

Abstract
The bacterial enzyme nitrogenase achieves the reduction of dinitrogen (N2) to ammonia (NH3) utilizing electrons, protons, and energy from the hydrolysis of ATP. Building on earlier foundational knowledge, recent studies provide molecular-level details on how the energy of ATP hydrolysis is utilized, the sequencing of multiple electron transfer events, and the nature of energy transduction across this large protein complex. Here, we review the state of knowledge about energy transduction in nitrogenase.
Graphical Abstract
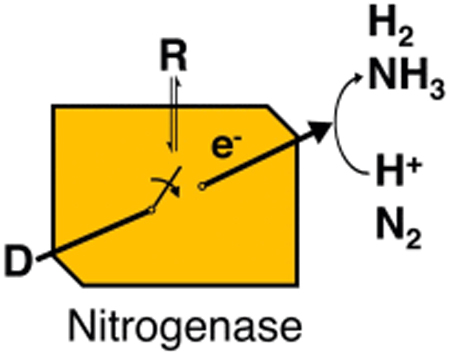
Introduction
The bacterial enzyme nitrogenase, which is known to occur in Mo, V, and Fe forms [1–5], reduces dinitrogen (N2) to ammonia (NH3) utilizing protons, electrons, and the hydrolysis of ATP. Since the discovery of these enzymes, there has been great interest in understanding their mechanism in order to gain insights into how nature solved the problem of reducing one of the most stable molecules, N2, at ambient temperatures and pressure. Recent progress provides significant new insights into the N2 binding event and how the reduction reaction is achieved [6•,7–10]. Here, we review new findings that are unravelling the complexities of how electrons traverse the nitrogenase two-component system to achieve the reduction of bound N2 and how energy is transduced from the hydrolysis of nucleotides in the process.
Nitrogenases comprised two proteins, one being a reductase component known as the Fe protein, that contains a single 4Fe-4S cluster and two ATP binding sites. A second catalytic protein, known as the MoFe-protein, VFe-protein or FeFe-protein, houses an electron transfer P cluster as well as the active-site metal cofactor (FeMo-co, FeV-co, and FeFe-co) (Figure 1). The Mo-nitrogenase system is the focus of this review, and insights into this system are likely to be applicable to the other systems. We know that an Fe protein, with two bound ATP molecules, associates with one-half of the MoFe protein, and that this association initiates a series of events that result in the transfer of an electron from the Fe protein to FeMo-co, the hydrolysis of two ATP molecules to two ADP and two Pi, release of phosphate, and finally the dissociation of the Fe protein (oxidized and with two bound ADP) from the MoFe protein (reduced by one electron) [1,11]. This cycle of events is repeated in order to accumulate electrons on the active site, with four cycles needed to achieve the state that binds N2 and eight cycles to achieve the complete reduction of N2 and release of one H2. The order and nature of the events that occur during this transient association of the two component proteins, often referred to as the Fe protein cycle, is one of the great mysteries of nitrogenase function. Studies during the last several years have added to the considerable body of knowledge about the Fe protein cycle [1], filling in and refining our understanding of nitrogenase function.
Figure 1.

Nitrogenases. Schematic diagram for the three forms of nitrogenase, where M is Mo, V, or Fe (a). Shown are the structures for the active site FeMo-cofactor [33] and FeV-cofactor [34] determined by X-ray crystallography and a proposed structure for the FeFe-cofactor (b). It is noteworthy that some recent work is indicating that changes in the structure of FeMo-co and FeV-co can occur upon addition of electrons and/or upon binding of substrates and inhibitors, suggesting structural dynamics of the cofactor is part of the mechanism.
ATP hydrolysis and electron transfer
Recent studies have contributed four significant insights into the key events that occur in the Fe protein cycle, including that: firstly electron transfer occurs ahead of ATP hydrolysis [12••]; secondly the overall rate limiting step is not component protein dissociation, but rather Pi release [13••]; thirdly the electron transfer events are staged to occur in the order, conformationally gated P cluster → FeMo-co first, followed by rapid Fe protein → Pox (together referred to as ‘deficit-spending’ electron transfer) [14]; and finally the two halves of nitrogenase MoFe protein function through negative cooperativity, with each side influencing activity on the opposite side [15••].
The earliest studies of nitrogenase revealed that the hydrolysis of 2 ATP to 2 ADP + 2 Pi was coupled to the transfer of one electron from the Fe protein into the MoFe protein [16–18]. This prompted the widely accepted model wherein the energy from hydrolysis of ATP was being used to drive electron transfer in nitrogenase [1]. Studies over the last few years have upended this model by showing that ATP hydrolysis occurs after the electron transfer events [12••]. Indeed, an early theoretical study by Beratan et al. [19] concluded that ATP hydrolysis energy should come at the end of the cycle, to drive the proteins apart. That model is consistent with the new experimental studies placing electron transfer ahead of ATP hydrolysis. The key steps of the new model are summarized in Figure 2. It shows that ATP binding to the Fe protein is essential to activating Fe protein for binding to the MoFe protein, while the energy of hydrolysis of ATP is used to weaken the interaction between the Fe protein and the MoFe protein for dissociation at the end of the cycle, with Pi release being the rate limiting step [13••].
Figure 2.

Scheme for the key events during electron transfer between the nitrogenase Fe protein and the MoFe protein. The MoFe protein is shown as one αß pair (right side) containing a P cluster (P) and FeMo-co (M). The Fe protein dimer (left side), with one [4Fe-4S] cluster (R or ox) and two bound ATP, associates with the MoFe protein (top). This association initiates a gated electron transfer event from the P cluster to FeMo-co with a rate constant (kET) of 140 s−1 at 25°C, with rapid follow up electron transfer from the Fe protein to the oxidized P cluster. The next event is ATP hydrolysis with a rate constant kATP of 70 s−1, followed by Pi release with a rate constant kPi of 16s−1, and then the fast dissociation (kdiss) of the two proteins.
On the basis of continuum electrostatics analysis, it was earlier proposed that ATP binding to the Fe protein energized the 4Fe-4S cluster for electron transfer by desolvation of the 4Fe-4S cluster caused by docking of the Fe protein with the MoFe protein, and that the protein-protein complex is made more stable by about the same amount [19]. ATP binding to the Fe protein also drives the 4Fe-4S cluster several Angstroms closer to the protein surface [20•,21–23] and strengthens its electronic coupling to the P cluster of the MoFe protein. It is possible that the docking of the Fe protein to the MoFe protein induces a conformational change within the MoFe protein that opens the gate for electron transfer from the P cluster to FeMo-co. The docking may further accelerate electron tunneling from the Fe protein to the MoFe protein beyond simple distance shortening. Alternatively, the gating may also be accomplished by a Marcus free energy effect created by changing solvent exposure or cofactor structure (or both effects may be at play). Given the osmotic pressure dependence of the gating process, it is likely that water molecules at this dynamical interface play a role in electron flow between the cofactors (as well as through their influence on coupling or redox potentials).
Deficit-spending ET
One of the most intriguing aspects of electron transfer within nitrogenase is the order of the two electron transfer events involving the FeS cluster of the Fe protein, the P cluster, and FeMo-co. Recent work showed that following the association of the Fe protein with the MoFe protein, one electron is transferred from the Fe protein to the MoFe protein in a process that is conformationally gated [24]. From structures of nitrogenase complexes of the two component proteins [25,26], it is clear that the P cluster in the MoFe protein is involved as an intermediate in the transfer of the electron to the FeMo-cofactor [27,28]. Early models assumed that the Fe protein would first transfer an electron to the P cluster [1]. A problem with this model is that the resting P cluster (designated as PN) has all Fe atoms in the ferrous oxidation state [29]. Since there are no known examples of reduction of enzyme FeS clusters below the ferrous oxidation state, instead, gated P cluster to FeMo-co electron transfer occurs first, followed by rapid reduction of the oxidized P cluster by the Fe protein. The ‘deficit-spending’ electron transfer model was given support by studies on MoFe proteins using amino acid substitutions that were consistent with such a model (Figure 3) [14].
Figure 3.

Deficit spending ET in nitrogenase. Shown is the Fe protein (left) and 1/2 of the MoFe protein (right) with the metal clusters R for the 4Fe-4S cluster, P for the 8Fe-7S cluster, and M for FeMo-cofactor. The first ET event is proposed to be from the P cluster to the M cluster, while the second electron transfer event is from the Fe protein to the oxidized P cluster.
While a reasonable model, the details of how this ‘deficit spending’ process would be achieved remain unknown. To a first approximation, the ‘deficit-spending’ electron transfer model would require that Fe protein docking to the MoFe protein activates electron transfer from the P cluster to the FeMo-cofactor. Given that the Fe protein docking interface is far from the P cluster [25,26], the activation of electron transfer from the P cluster initiated at the protein-protein interface, must be achieved through protein conformational changes that are propagated deep in the MoFe protein. Such changes could activate electron transfer by changing parameters to favor the electron transfer event. Parameters that seem most likely to be changed would be the driving force for the electron transfer (the difference in the midpoint reduction potential Eo′ for the donor and acceptor) or the coupling between the donor and acceptor [30,31]. A model of this kind can be refined by computation to examine possible conformational changes that could be used as ‘levers’ to communicate from the Fe protein-MoFe protein docking interface to control the electron transfer events at the P cluster.
Examination of the environment of the P cluster suggests models to explain the transduction of mechanical energy from protein–protein docking to electrochemical energy to drive electron transfer, through changes in the ligation or environments of the P cluster [32] or FeMo-co.
Half-sites reactivity in nitrogenase
The complexity of the overall energy transduction landscape in nitrogenase is enhanced by a recent unexpected finding showing negative cooperativity between the two symmetric halves of nitrogenase [15••]. Quench-flow studies find during pre-steady state, that the entire nitrogenase complex only shows the hydrolysis of 2 ATP (not 4) and the transfer of one electron (not two) in the initial time after mixing (Figure 4), contrary to the expectation if the two halves of the complex functioned independently. Even though a fully charged Fe protein is already bound on the opposite end of the MoFe protein, only the first side proceeds through the ET and ATP hydrolysis cycle [15••]. The second side is partially ‘held up’ by ‘negative cooperative’ control across the entire complex. The effective outcome of this control is that only after the first side has completed the Fe protein cycle is the second side partially released to proceed through a full Fe protein cycle. It is not clear if this back and forth mechanism continues for many cycles or if the system proceeds stochastically at longer times. Coarse grained modeling, based on a Gaussian network, reveal connection between the Fe protein on one end, through the MoFe protein, to the Fe protein on the other end [15••]. Thus, the beginnings of a mechanism for coupling of events on the two opposite ends of the nitrogenase complex can be envisioned, although why such communication occurs remains unknown. It seems reasonable that the communication across the complex is part of the energy transduction in the system. Resolving the nature and potential advantages of this cross-protein communication will be an important topic for future research, as it may reveal how nitrogenase manipulates free-energy to control electron transfer and to accomplish a challenging multi-electron substrate reduction.
Figure 4.

Negative cooperativity in nitrogenase. The left half of the complex is shown after ET, ATP hydrolysis, and Pi release. The right side is held up with the Fe protein bound, but with ET and ATP hydrolysis suppressed through negative cooperativity.
Conclusions and prospects
Understanding how nitrogenase couples the energy from ATP binding and hydrolysis to catalyze electron transfer and substrate binding/reduction continues to be a significant challenge. Building on decades of foundational work [1,11], recent studies have created the framework for an understanding of the Fe protein cycle. It is now clear that the energy from ATP hydrolysis is used at the end of the cycle [12••], likely to drive the dissociation of the Fe protein from the MoFe protein. Further, it is clear that the events associated with Pi release after ATP hydrolysis are rate limiting for the entire cycle [13••]. Recent studies also support a deficit-spending electron transfer model, wherein the binding of the Fe protein to the MoFe protein stimulates gated electron transfer from the P cluster to FeMo-co, with follow-up electron transfer from the Fe protein to the oxidized P cluster [14]. Significant work remains to develop a molecular-level understanding of how Fe protein binding activates the electron transfer reactions and how ATP hydrolysis and Pi release destabilize the complex allowing spent Fe protein to dissociate. Finally, the observation of negative cooperativity associated with conformational coupling across the nitrogenase complex [15••] opens an important new frontier for furthering our understanding of energy transduction in nitrogenase.
Acknowledgements
This work was supported as part of the Biological Electron Transfer and Catalysis (BETCy) Energy Frontier Research Center (EFRC) funded by the United States Department of Energy (U.S. DOE), Office of Science, Basic Energy Sciences (DE-SC0012518). S.R. was supported by the U.S. DOE, Office of Science, Basic Energy Sciences, Division of Chemical Sciences, Geosciences, and Bio-Sciences (DE-AC05–76RL01830/FWP6647). B.M.H. was supported by the National Institutes of Health (GM111097). J.W.P. and L.C.S. were supported by the National Science Foundation (MCB 1330807).
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Burgess BK, Lowe DJ: Mechanism of molybdenum nitrogenase. Chem Rev 1996, 96:2983–3012. [DOI] [PubMed] [Google Scholar]
- 2.Eady RR: Structure–function relationships of alternative nitrogenases. Chem Rev 1996, 96:3013–3030. [DOI] [PubMed] [Google Scholar]
- 3.Mus F, Alleman AB, Pence N, Seefeldt LC, Peters JW: Exploring the alternatives of biological nitrogen fixation. Metallomics 2018, 10:523–538. [DOI] [PubMed] [Google Scholar]
- 4.Hales BJ: Alternative nitrogenase. Adv Inorg Biochem 1990, 8:165–198. [PubMed] [Google Scholar]
- 5.Joerger RD, Bishop PE: Bacterial alternative nitrogen fixation systems. Crit Rev Microbiol 1988, 16:1–14. [DOI] [PubMed] [Google Scholar]
- 6•.Hoffman BM, Lukoyanov D, Yang Z-Y, Dean DR, Seefeldt LC: Mechanism of nitrogen fixation by nitrogenase: the next stage. Chem Rev 2014, 114: 4041–4062.Review summarizing the suite of studies revealing molecular insights into the mechanism of N2 binding and H2 evolution at the nitrogenase active site.
- 7.Lukoyanov D, Khadka N, Dean DR, Raugei S, Seefeldt LC, Hoffman BM: Photoinduced reductive elimination of H2 from the nitrogenase dihydride (Janus) state involves a FeMo-cofactor-H2 intermediate. Inorg Chem 2017, 56:2233–2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lukoyanov D, Khadka N, Yang Z-Y, Dean DR, Seefeldt LC, Hoffman BM: Reversible photoinduced reductive elimination of H2 from the nitrogenase dihydride state, the E4(4H) Janus intermediate. J Am Chem Soc 2016, 138:1320–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lukoyanov D, Khadka N, Yang Z-Y, Dean DR, Seefeldt LC, Hoffman BM: Reductive elimination of H2 activates nitrogenase to reduce the N≡N triple bond: characterization of the E4(4H) Janus intermediate in wild-type enzyme. J Am Chem Soc 2016, 138:10674–10683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lukoyanov D, Yang Z-Y, Khadka N, Dea n DR, Seefeldt LC, Hoffman BM: Identification of a key catalytic intermediate demonstrates that nitrogenase is activated by the reversible exchange of N2 for H2. J Am Chem Soc 2015, 137:3610–3615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thorneley RNF, Lowe DJ: Kinetics and mechanism of the nitrogenase enzyme In Molybdenum Enzymes. Edited by Spiro TG. Wiley-Interscience Publications; 1985:221–284. [Google Scholar]
- 12.••.Duval S, Danyal K, Shaw S, Lytle AK, Dean DR, Hoffman BM, Antony E, Seefeldt LC: Electron transfer precedes ATP hydrolysis during nitrogenase catalysis. Proc Natl Acad Sci U S A 2013, 110:16414–16419.Using rapid quench flow methods, the authors establish that ATP hydrolysis occurs after the electron transfer events in nitrogenase.
- 13.••.Yang Z-Y, Ledbetter R, Shaw S, Pence N, Tokmina-Lukaszewska M, Eilers B, Guo Q, Pokhrel N, Cash VL, Dean DR et al. : Evidence that the Pi release event is the rate-limiting step in the nitrogenase catalytic cycle. Biochemistry 2016, 55:3625–3635.Using rapid quench techniques, the authors establish that the rate limiting step in the nitrogenase Fe protein cycle is associated with the Pi release step, not the dissociation of the two nitrogenase component proteins as was previously held.
- 14.Danyal K, Dean DR, Hoffman BM, Seefeldt LC: Electron transfer within nitrogenase: evidence for a deficit-spending mechanism. Biochemistry 2011, 50:9255–9263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.••.Danyal K, Shaw S, Page TR, Duval S, Horitani M, Marts AR, Lukoyanov D, Dean DR, Raugei S, Hoffman BM et al. : Negative cooperativity in the nitrogenase Fe protein electron delivery cycle. Proc Natl Acad Sci U S A 2016, 113:E5783–E5791.The authors provide evidence that the two halves of nitrogenase show negative cooperative behavior, with one side suppressing the activity on the other side.
- 16.McNary JE, Burris RH: Energy requirements for nitrogen fixation by cell-free preparations from Clostridium pasteurianum. J Bacteriol 1962, 84:598–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hageman RV, Orme-Johnson WH, Burris RH: Role of magnesium adenosine 5’-triphosphate in the hydrogen evolution reaction catalyzed by nitrogenase from Azotobacter vinelandii. Biochemistry 1980, 19:2333–2342. [DOI] [PubMed] [Google Scholar]
- 18.Mortenson LE: Ferredoxin and ATP, requirements for nitrogen fixation in cell-free extracts of Clostridium pasteurianum. Proc Natl Acad Sci U S A 1964, 52:272–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kurnikov IV, Charnley AK, Beratan DN: From ATP to electron transfer: electrostatics and free-energy transduction in nitrogenase. J Phys Chem B 2001, 105:5359–5367. [Google Scholar]
- 20•.Tezcan FA, Kaiser JT, Howard JB, Rees DC: Structural evidence for asymmetrical nucleotide interactions in nitrogenase. J Am Chem Soc 2015, 137:146–149.Authors use structures of nitrogenase protein complexes to provide insights into the nature of complex formation.
- 21.Owens CP, Katz FEH, Carter CH, Luca MA, Tezcan FA: Evidence for functionally relevant encounter complexes in nitrogenase catalysis. J Am Chem Soc 2015, 137:12704–12712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tezcan FA, Kaiser JT, Mustafi D, Walton MY, Howard JB, Rees DC: Nitrogenase complexes: multiple docking sites for a nucleotide switch protein. Science 2005, 309:1377–1380. [DOI] [PubMed] [Google Scholar]
- 23.Rees DC, Tezcan AF, Haynes CA, Walton MY, Andrade S, Einsle O, Howard JB: Structural basis of biological nitrogen fixation. Philos Trans A Math Phys Eng Sci 2005, 363:971–984. [DOI] [PubMed] [Google Scholar]
- 24.Danyal K, Mayweather D, Dean DR, Seefeldt LC, Hoffman BM: Conformational gating of electron transfer from the nitrogenase Fe protein to MoFe protein. J Am Chem Soc 2010, 132:6894–6895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schindelin H, Kisker C, Schlessman JL, Howard JB, Rees DC: Structure of ADP x AIF4(−)-stabilized nitrogenase complex and its implications for signal transduction. Nature 1997, 387:370–376. [DOI] [PubMed] [Google Scholar]
- 26.Chiu H, Peters JW, Lanzilotta WN, Ryle MJ, Seefeldt LC, Howard JB, Rees DC: MgATP-bound and nucleotide-free structures of a nitrogenase protein complex between the Leu 127 Delta-Fe-protein and the MoFe-protein. Biochemistry 2001, 40:641–650. [DOI] [PubMed] [Google Scholar]
- 27.Chan JM, Christiansen J, Dean DR, Seefeldt LC: Spectroscopic evidence for changes in the redox state of the nitrogenase P-cluster during turnover. Biochemistry 1999, 38:5779–5785. [DOI] [PubMed] [Google Scholar]
- 28.Smith BE, Lowe DJ, Chen GX, O’Donnell MJ, Hawkes TR: Evidence on intramolecular electron transfer in the MoFe protein of nitrogenase from Klebsiella pneumoniae from rapid-freeze electron-paramagnetic-resonance studies of its oxidation by ferricyanide. Biochem J 1983, 209:207–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yoo SJ, Angove HC, Papaefthymiou V, Burgess BK, Münck E: Mössbauer study of the MoFe protein of nitrogenase from Azotobacter vinelandii using selective 57Fe enrichment of the M-centers. J Am Chem Soc 2000, 122:4926–4936. [Google Scholar]
- 30.Migliore A, Polizzi NF, Therien MJ, Beratan DN: Biochemistry and theory of proton-coupled electron transfer. Chem Rev 2014, 114:3381–3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Winkler JR, Gray HB: Electron flow through metalloproteins. Chem Rev 2014, 114:3369–3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peters JW, Stowell MHB, Soltis SM, Finnegan MG, Johnson MK, Rees DC: Redox-dependent structural changes in the nitrogenase P-cluster. Biochemistry 1997, 36:1181–1187. [DOI] [PubMed] [Google Scholar]
- 33.Kim J, Rees DC: Structural models for the metal centers in the nitrogenase molybdenum-iron protein. Science 1992, 257:1677–1682. [DOI] [PubMed] [Google Scholar]
- 34.Sippel D, Einsle O: The structure of vanadium nitrogenase reveals an unusual bridging ligand. Nat Chem Biol 2017,13:956–960. [DOI] [PMC free article] [PubMed] [Google Scholar]