored (ΔE = 5.65. 3.96, 2.56, and 4.70 kcal/mol, respectively). Indeed, the reaction of 2a with all oxygen nucleophiles provided more stable regioisomers as the major products. This can be explained by formation and ring opening of aziridinium ions in thermodynamic equilibrium that is achieved by a weak or neutral oxygen nucleophile. With the oxygen nucleophile in steric demand, the back reaction of less stable (S)-ring opening product for the formation of aziridinium ion is less feasible, and thus regiochemistry of ring opening is controlled by activation barrier. The steric effect of oxygen nucleophiles on regiochemsitry is well reflected in both the experimental and theoretical results. Under basic reaction condition, the ring opening reaction of aziridinium ion with oxygen nucleophiles produced the less stable but kinetically favorable (R)-isomer as the major product. This experimental finding demonstrates that the steric factor is more significant in controlling ring opening of aziridinium ions with oxygen nucleophiles as the activation barrier for the back formation of aziridinium ions from the less stable ring opening product containing a poor leaving group becomes too high to overcome.
Conclusion
Optically active β-halo amines and aziridinium ions were prepared starting from β-alaniols and characterized and experimentally and theoretically studied for the nucleophilic substitution reactions. Ring opening of aziridinium ions by halides was highly regioselective and occurred preferably at the more hindered carbon to provide more stable β-halo amines. The high regioselectivity experimentally observed in the ring opening reactions in an SN2 pathway was rationalized by optimized geometries and relative stability of aziridinium ions, transition states, and the ring opening products using DFT and DLPNO-CCSD(T) methods. Ring opening at the less hindered carbon of aziridinium ions by cyanide, hydride, thiolate, amine and succinimide as relatively stronger nucleophiles was shown to be kinetically more favorable and provided less stable ring opening products as the major regioisomers. Azide and oxygen nucleophiles reacted at the more hindered carbon of aziridinium ions leading to the formation of thermodynamically favored β-amino azide, β-amino alcohol, β-amino ether, and β-amino nitrate analogues as the major ring opening products. One-pot halogenation of primary β-amino alcohol and nucleophilic ring opening of aziridinium ions was demonstrated to provide β-amino nitrile, β-amino alcohol, and vicinal diamine derivatives in excellent stereoselectivity (>99% ee).
Experimental Section
General Information.
1H and 13C NMR spectra were obtained using a Bruker 300 instrument and chemical shifts are reported in ppm on the δ scale relative to TMS or solvent. Electrospray iodization (ESI) high resolution mass spectra (HRMS) were obtained on JEOL double sector JMS-AX505HA mass spectrometer (University of Notre Dame, IN). Structure determination of compound 5a was conducted using SMART V5.054 (Bruker Analytical X-ray Systems, Madison, WI) at the X-Ray Crystallographic Laboratory (Department of Chemistry, University of Minnesota). Analytical chiral HPLC was performed on an Agilent 1200 (Agilent, Santa Clara, CA) equipped with a diode array detector and a column (Chiralpak® AD-H, 4.6 × 150 mm, 80Å). Enantiomeric excess of optically active compounds (20 μL, 1 mg of sample in 1 mL of hexanes) was determined by chiral HPLC (isocratic, 230 nm) using the following chromatographic conditions: method A (1/99 = i-PrOH/Hexanes at a flow rate of 0.5 mL/min, 30 min); method B (1/99 = i-PrOH/Hexanes at a flow rate of 1 mL/min, 15 min); method C (10/90 = i-PrOH/Hexanes at a flow rate of 0.5 mL/min, 60 min); method D (8/92 = i-PrOH/Hexanes at a flow rate of 0.5 mL/min, 160 min); method E (0.2/99.8 = i-PrOH/Hexanes at a flow rate of 0.5 mL/min, 15 min); method F (3/97 = i-PrOH/Hexanes at a flow rate of 1 mL/min, 15 min). Optical rotation was determined using JASCO P-2000 polarimeter.
(2S)-2-{bis[(4-nitrophenyl)methyl]amino}propan-1-ol ((S)-1b).
To a stirred solution of (S)-alaninol (100 mg, 1.33 mmol) in CH3CN (2 mL) was added K2CO3 (405 mg, 2.93 mmol) at 0 oC. To the resulting mixture was added dropwise a solution of 4-nitrobenzyl bromide (605 mg, 2.80 mmol) in CH3CN (1 mL) at 0 oC. The reaction mixture was gradually warmed to room temperature and stirred for 24 h. The reaction mixture was filtered and concentrated in vacuo. The residue was purified by column chromatography on silica gel (60–230 mesh) eluted with 30% ethyl acetate in hexanes to afford pure (S)-1b (254 mg, 55%) as a yellow waxy solid. [α]25D = − 25.0o (c = 1.0, CHCl3). 1H NMR (CDCl3, 300 MHz) δ 1.05 (d, J = 6.6 Hz, 3H), 2.48 (s, br, 1H), 2.87–3.02 (m, 1H), 3.45 (dd, J = 10.5, 4.2 Hz, 1H), 3.54–3.63 (m, 3H), 3.85 (d, J = 14.1 Hz, 2H), 7.48 (d, J = 8.1 Hz, 4H), 8.13 (d, J = 8.1 Hz, 4H); 13C NMR (CDCl3, 75 MHz) δ 9.6 (q), 53.1 (t), 55.5 (d), 63.4 (t), 123.8 (d), 129.5 (d), 147.0 (s), 147.2 (s). HRMS (Positive ion ESI) Calcd for C17H20N3O5 [M + H]+ m/z 346.1397. Found: [M + H]+ m/z 346.1399.
2-{bis[(4-nitrophenyl)methyl]amino}propan-1-ol ((rac)-1b).
To a stirred solution of (rac)-alaninol (300 mg, 3.99 mmol) in CH3CN (5 mL) was added K2CO3 (1.21 g, 8.79 mmol) at 0 oC. To the mixture was added dropwise a solution of 4-nitrobenzyl bromide (1.90 g, 8.79 mmol) in CH3CN (10 mL) at 0 oC. The reaction mixture was gradually warmed to room temperature and stirred for 24 h. The reaction mixture was filtered and concentrated in vacuo. The residue was purified by column chromatography on silica gel (60–230 mesh) eluted with 40% ethyl acetate in hexanes to afford pure (rac)-1b (900 mg, 65.3%) as a yellow waxy solid.
(2S)-2-{4-[(4-methylbenzene)sulfonyl]piperazin-1-yl}propan-1-ol ((S)-1c).
To a stirred solution of (S)-alaninol (108 mg, 1.44 mmol) in CH3CN (8 mL) was added Na2CO3 (1.52 g, 14.4 mmol) at 0 oC. To the mixture was added dropwise a solution of tritosyldiethanolamine15 (817 mg, 1.44 mmol) in CH3CN (6.5 mL) at 0 oC. The reaction mixture was refluxed for 26 h. The reaction mixture was filtered, and the filtrate was concentrated in vacuo. The residue was purified by column chromatography on silica gel (60–230 mesh) eluted with 2% CH3OH in CH2Cl2 to afford pure (S)-1c (259 mg, 60%) as a white waxy solid. [α]25D = − 15.6° (c = 1.0, CHCl3). 1H NMR (CDCl3, 300 MHz) δ 0.90 (d, J = 6.6 Hz, 3H), 1.60 (s, br, 1H), 2.40– 2.54 (m, 5H), 2.70–2.89 (m, 3H), 2.90–3.10 (m, 4H), 3.28 (dd, J = 10.2, 10.2 Hz, 1H), 3.38 (dd, J = 11.1, 5.4 Hz, 1H), 7.35 (d, J = 7.8 Hz, 2H), 7.64 (d, J = 8.1 Hz, 2H); 13C NMR (CDCl3, 75 MHz) δ 9.7 (q), 21.5 (q), 46.4 (t), 47.0 (t), 60.2 (d), 62.1 (t), 127.8 (d), 129.7 (d), 132.2 (s), 143.8 (s). HRMS (Positive ion ESI) Calcd for C14H23N2O3S [M + H]+ m/z 299.1424. Found: [M + H]+ m/z 299.1433.
2-{4-[(4-methylbenzene)sulfonyl]piperazin-1-yl}propan-1-ol ((rac)-1c).
To a stirred solution of (rac)-alaninol (200 mg, 2.66 mmol) in CH3CN (3 mL) was added Na2CO3 (2.82 g, 26.6 mmol) at 0 oC. To the resulting mixture was added dropwise a solution of tritosyldiethanolamine (1.66 g, 2.93 mmol) in CH3CN (5 mL) at 0 oC. The reaction mixture was refluxed for 22 h. The reaction mixture was filtered and concentrated in vacuo. The residue was purified by column chromatography on silica gel (60–230 mesh) eluted with 2% CH3OH in CH2Cl2 to afford pure (rac)-1c (250 mg, 31.5%) as a white waxy solid. 1H and 13C NMR data were essentially identical to those of (R)-1c as described above.
Dibenzyl[(2R)-2-fluoropropyl]amine ((R)-3a).
To a solution of (S)-1a9 (50 mg, 0.196 mmol) in CH2Cl2 (3 mL) in an ice-bath maintained at 0 oC was added dropwise (diethylamino)sulfur trifluoride (DAST, 37.9 mg, 0.235 mmol) in CH2Cl2 (2 mL) over 10 min. The resulting mixture was stirred for 4 h at 0 °C. The ice bath was removed, and the reaction mixture was warmed to room temperature and stirred for 20 h. Then the reaction mixture was treated with 10% Na2CO3 aqueous solution (10 mL) and extracted with CH2Cl2 (2 × 10 mL) to provide crude mixture (49 mg), which was sequentially purified by silica gel column chromatography eluted with 3% ethyl acetate in hexanes to afford (R)-3a in an inseparable mixture with (S)-4a (35.6 mg, 71%, 3a/4a = 5:1, as determined by 1H NMR) as a yellow oil. 1H NMR (CDCl3, 300 MHz) δ 1.26 (dd, J = 24.0, 6.3 Hz, 3H), 2.51–2.78 (m, 2H), 3.72 (s, 4H), 4.83 (dm, J = 49.2, 1H), 7.23–7.40 (m, 10H); 13C NMR (CDCl3, 75 MHz) δ 19.3 (dq, J = 22.5 Hz), 58.5 (dt, J = 22.5 Hz), 59.1 (t), 89.9 (dd, J = 165.0 Hz), 127.0 (d), 128.2 (d), 128.8 (d), 139.5 (s). HRMS (ESI) Calcd for C17H21FN: [M + H]+ m/z 258.1653. Found: [M + H] + m/z 258.1642.
Dibenzyl[(2R)-2-chloropropyl]amine ((R)-5a)10
To a solution of (S)-1a9 (50 mg, 0.196 mmol) and PPh3 (61.7 mg, 0.235 mmol) in CHCl3 (5 mL) maintained at 0 oC was added NCS (31.4 mg, 0.235 mmol). The resulting mixture was stirred for 4 h at 0 °C. The ice bath was removed, and the reaction mixture was warmed to room temperature and stirred for 20 h before evaporated to dryness. The residue was purified by silica gel column chromatography eluted with 5% ethyl acetate in hexanes to afford (R)-5a (38 mg, 70%) as a white solid. M.P. 37.1–40.1 oC. [α]26D = + 16.5° (c = 1.0, CHCl3). [Lit.10 [α]22D = + 19.3° (c = 0.8, CHCl3)]. 1H NMR (CDCl3, 300 MHz) δ 1.44 (d, J = 6.6 Hz, 3H), 2.66 (dd, J = 13.2, 7.8 Hz, 1H), 2.82 (dd, J = 6.3, 3.0 Hz, 1H), 3.59 (d, J = 13.5 Hz, 2H), 3.71 (d, J = 13.5 Hz, 2H), 4.00–4.06 (m, 1H), 7.24–7.40 (m, 10H); 13C NMR (CDCl3, 75 MHz) δ 23.1 (q), 55.5 (d), 59.2 (t), 62.1 (t), 127.1 (d), 128.3 (d), 128.9 (d), 139.1 (s). 1H and 13C NMR data were essentially identical to those of (R)-5a previously reported.10
Dibenzyl[(2R)-2-bromopropyl]amine ((R)-6a).8
To a solution of (S)-1a (50 mg, 0.196 mmol) and PPh3 (61.6 mg, 0.235 mmol) in CHCl3 (5 mL) maintained at 0 oC was added NBS (45.3 mg, 0.235 mmol). The resulting mixture was stirred for 4 h at 0 °C. The ice bath was removed, and the reaction mixture was warmed to room temperature and stirred for 20 h before evaporated to dryness. The residue was purified by silica gel column chromatography eluted with 5% ethyl acetate in hexanes to afford (R)-6a (54 mg, 86%) as a white solid. M.P. 37.3–38.3 oC [α]26D = + 15.1° (c = 1.0, CHCl3). 1H NMR (CDCl3, 300 MHz) δ 1.67 (d, J = 6.6 Hz, 3H), 2.75 (dd, J = 13.2, 8.1 Hz, 1H), 2.94 (dd, J = 13.2, 6.0 Hz,, 1H), 3.61 (d, J = 13.5 Hz, 2H), 3.72 (d, J = 13.5 Hz, 2H), 4.09–4.16 (m, 1H), 7.30–7.44 (m, 10H); 13C NMR (CDCl3, 75 MHz) δ 24.0 (q), 47.9 (d), 59.1 (t), 62.7 (t), 127.2 (d), 128.3 (d), 129.0 (d), 139.1 (s). 1H and 13C NMR data were essentially identical to those of (R)-6a previously reported.8
Dibenzyl[(2R)-2-iodopropyl]amine ((R)-7a).
To a solution of (S)-1a9 (50 mg, 0.196 mmol) and PPh3 (61.6 mg, 0.235 mmol) in CHCl3 (6 mL) maintained at 0 oC was added imidazole (16 mg, 0.235 mmol) and I2 (59.6 mg, 0.235 mmol). The resulting mixture was stirred for 4 h at 0 °C. The ice bath was removed and the reaction mixture was warmed to room temperature and stirred for 20 h before evaporated to dryness. The residue was purified by silica gel column chromatography eluted with 5% ethyl acetate in hexanes to afford (R)-7a as an inseparable mixture with (S)-8a (52 mg, 72.6%, 7a/8a = 6.3:1, as determined by 1H NMR) as a light yellow oil. 1H NMR (CDCl3, 300 MHz) δ 1.85 (d, J = 7.2 Hz, 3H), 2.59 (dd, J = 13.5, 8.1 Hz, 1H), 2.94 (dd, J = 13.2, 6.9 Hz, 1H), 3.50–3.74 (m, 4H), 4.11–4.18 (m, 1H), 7.25–7.45 (m, 10H); 13C NMR (CDCl3, 75 MHz) δ 25.9 (q), 26.7 (d), 58.8 (t), 64.3 (t), 127.2 (d), 128.3 (d), 129.1 (d), 139.0 (s). HRMS (ESI) Calcd for C17H22NO: [M − I + H2O]+ m/z 256.1696. Found: [M − I + H2O] + m/z 256.1682.
[(2R)-2-bromopropyl]bis[(4-nitrophenyl)methyl]amine ((R)-6b).
To a solution of (S)-1b (500 mg, 1.23 mmol) and PPh3 (386 mg, 1.47 mmol) in CHCl3 (10 mL) maintained at 0 oC was added NBS (262 mg, 1.47 mmol). The resulting mixture was stirred for 4 h at 0 °C. The ice bath was removed, and the reaction mixture was warmed to room temperature and stirred for 20 h and evaporated to dryness. The residue was purified by silica gel column chromatography eluted with 15% ethyl acetate in hexanes to afford (R)-6b (314 mg, 63%) as a yellow solid. M.P. 125.1–127.8 oC. [α]26D = − 9.1° (c = 1.0, CHCl3). 1H NMR (CDCl3, 300 MHz) δ 1.64 (d, J = 6.9 Hz, 3H), 2.69 (dd, J = 13.8, 6.0 Hz, 1H), 2.86 (dd, J = 13.8, 8.1 Hz, 1H), 3.69 (d, J = 14.4 Hz, 2H), 3.78 (d, J = 14.4 Hz, 2H), 4.11–4.18 (m, 1H), 7.58 (d, J = 8.7 Hz, 2H), 8.20 (d, J = 8.7 Hz, 2H); 13C NMR (CDCl3, 75 MHz) δ 23.9 (q), 46.8 (d), 58.4 (t), 62.8 (t), 123.7 (d), 129.5 (d), 146.2 (s), 147.4 (s). HRMS (Positive ion ESI) Calcd for C17H20N3O5: [M − Br + H2O]+ m/z 346.1397. Found: [M − Br + H2O] + m/z 346.1375.
(2-bromopropyl)bis[(4-nitrophenyl)methyl]amine ((rac)-6b).
To a solution of (rac)-1b (400 mg, 1.158 mmol) and PPh3 (455.6 mg, 1.737 mmol) in CHCl3 (6 mL) maintained at 0 oC was added NBS (309.3 mg, 1.737 mmol). The resulting mixture was stirred for 4 h at 0 °C. The ice bath was removed, and the reaction mixture was warmed to room temperature and stirred for 20 h and evaporated to dryness. The residue was purified by silica gel column chromatography eluted with 15% ethyl acetate in hexanes to afford (rac)-6b (360 mg, 76.7%) as a yellow solid. 1H and 13C NMR data were essentially identical to those of (R)-6b as described above.
1-[(2R)-2-bromopropyl]-4-[(4-methylbenzene)sulfonyl]piperazine ((R)-6c)).
To a solution of (S)-1c (180 mg, 0.603 mmol) and PPh3 (189.9 mg, 0.724 mmol) in CHCl3 (5 mL) at 0 oC was added NBS (139.6 mg, 0.784 mmol) and stirred for 4 h while being maintained at 0 °C. The ice bath was removed, and the reaction mixture was warmed to room temperature and stirred for 20 h and evaporated to dryness. The residue was purified by silica gel column chromatography eluted with 20% ethyl acetate in hexanes to afford (R)-6c (106 mg, 44%) as a light yellow solid. M.P. 102.0–104.3 oC. [α]26D = − 26.8° (c = 1.0, CHCl3). 1H NMR (CDCl3, 300 MHz) δ 1.64 (d, J = 6.3 Hz, 3H), 2.44 (s, 3H), 2.59 (s, 4H), 2.74 (dd, J = 13.2, 7.2 Hz, 2H), 3.03 (s, 4H), 4.04–4.06 (m, 1H), 7.33 (d, J = 7.8 Hz, 2H), 7.63 (d, J = 8.1 Hz, 2H); 13C NMR (CDCl3, 75 MHz) δ 21.5 (q), 24.0 (q), 46.0 (t), 46.3 (d), 52.5 (t), 66.3 (t), 127.8 (d), 129.7 (d), 132.6 (s), 143.7 (s). HRMS (ESI) Calcd for C14H23N2O3S [M + H]+ m/z 299.1424. Found: [M + H]+ m/z 299.1428.
1-(2-bromopropyl)-4-[(4-methylbenzene)sulfonyl]piperazine ((rac)-6c)).
To a solution of (rac)-1c (200 mg, 0.650 mmol) and PPh3 (255.7 mg, 0.975 mmol) in CHCl3 (3 mL) at 0 oC was added NBS (173.6 mg, 0.975 mmol) and stirred for 4 h at 0 °C. The ice bath was removed, and the reaction mixture was warmed to room temperature and stirred for 20 h and evaporated to dryness. The residue was purified by silica gel column chromatography eluted with 20% ethyl acetate in hexanes to afford (rac)-6c (140 mg, 59.6%) as a light yellow solid. 1H and 13C NMR data were essentially identical to those of (R)-6c previously reported.
General synthesis of isolable aziridinium ions.
To a solution of β-halo amine in CDCl3 (0.8 mL) at – 10 °C was added silver tetrafluoroborate (1 equiv) or silver triflate (5 equiv). The resulting mixture was continuously stirred at – 10 °C, while the reaction progress was monitored using TLC. After completing of the reaction, silver halide was filtered, and the resulting solution was immediately characterized by 1H and 13C NMR and HMQC.
(2S)-1,1-dibenzyl-2-methylaziridin-1-ium tetrafluoroboranuide ((S)-2aa).
The general procedure was followed for the reaction of (R)-5a (40 mg, 0.15 mmol) and AgBF4 (28.4 mg, 0.15 mmol) in CDCl3 (0.8 mL) for 1 h. After completion of the reaction, silver chloride was filtered, and the filtrate was immediately characterized by NMR and optical rotation. [α]26D = + 22.8° (c = 1.0, CHCl3). 1H NMR (CDCl3, 300 MHz) δ 1.77 (d, J = 6.3 Hz, 3H), 3.17 (dd, J = 8.4, 3.9 Hz, 1H), 3.27 (dd, J = 7.2, 3.9 Hz, 1H), 3.51–3.58 (m, 1H), 4.12 (d, J = 13.5 Hz, 1H), 4.19 (d, J = 14.4 Hz, 1H), 4.33 (d, J = 9.9 Hz, 1H), 4.38 (d, J = 10.8 Hz, 1H), 7.21 (d, J = 6.3 Hz, 2H), 7.30–7.47 (m, 8H); 13C NMR (CDCl3, 75 MHz) δ 11.6 (q), 42.9 (t), 47.8 (d), 56.1 (t), 61.0 (t), 128.3 (s), 129.2 (s), 129.6 (d), 129.7 (d), 130.0 (d), 130.1 (d), 130.6 (d), 131.0 (d).
(2S)-1,1-dibenzyl-2-methylaziridin-1-ium trifluoromethanesulfonate ((S)-2ab).
The general procedure was followed for the reaction of (R)-5a (40 mg, 0.15 mmol) and AgOTf (187.8 mg, 0.75 mmol) in CDCl3 (0.8 mL) for 25 min. After completion of the reaction, silver chloride was filtered, and the filtrate was immediately characterized by NMR. [α]26D = + 16.0° (c = 1.0, CHCl3). 1H NMR (CDCl3, 300 MHz) δ 1.77 (d, J = 6.0 Hz, 3H), 3.25–3.38 (m, 2H), 3.61–3.72 (m, 1H), 4.14 (d, J = 14.1 Hz, 1H), 4.19 (d, J = 14.1 Hz, 1H), 4.30 (d, J = 5.7 Hz, 1H), 4.35 (d, J = 5.1 Hz, 1H), 7.22 (d, J = 6.6 Hz, 2H), 7.30–7.48 (m, 8H); 13C NMR (CDCl3, 75 MHz) δ 11.8 (q), 43.4 (t), 48.4 (d), 55.8 (t), 61.2 (t), 120.5 (q, J = 322.5 Hz), 128.5 (s), 129.2 (s), 129.4 (d), 129.9 (d), 130.3 (d), 130.8 (d).
General Procedure for nucleophilic reaction of 6 with NaCN.
To a stirred solution of (R)-6 (0.1 mmol) in solvent was added NaCN (0.12 mmol). The reaction mixture was stirred at room temperature. The resulting mixture was evaporated and dissolved in CH2Cl2 and filtered, and the filtrate was concentrated in vacuo. The residue was purified by silica gel column chromatography eluted with 2–4% ethyl acetate in hexanes to afford (S)-9 and (R)-10.
(S)-3-(dibenzylamino)butanenitrile ((S)-9aa) and (R)-3-(dibenzylamino)-2-methylpropanenitrile ((R)-10aa).
From reaction of 6a in CH3CN.
To the stirred solution of (R)-6a (60 mg, 0.188 mmol) in CH3CN (1.5 mL) was added NaCN (18.4 mg, 0.38 mmol). The reaction mixture was stirred at room temperature for 4 d. The resulting mixture was evaporated and dissolved in CH2Cl2 and filtered. The filtrate was concentrated and dried in vacuo. A mixture of (S)-9aa and (R)-10aa (49.7 mg, 100%) was obtained. The regioisomeric mixture was separated by preparative thin layer column chromatography (Prep-TLC, SiO2) eluted with 2–4% ethyl acetate in hexanes to provide (S)-9aa (40.8 mg, 82.2%) as a white solid and (R)-10aa (2.9 mg, 5.8%) as a colorless oil.
(S)-9aa:
M.P. 95.6–98.5 oC. [α]26D = + 4.0° (c = 1.0, CHCl3). 1H NMR (CDCl3, 300 MHz) δ 1.21 (d, J = 6.9 Hz, 3H), 2.36 (dd, J = 16.5, 6.6 Hz, 1H), 2.56 (dd, J = 16.8, 8.1 Hz, 1H), 3.22–3.28 (m, 1H), 3.55 (d, J = 13.8 Hz, 2H), 3.70 (d, J = 13.8 Hz, 2H), 7.26–7.45 (m, 10H); 13C NMR (CDCl3, 75 MHz) δ 14.0 (q), 21.9 (t), 50.4 (d), 53.4 (t), 118.8 (s), 127.2 (d), 128.4 (d), 128.6 (d), 139.1 (s). HRMS (Positive ion ESI) Calcd for C18H21N2 [M + H]+ m/z 265.1699. Found: [M + H]+ m/z 265.1707. Chiral HPLC (method A): tR = 15.4 min (S, major), tR = 16.8 min (R, minor), 97.4% e.e.
(R)-10aa:
[α]25D = − 17.2° (c = 0.15, CHCl3). 1H NMR(CDCl3, 300 MHz) δ 1.19 (d, J = 6.6 Hz, 3H), 2.52 (dd, J = 12.0, 5.7 Hz, 1H), 2.63–2.84 (m, 2H), 3.59 (d, J = 13.5 Hz, 2H), 3.69 (d, J = 13.5 Hz, 2H), 7.26–7.41 (m, 10H); 13C NMR (CDCl3, 75 MHz) δ 15.8 (q), 25.0 (t), 56.9 (d), 59.0 (d), 127.3 (d), 128.4 (d), 128.9 (d), 138.6 (s). HRMS (Positive ion ESI) Calcd for C18H21N2 [M + H]+ m/z 265.1699. Found: [M + H]+ m/z 265.1728. Chiral HPLC (method B): tR = 4.6 min (R, major), tR = 5.9 min (S, minor), >99% e.e.
From reaction of (R)-6a in DMSO.
To a solution of (R)-6a (60 mg, 0.188 mmol) in DMSO (2 mL) was added NaCN (18.4 mg, 0.38 mmol). The reaction mixture was continuously stirred at room temperature for 30 min. The resulting mixture was treated with H2O (25 mL) and extracted with diethyl ether (3 × 25 mL). The combined organic layer was treated with MgSO4 and concentrated in vacuo. A mixture of (S)-9aa and (R)-10aa (49 mg, 98.8%) was obtained. The regioisomeric mixture was separated by Prep-TLC (SiO2) eluted with 2–4% ethyl acetate in hexanes to provide (S)-9aa (37.8 mg, 76.2 %) as a white solid and (R)-10aa (3.5 mg, 7.1%) as a colorless oil.
(S)-9aa:
[α]26D = + 3.5° (c = 1.0, CHCl3). 1H and 13C NMR data of (S)-9aa was identical to those of (S)-9aa obtained from the reaction described above. Chiral HPLC (method A): tR = 15.2 min (S, major), tR = 16.9 min (R, minor), 97.5% e.e.
(R)-10aa:
[α]26D = − 13.2° (c = 0.18, CHCl3). 1H and 13C NMR data of (S)-10aa was identical to those of (S)-10aa obtained from the reaction described above. Chiral HPLC (method B): tR = 4.6 min (R, major), tR = 5.9 min (S, minor), 97.9% e.e.
From reaction of (R)-6a in CH3CN/H2O.
To a stirred solution of (R)-6a (60 mg, 0.188 mmol) in CH3CN/H2O (1.25 mL/0.25 mL) was added NaCN (18.4 mg, 0.38 mmol). The reaction mixture was stirred at room temperature for 10 min. The resulting mixture was evaporated and dissolved in CH2Cl2 and filtered. The filtrate was concentrated and dried in vacuo. A mixture of (S)-9aa and (R)-10aa (49.3 mg, 99.3%) was obtained. The regioisomeric mixture was separated by Prep-TLC (SiO2) eluted with 2–4% ethyl acetate in hexanes to provide (S)-9aa (39.6 mg, 79.8%) as a white solid and (R)-10aa (3.5 mg, 7.1%) as a colorless oil.
(S)-9aa:
[α]26D = + 3.0° (c = 1.0, CHCl3). Chiral HPLC (method A): tR = 15.2 min (S, major), tR = 17.0 min (R, minor), 97.9% e.e. 1H and 13C NMR data of (S)-9aa was identical to the one of (S)-9aa obtained from the above reaction.
(R)-10aa:
[α]26D = − 15.1° (c = 0.18, CHCl3). Chiral HPLC (method B): tR = 4.7 min (R, major), tR = 6.0 min (S, minor), 98.6% e.e. 1H and 13C NMR data of (S)-10aa was identical to those of (S)-10aa obtained from the reaction described above.
3-(dibenzylamino)butanenitrile ((rac)-9aa)16 and 3-(dibenzylamino)-2-methylpropanenitrile ((rac)-10aa).
To a solution of (rac)-6a (150 mg, 0.471 mmol) in CH3CN/H2O (2.5 mL/0.5 mL) was added NaCN (46 mg, 0.95 mmol). The reaction mixture was continuously stirred at room temperature for 10 min. The resulting mixture was evaporated and dissolved in CH2Cl2 and filtered. The filtrate was concentrated and dried in vacuo. A mixture of (rac)-9aa and (rac)-10aa (123.0 mg, 98%) was obtained. The regioisomeric mixture was separated by Prep-TLC (SiO2) eluted with 2–4% ethyl acetate in hexanes to provide (rac)-9aa (93.4mg, 75.3%) as a white solid and (rac)-10aa (9.6 mg, 7.7%) as a colorless oil. 1H and 13C NMR of (rac)-9aa and (rac)-10aa were identical to those of (S)-9aa and (S)-10aa obtained from the reaction described above. Chiral HPLC: tR = 15.4 min (S) and 16.8 min (R) (method A) for 9a and tR = 4.6 min (R) and 5.9 min (S) (method B) for 10aa.
(3S)-3-{bis[(4-nitrophenyl)methyl]amino}butanenitrile ((S)-9b).
To a solution of (R)-6b (30 mg, 0.074 mmol) in DMSO (1.5 mL) was added NaCN (7.2 mg, 0.15 mmol). The reaction mixture was continuously stirred at room temperature for 4 h. The resulting mixture was treated with H2O (25 mL) and extracted with diethyl ether (3 × 25 mL). The combined organic layer was dried over MgSO4 and concentrated in vacuo to provide (S)-9b which was purified by column chromatography eluting 25% ethyl acetate in hexane to afford pure 9b (24.3 mg, 92.8%) as a colorless oil. [α]26D = − 40.2° (c = 1.0, CHCl3). 1H NMR (CDCl3, 300 MHz) δ 1.23 (d, J = 6.6 Hz, 3H), 2.45 (dd, J = 16.8, 5.4 Hz, 1H), 2.61 (dd, J = 17.1, 9.3 Hz, 1H), 3.14–3.19 (m, 1H), 3.65 (d, J = 14.7 Hz, 2H), 3.81 (d, J = 14.7 Hz, 2H), 7.61 (d, J = 8.7 Hz, 2H), 8.20 (d, J = 8.6 Hz, 2H); 13C NMR (CDCl3, 75 MHz) δ 13.8 (q), 22.5 (t), 51.2 (d), 53.1 (t), 118.1 (s), 123.9 (d), 129.3 (d), 146.1 (s), 147.5 (s). HRMS (Positive ion ESI) Calcd for C18H19N4O4 [M + H]+ m/z 355.1401. Found: [M + H]+ m/z 355.1401. Chiral HPLC (method C): tR = 49.0 min (R, minor) and 51.2 min (S, major), >99% ee.
3-{bis[(4-nitrophenyl)methyl]amino}butanenitrile ((rac)-9b).
To a solution of (rac)-6b (50 mg, 0.122 mmol) in CH3CN/H2O (1.25 mL/0.25 mL) was added NaCN (12 mg, 0.245 mmol). The reaction mixture was continuously stirred at room temperature for 8 h. The resulting mixture was treated with H2O (25 mL) and extracted with CH2Cl2 (3 × 25 mL). The combined organic layer was treated with MgSO4 and concentrated in vacuo to provide (rac)-9b (41 mg, 92.5 %) as a colorless oil. 1H and 13C NMR of (rac)-9b was identical to (S)-9b. Chiral HPLC (method C): tR = 49.0 min (R) and 51.7 min (S)
(3S)-3-{4-[(4-methylbenzene)sulfonyl]piperazin-1-yl}butanenitrile ((S)-9c).
To a solution of (R)-6c (30 mg, 0.083 mmol) in CH3CN (1.25 mL) and H2O (0.25 mL) was added NaCN (8.1 mg, 0.17 mmol). The reaction mixture was continuously stirred at room temperature for 30 min. The resulting mixture was evaporated to dryness and the residue was treated with H2O (15 mL) and extracted with CH2Cl2 (3 × 15 mL). The combined organic layer was treated with MgSO4 and concentrated under vacuum to provide (S)-9c which was purified by column chromatography eluting 10% ethyl acetate in hexane to afford pure (S)-9c (25.1 mg, 98.5%) as a white solid. M.P. 101.4–103.9 oC. [α]26D = + 3.32° (c = 1.0, CHCl3). 1H NMR (CDCl3, 300 MHz) δ 1.13 (d, J = 6.6 Hz, 3H), 2.31–2.43 (m, 5H), 2.60–2.62 (m, 4H), 2.91–3.09 (m, 4H), 7.32 (d, J = 8.1 Hz, 2H), 7.62 (d, J = 8.1 Hz, 2H); 13C NMR (CDCl3, 75 MHz) δ 15.0 (q), 21.5 (d), 21.7 (t), 46.2 (t), 47.6 (t), 56.0 (d), 118.3 (s), 127.8 (d), 129.7 (d), 132.3 (s), 143.8 (s). HRMS (Positive ion ESI) Calcd for C15H22N3O2S [M + H]+ m/z 308.1427. Found: [M + H]+ m/z 308.1424. Chiral HPLC (method D): tR = 85.7 min (R, minor) and 89.4 min (S, major), >99% ee.
3-{4-[(4-methylbenzene)sulfonyl]piperazin-1-yl}butanenitrile ((rac)-9c).
To a solution of (rac)-6c (25 mg, 0.069 mmol) in CH3CN (1.25 mL) and H2O (0.25 mL) was added NaCN (6.8 mg, 0.138 mmol). The reaction mixture was continuously stirred at room temperature for 2 h. The resulting mixture was evaporated to dryness and the residue was treated with H2O (15 mL) and extracted with CH2Cl2 (3 × 15 mL). The combined organic layer was treated with MgSO4 and concentrated under vacuum to afford (rac)-9c (21 mg, 98.6%) a white solid. 1H and 13C NMR of (rac)-9c was identical to (S)-9c. Chiral HPLC (method D): tR = 85.7 min (R) and 90.1 min (S).
(S)-3-(dibenzylamino)butanenitrile ((S)-9aa)
From one-pot reaction of (S)-1a with NaCN (Table 4, entry 1).
To a solution of (S)-1a (100 mg, 0.392 mmol) and PPh3 (123.3 mg, 0.470 mmol) in CH3CN (5 mL) at 0 oC was added NBS (83.6 mg, 0.470 mmol) over 5 min. The resulting mixture was stirred for 4 h at 0 °C. The ice bath was removed, and the reaction mixture was warmed to room temperature and stirred for 1 h. NaCN (23 mg, 0.470 mmol) was added to the reaction mixture followed by the addition of H2O (0.5 mL). The reaction mixture was stirred for 1 h at room temperature. The residue was purified by column chromatography on silica gel (60–230 mesh) eluting with 5% ethyl acetate in hexanes to afford (S)-9aa (76.2 mg, 73.6%) as a white solid. [α]26D = + 3.13° (c = 1.0, CHCl3). 1H and 13C NMR data were essentially identical to those described above. Chiral HPLC (method A): tR = 15.0 min (S, major) and 16.8 min (R, minor), >99% ee.
From synthesis of authentic sample for structural determination (Scheme 3).
To a solution of 18 (60 mg, 0.33 mmol) in CH3Cl (3 mL) at 0 oC was added dropwise trifluoroacetic acid (3 mL). The resulting mixture was stirred for 3 h at room temperature. The reaction mixture was concentrated to dryness to provide (3S)-3-aminobutanenitrile as a waxy yellowish solid. The compound was directly used for the next step without purification. To a solution of (3S)-3-aminobutanenitrile (38 mg, 0.24 mmol) and K2CO3 (220.8 mg, 1.6 mmol) in CH3CN (1 mL) was added benzyl bromide (107.8 mg, 0.63 mmol). The resulting mixture was stirred at 60 oC for 24 h. The resulting mixture was filtered and concentrated in vacuo. The residue was purified by column chromatography on silica gel (60–230 mesh) eluting with 3–5% ethyl acetate in hexanes to afford (S)-9aa (53.1 mg, 83.8%) as a white solid. [α]26D = + 4.6° (c = 1.0, CHCl3). 1H and 13C NMR data were essentially identical to those described above. Chiral HPLC (method A): tR = 7.8 min (S, major) and 8.6 min (R, minor), >99% ee.
dibenzyl(propan-2-yl)amine (9ab)24 and dibenzyl(propyl)amine (10ab)25.
To the stirred solution of (R)-6a (60 mg, 0.188 mmol) in CH3CN (1.25 mL) was added NaBH4 (14.3 mg, 0.377 mmol), and the reaction mixture was stirred at room temperature for 15 min. The resulting mixture was evaporated to dryness. The residue was treated with buffer (Et3N/AcOH, 0.05M, pH 6, 15 mL) and then extracted with DCM (3 × 15 mL). The combined organic layer was dried over MgSO4, filtered, and concentrated in vacuo to provide a regioisomeric mixture of 9ab and 10ab (43.5 mg, 96.5%) as colorless oil which was further purified by column chromatography on silica gel (60–220 mesh) eluting with 5% ethyl acetate in hexane to provide an inseparable mixture of 9ab and 10ab (41.8 mg, 92.7%, 9ab/10ab = 2.4/1, as determined by 1H NMR) as a colorless oil. 1H NMR (CDCl3, 300 MHz) δ 0.89 (t, J = 7.2 Hz, 3H), 1.09 (d, J = 6.6 Hz, 6H), 1.53–1.60 (m, 2H), 2.41 (t, J = 7.2 Hz, 2H), 2.91–3.00 (m, 1H), 3.59 (s, 8H), 7.21–7.43 (m, 20H); 13C NMR (CDCl3, 75 MHz) δ 11.8 (q), 17.6 (q), 20.2 (t), 48.2 (d), 53.2 (t), 55.4 (t), 58.3 (t), 126.6 (d), 126.7 (d), 128.1 (d), 128.3 (d), 128.5 (d), 128.8 (d), 140.1 (s), 141.0 (s).
[(2S)-1-azidopropan-2-yl]dibenzylamine ((S)-9ac) and [(2R)-2-azidopropyl]dibenzylamine ((R)-10ac).
To the stirred solution of (R)-6a (60 mg, 0.188 mmol) in CH3CN (1.25 mL) was added the solution of NaN3 (24.5 mg, 0.377 mmol) in H2O (0.25 mL), and the reaction mixture was stirred at room temperature for 10 min. The resulting mixture was evaporated and dissolved in CH2Cl2 and filtered. The filtrate was concentrated and dried in vacuo to provide a regioisomeric mixture of (S)-9ac and (R)-10ac (52.3 mg, 98.9%) as colorless oil which was further purified by column chromatography on silica gel (60–220 mesh) eluting with 5% ethyl acetate in hexane to provide a mixture of (S)-9ac and (R)-10ac (50.7 mg, 95.8%, 9ac/10ac = 1/2, as determined by 1H NMR). 1H NMR showed the ratio of (S)-9ac to (R)-10ac is 1/2. 1H NMR (CDCl3, 300 MHz) δ 1.10–1.15 (m, 6H), 2.47 (dd, J = 13.2, 5.1 Hz, 1H), 2.64 (dd, J = 13.2, 7.8 Hz, 1H), 3.05–3.17 (m, 2H), 3.44 (dd, J = 12.3, 7.2 Hz, 1H), 3.52–3.62 (m, 5H), 3.71 (d, J = 13.5 Hz, 2H), 3.79 (d, J = 13.8 Hz, 2H), 7.24–7.46 (m, 20H); 13C NMR (CDCl3, 75 MHz) δ 11.8 (q), 18.0 (q), 53.0 (d), 53.8 (t), 54.2 (t), 56.1 (d), 59.2 (t), 59.7 (t), 127.0 (d), 127.1 (d), 128.3 (d), 128.7 (d), 128.9 (d), 139.0 (s), 139.8 (s). HRMS (ESI) Calcd for C17H21N4 [M + H]+ m/z 281.1761. Found: [M + H]+ m/z 281.1757.
[(2S)-2-(dibenzylamino)propyl](propyl)amine ((S)-9ad) and [(2R)-1-(dibenzylamino)propan-2-yl](propyl)amine ((R)-10ad).
To the stirred solution of (R)-6a (60 mg, 0.188 mmol) in CH3CN (0.75 mL) was added propan-1-amine (0.75 mL), and the reaction mixture was stirred at room temperature for 4 h. The resulting mixture was evaporated to dryness. The residue was treated with buffer (Et3N/AcOH, 0.05M, pH 6, 15 mL) and then extracted with DCM (3 × 15 mL). The combined organic layer was dried over MgSO4, filtered, and concentrated in vacuo to provide a regioisomeric mixture of (S)-9ad and (R)-10ad (55.3 mg, 99.2%) as colorless oil which was further purified by column chromatography on silica gel (60–220 mesh) eluting with 5% methanol in DCM to 30% methanol in DCM containing 1% Et3N to provide a mixture of (S)-9ad and (R)-10ad (53.6 mg, 96.2%, 9ad/10ad = 2.1/1, as determined by 1H NMR) as a colorless oil. 1H NMR (CDCl3, 300 MHz) δ 0.82 (t, J = 7.5 Hz, 3H), 0.89 (t, J = 7.2 Hz, 3H), 1.04–1.08 (m, 6H), 1.44–1.56 (m, 4H), 2.15–2.24 (m, 3H), 2.39–2.44 (m, 1H), 2.49–2.57 (m, 2H), 2.66–2.80 (m, 3H), 3.07–3.16 (m, 1H), 3.40 (d, J = 13.2 Hz, 2H), 3.54 (d, J = 13.5 Hz, 2H), 3.66 (d, J = 13.5 Hz, 2H), 3.75 (d, J = 13.5 Hz, 2H), 5.92 (s, br, 2H), 7.22–7.34 (m, 20H); 13C NMR (CDCl3, 75 MHz) δ 10.6 (q), 11.4 (q), 11.6 (q), 16.8 (q), 21.8 (t), 22.1 (t), 47.7 (t), 50.0 (t), 51.1 (d), 51.3 (d), 51.5 (t), 53.5 (t), 59.4 (t), 59.7 (t), 127.2 (d), 128.1 (d), 128.5 (d), 129.0 (d), 139.0 (s), 139.7 (s). HRMS (Positive ion ESI) Calcd for C20H29N2 [M + H]+ m/z 297.2331. Found: [M + H]+ m/z 297.2333.
dibenzyl[(2S)-1-(propylsulfanyl)propan-2-yl]amine ((S)-9ae) and dibenzyl[(2R)-2-(propylsulfanyl)propyl]amine ((R)-10ae).
To the stirred solution of (R)-6a (60 mg, 0.188 mmol) in CH3CN (0.75 mL) was added propane-1-thiol (0.75 mL), and the reaction mixture was stirred at room temperature for 10 min. After which, 1M NaOH aqueous solution (0.38 mL) was added and the resulting mixture was stirred at room temperature for 4 h. The resulting mixture was evaporated to dryness. The residue was treated with buffer (Et3N/AcOH, 0.05M, pH 6, 15 mL) and then extracted with DCM (3 × 15 mL). The combined organic layer was dried over MgSO4, filtered, and concentrated in vacuo to provide a regioisomeric mixture of (S)-9ae and (R)-10ae (56.2 mg, 95.1%) as colorless oil which was further purified by column chromatography on silica gel (60–220 mesh) eluting with 4% ethyl acetate in hexane to provide the mixture of (S)-9ae and (R)-10ae (53.4 mg, 90.4%, 9ae/10ae = 5.7/1, as determined by 1H NMR) as a colorless oil. 1H NMR (CDCl3, 300 MHz) δ 0.93–1.01 (m, 6H), 1.17 (d, J = 6.3 Hz, 3H), 1.27 (d, J = 6.6 Hz, 3H), 1.49–1.60 (m, 4H), 2.31–2.48 (m, 6H), 2.85–3.00 (m, 4H), 3.47–3.55 (m, 4H), 3.72 (d, J = 14.1 Hz, 4H), 7.22–7.47 (m, 20H); 13C NMR (CDCl3, 75 MHz) δ 13.4 (q), 13.5 (q), 13.7 (q), 19.7 (q), 22.9 (t), 23.3 (t), 32.4 (t), 34.5 (t), 36.3 (t), 38.1 (d), 52.7 (d), 53.4 (t), 59.1 (t), 60.4 (t), 126.8 (d), 127.0 (d), 128.2 (d), 128.7 (d), 129.0 (d), 139.5 (s), 140.3 (s). HRMS (Positive ion ESI) Calcd for C20H28NS [M + H]+ m/z 314.1942. Found: [M + H]+ m/z 314.1937.
(2S)-2-(dibenzylamino)propan-1-ol ((S)-9af)11 and (2R)-1-(dibenzylamino)propan-2-ol ((R)-10af)7b.
From reaction of (R)-6a and H2O with NaOH.
To the stirred solution of (R)-6a (60 mg, 0.188 mmol) in CH3CN (0.75 mL) was added H2O (0.75 mL), and the reaction mixture was stirred at room temperature for 10 min. After which, 1M NaOH aqueous solution (0.38 mL) was added and the resulting mixture was stirred at room temperature for 3 h. The resulting mixture was evaporated to dryness. The residue was treated with buffer (Et3N/AcOH, 0.05M, pH 6, 15 mL) and then extracted with DCM (3 × 15 mL). The combined organic layer was dried over MgSO4, filtered, and concentrated in vacuo to provide a regioisomeric mixture of (S)-9af and (R)-10af (47.3 mg, 98.3%) as colorless oil which was further purified by column chromatography on silica gel (60–220 mesh) eluting with 8% ethyl acetate in hexane to provide the mixture of (S)-9af and (R)-10af (44.4 mg, 92.3%, 9af/10af = 1.2/1, as determined by 1H NMR). 1H NMR (CDCl3, 300 MHz) δ 1.00 (d, J = 6.6 Hz, 3H), 1.08 (d, J = 6.0 Hz, 3H), 2.43 (d, J = 6.6 Hz, 2H), 2.98–3.05 (m, 1H), 3.30 (s, 1H), 3.35–3.44 (m, 7H), 3.85–3.91 (m, 5H), 7.26–7.38 (m, 20H); 13C NMR (CDCl3, 75 MHz) δ 8.7 (q), 20.0 (q), 53.0 (t), 54.3 (d), 58.5 (t), 61.5 (t), 62.8 (t), 63.2 (d), 127.2 (d), 127.3 (d), 128.5 (d), 128.6 (d), 129.0 (d), 129.1 (d), 138.6 (s), 139.3 (s).
From reaction of (R)-6a and H2O without NaOH.
To the stirred solution of (R)-6a (60 mg, 0.188 mmol) in CH3CN (0.75 mL) was added H2O (0.75 mL), and the reaction mixture was stirred at room temperature for 5 h. The resulting mixture was evaporated to dryness. The residue was treated with buffer (Et3N/AcOH, 0.05M, pH 6, 15 mL) and then extracted with DCM (3 × 15 mL). The combined organic layer was dried over MgSO4, filtered, and concentrated in vacuo to provide a regioisomeric mixture of (S)-9af and (R)-10af (45.1 mg, 93.8%) as colorless oil which was further purified by column chromatography on silica gel (60–220 mesh) eluting with 5% ethyl acetate in hexane to provide a mixture of (S)-9af and (R)-10af (43.6 mg, 90.6%, 9af/10af = 1/8.6, as determined by 1H NMR) as a colorless oil. 1H NMR (CDCl3, 300 MHz) δ 1.00 (d, J = 6.3 Hz, 3H), 1.08 (d, J = 6.0 Hz, 3H), 2.43 (d, J = 6.3 Hz, 2H), 2.98–3.05 (m, 1H), 3.26–3.52 (m, 8H), 3.82–3.89 (m, 5H), 7.27–7.33 (m, 20H); 13C NMR (CDCl3, 75 MHz) δ 8.7 (q), 19.9 (q), 52.9 (t), 54.2 (d), 58.5 (t), 61.4 (t), 62.7 (t), 63.1 (d), 127.2 (d), 127.3 (d), 128.5 (d), 128.6 (d), 129.0 (d), 129.1 (d), 138.6 (s), 139.3 (s).
dibenzyl[(2S)-1methoxypropan-2-yl]amine ((S)-9ag) and dibenzyl[(2R)-2-mothoxypropyl]amine ((R)-10ag).
From reaction of (R)-6a and MeOH with NaOH.
To the stirred solution of (R)-6a (60 mg, 0.188 mmol) in CH3CN (0.75 mL) was added MeOH (0.75 mL), and the reaction mixture was stirred at room temperature for 10 min. After which, 1M NaOH aqueous solution (0.38 mL) was added and the resulting mixture was stirred at room temperature for 1.5 h. The resulting mixture was evaporated to dryness. The residue was treated with buffer (Et3N/AcOH, 0.05M, pH 6, 15 mL) and then extracted with DCM (3 × 15 mL). The combined organic layer was dried over MgSO4, filtered, and concentrated in vacuo to provide a regioisomeric mixture of (S)-9ag and (R)-10ag (49.8 mg, 98.4%) as colorless oil which was further purified by column chromatography on silica gel (60–220 mesh) eluting with 5% ethyl acetate in hexane to provide the mixture of (S)-9ag and (R)-10ag (49.0 mg, 96.8%, 9ag/10ag = 1.6/1, as determined by 1H NMR) as a colorless oil. For preparation of analytical samples, the regioisomeric mixture was separated by Prep-TLC (SiO2) eluted with 1–2% ethyl acetate in hexanes.
(S)-9ag.
[α]25D = − 4.9° (c = 0.5, CHCl3). 1H NMR (CDCl3, 300 MHz) δ 1.08 (d, J = 6.6 Hz, 3H), 3.02 (q, J = 6.6 Hz, 1H), 3.30 (s, 3H), 3.33–3.36 (m, 1H), 3.54–3.62 (m, 3H), 3.72 (d, J = 13.8 Hz, 2H), 7.19–7.32 (m, 6H), 7.40 (d, J = 7.2 Hz, 4H); 13C NMR (CDCl3, 75 MHz) δ 12.1 (q), 52.1 (q), 54.1 (t), 58.8 (d), 75.8 (t), 126.7 (d), 128.1 (d), 128.5 (d), 140.7 (s). HRMS (Positive ion ESI) Calcd for C18H24NO [M + H]+ m/z 270.1858. Found: [M + H]+ m/z 270.1867.
(R)-10ag.
[α]25D = + 13.0° (c = 0.5, CHCl3). 1H NMR (CDCl3, 300 MHz) δ 1.10 (d, J = 6.0 Hz, 3H), 2.41 (dd, J = 12.9, 6.0 Hz, 1H), 2.58 (dd, J = 13.2, 5.7 Hz, 1H), 3.31 (s, 3H), 3.45 (q, J = 14.7 Hz, 1H), 3.54 (d, J = 13.5 Hz, 2H), 3.69 (d, J = 13.8 Hz, 2H), 7.21–7.39 (m, 10H); 13C NMR (CDCl3, 75 MHz) δ 17.9 (q), 56.3 (q), 58.9 (t), 59.3 (t), 75.9 (d), 126.8 (d), 128.1 (d), 128.9 (d), 139.7 (s). HRMS (Positive ion ESI) Calcd for C18H24NO [M + H]+ m/z 270.1858. Found: [M + H]+ m/z 270.1865.
From reaction of (R)-6a and MeOH without NaOH.
(R)-6a (60 mg, 0.188 mmol) was dissolved in MeOH (1.5 mL), and the reaction mixture was refluxed for 1 h. The resulting mixture was evaporated to dryness. The residue was treated with buffer (Et3N/AcOH, 0.05M, pH 6, 15 mL) and then extracted with DCM (3 × 15 mL). The combined organic layer was dried over MgSO4, filtered, and concentrated in vacuo to provide a regioisomeric mixture of (S)-9ag and (R)-10ag (49.6 mg, 98.0%) as colorless oil which was further purified by column chromatography on silica gel (60–220 mesh) eluting with 5% ethyl acetate in hexane to provide the mixture of (S)-9ag and (R)-10ag (48.8 mg, 96.4%, 9ag/10ag = 1/6.3, as determined by 1H NMR) as a colorless oil. 1H NMR (CDCl3, 300 MHz) δ 1.09–1.13 (m, 6H), 2.44 (dd, J = 13.2, 6.3 Hz, 1H), 2.60 (dd, J = 12.9, 5.4 Hz, 1H), 3.01–3.10 (m, 1H), 3.32–3.36 (m, 7H), 3.38–3.77 (m, 10H), 7.23–7.41 (m, 20H); 13C NMR (CDCl3, 75 MHz) δ 12.1 (q), 17.9 (q), 52.1 (q), 54.1 (t), 56.4 (q), 58.9 (t), 59.3 (t), 75.8 (t), 75.9 (d), 126.7 (d), 126.9 (d), 128.2 (d), 128.6 (d), 128.9 (d), 139.8 (s), 140.7 (s).
dibenzyl[(2S)-1-propoxypropan-2-yl]amine ((S)-9ah) and dibenzyl[(2R)-2-propoxypropyl]amine ((R)-10ah).
To the stirred solution of (R)-6a (60 mg, 0.188 mmol) in CH3CN (0.75 mL) was added propan-1-ol (0.75 mL), and the reaction mixture was refluxed for 3 h. The resulting mixture was evaporated to dryness. The residue was treated with buffer (Et3N/AcOH, 0.05M, pH 6, 15 mL) and then extracted with DCM (3 × 15 mL). The combined organic layer was dried over MgSO4, filtered, and concentrated in vacuo to provide a regioisomeric mixture of (S)-9ah and (R)-10ah (51.8 mg, 92.3%) as colorless oil which was further purified by column chromatography on silica gel (60–220 mesh) eluting with 5% ethyl acetate in hexane to provide the mixture of (S)-9ah and (R)-10ah (50.6 mg, 90.2%, 9ah/10ah = 1/5.9, as determined by 1H NMR) as a colorless oil. 1H NMR (CDCl3, 300 MHz) δ 0.89–0.97 (m, 6H), 1.08–1.12 (m, 6H), 1.53–1.63 (m, 4H), 2.43 (dd, J = 12.9, 6.0 Hz, 1H), 3.40 (dd, J = 12.9, 5.7 Hz, 1H), 3.01–3.07 (m, 1H), 3.32–3.47 (m, 4H), 3.53–3.73 (m, 11H), 7.21–7.42 (m, 20H); 13C NMR (CDCl3, 75 MHz) δ 10.7 (q), 12.4 (q), 18.5 (q), 23.0 (t), 23.4 (t), 52.3 (d), 54.1 (t), 59.2 (t), 59.3 (t), 70.6 (t), 72.8 (t), 73.7 (t), 74.3 (d), 126.6 (d), 126.8 (d), 128.1 (d), 128.6 (d), 128.9 (d), 139.9 (s), 140.9 (s). HRMS (Positive ion ESI) Calcd for C20H28NO [M + H]+ m/z 298.2171. Found: [M + H]+ m/z 298.2176.
(2R)-1-(dibenzylamino)propan-2-ol ((R)-9af).11
From one-pot reaction using AgCN/H2O (Table 4, entry 2).
To a solution of (S)-1a (100 mg, 0.392 mmol) and PPh3 (123.3 mg, 0.470 mmol) in CH3CN (5 mL) at 0 oC was added NBS (83.6 mg, 0.470 mmol) over 5 min. The resulting mixture was stirred for 4 h at 0 °C. The ice bath was removed, and the reaction mixture was warmed to room temperature and stirred for 1 h. Then AgCN (62.9 mg, 0.470 mmol) was added to the reaction mixture followed by the addition of H2O (0.5 mL). The reaction mixture was allowed to stir at room temperature for 24 h and evaporated to dryness. The residue was purified by column chromatography on silica gel (60–230 mesh) eluting with 10% ethyl acetate in hexanes to afford (R)-9af (70 mg, 70%) as a colorless oil. [α]25D = – 75.1° (c = 1.0, CHCl3). [Lit.11 [α]20D = – 92.2° (c = 1.0, CHCl3)]. 1H NMR (CDCl3, 300 MHz) δ 1.10 (d, J = 6.3 Hz, 3H), 2.45 (d, J =6.9 Hz, 2H), 3.29 (s, 1H), 3.43 (d, J = 13.5 Hz, 2H), 3.88 (d, J = 13.5 Hz, 2H), 7.26–7.40 (m, 10H); 13C NMR (CDCl3, 75 MHz) δ 20.0 (q), 58.5 (t), 61.4 (t), 63.2 (d), 127.3 (d), 128.5 (d), 129.1 (d), 138.6 (s). 1H and 13C NMR data were essentially identical to those previously reported.11 Chiral HPLC (method E): tR = 6.6 min (S, minor) and 8.0 min (R, major), 90% ee.
From acidification of (R)-12 (Table 4).
(R)-12 (30 mg, 0.010 mmol) was treated dropwise with 4M HCl in 1,4-dioxane (5 mL) at 0 °C. The reaction mixture was heated to 50 °C and stirred for 2 d. H2O (20 mL) and 2M NaOH (aq) (10 mL) were added to the reaction mixture. The resulting mixture was extracted with ethyl acetate (3 × 30 mL). The combined organic layers were dried over MgSO4, filtered, and concentrated to the dryness in vacuo. The residue was purified by column chromatography on silica gel (60–230 mesh) eluting with 10% ethyl acetate in hexanes to afford (R)-9af (16 mg, 61%) as a colorless oil. [α]25D = – 65.6° (c = 0.8, CHCl3). Chiral HPLC (method E): tR = 6.7 min (S) and 8.3 min (R). Chiral HPLC (method E): tR = 6.6 min (S, minor) and 8.0 min (R, major), 81% ee. 1H and 13C NMR data of (R)-9af were essentially identical to those described above.
1-[(2S)-2-(dibenzylamino)propyl]pyrrolidine-2,5-dione ((S)-11).
From one-pot reaction using 1M NaOH (Table 4, entry 3).
To a solution of (S)-1a (100 mg, 0.392 mmol) and PPh3 (123.3 mg, 0.470 mmol) in CH3CN (5 mL) at 0 oC was added NBS (83.6 mg, 0.470 mmol) over 5 min. The resulting mixture was stirred for 4 h at 0 °C. The ice bath was removed, and the reaction mixture was warmed to room temperature and stirred for 1 h. 1M NaOH (0.5 mL) was added to the reaction mixture. The reaction mixture was stirred at room temperature for 2 h. The residue was purified by column chromatography on silica gel (60–230 mesh) eluting with 15% ethyl acetate in hexanes to afford (S)-11 (77 mg, 58%) as a colorless oil. [α]26D = + 99.3° (c = 1.0, CHCl3). 1H and 13C NMR data were essentially identical to those described above Chiral HPLC (method F): tR = 10.4 min (S, major) and 9.4 min (R, minor), >99% ee.
From one-pot reaction using Ag2CO3 (Table 4, entry 4).
To a solution of (S)-1a (100 mg, 0.392 mmol) and PPh3 (123.3 mg, 0.470 mmol) in CH3CN (5 mL) at 0 oC was added NBS (83.6 mg, 0.470 mmol) over 5 min. The resulting mixture was stirred for 4 h at 0 °C. The ice bath was removed, and the reaction mixture was warmed to room temperature and stirred for 1 h. Ag2CO3 (129.6 mg, 0.470 mmol) was added to the reaction mixture. The reaction mixture was allowed to stir at room temperature for 24 h. The residue was purified by column chromatography on silica gel (60–230 mesh) eluting with 15% ethyl acetate in hexanes to afford (S)-11 (69 mg, 52%) as a colorless oil. [α]26D = + 89.7° (c = 1.0, CHCl3). 1H NMR (CDCl3, 300 MHz) δ 1.12 (d, J = 6.6 Hz, 3H), 2.44–2.59 (m, 4H), 3.12–3.16 (m, 2H), 3.29 (d, J = 13.2 Hz, 2H), 3.72–3.91 (m, 3H), 7.22–7.38 (m, 10H); 13C NMR (CDCl3, 75 MHz) δ 11.1 (q), 28.0 (t), 41.3 (t), 51.1 (d), 53.0 (t), 126.9 (d), 128.1 (d), 129.1 (d), 140.1 (s). 177.0 (s). HRMS (Positive ion ESI) Calcd for C21H25N2O2 [M + H]+ m/z 337.1911. Found: [M + H]+ m/z 337.1911. Chiral HPLC (method F): tR = 10.4 min (S, major) and 9.4 min (R, minor), >99% ee.
For structural determination prepared as the authentic sample (Scheme 3).
To a solution of 17 (30 mg, 0.19 mmol) and K2CO3 (131.1 mg, 0.95 mmol) in CH3CN (3 mL) was added benzyl bromide (65.7 mg, 0.38 mmol). The resulting mixture was warmed to 60 oC and continuously stirred for 40 h. The resulting mixture was concentrated in vacuo, and the residue was purified by column chromatography on silica gel (60–230 mesh) eluting with 15% ethyl acetate in hexanes to afford (S)-11 (43.4 mg, 68%) as a colorless oil. [α]26D = + 83.2° (c = 1.0, CHCl3). 1H and 13C NMR spectra of the compound were essentially identical to (S)-13 reported above. Chiral HPLC (method F): tR = 10.4 min (S, major) and 9.4 min (R, minor), >99% ee.
1-[2-(dibenzylamino)propyl]pyrrolidine-2,5-dione((rac)-11).
To a solution of (rac)-1a (100 mg, 0.392 mmol) and PPh3 (123.3 mg, 0.470 mmol) in CH3CN (5 mL) at 0 oC was added NBS (83.6 mg, 0.470 mmol) over 5 min. The resulting mixture was stirred for 4 h at 0 °C. The ice bath was removed, and the reaction mixture was warmed to room temperature and stirred for 1 h. Then 1M NaOH (0.5 mL) was added to the reaction mixture. The reaction mixture was stirred at room temperature for 2 h. The residue was purified by column chromatography on silica gel (60–230 mesh) eluting with 15% ethyl acetate in hexanes to afford (rac)-11 (50 mg, 37.7%) as a colorless oil. 1H and 13C NMR of (rac)-11 was identical to (S)-11. Chiral HPLC (method F): tR = 9.4 (R) and 10.6 min (S).
(2R)-1-(dibenzylamino)propan-2-yl nitrate ((R)-12).
To a solution of (S)-1a (100 mg, 0.392 mmol) and PPh3 (123.3 mg, 0.470 mmol) in CH3CN (5 mL) at 0 oC was added NBS (83.6 mg, 0.470 mmol) over 5 min. The resulting mixture was stirred for 4 h at 0 °C. The ice bath was removed, and the reaction mixture was warmed to room temperature and stirred for 1 h. Then AgNO3 (80 mg, 0.470 mmol) was added to the reaction mixture followed by the addition of H2O (0.5 mL). The reaction mixture was allowed to stir at room temperature for 1 h. The residue was purified by column chromatography on silica gel (60–230 mesh) eluting with 3% ethyl acetate in hexanes to afford (R)-12 (80 mg, 67%) as a colorless oil.
[α]26D = + 25.1° (c = 1.0, CHCl3). 1H NMR (CDCl3, 300 MHz) δ 1.27 (d, J = 6.6 Hz, 3H), 2.58 (dd, J = 13.8, 5.4 Hz, 1H), 2.78 (dd, J = 13.8, 7.2 Hz, 1H), 3.64 (d, J = 13.8 Hz, 2H), 3.71 (d, J = 13.5 Hz, 2H), 5.23–5.25 (m, 1H), 7.28–7.38 (m, 10H); 13C NMR (CDCl3, 75 MHz) δ 16.8 (q), 56.8 (t), 59.3 (t), 78.9 (d), 127.3 (d), 128.4 (d), 128.9 (d), 138.8 (s). HRMS (Positive ion ESI) Calcd for C17H21N2O3 [M + H]+ m/z 301.1547. Found: [M + H]+ m/z 301.1562. Chiral HPLC (method E): tR = 8.7 min (S, minor) and 9.2 min (R, major), >99% ee.
1-(dibenzylamino)propan-2-yl nitrate ((rac)-12).
To a solution of (rac)-6a (50 mg, 0.106 mmol) in CH3CN (1 mL) was added AgNO3 (32.7 mg, 0.192 mmol) followed by the addition of H2O (0.1 mL). The reaction mixture was stirred at room temperature overnight. The residue was purified by column chromatography on silica gel (60–230 mesh) eluting with 5% ethyl acetate in hexanes to afford (rac)-12 (30 mg, 59%) as a colorless oil. 1H and 13C NMR of (rac)-12 was identical to (S)-12. Chiral HPLC (method E): tR = 8.7 min (S) and 9.3 min (R).
tert-Butyl N-[(2S)-1-(methanesulfonyloxy)propan-2-yl]carbamate (14).14
To a solution of 1312 (300 mg, 1.71 mmol) and triethylamine (207.1 mg, 2.05 mmol) in CH2Cl2 (10 mL) at 0 °C was added methanesulfonyl chloride (216.4 mg, 1.89 mmol) over 5 min. The resulting mixture was stirred for 3 h at 0 °C. The reaction mixture was concentrated to dryness, treated with H2O (30 mL), and extracted with CH2Cl2 (3 × 20 mL). The organic layers were combined, dried over MgSO4, filtered, and evaporated. The residue was purified by column chromatography on silica gel (60–220 mesh) eluting 5% ethyl acetate in hexanes to provide (S)-14 (406 mg, 93.8%) as a waxy solid. [α]26D = – 26.4° (c = 1.0, CHCl3). 1H NMR (CDCl3, 300 MHz) δ 1.14 (d, J = 6.9 Hz, 3H), 1.35 (s, 9H), 2.96 (s, 3H), 3.80–3.95 (m, 1H), 4.07–4.09 (m, 2H), 4.84 (d, J = 6.9 Hz, 1H); 13C NMR (CDCl3, 75 MHz) δ 17.0 (q), 28.2 (q), 37.2 (d), 45.4 (t), 72.1(t), 79.6 (s), 155.1 (s). The compound was not very stable and directly used for the next step without further purification.
tert-butyl N-[(2S)-1-cyanopropan-2-yl]carbamate (15).13
To a solution of (S)-14 (150 mg, 0.59 mmol) in DMF (1.5 mL) was added NaCN (43.6 mg, 0.89 mmol) at room temperature. The reaction mixture was stirred at 90 °C for 1 h. The resulting mixture was washed by H2O (50 ml) and extracted with ethyl acetate (3 × 20 mL). The organic layers were combined, dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by column chromatography on silica gel (60–220 mesh) eluting 10% ethyl acetate in hexanes to provide (S)-15 (93.6 mg, 86%) as a white solid. [α]26D = − 81.6° (c = 1.0, CHCl3). [Lit.13 [α]20D = − 89.3° (c = 0.96, CHCl3)]. 1H NMR (CDCl3, 300 MHz) δ 1.28 (d, J = 6.9 Hz, 3H), 1.40 (s, 9H), 2.50 (dd, J = 16.8, 4.2 Hz, 1H), 2.71 (dd, J = 16.5, 4.8 Hz, 1H), 3.85–3.98 (m, 1H), 4.81 (d, J = 5.4 Hz, 1H); 13C NMR (CDCl3, 75 MHz) δ 19.4 (q), 25.1 (t), 28.3 (q), 43.2 (d), 80.0 (s), 117.4 (s), 154.8 (s). 1H and 13C NMR data of (S)-15 were essentially identical to those previously reported.13
tert-butyl N-[(2S)-1-(2,5-dioxopyrrolidin-1-yl)propan-2-yl]carbamate (16).14
To a solution of (S)-14 (150 mg, 0.59 mmol) in DMF (3 mL) was added K2CO3 (162.8 mg, 1.18 mmol) and succinimide (58.5 mg, 0.59 mmol) at room temperature. The reaction mixture was stirred at 90 °C for 1 h. The resulting mixture was washed by H2O (50 ml) and extracted with ethyl acetate (3 × 20 mL). The organic layers were combined, dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by column chromatography on silica gel (60–220 mesh) eluting 15% ethyl acetate in hexanes to provide (S)-16 (90.7 mg, 60%) as a white solid. [α]26D = + 19.1° (c = 1.0, CHCl3). [α]26D = +18.3° (c = 0.9, CHCl3). M.P. 117.2–120.5 oC.1H NMR (CDCl3, 300 MHz) δ 1.11 (d, J = 6.6 Hz, 3H), 1.34 (s, 9H), 2.64–2.70 (m, 4H), 3.39–3.50 (m, 2H), 3.90–4.05 (m, 1H), 4.65 (d, J = 7.5 Hz, 1H); 13C NMR (CDCl3, 75 MHz) δ 18.5 (q), 28.3 (q), 44.5 (d), 44.9 (t), 79.1 (s), 155.6 (s), 177.6 (s). HRMS (Positive ion ESI) Calcd for C12H20N2O4Na [M + H]+ m/z 279.1315. Found: [M + H]+ m/z 279.1326.
1-[(2S)-2-aminopropyl]pyrrolidine-2,5-dione (17).
To a flask containing 16 (60 mg, 0.23 mmol) was added 4 M HCl in 1,4-dioxane (2 mL) at 0 oC. The resulting mixture was stirred at room temperature for 18 h. Ethyl ether (20 mL) was added to the mixture, and the resulting mixture was stirred for 15 min. The resulting mixture was filtered, and the slurry on the filter funnel was immediately treated with H2O (30 mL). The aqueous solution was concentrated in vacuo. The residue was purified by column chromatography on silica gel (60–220 mesh) eluting 5% ethyl acetate in hexanes to provide product (S)-17 (37.8 mg, 84.1%) as a white solid. [α]26D = + 16.4. M.P. 226.3–228.9 oC.1H NMR (D2O, 300 MHz) δ 1.15 (d, J = 5.7 Hz, 3H), 2.68 (s, 4H), 3.45–3.70 (m, 3H); 13C NMR (D2O, 75 MHz) δ 15.6 (t), 28.1 (q), 41.4 (d), 46.6 (t), 181.4 (s). HRMS (Positive ion ESI) Calcd for C18H19N4O4 [M + H]+ m/z 157.1000. Found: [M + H]+ m/z 157.0972.
Calculation details.
Geometry optimizations were performed at the Density Functional Theory (DFT) level of theory using Perdew-Burke-Erzenhof parameter-free exchange-correlation functional (PBE0).18 All atoms were described by correlation-consistent basis sets of triple-ζ quality (cc-pVTZ). Basis set for heavy iodine was augmented by effective core potential (so-called cc-pVTZ-pp). In all cases, no symmetry restrictions were applied. All calculated structures were found to be minima on PES (no negative elements of the Hessian matrix and, consequently, no imaginary frequencies) unless otherwise noted. Transition states were found to possess only one imaginary frequency, corresponding to the transition between reactants and products intended. The nature of transition states was probed through IRC (intrinsic reaction coordinate) technique that led directly to target products or reactants. All computations were performed by Firefly 8.1.0 program package.19
In the next step, optimized geometries were used to get insight into the electronic structure of our target systems in terms of natural bond orbitals (NBO).20 Bond orders quoted are those from the Wiberg formulation21 (so-called Wiberg bond indexes) incorporated in the NBO analysis. All computations were performed with NBO 6.0 program,22 using the converged wavefunctions generated by Firefly program.
In order to get more reliable evaluation on reaction energetics, single-point calculations (for DFT-optimized geometries) were performed for all minimum structures and transition states with help of highly-accurate single-reference corrBrienelated method DLPNO-CCSD(T)15 (domain-based local pair natural orbitals coupled-cluster with single, double and perturbative triple corrections) with the same basis sets. This method was recently proposed by the group Neese and found to be very accurate, providing accuracy comparable with that of CCSD(T), which has reputation of serving as benchmarking method. To accelerate these calculations, the resolution-of-identity (RI) algorithm was applied using the chain-of-sphere approach (RIJCOSX).23 All these high-level calculations were carried out by using ORCA program suite (version 3.1.0).
X-ray Crystal Determination of (R)-5a.
Crystals for structure determination were obtained by slow evaporation of 5a from a mixture of CH2Cl2/hexanes. The structure was solved using SIR-92 and refined using Bruker SHELXTL. Crystallographic data for 5a have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication NO. CCDC 999697. Copies of the data can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK, (fax: +44-(0)1223–336033 or e-mail: deposit@ccdc.cam.ac.uk).
Supplementary Material
Footnotes
Supporting Information. A copy of 1H and 13C NMR spectra of all new compounds and HPLC chromatograms of all chiral compounds and a Crystallographic Information Framework (CIF) file for X-ray structure of compound 5a. This material is available free of charge via the Internet.
References
- 1.Metro T-X; Duthion B; Pardo DG; Cossy, J. Chem. Soc. Rev. 2010, 39, 89. [DOI] [PubMed] [Google Scholar]
- 2.(a) Jarvis SBD; Charette AB Org. Lett. 2011, 13, 3830. [DOI] [PubMed] [Google Scholar]; (b) Catak S; D’hooghe M; De Kimpe N; Waroquier M; Van Speybroeck VJ Org. Chem. 2010, 75, 885. [DOI] [PubMed] [Google Scholar]; (c) Yun SY; Catak S; Lee WK; D’hooghe M; De Kimpe N; Van Speybroeck V; Waroquier M; Kim Y; Ha Y-J Chem. Commun. 2009, 2508. [DOI] [PubMed] [Google Scholar]; (d) Hamilton GL; Kanai T; Toste FD J. Am. Chem. Soc. 2008, 130, 14984. [DOI] [PubMed] [Google Scholar]; (e) Couturier C; Blanchet J; Schlama T; Zhu J Org. Lett, 2006, 8, 2183. [DOI] [PubMed] [Google Scholar]; (f) O’Brien P; Towers TD J. Org. Chem. 2002, 67, 304. [DOI] [PubMed] [Google Scholar]; (g) Chuang T-H; Sharpless KB Org. Lett. 2000, 2, 3555. [DOI] [PubMed] [Google Scholar]; (h) Katagiri T; Takahashi M; Fujiwara Y; Ihara H; Uneyama K J. Org. Chem. 1999, 64, 7323. [Google Scholar]
- 3.(a) Polavarapu A; Stillabower JA; Stubblefield SGW; Taylor WM; Baik M-H J. Org. Chem. 2012, 77, 5914. [DOI] [PubMed] [Google Scholar]; (b) Noll DM; Mason TM; Miller PS Chem. Rev. 2006, 106, 277. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Dutta S; Abe H; Aoyagi S; Kibayashi C; Gates KS J. Am. Chem. Soc. 2005, 127, 15004. [DOI] [PubMed] [Google Scholar]; (d) Ringdahl B; Mellin C; Ehlert FJ; Roch M; Rice KM; Jenden DJ J. Med. Chem. 1990, 33, 281. [DOI] [PubMed] [Google Scholar]
- 4.(a) Leonard NJ; Jann K J. Am. Chem. Soc. 1960, 82, 6418. [Google Scholar]; (b) Leonard NJ; Jann K J. Am. Chem. Soc. 1962. 84, 4806. [Google Scholar]; (c) Leonard NJ; Brady LE J. Org. Chem. 1965, 30, 817. [DOI] [PubMed] [Google Scholar]
- 5.(a) Yoon D-H; Kang P; Lee WK; Kim Y; Ha H-J Org. Lett. 2012, 14, 429. [DOI] [PubMed] [Google Scholar]; (b) Oxenford SJ; Moore SP; Carbone G; Barker G; O’Brien P; Shipton MR; Gilday J; Campos KR Tetrahedron Asymm. 2010, 21, 1563. [Google Scholar]; (c) Piotrowska DG; Wróblewski AE Tetrahedron 2009, 65, 4310. [Google Scholar]; (d) Guan H; Saddoughi SA; Shaw AP; Norton JR Organometallics 2005, 24, 6358. [Google Scholar]; (e) Carter C; Fletcher S; Nelson A Tetrahedron Asymm. 2003, 14, 1995. [Google Scholar]; (f) Quanying L, Marchington AP; Boden N; Rayner CM J. Chem. Soc. Perkin Trans. 1, 1997, 511. [Google Scholar]
- 6.(a) Aziridines and epoxides in organic synthesis; Yudin AK, Ed; Wiley-VCH, Weinheim: 2006. [Google Scholar]; (b) Padwa A; Murphree SS ARKIVOC 2006, (iii), 6. [Google Scholar]; (c) Singh GS; D’hooghe M; De Kimpe N Chem. Rev. 2007, 107, 2080. [DOI] [PubMed] [Google Scholar]; (d) Callebaut G; Meiresonne T; De Kimpe N; Mangelinckx S Chem. Rev. 2014, 114, 7954. [DOI] [PubMed] [Google Scholar]; (e) Degennaro L; Trinchera P; and Luisi R Chem. Rev. 2014, 114, 7881. [DOI] [PubMed] [Google Scholar]; (f) Catak S; D’hooghe M; Verstraelen T; Hemelsoet K; Van Nieuwenhove A; Ha H-J; Waroquier M; De Kimpe N; Van Speybroeck V J. Org. Chem. 2010, 75, 4530. [DOI] [PubMed] [Google Scholar]; (g) Stanković S; D’hooghe M; Catak S; Eum H; Waroquier M; Van Speybroeck V; De Kimpe N; Ha H-J Chem. Soc. Rev. 2012, 41, 643. [DOI] [PubMed] [Google Scholar]
- 7.(a) Chong H-S; Sun X; Zhong Y; Bober K; Lewis MR; Liu D; Ruthengael VC; Sin I; Kang CS Eur. J. Org. Chem. 2014, 1305. [Google Scholar]; (b) Chong H-S; Chen Y Org. Lett. 2013, 15, 5912. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chong H-S; Song HA; Kang CS; Le T; Sun X; Dadwal M; Lee H; Lan X; Chen Y; Dai A Chem Commun. 2011, 47, 5584. [DOI] [PubMed] [Google Scholar]; (d) Chong H-S; Song HA; Dadwal M; Sun X; Sin I; Chen Y J. Org. Chem. 2010, 75, 219. [DOI] [PubMed] [Google Scholar]
- 8.(a) Nagle AS; Salvatore RN; Chong B-D; Jung KW Tetrahedron Lett. 2000, 41, 3011. [Google Scholar]; (b) Dakanali M; Tsikalas GK; Krautscheid H; Katerinopoulos HE Tetrahedron Lett. 2008, 49, 1648. [Google Scholar]; (c) Wefelscheid UK; Woodward S J. Org. Chem. 2009, 74, 2254. [DOI] [PubMed] [Google Scholar]; (d) Weber K; Kuklinski S; Gmeiner P Org. Lett, 2000, 2, 647. [DOI] [PubMed] [Google Scholar]; (e) D’hooghe M; Catak S; Stankovic S; Waroquier M; Kim Y; Ha H-J; Van Speybroeck V; De Kimpe N Eur. J. Org. Chem. 2010, 4920. [Google Scholar]
- 9.Beaulieu PL; Wernic D J. Org. Chem. 1996, 61, 3635. [DOI] [PubMed] [Google Scholar]
- 10.Van Beijnen AJM; Nolte RJM; Naaktgeboren AJ; Zwikker JW; Drenth W; Hezemans AMF Macromolecules, 1983, 16, 1679. [Google Scholar]
- 11.Métro T-X; Pardo DG; Cossy J J. Org. Chem. 2007, 72, 6556. [DOI] [PubMed] [Google Scholar]
- 12.Douat-Casassus C; Pulka K; Claudon P; Guichard G Org. Lett. 2012, 14, 3130. [DOI] [PubMed] [Google Scholar]
- 13.Sohtome Y; Shin B; Horitsugi N; Takagi R; Noguchi K; Nagasawa K Angew. Chem. Int. Ed. 2010. 49, 7299. [DOI] [PubMed] [Google Scholar]
- 14.Goldstein DA; Gong L; Michoud C; Palmer WS; Sidduri A PCT/EP/2007/059040. March 13, 2008. [Google Scholar]
- 15.(a) Liakos DG; Sparta M; Kesharwani MK; Martin JML; Neese F J. Chem. Theory Comput. 2015, 11, 1525. [DOI] [PubMed] [Google Scholar]; (b) Riplinger C; Sandhoefer B; Hansen A; Neese F J. Chem. Phys. 2013, 139, 134101. [DOI] [PubMed] [Google Scholar]
- 16.McMurry TJ; Brechbiel M; Wu C; Gansow OA Bioconjugate Chem. 1993, 4, 236. [DOI] [PubMed] [Google Scholar]
- 17.Hesek D; Lee M; Noll BC; Fisher JF; Mobashery S J. Org. Chem. 2009, 74, 2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Perdew JP; Burke K; Ernzerhof M Phys. Rev. Lett. 1997, 78, 1396. [DOI] [PubMed] [Google Scholar]; (b) Perdew JP, Burke K; Ernzerhof M Phys. Rev. Lett. 1996, 77, 3865. [DOI] [PubMed] [Google Scholar]
- 19.Granovsky AA Firefly version 8.1.0, http://classic.chem.msu.su/gran/firefly/index.html.
- 20.(a) Weinhold F; Landis CR Valency and Bonding: A Natural Bond Orbital Donor − Acceptor Perspective; Cambridge University Press: Cambridge, U. K., 2005. [Google Scholar]; (b) Reed AE; Curtiss LA; Weinhold F Chem. Rev. 1988, 88, 899. [Google Scholar]
- 21.Wiberg KB Tetrahedron 1968, 24, 1083. [Google Scholar]
- 22.(a) Glendening ED; Badenhoop JK; Reed AE; Carpenter JE; Bohmann JA; Morales CM; Landis CR; Weinhold F NBO 6.0, 2013, University of Wisconsin, Madison. [Google Scholar]; (b) Glendening ED; Landis CR; Weinhold F J. Comp. Chem. 2013, 34, 1429. [DOI] [PubMed] [Google Scholar]
- 23.Neese F; Wennmohs F; Hansen A; Becker U Chem. Phys. 2009, 356, 98. [Google Scholar]
- 24.Zhang M; Flynn DL; Hanson PR J. Org. Chem. 2007, 72, 3194. [DOI] [PubMed] [Google Scholar]
- 25.Tokizane M; Sato K; Sakami Y; Imori Y; Matsuo C; Ohta T; Ito Y Synthesis 2010, 1, 36. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.