

Abstract
Newborn screening for cystic fibrosis (CF) offers the opportunity for early medical and nutritional intervention that can lead to improved outcomes. Management of the asymptomatic infant diagnosed with CF through newborn screening, prenatal diagnosis, or sibling screening is different from treatment of the symptomatically diagnosed individual. The focus of management is on maintaining health by preventing nutritional and respiratory complications. The CF Foundation convened a committee to develop recommendations based on a systematic review of the evidence and expert opinion. These guidelines encompass monitoring and treatment recommendations for infants diagnosed with CF and are intended to help guide families, primary care providers, and specialty care centers in the care of infants with CF. (J Pediatr 2009;155:S73–93).
Symptomatic diagnosis of cystic fibrosis (CF) is associated with short- and long-term complications including failure to thrive, stunting, wasting, vitamin and mineral deficiencies, recurrent pulmonary infections associated with decreased lung function, and recurrent hospitalizations. Early identification of CF by newborn screening (NBS), prenatal diagnosis, or family history offers the opportunity to delay and potentially prevent many of these complications through early treatment. Although many treatment issues are the same for individuals with CF regardless of when they are diagnosed, some are unique to the population of newborn infants who may not have overt symptoms of the disease before referral to the CF care center. The CF Foundation convened a committee of experts to develop guidelines for care based on currently available evidence for this distinct population of newly diagnosed infants.
Methods
The CF Foundation convened a group of experts to identify issues in the care of infants with CF and commissioned an evidence review from Johns Hopkins University. Details of this evidence review, including methods and results, are provided in the accompanying article.1 Committee members assessed this evidence in developing recommendation statements and, where possible, made evidence-based recommendations. Recommendations were graded using the United States Preventive Health Services Task Force (USPSTF) grading system.2 Recommendations from published guidelines were used if available and appropriate. The committee made consensus recommendations for topics not included in the evidence review, for topics where prior guidelines were available, and for topics for which there was limited or no evidence but the potential net benefit was assessed as at least moderate (Table I).
Table I.
Treatment Recommendations for Infants with Cystic Fibrosis
| Strength of evidence graded using the USPSTF grading system (2): | |||||||
|---|---|---|---|---|---|---|---|
| Certainty of Net Benefit |
Estimate of Net Benefit (Benefit minus Harms) |
||||||
| Substantial | Moderate | Small | Zero/negative | ||||
| High | A | B | C | D | |||
| Moderate | B | B | C | D | |||
| Low | I (insufficient evidence) | ||||||
| Question # | Recommendation | Strength of Evidence | |||||
| Initial Diagnosis: | |||||||
| 1 | The CF Foundation recommends that treatment for infants diagnosed with CF by NBS should be done at an accredited CF care center, with the goal of an initial visit within 24–72 hours of diagnosis (1–3 working days in absence of overt symptoms). | Consensus recommendation | |||||
| Nutritional recommendations: | |||||||
| Pancreatic Function and Pancreatic Enzymes | |||||||
| 2 | For infants with CF under two years of age, the CF Foundation recommends that pancreatic functional status should be measured by fecal elastase or coefficient of fat absorption in all individuals. | Certainty: Low; Benefit: Substantial Consensus recommendation | |||||
| 3 | For infants with CF under two years of age, the CF Foundation recommends that pancreatic enzyme replacement therapy should be started. | Certainty: Low; Benefit: Substantial Consensus recommendation | |||||
| ● In all infants with two CFTR mutations associated with PI. ● In all infants with fecal elastase < 200 μg/g or CFA < 85% (in infants < 6 months of age), or other objective evidence of PI. ● In infants with unequivocal signs or symptoms of malabsorption, while awaiting confirmatory test results. | |||||||
| 4 | For infants with CF under two years of age, the CF Foundation recommends that pancreatic enzyme therapy should not be started in infants with one or two CFTR mutations associated with pancreatic sufficiency unless: | Consensus Recommendation |
|||||
| ● an objective test of pancreatic function indicates fat malabsorption; or ● the infant has unequivocal signs or symptoms of malabsorption, while awaiting confirmatory test results. |
|||||||
| 5 | For infants with CF under two years of age, the CF Foundation recommends that pancreatic enzyme replacement therapy be initiated at a dose of 2,000– 5,000 lipase units at each feeding, adjusted up to a dose of no greater than 2,500 lipase units per kg per feeding with a maximum daily dose of 10,000 lipase units per kg. | Certainty: Low; Benefit: Substantial Consensus Recommendation Recommended in the CF Foundation Consensus Report on Nutrition for Pediatric Patients (35) and the European Consensus on Nutrition in Patients with CF (36) | |||||
| 6 | For infants with CF under two years of age, as for patients of all ages, the CF Foundation recommends that generic, non-proprietary PERT should not be used. | Certainty: Low; Benefit: Moderate: Consensus Recommendation Recommended in the CF Foundation Evidence-based Practice Recommendations for Nutrition (Consensus recommendation) (8) | |||||
| Feedings, Vitamins and Micronutrients | |||||||
| 7 | For infants with CF under two years of age, the CF Foundation recommends human milk as the initial type of feeding. | Certainty: Moderate; Benefit: Substantial Grade: B recommendation | |||||
| 8 | For infants with CF under two years of age, the CF Foundation recommends that if infants are fed formula, standard infant formulas (as opposed to hydrolyzed protein formulas) should be used. | Certainty: Low; Benefit: moderate Consensus recommendation | |||||
| 9 | For infants with CF under two years of age, the CF Foundation recommends that calorie-dense feedings should be used if weight loss or inadequate weight gain is identified. | Certainty: Low; Benefit: Moderate Consensus recommendation | |||||
| 10 | For infants with CF under two years of age, the CF Foundation recommends that positive feedings behaviors should be encouraged, such as by the provision of educational resources. | Certainty: Low; Benefit: Moderate Consensus recommendation | |||||
| 11 | For children aged 1 to 12 years with growth deficits, the CF Foundation recommends that intensive treatment with behavioral intervention in conjunction with nutritional counseling be used to promote weight gain. | Consensus recommendation Recommended in the CF Foundation Evidence-based Practice Recommendations for Nutrition (Grade B) (8) | |||||
| 12 | For infants with CF under two years of age, the CF Foundation recommends that multivitamins designed to provide at least the recommended levels of vitamins A, D, E and K for patients with CF should be prescribed, beginning shortly after diagnosis. | Certainty: Low; Benefit: Moderate Consensus Recommendation Recommended in the CF Foundation Consensus Report on Nutrition for Pediatric Patients (35) | |||||
| 13 | For infants with CF under two years of age, the CF Foundation recommends that blood levels of fat-soluble vitamins should be measured approximately two months after starting vitamin supplementation and annually thereafter; measure more frequently if values are abnormal. | Consensus recommendation | |||||
| 14 | For infants with CF under two years of age, the CF Foundation recommends that a trial of zinc supplementation (1 mg elemental zinc/kg/day in divided doses for six months) may be given to some infants who are not adequately growing despite adequate caloric intake and pancreatic enzyme replacement therapy. | Certainty: Low; Benefit: Moderate Consensus Recommendation Recommended in the CF Foundation Consensus Report on Nutrition for Pediatric Patients (35) | |||||
| 15 | For infants with CF under two years of age, the CF Foundation recommends supplementation with 1/8 teaspoon table salt per day starting at diagnosis, increasing to 1⁄4 teaspoon of table salt per day at 6 months of age. | Certainty: Low; Benefit: Moderate Consensus recommendation | |||||
| 16 | Patients aged 6 months to 2 years whose community water supply contains less than 0.3 ppm should be supplemented with fluoride 0.25 mg/dl. | Consensus recommendation Recommended in the Center for Disease Control and Prevention Guidelines (72) | |||||
| 17 | For infants with CF under two years of age, the CF Foundation concludes that there is insufficient evidence to recommend for or against supplementation with linoleic acid. | Certainty: Low; Benefit: Small Grade: I recommendation | |||||
| 18 | For infants with CF under two years of age, the CF Foundation concludes that there is insufficient evidence to recommend for or against supplementation with docosahexaenoic acid. | Certainty: Low; Benefit: Unknown Grade: I recommendation | |||||
| Pulmonary Recommendations: | |||||||
| 19 | For infants with CF under two years of age, the CF Foundation recommends that a smoke-free environment be provided and that all caregivers are informed that cigarette smoke exposure harms children with CF. | Consensus recommendation | |||||
| Airway Clearance: | |||||||
| 20 | For infants with CF under two years of age, the CF Foundation recommends that airway clearance therapy be initiated in the first few months of life. | Certainty: Low; Benefit: Moderate Consensus recommendation | |||||
| 21 | For infants with CF under two years of age, the CF Foundation recommends use of albuterol before percussion and postural drainage. | Certainty: Low; Benefit: Moderate Consensus recommendation | |||||
| 22 | For infants with CF under two years of age, the CF Foundation recommends that the head-down position should not be used for percussion and postural drainage. | Certainty: Low; Benefit: Moderate Consensus recommendation | |||||
| Infection Control, Surveillance and Treatment: | |||||||
| 23 | For infants with CF under two years of age, the CF Foundation recommends that newly diagnosed patients should be separated from other patients cared for in CF clinics until adequate infection control education has been provided to and is understood by the caregivers. | Certainty: Low; Benefit: Moderate Consensus recommendation | |||||
| 24 | Infection control measures should be implemented in compliance with CF Foundation recommendations to minimize transmission of bacterial infections to infants. | Consensus Recommendation Recommended in the CF Foundation Consensus Conference on Infection Control (95) | |||||
| 25 | Annual influenza vaccination is recommended for infants with CF > 6 months of age, all household members, and all healthcare providers caring for these infants. Household contacts and out-of-home caregivers of children with CF < 6 months of age also should receive annual influenza vaccine. | Consensus Recommendation. Recommended in American Academy of Pediatrics Guidelines(97) and CF Foundation Consensus Conference on Infection Control (95) | |||||
| 26 | For infants with CF under two years of age, the CF Foundation recommends that use of palivizumab be considered for prophylaxis of respiratory syncytial virus. | Certainty: Low; Benefit: Moderate Consensus recommendation | |||||
| 27 | For infants with CF under two years of age, the CF Foundation recommends that oropharyngeal cultures should be performed at least quarterly. | Certainty: Low; Benefit: Moderate Consensus recommendation | |||||
| 28 | For infants with CF under two years of age, the CF Foundation recommends that bronchoscopy and bronchoalveolar lavage be considered in infants with symptoms or signs of lung disease, particularly those who fail to respond to appropriate intervention. | Consensus recommendation | |||||
| 29 | For infants with CF under two years of age, the CF Foundation recommends against the prophylactic use of oral antistaphylococcal antibiotics in asymptomatic infants. | Certainty: Low; Benefit: Zero-negative Recommended in the CF Foundation Pulmonary Guidelines on Chronic Medications (88) | |||||
| 30 | For infants with CF under two years of age, the CF Foundation concludes that there is insufficient evidence to recommend for or against active attempts to eradicate Staphylococcus aureus in asymptomatic infants. | Certainty: Low; Benefit: Unknown Grade: I recommendation | |||||
| 31 | For infants with CF under two years of age, the CF Foundation concludes that there is insufficient evidence to recommend for or against active attempts to eradicate methicillin-resistant Staphylococcus aureus (MRSA) in asymptomatic infants. | Certainty: Low; Benefit: Unknown Grade: I recommendation | |||||
| 32 | For infants with CF under two years of age, the CF Foundation recommends against the use of chronic antibiotics for prophylaxis to prevent Pseudomonas aeruginosa. | Certainty: Low; Benefit: Moderate Consensus recommendation | |||||
| 33 | For infants with CF under two years of age, the CF Foundation recommends that new acquisition of Pseudomonas eruginosa, defined as initial acquisition or new acquisition after ‘successful’ eradication therapy, should be treated with antipseudomonal antibiotics and increased airway clearance, regardless of the presence or absence of symptoms. | Certainty: Low; Benefit: Moderate Consensus recommendation | |||||
| 34 | For infants with CF under two years of age, the CF Foundation recommends that infants who remain persistently colonized with Pseudomonas. aeruginosa after two attempts at eradication be treated chronically with alternate month tobramycin solution for inhalation. | Certainty: Low; Benefit: Moderate Consensus recommendation | |||||
| Diagnostic Testing: | |||||||
| 35 | For infants with CF under two years of age, the CF Foundation concludes that there is insufficient evidence to recommend for or against use of pulse oximetry routinely as an adjunctive tool to detect lung disease. | Certainty: Low; Benefit: Small Grade: I recommendation | |||||
| 36 | For infants with CF under two years of age, the CF Foundation recommends that pulse oximetry measurements be obtained in the infant with CF with acute respiratory symptoms. | Certainty: Low; Benefit: Substantial Consensus recommendation | |||||
| 37 | For infants with CF under two years of age, the CF Foundation recommends that a baseline chest x-ray should be obtained within the first 3 – 6 months and once again within the first two years of life. | Certainty: Low; Benefit: Moderate Consensus recommendation | |||||
| 38 | For infants with CF under two years of age, the CF Foundation recommends against the use of chest CT scans for routine surveillance. | Certainty: Low; Benefit: Zero-negative Consensus recommendation | |||||
| 39 | For infants with CF under two years of age, the CF Foundation recommends that chest CT scans be considered in infants with symptoms or signs of lung disease who fail to respond to appropriate interventions. | Consensus recommendation | |||||
| 40 | For infants with CF under two years of age, the CF Foundation recommends that infant PFTs be considered as an adjunctive tool to monitor respiratory status. | Certainty: Moderate; Benefit: Small Grade: C recommendation | |||||
| Chronic Pulmonary Therapies: | |||||||
| 41 | For infants with CF under two years of age, the CF Foundation recommends that dornase alfa (recombinant human DNase) may be used in symptomatic infants. | Certainty: Low; Benefit: Moderate Consensus recommendation | |||||
| 42 | For infants with CF under two years of age, the CF Foundation recommends that 7% hypertonic saline may be used in symptomatic infants. | Certainty: Low; Benefit: Moderate Consensus recommendation | |||||
| 43 | For infants with CF under two years of age, the CF Foundation concludes that there is insufficient evidence to recommend for or against the routine use of chronic azithromycin in patients colonized with Pseudomonas. | Certainty: Low; Benefit: Unknown Grade: I recommendation | |||||
| 44 | For infants with CF under the age of two years without airway reactivity or asthma, the CF Foundation does not recommend use of inhaled corticosteroids to improve lung function or reduce exacerbations. | Certainty: Low; Benefit: Zero-negative Consensus recommendation | |||||
A draft of the guidelines was posted on a secure web site for comment from CF Center care teams (physicians and ancillary care providers) and was revised as appropriate. As per the CF Foundation guidelines process, these guidelines will be assessed within 3 years to determine if revisions are necessary.
Guidelines for the Care of Newly Diagnosed Infants with CF
These guidelines are intended to be used by families, primary care providers, and care centers. Even though a framework has been developed to standardize optimal care, it is expected that care will be individualized to the needs of patients and families. Although the guidelines were developed in response to the expansion of NBS programs in the United States, they also apply to infants diagnosed prenatally or due to a family history. Infants diagnosed with CF due to meconium ileus at birth or due to a symptomatic presentation should be treated in a similar manner once their acute medical needs are addressed. Infants diagnosed with CF through NBS are often labeled ‘‘pre-symptomatic,’’ ‘‘asymptomatic,’’ or ‘‘subclinically affected.’’ However, many have clinical manifestations by 1 month of age.3 Most infants diagnosed through CF NBS are at risk for some complication of the disease including hypoelectrolytemia, pancreatic insufficiency (PI), and lung disease.4
Diagnostic and treatment recommendations are listed at the end of each section and are summarized in Table I.
Initial Visit
Infants diagnosed with CF through NBS often appear to be totally healthy to the parents, and the diagnosis probably will be unexpected. Thus, the psychosocial impact of the diagnosis of CF on the family must be carefully addressed at the initial visit. Infants diagnosed with CF through NBS should be treated at an accredited CF care center, with the goal of an initial visit within 24 to 72 hours of diagnosis (1 to 3 working days in the absence of overt symptoms). At the initial visit to the care center, there should be adequate time for the family to receive comprehensive education regarding CF care. The duration of the visit may need to be as long as 2 hours of direct face-to-face time between the care team and family; however, the length of the visit and the amount of the information addressed needs to be customized to each family. The most important issues to be discussed (based on expert opinion) are listed in Table II.
Table II.
Medical Issues to Discuss with the Family at the Initial Visit at the CF Center
| • Assess emotional and educational status of the family |
| • Explain how we know the infant has CF |
| • Explain basic genetic concepts |
| • Convey the most difficult facts about the disease: |
| ○ Currently CF is a life-limiting disorder |
| ○ Most males are infertile |
| ○ CF is a chronic condition requiring ongoing daily care |
| • Provide a general description of CF symptoms and what causes them |
| • Introduce the care team concept |
| ○ Parents and primary care provider are part of care team |
| ○ CF Foundation as part of the team |
| • Emphasize the need to get CF information from reliable sources: |
| ○ CF Care team |
| ○ CF Foundation’s website (www.cff.org) |
| ○ Explain that incorrect and outdated information about CF is common (e.g. from friends and family, in books, and on the web) |
| • Give the family hope |
| ○ Life expectancy has been steadily increasing |
| ○ Many new treatments are actively being studied |
| ■ These are likely to be of direct benefit to your child in his or her life |
| ■ This is why prevention is especially important now |
| ■ This is why we need to follow this child in our clinic; consider giving the family a copy of the Monitoring and Care Recommendations (see Table III) |
| • Describe how to contact the CF care center with questions or concerns; schedule the next visit before the family leaves |
Disbelief, anger, or anxiety about the new diagnosis may be present and retention of information may be a challenge.5 Families should be encouraged to invite extended family members and other support persons to attend the initial visit to assist in listening to and remembering information, as well as to provide emotional support. Giving basic information in the clearest of terms and conveying the information in a sensitive, empathetic, and positive manner are key components of the visit.6 Information should be provided in varied formats (eg, verbal, written and audiovisual) and be reviewed at subsequent visits. Introduction of other CF clinicians, namely the nurse, registered dietitian, respiratory therapist, and social worker, should occur at 1 of the first 2 visits. This allows for the key components of nutrition and airway clearance to be taught and reinforced, as well as for the development of relationships with team members. A genetic counselor should meet with the family at the initial visit or at another time in the first 2 months after diagnosis to discuss in greater depth how mutations in the CF transmembrane conductance regulator (CFTR) gene cause CF and the implications for other family members. The pivotal role that both the parents and primary care provider (PCP) play as part of the CF team should be emphasized at the early visits. The positive outlook for newly diagnosed infants should be reinforced and a sense of hope instilled. Last, information describing how to contact the CF center if questions or concerns arise before the next visit should be shared with the family.
Recommendation #1 is in Table I.
Coordination of Care with Primary Care Providers
A collaborative care model should be the goal, with regular and open tri-lateral communication among the family, the PCP and the CF Center. Families will be making numerous visits to their PCP and CF Center during the first 2 years of life: standard pediatric visits are at age 1 to 2 weeks and at 2, 4, 6, 9, and 12 months in the first year of life; CF Center visits should be once monthly during the first 6 months, and every 1 to 2 months in the second 6 months of life (Table III). These visits should complement each other, in that the expertise provided at primary and tertiary care sites differ. A written form to delineate appropriate contacts for parents as problems and questions arise can facilitate efficient communication and care (Figure 1). Insurers need to recognize that despite frequent visits early in life, the cost of care of infants diagnosed with CF through NBS is lower than for those diagnosed following the onset of symptoms.7 Communication between the PCP and CF Center is critical to ensure that parents do not get conflicting messages, especially since many CF care goals are different than those of standard pediatric care (eg, an emphasis on the need for the child with CF to be slightly chubby versus concerns about obesity in the general population). Care providers should convey to each other concerns about issues such as but not limited to poor weight gain, cough or wheezing, change of medications or treatments, lack of adherence to the prescribed regimen, difficulties in administering prescribed medicines or treatments, worrisome infections in other family members, and immunization status. In addition, critical events such as admission to the hospital or change in medications should be documented in the medical record and shared. The most important issues for PCPs to know about CF at present (based on expert opinion) are outlined in Table IV.
Table III.
Routine monitoring and care recommendations for the infant diagnosed with CF
| AGE AT VISIT → | Day of Sweat test |
24–48 hrsof dx |
1 wk later or age 1 mo |
2 mo* | 3 mo* | 4 mo* | 5 mo* | 6 mo | 8 mo | 10 mo |
1 year |
Every 2–3 months in the second year of ife |
24 mo | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ↓INTERVENTIONS | Note: Some Centers may plan additional routine visits at 7, 9 and 11 months |
||||||||||||||
| DATE DONE→ | |||||||||||||||
| CARE ISSUES | |||||||||||||||
| Discuss diagnosis | Either visit | C | C | C | |||||||||||
| Encourage human milk feeding | Either visit | ||||||||||||||
| Start PERT1 | Either visit | C | C | C | C | C | C | C | C | C | C | C | |||
| Start salt supplementation | 1/8. tsp salt | ¼ tsp | |||||||||||||
| Start vitamins designed for CF patients | Either visit | ||||||||||||||
| History and physical with weight, length, OFC | Either visit | ||||||||||||||
| Teach / initiate P&PD | |||||||||||||||
| Assess weight gain, caloric intake and PERT dose | |||||||||||||||
| DIAGNOSTIC TESTING | |||||||||||||||
| Sweat test | C | All 10 sibs | |||||||||||||
| Pancreatic functional status testing4 | At one of these visits | C | C | C | C | C | c | C | C | C | C | C | |||
| Respiratory culture 5 | |||||||||||||||
| Chest x-ray | At one of these visits | ||||||||||||||
| Vitamin levels A, D, E 6 | At one of these visits | ||||||||||||||
| Serum electrolytes BUN7, creatinine | |||||||||||||||
| Complete blood count | |||||||||||||||
| AST/ALT/GGT/ bili , abumin, ALP 7 | |||||||||||||||
| EDUCATION | |||||||||||||||
| Infection Control | |||||||||||||||
| Fill out “Who to call - where to Go” sheet | Either visit | ||||||||||||||
| CFF Patient Registry consent | Either visit | ||||||||||||||
| Discuss clinical research | C | C | C | C | C | ||||||||||
| Feeding Behavior Anticipatory Guidance | Either visit | Either visit | At 2 of these visits | ||||||||||||
| Referrals to community food resources | C | C | C | C | C | C | C | C | C | C | C | C | |||
| Review ACT technique | |||||||||||||||
| Tobacco smoke exposure avoidance education | |||||||||||||||
| Genetic counseling | At one of these visits | C | C | ||||||||||||
CF, cystic fibrosis; PERT, pancreatic enzyme replacement therapy; PCP, primary care provider; X, do at this visit; C, consider doing at this visit.
In some circumstances, care may be shared with PCP; infants growing poorly may need to be seen more often; some stable infants can be seen every 6 weeks.
Start PERT if patient has symptoms, fecal elastase <200 μg/g, coefficient of fat absorption <85%, or 2 CFTR mutations associated with PI.
Many centers include oximetry; pulse oximetry should be performed in infants with acute respiratory symptoms.
Routine immunizations should be given by the primary care provider; Palivizumab may be given in appropriate season (see text); influenza vaccine should be given in the appropriate season after 6 months of age.
Recheck a measure of pancreatic phenotype, such as fecal elastase, if PS pts have weight loss or GI symptoms.
Respiratory cultures may be performed more frequently if patient has symptoms.
Vitamin levels are optimally checked 1 to 2 months after starting supplements; ensure that fluoride intake is adequate or is supplemented.
BUN, blood urea nitrogen; AST, alanine aminotransferase; ALT, aspartate aminotransferase; GGT, gamma-glutamyl transferase; bili, bilirubin; ALP, alkaline phosphatase.
Figure 1.

For parents: Who to call and where to go to care for your infant with CF. Infants with CF need basic medical care as well as CF care. Your primary care provider will take care of some of your child’s health needs; the CF team will provide care specifically related to CF. Sometimes you will make a phone call with questions and other times you will need to bring your baby in to be seen. After you and your CF care team fill out this checklist, they will share it with your primary care provider so that everyone knows the care plan.
Table IV.
CF-Specific Issues for Primary Care Providers
| • Infants with CF need supplemental salt and older children with CF should be taught to salt their food |
| • The goal for infants with CF is to be at or above the 50th percentile weight-for-length (‘‘slightly chubby’’) |
| ○ We encourage a high-fat diet, including use of whole milk |
| ○ Inform the CF Center if there is any lack of weight gain or weight loss |
| • Early symptoms can be subtle; call the CF Center for any pulmonary or GI symptoms such as: |
| ○ Cough or wheezing |
| ■ Antibiotics are used with a lower threshold in patients with CF and for a longer period of time than in other children |
| ○ Loose stools or abdominal pain |
| • Life expectancy is steadily increasing, but this depends on daily preventive care |
| ○ Convey hope: we expect these infants to lead full adult lives |
| ○ Inquire about adherence to the prescribed CF regimen at each primary care visit |
| ○ Reinforce limit-setting (especially important in children who require daily treatments but may be perceived as vulnerable) |
| • The diagnosis creates psychosocial challenges |
| ○ We strongly discourage children with CF from person-to-person interactions with others with CF to prevent cross-infection |
| ○ CF places the entire family under stress |
| ■ Ask how unaffected siblings feel about CF |
| ■ Share insights on family functioning with CF Center team |
Beyond the Initial Diagnosis
After diagnosis, the goals are to maintain normal growth and development and to delay the onset of pulmonary disease. Recommendations for monitoring and treating infants diagnosed with CF through NBS were developed with these goals in mind (Tables I and III).
Rationale for Early Nutritional Treatment.
The goal of nutritional treatment of infants diagnosed with CF in the newborn period is normal growth. Special attention to growth and nutrition early in the first year of life is essential because it is a time of extraordinary metabolic need; healthy infants double their birth weight by 4 months of age and triple it by 1 year. The CF Foundation recommends that children reach a weight-for-length status of the 50th percentile by 2 years of age,8 though achieving this goal earlier in infancy is likely to be beneficial. Data from the CF Foundation Patient Registry indicate that higher body mass index (BMI) percentiles at 2 years of age are strongly associated with better pulmonary function later in childhood (Figure 2). Epidemiologic data suggest that improving the nutritional status of young children is associated with better pulmonary outcomes.9 Infants diagnosed by CF NBS may appear healthy but can be malnourished.3,10,11 Infants who do not achieve expected gains in weight and length, or those who are less than the 25th percentile on the National Center for Health Statistics/Centers for Disease Control 2000 growth chart for weight-for-length, are at nutritional risk and should be followed very closely, with appropriate evaluations and interventions made (Figure 3).
Figure 2.
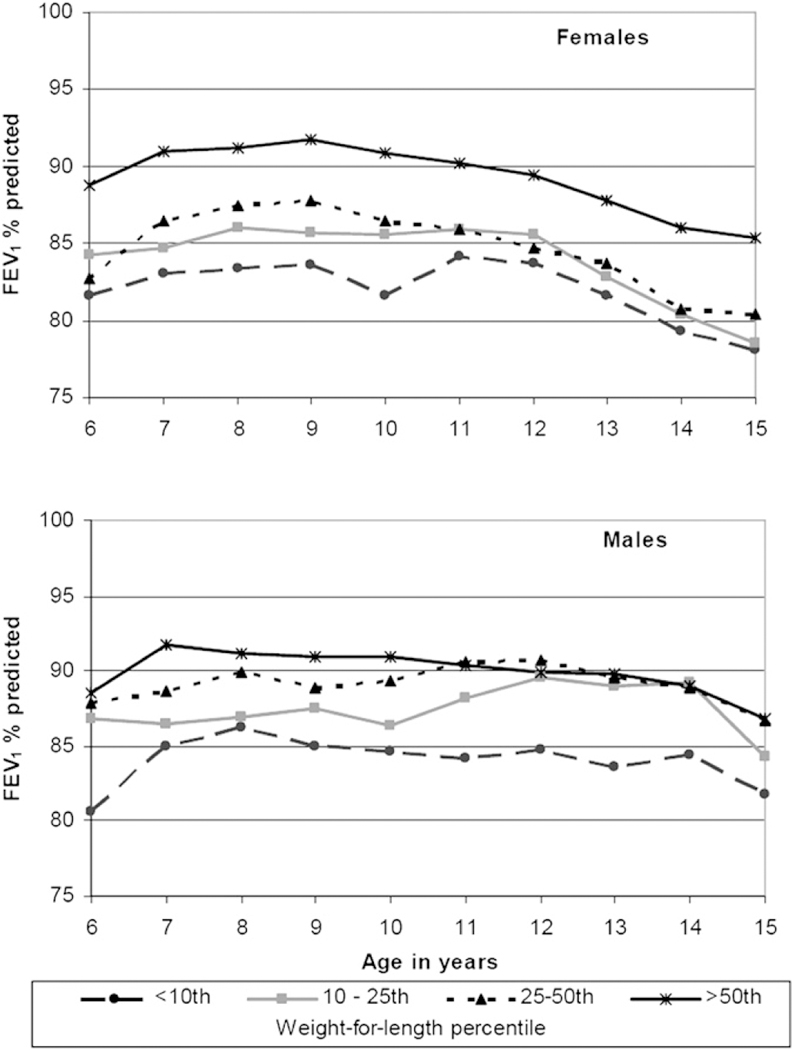
Analysis of CF Foundation Patient Registry data from 2006: Forced expiratory volume in 1 second (FEV1) in childhood in pancreatic insufficient patients with CF, stratified by their weight-for-length percentile at age 2 years. Reprinted with permission from Stallings et al,8 Evidence-based practice recommendations for nutrition-related management of children and adults with cystic fibrosis and pancreatic insufficiency: results of a systematic review, Journal of the American Dietetic Association, May 2008. 2008 American Dietetic Association. Published by Elsevier Inc.
Figure 3.

Evaluation of infants with weight loss or inadequate weight gain (based on consensus opinion).
Diagnosis and Treatment of Pancreatic Insufficiency (PI).
Pancreatic dysfunction, seen in most individuals with CF, evolves over the first year of life. Patients with pancreatic sufficiency (PS) have a significantly longer median life-span than patients with PI.12 PI is present at birth in 60% of infants diagnosed with CF through NBS and approximately 90% of infants have PI at 1 year of age.13
We found 9 studies that addressed the use of several different objective measures of pancreatic function phenotype in infants.14–22 No test was demonstrated to be better than another, though some are not useful in infants or are no longer commercially available. One study compared fecal fat content, expressed as coefficient of fat absorption (CFA), and found it to be as useful as the reference standard pancreatic stimulation test in infants diagnosed with CF through NBS.15 Of note, all infants have physiologic fat malabsorption early in life; a CFA ≥ 85% is normal for infants under 6 months of age, whereas the normal value for patients above this age is ≥ 93%. The fecal elastase (FE) assay is highly predictive of PI23; healthy infants, even if premature, demonstrate FE levels within the normal adult range by two weeks of age.24,25 A study showed that a group of infants diagnosed with CF through NBS, who had 2 CFTR mutations associated with PI, demonstrated low FE and measurable steatorrhea at 1 year of age, though values fluctuated in some patients before that time.14
Recommendation # 2 is in Table I.
Pancreatic Enzyme Replacement Therapy (PERT).
There is a strong association of pancreatic phenotype and genotype.26 PERT should be started if the patient is known to have 2 CFTR mutations associated with PI or objective evidence of PI. PERT should not be started in infants with a CFTR mutation known to be associated with the PS phenotype (Table V), unless there are unequivocal signs or symptoms of malabsorption; an objective measure of pancreatic function should be obtained to corroborate the clinical impression. Pancreatic insufficiency is not present in all infants at the time of diagnosis through NBS but evolves during infancy.13,14 Fecal elastase is easy to repeat and may need to be checked more than once during the first year of life, especially if infants develop gastrointestinal symptoms or have inadequate weight gain.
Table V.
CFTR mutations consistently associated with pancreatic insufficient and pancreatic sufficient Phenotypes
| Usually PI-associated CFTR mutations |
Usually PS-associated CFTR mutations |
|
|---|---|---|
| F508del | Y122X | R117H |
| G542X | 1898+5G>T | R347P† |
| G551D | 3120+1G>A | 3849+10kbC>T |
| N1303K | E822X | A455E |
| W1282X | 2751+2T>A | R334W† |
| R553X | 296+1G>C | G178R |
| 621+1G>T | R1070Q-S466X* | R352Q |
| 1717–1G>A | R1158X | R117C |
| R1162X | W496X | 3272–26A>G |
| I507del | 2789+5G>A | 711+3A>G |
| 394del1TT | 2184insA | D110H |
| G85E* | 1811+1.6kbA>G | D565G |
| R560T | 1898+1G>A | G576A |
| 1078delT | 2143delT | D1152H |
| 3659delC | 1811+1.6kbA>G | L206W |
| 1898+1G>T | R1066C | V232D |
| 711+1G>T | Q890X | D1270N |
| 2183AA>G | 2869insG | |
| 3905insT | K710X | |
| S549N | 1609delCA | |
| 2184delA | ||
PI, pancreatic insufficient; PS, pancreatic sufficient.
Reprinted with permission from Castellani et al,26 Consensus on the use and interpretation of cystic fibrosis mutation analysis in clinical practice, Journal of Cystic Fibrosis, May 2008.
© 2008 Eurpopean Cystic Fibrosis Society. Published by Elsevier B.V.
May also be associated with PS.
May also be associated with PI.
Patients with laboratory evidence of PI should be started on PERT even in the absence of signs or symptoms of fat malabsorption. We did not find any studies that assessed the benefit of starting PERT immediately versus waiting for the appearance of symptoms; however, the dangers of nutritional deficiency and the negative long-term behavioral consequences of the potential association of feedings with abdominal pain argue strongly in favor of proactive treatment. We found 2 studies that confirmed that PERT reduces fat malabsorption in infants (one used CFA27 and one used C13 breath tests28 as the outcome measure; both used a before-after design). PERT should be given with breast milk and formulas, including elemental and medium chain triglyceride (MCT)-containing formulas and all foods.27,29
To date, no studies have been performed in infants to determine the optimal dose of PERT.8 Until reliable data are available, dosing based on the historic recommendations must suffice: start PERT at a dose of 2000 to 5000 lipase units for each feeding (usually described as 120 mL; although newborn infants initially may consume less than 120 mL per feeding, PERT should still be initiated). As the infant grows and the volume of intake increases, adjust the dose to no greater than 2500 lipase units per kilogram per feeding with a maximum daily dose of 10 000 lipase units per kg per day.30 Review of CF Foundation Patient Registry data shows that average enzyme dose may be inappropriately at the low end of the weight-based dosing range early in life (Figure 4). Enzyme dose and rate of weight gain in relation to caloric intake should be evaluated at each visit since the per kilogram dose of PERT and the volume of intake will rapidly increase in the first few months of life. Doses should not be increased beyond the upper limit of the recommended range because children are at risk of developing fibrosing colonopathy31–33 and infants may be at even higher risk because of their more permeable gut. Families should be instructed not to adjust the PERT dose on their own, but always in consultation with the CF care center team.
Figure 4.
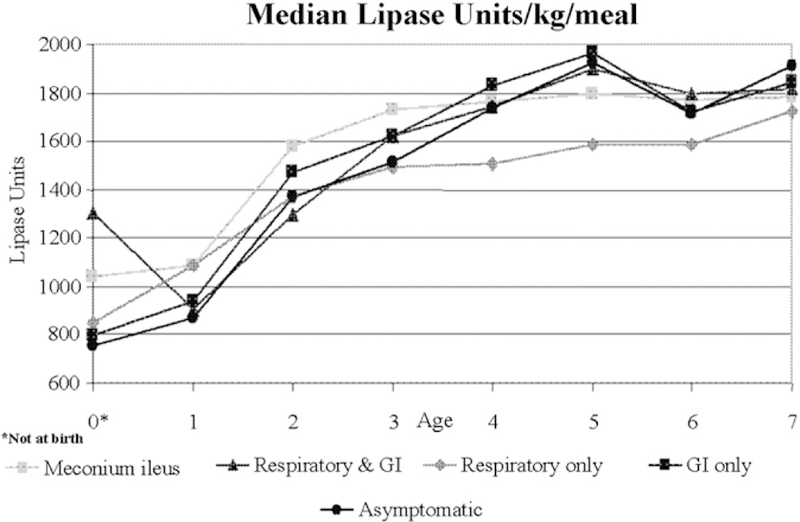
Enzyme dose by age, CF Foundation Patient Registry, 2005.
The use of generic or proprietary PERT was also considered. We identified one case report that described a therapeutic failure in an infant when generic PERT was used.34 As per the recent CF Foundation Evidence-Based Practice Recommendations for Nutrition,8 generic PERT should not be used. The care team should remain vigilant that a generic product has not been substituted for a prescribed proprietary product.
Recommendations # 3 through 6 are in Table I.
Types of Feedings.
The basic principles of infant feeding for healthy term babies apply to feeding infants with CF. Well-designed clinical trials are not available that define which type of milk feeding (human or formula) or what type of diet should be recommended for the infant and toddler with CF.
The advantages of human milk feeding and potentially beneficial constituents of human milk for healthy infants are widely recognized. Nutritional guidelines published by the CF Foundation,35 the European Consensus on Nutrition in Patients with CF,36 and the United Kingdom CF Trust37 recommend human milk feeding. A prospective cohort study found no difference in weight or length between exclusively human milk–fed infants with CF and those who were exclusively formula fed.38 Three retrospective studies provided evidence for the benefit of human milk compared with formula feeding in infants with CF.13,39,40 One identified lower digestible protein intake in human milk–fed infants, but there was no difference in anthropometric measures, blood urea nitrogen, or serum albumin when compared with formula-fed infants at 7 weeks.13 Another study found that human milk–fed infants had higher weight and height z-scores than those who were formula fed.40 Two cohort studies (one prospective and one retrospective) suggested that human milk provides pulmonary or other medical benefits to patients with CF.38,41
There is limited evidence to address whether formula-fed babies with CF and PI should consume standard infant formula or a hydrolyzed protein formula containing MCT oil. In a recent, randomized prospective study, standard formula was compared against a hydrolyzed formula to assess the nutritional benefits, absorption, and tolerance of feedings.42 This study concluded that use of a hydrolyzed formula for infants with CF and PI did not provide additional nutritional or other health benefits when compared with standard infant formula. Conflicting results were reached in 2 older studies. One nonrandomized study demonstrated significantly improved anthropometric measures in infants fed the hydrolysate,43 and another showed a significant improvement in fat and nitrogen absorption when fed a semi-elemental diet without PERT.44 We conclude that if the infant is fed formula, standard infant formulas (as opposed to hydrolyzed protein formulas) should be used.
Infancy is a time of high metabolic demand. Patients with CF are encouraged to eat a high-calorie diet throughout their lives. No data are available to guide us in the proactive use of fortified human milk, high-calorie formulas, or high-calorie complementary foods (solids) in infants and toddlers who are growing well. Nevertheless, fortified human milk, calorie-dense formulas, or complementary foods should be used if weight loss or inadequate weight gain is identified (Figure 3). For children with growth deficits, the CF Foundation recommends the use of enteral supplements in addition to usual dietary intake to improve the rate of weight gain8; the concept of enteral feedings should be introduced early as a component of CF care (Figure 3). Complementary foods should be added to the diet as recommended by the American Academy of Pediatrics (AAP).45 The CF Center dietitian should guide parents toward foods that will enhance weight gain. Meat, a good source of iron and zinc, may be recommended as a first food for infants consuming human milk.45
The CF care team should ensure that families in need have access to adequate food for breast-feeding mothers, formula for infants, and food for toddlers. Referrals should be made to appropriate community-based resources such as The Special Supplemental Nutrition Program for Women, Infants, and Children (WIC) and Food Stamp programs as needed.
Recommendations # 7 through 9 are in Table I.
Behavioral Feeding Issues.
One of the benefits to newborn screening is the ability to start working with families during infancy to prevent the behavioral feeding problems commonly reported for preschool46–49 and school-age children50,51 with CF. As is true for all children, the most beneficial breast or bottle feeding environment is one that has limited noise, light, and other distractions. It is also important for infants to be held during all feedings to allow for adequate supervision and opportunity for crucial parent-child bonding. Poor early bonding may be associated with decreased health status later in childhood.52
Between the ages of 4 and 6 months, solid foods are gradually introduced and self-feeding skills subsequently develop. One challenge to introducing new foods into a child’s diet is neophobia, or reluctance to try new foods (eg, spitting food out, turning head, and becoming upset). Although neophobia is a typical developmental process for all children, parents of infants and toddlers with CF report higher rates of children’s unwillingness to try new foods, having a poor appetite, and preferring to drink rather than eat.49 To manage neophobia, new foods should be presented up to 10 to 12 times before determining that a child does not like them.53 It is common, however, for parents of young children to present a food only 3 to 5 times,54 which is not adequate for children to eventually accept it. Because parental attention is a particularly motivating factor during infancy and toddlerhood, parents should be encouraged to consistently and specifically compliment appropriate eating behaviors (eg, trying a new food, taking a bite, and eating independently), and to pay minimal attention to behavior not compatible with eating (ie, refusing food). The CF care team should provide written educational materials to families about positive feeding interactions (Table III).
Infants and toddlers with CF have longer mealtimes than their peers without CF, yet still do not meet the CF Foundation’s dietary recommendations for increased energy intake.49 As the duration of mealtimes increases, difficult behaviors also occur more frequently.55 One strategy to address behavior problems is to limit mealtimes to 15 minutes for toddlers and use snack times as mini-meals. One before-after study of 4 children with CF ages 10 to 42 months showed improvements in caloric intake after parents used a structured behavioral program.56 Monitoring food and energy intake is key to assisting parents in meeting the CF Foundation’s dietary recommendations. In fact, when parents were provided with information about the Dietary Reference Intake (DRI) and the above behavioral strategies, their toddlers with CF aged 18 to 48 months attained the CF Foundation’s energy recommendation of 120% of the DRI and had greater than expected weight and height gains up to 12 months after treatment.57–59
Recommendations # 10 and 11 are in Table I.
Micronutrients
Fat-Soluble Vitamins.
Symptomatic vitamin A60 and vitamin E deficiency61 has been reported in patients with CF. Many newly diagnosed infants have low levels of one or more fat-soluble vitamins.10,62 Some groups of infants are at higher risk of having low vitamin levels, such as those who are homozygous for F508del (also known as ΔF508) and those with hypoalbuminemia or elevated alkaline phosphatase at the time of diagnosis.10 Low levels of vitamin E are associated with decreased cognitive performance.63 Because of the prevalence of fat-soluble vitamin deficiency and the resulting clinical impact, blood levels of these vitamins should be measured regularly (Table III) and all infants with CF should receive standard, age-appropriate non–fat-soluble vitamins and vitamins A, D, E, and K as recommended in the CF Foundation Consensus Report on Nutrition for Pediatric Patients (Table VI).35
Table VI.
Fat-Soluble Vitamin and Zinc Content of Multivitamins1
| AGE (volume-based dose) |
CFF Consensus Report2 |
SourceCF Pediatric Drops |
AquADEKs Pediatric Liquid |
Vitamax Pediatric Drops |
Enfamil Poly-Vi-Sol Drops |
|---|---|---|---|---|---|
| Vitamin A (IU) (Retinol and Beta-Carotene) | |||||
| 0 – 12 months (1 ml) | 1,500 | 4627 75% as beta-carotene | 5,751 87% as beta-carotene | 3,170 100% retinol palmitate | 1,500 100% retinol palmitate |
| 1 – 3 years (2 ml) | 5,000 | 9254 75% as beta-carotene | 11,502 87% as beta-carotene | 6,340 100% retinol palmitate | 3,000 100% retinol palmitate |
| Vitamin E (IU) | |||||
| 0 – 12 months (1 ml) | 40 – 50 | 50 | 503 | 50 | 5 |
| 1 – 3 years (2 ml) | 80 – 150 | 100 | 1003 | 100 | 10 |
| Vitamin D (IU) | |||||
| 0 – 12 months (1 ml) | 400 | 500 | 400 | 400 | 400 |
| 1 – 3 years (2 ml) | 400 – 800 | 1000 | 800 | 800 | 800 |
| Vitamin K (mcg) | |||||
| 0 – 12 months (1 ml) | 300 to 500 | 400 | 400 | 300 | 0 |
| 1 – 3 years (2 ml) | 300 to 500 | 800 | 800 | 600 | 0 |
| Zinc (mg) | |||||
| 0 – 12 months (1 ml) | - | 5 | 5 | 7.5 | 0 |
| 1 – 3 years (2 ml) | - | 10 | 10 | 15 | 0 |
Table reproduced with permission of SourceCF Inc., a subsidiary of Eurand Pharmaceuticals, Inc.
The content of this Table is as of January 2009. Products also contain a full range of water-soluble vitamins. For a copy of the full table go to: www.SourceCF.com.
Reference #(35) (Table 7).
Also contains mixed tocopherols.
Most patients who are vitamin deficient can be treated adequately with the doses of fat-soluble vitamins recommended in the CF Foundation Consensus Report on Nutrition for Pediatric Patients.35,63
Low levels of Vitamin K are seen in patients with CF not taking appropriate vitamin supplementation.64 Currently available vitamin preparations designed for patients with CF contain more vitamin K than in the past. Prothrombin time is prolonged in severe vitamin K deficiency but the test requires a large amount of blood. PIVKA (Proteins Induced in Vitamin K Absence) is more sensitive, but beyond the immediate neonatal period, standard values for healthy infants in the first 2 years of life are not available (Peter Durie, personal communication). If PIVKA laboratory reference standards or a laboratory test that is practical become available, vitamin K status should be measured. Prothrombin time should be measured in infants with liver disease.
Recommendations # 12 and 13 are in Table I.
Zinc.
Infants diagnosed with CF through NBS who are not treated with pancreatic enzyme supplements have excessive fecal losses of zinc that correlate with fecal fat losses65 and fractional absorption of zinc improves when PERT is given.66 However, we did not find any primary studies that addressed zinc supplementation in infants with CF. Plasma zinc level is an inadequate measure of zinc sufficiency; therefore the CF Foundation Consensus Report on Nutrition for Pediatric Patients recommended that zinc levels should not be measured and that a trial of zinc supplementation (1 mg elemental zinc/kg/day in divided doses for 6 months) may be given to patients who are not adequately growing despite adequate calorie intake and pancreatic enzyme replacement therapy.35
Recommendation #14 is in Table I.
Sodium.
Infants with CF lose large amounts of sodium in their sweat.67 Although human milk and infant formulas provide adequate sodium for full-term non-CF infants, they may not meet the needs of infants who have CF. Baby foods contain no added salt, making the older infant vulnerable to inadequate sodium intake.68 Due to increased epithelial sodium losses, CF infants are at risk for hyponatremic, hypochloremic dehydration with metabolic alkalosis, which can be asymptomatic or can be characterized by anorexia, often with failure to thrive as well as fever, vomiting, irritability, and weakness.69 The DRI recommendation for total daily sodium intake for healthy infants is 5 mEq from birth to 6 months of age and 16 mEq from 7 to 12 months of age,70 which may be insufficient for a baby with CF. Additional sodium may be necessary for infants exposed to high environmental temperatures, including warm homes, over-bundling, or those living in a hot climate. Infants experiencing vomiting or diarrhea may require more than the maintenance amounts of sodium. The CF Foundation Consensus Report on Nutrition for Pediatric Patients35 recommended that babies with CF should have supplemental sodium. Historically, a daily dose of one-eighth teaspoon of table salt per day, which contains approximately 12.5 mEq of sodium, has been recommended. Sodium chloride solutions are available and can be dispensed more accurately. There is insufficient evidence to change this recommendation, although to be consistent with the age-related increase in DRI, this supplemental amount should be increased in the second half of the first year to one-quarter teaspoon of salt (25.2 mEq sodium) per day, not to exceed 4 mEq/kg/day. Premeasured doses or measuring spoons, not general household spoons, should be used. The salt can be divided among the day’s feedings, but parents should be instructed not to exceed the total daily dose. Serum sodium levels do not accurately reflect total body sodium; hyponatremia, if present, probably indicates significant total body sodium depletion. One prospective study, published in abstract form only, suggested that urinary sodium in relation to creatinine can be used to assess sodium status in CF infants.71 If infants are fed human milk, some expressed milk or supplemental formula feedings may be needed to provide a vehicle for salt supplements.
Recommendation # 15 is in Table I.
Fluoride.
Fluoride is a mineral that is critical for the prevention of dental caries. Vitamins that are formulated for patients with CF do not contain fluoride. Patients aged 6 months to 2 years whose community water supply contains less than 0.3 parts per million should be supplemented with 0.25 mg/day regardless of mode of feeding.72 The primary care provider may be the best source of information about fluoridation of the local water supply.
Recommendation# 16 is in Table I.
Other Antioxidants and Fatty Acids.
The newborn period may represent a unique opportunity to prevent lung damage. Some have speculated that this could be accomplished by supplementing patients with antioxidants73 such as selenium or coenzyme Q10 or other micronutrients such as beta-carotene and essential fatty acids. There is poor evidence either for diagnosis or treatment of selenium deficiency. Patients with CF have been reported to have low serum levels of coenzyme Q10 and beta-carotene. Although supplementation can normalize these levels,73 no deficiency state has been described and no evidence of clinical benefit has been shown. Low serum triene:tetraene ratios, reflecting essential fatty acid (EFA) deficiency, have been observed, and supplementation with linoleic acid can lead to normal serum levels in infants and older patients.62,74 In the one study we found that addressed the effect of dietary supplementation with EFA in infants, linoleic-rich formula led to better length but not better weight.75 Directing parents to supplement liquids with oils rich in linoleic acid has the benefit of additional calories but also has the potential risk of lipid aspiration in very young infants. There is also a theoretical risk of an increase in the arachidonic acid pathway leading to an increase in inflammatory mediators in individuals with CF; low triene-to-tetraene ratio potentially may reflect this shunting. Because the omega-3 essential fatty acid docosa-hexaenoic acid (DHA) is not metabolized to arachidonate, it has been proposed that DHA supplementation could decrease inflammation in CF. A recent Cochrane review addressed the use of omega-3 fatty acids in people with CF of all ages and the authors concluded that there was insufficient evidence to recommend routine use of these supplements.76 A clinical trial of use of a highly DHA-enriched formula, containing DHA at a level greater than is in currently available infant formulas, is ongoing but results are not yet available.77
Recommendations # 17 and 18 are in Table I.
Pulmonary Interventions.
Despite major advances in care, CF remains a life-shortening disease, most often as a consequence of recurrent respiratory infection, obstruction and inflammation. Studies using infant pulmonary function tests (PFTs), chest x-rays, computed tomography (CT), and bronchoalveolar lavage demonstrate that CF lung disease begins early and often in the first few months of life, prior to obvious symptoms.78–81 Although prevention of lung disease is a major goal of CF care, evidence to support guidelines for early prevention or treatment of pulmonary disease in infants with CF is sparse. However, one firm recommendation may be made: Exposure to environmental tobacco smoke should be avoided. This may be particularly true in CF, as environmental tobacco smoke is known to have a deleterious effect on lung health in patients with CF.82,83
Recommendation #19 is in Table I.
Airway Clearance.
In the CF Foundation Pulmonary Guidelines on Airway Clearance Therapies,84 airway clearance therapy is recommended for all patients with CF (consensus recommendation).
The evidence-based clinical practice guidelines on non-pharmacologic airway clearance therapies published by the American College of Chest Physicians also recommended airway clearance therapy as an effective technique to increase mucus clearance but noted that the effect of each treatment was relatively modest and the long-term benefits are unproven.85 Seventeen of the 25 studies reviewed included patients with CF, but none were infants. There is no evidence evaluating the utility of routine daily percussion and postural drainage (P&PD) in infants, and scant evidence of the positive effects of acute treatment. One single visit, before-after study of 13 infants administered a bronchodilator followed by P&PD showed significant decreases in total resistive work of breathing and pulmonary resistance, but minute ventilation was unchanged.86 Another randomized trial showed more improvement in lung function in a group of infants given P&PD in combination with albuterol and n-acetyl cysteine than those given P&PD alone.87 The CF Pulmonary Guidelines for Chronic Medications for Maintenance of Lung Health88 reviewed the use of β2-adrenergic receptor agonists in patients over 6 years of age and found the level of evidence to be good and the net benefit of treatment to be moderate, and recommended chronic use of these medicines (Grade B). In considering the unanswered question of when to initiate airway clearance, the CF Foundation Pulmonary Guidelines on Airway Clearance Therapies84 for patients with CF of all ages stated that despite the paucity of evidence of the benefit of airway clearance therapies (ACT) in infants with CF, ‘‘the presence of lung disease early in life is well-established and the committee feels that airway clearance, likely in the form of percussion and postural drainage, should be instituted in the first few months of life. The committee believes that there is potential benefit and little harm in teaching ACT to parents early and encouraging airway clearance to be part of the child’s daily routine. In most cases, the form of ACT for infants will be P&PD.’’
Recommendations # 20 and 21 are in Table I.
Two crossover trials have addressed the effects of head-down positioning on esophageal acid during P&PD in infants with CF.89,90 In one study,89 infants with CF had more reflux episodes than non-CF infants, and episodes were more frequent during P&PD than at baseline, although the percent of time and duration of reflux episodes was no different between the two groups. Head-down supine and prone positioning was associated with more reflux episodes than head-down left lateral decubitus positioning. A second study did not find any significant difference in the duration of lower esophageal pH falls comparing P&PD delivered in the head-down position to that delivered without head-down position, although the mean number of reflux episodes was higher in the head-down group.90 Parents of participants in one of the cross-over studies were given the option of having their child randomized to a long-term study of either standard (head-down) or modified physiotherapy (no head-down positions). Seven of 10 participants in each group were available for 21⁄2 - and 5-year follow-up.91 The methods are unclear, making the results of this study hard to interpret; however, those given modified physiotherapy had fewer days with upper respiratory tract symptoms, fewer days of antibiotics, and better pulmonary function tests and chest x-ray scores. Another article describing the same study population reported on the impact of position on reflux, arousal states, heart rate and oxygen saturation with differing results described.92
Recommendation # 22 is in Table I.
Infection Control, Surveillance, and Treatment.
The initiation of CF NBS in most states in the United States as well as in other countries has created a cohort of relatively healthy young infants and children intermingling with older individuals with CF in outpatient clinics. Patient-to-patient transmission of pathogens and clonal spread of pathogens within CF centers have been increasingly demonstrated over the past 20 years. Some studies have specifically documented the spread of pathogens to young children from older patients. In one study of 56 children detected by CF NBS in Australia, 4 children were infected with a multidrug-resistant mucoid strain of P aeruginosa and died before 7 years of age.93 This strain was shared among older, unrelated children attending the clinic as well as 3 other children in the NBS cohort. In addition, analysis of results from a clinical trial of CF NBS in Wisconsin demonstrated that intermingling of patients in a crowded CF center can be a risk factor for early acquisition of P aeruginosa.94 Infants newly diagnosed with CF should be seen separately (eg, first appointment of the clinic session or in another location) from other patients cared for in CF clinics until adequate infection control education has been provided to the caregivers. Educational materials on infection control should be provided to families immediately after diagnosis. All care activities should be implemented in compliance with the CF Foundation Infection Control Consensus recommendations.95 Appropriate hand hygiene and cough etiquette for healthcare providers, families, and patients should be taught and reinforced frequently (Table III). These principles should also be emphasized in non-healthcare settings such as the home and day care. Appropriate cleaning and disinfecting of devices for inhaled medications used in non-healthcare settings should be performed after every use to prevent acquisition of potential pathogens.95 Appropriate cleaning and disinfecting of devices for inhaled medications in healthcare settings should be implemented as described in the Healthcare Infection Control Practices Advisory Committee guidelines from the CDC.96
Recommendation #23 and 24 are in Table I.
Prevention of Viral Infections.
The AAP recommendations for standard vaccinations apply to infants with CF.97 The frequency of visits to the PCP for immunizations in the first 2 years of life offers an additional opportunity for intensive medical surveillance. Annual influenza vaccination is recommended for infants with CF ≥6 months of age, all household members, and all healthcare providers caring for these infants. Adherence to influenza vaccination should be monitored by the CF Center. If the vaccine is given by the CF Center, the PCP should be informed and vice versa. Household contacts and out-of-home caregivers of children with CF <6 months of age should also receive the annual influenza vaccine.
Recommendation # 25 is in Table I.
Respiratory syncytial virus (RSV) may have adverse effects on respiratory status in patients with CF.98 Palivizumab is a humanized monoclonal antibody that provides passive immunity against RSV. The AAP revised policy statement in December 2003 on the prevention of RSV infections states, ‘‘Palivizumab .prophylaxis should be considered for infants and children younger than 2 years with chronic lung disease …’’99 Two studies have addressed the use of palivizumab in infants with CF. A chart review of hospitalized infants found that fewer children who received palivizumab were hospitalized and their length of stay was shorter, although these differences did not reach statistical significance.100 None of the children who were hospitalized and had received palivizumab had RSV, whereas 42% of those who did not receive palivizumab but were hospitalized were RSV positive. A report of the Palivizumab Outcomes Registry found no hospitalizations over 24 hours in length in which RSV infection was confirmed in 91 patients with CF.101 Extrapolation of data from other populations suggests that there could be benefit from the use of RSV prophylaxis in infants with CF.
Recommendation #26 is in Table I.
Surveillance and Intervention for Bacterial Infection.
Routine surveillance of respiratory microbiology is important in CF management, and aggressive eradication regimens for newly acquired positive Pseudomonas cultures have become a routine part of care.102 Although early acquisition of Pseudomonas is clearly associated with worse short- and long-term outcomes,103–105 isolation of Pseudomonas or other respiratory pathogens is often not associated with a discrete change in clinical signs or symptoms.
As infants do not expectorate, cultures are generally obtained by oropharyngeal (OP) swab or endolaryngeal suction. Two studies have compared the ability of OP cultures with predict lower airway colonization.106,107 Both authors concluded that a positive OP culture does not reliably predict the presence of pathogens in the lower airways, but a negative culture is more helpful in ruling out lower airway infection than ruling it in. An ongoing study is comparing the outcomes at school age of infants monitored by OP cultures versus bronchoscopy, but results are not yet available (Claire Wainwright, personal communication). We conclude that OP cultures should be performed at least quarterly (Table III). The first culture should be obtained at the first or second visit following diagnosis. Cultures should be repeated more frequently in the presence of respiratory symptoms. Bronchoalveolar lavage may be used in individual cases as clinically indicated (Figure 5). Cultures should be processed in clinical microbiology laboratories affiliated with CF Foundation-accredited care centers using the methods described in the CF Foundation Consensus Conference on Infection Control recommendations.95
Figure 5.

Care of infants with a change in respiratory status (based on consensus opinion). Change in respiratory status identified: increased cough, tachypnea, wheezing, oxygen desaturation, crackles. Clinicians should assess whether these indicate a mild or moderate-to-severe exacerbation. In most cases, significant oxygen desaturation or the presence of crackles will indicate a moderate-to-severe exacerbation.
Recommendations # 27 and 28 are in Table I.
Staphylococcus Aureus.
Staphylococcus aureus and Hae-mophilus influenzae often colonize the respiratory tract before the emergence of Pseudomonas aeruginosa.108 According to the 2002 consensus guidelines from the UK CF Trust Antibiotics Group, ‘‘All CF infants less than 2 years of age should receive long term flucloxacillin from diagnosis’’;109 however a Cochrane systematic review of the use of prophylactic antistaphylococcal antibiotics found a reduced frequency of isolation of Staphylococcus but no significant differences in lung function, nutrition, number of hospitalizations, or number of courses of additional antibiotics.110 We identified one additional study that examined this same question,111 which, along with three112–114 of the four studies cited in the Cochrane Review, noted an increased risk of Pseudomonas acquisition in patients receiving anti-staphylococcal prophylaxis. Of note, these studies were performed before the era when eradication regimens for new acquisition of Pseudomonas have become standard care. Different results regarding increased risk of Pseudomonas acquisition may be due to use of broader versus narrower-spectrum anti-staphylococcal antibiotics, sample sizes and length of follow-up. These studies did not address the possibility of acquisition of methicillin-resistant Staphylococcus aureus (MRSA). The CF Foundation Pulmonary Guidelines on Chronic Medications88 recommended against the routine use of anti-staphylococcal antimicrobial prophylaxis due to concerns about increased emergence of P aeruginosa and lack of demonstrable long-term benefits.
One study evaluated acute treatment of asymptomatic patients found to have Staphylococcus aureus.115 Only three infants were included, and the success of eradication was measured at 1-week follow-up. We did not find any studies that addressed this question when the organism was methicillin-resistant. There is insufficient evidence to recommend active attempts to eradicate Staphylococcus aureus in asymptomatic infants.
Recommendations # 29 and 31 are in Table I.
Pseudomonas aeruginosa.
Early age at acquisition of Pseudomonas aeruginosa has been shown to adversely affect long-term pulmonary disease and survival,93,103,104 providing a rationale for early eradication regimens.116,117
Compared with infants without Pseudomonas on serial cultures, those with Pseudomonas on serial cultures had significantly more daily cough, lower chest x-ray scores, and higher levels of circulating immunoglobulins, suggesting chronic inflammation.105 We did not identify any studies of the clinical benefits of eradication of Pseudomonas in infants <2 years of age. A randomized placebo-controlled trial is currently underway in the United States (http://clinicaltrials.gov/). Full results from a recently completed eradication trial in infants that was conducted in Europe are not yet available, but preliminary results indicate that microbiologic eradication can be achieved equally with either a 28-day or a 56-day course of nebulized tobramycin solution for inhalation (TOBI).118 Trials of early eradication regimens in older CF patients have shown microbiologic benefit,119,120 and nonrandomized studies in older patients suggest clinical benefit.121
Because of the risk of more rapid progression of lung disease, and extrapolating from studies in older CF patients, we recommend that new acquisition of P aeruginosa, (defined as initial acquisition or new acquisition after ‘‘successful’’ eradication therapy) should be treated in infants <2 years of age regardless of whether symptoms are present or not. Acceptable treatment approaches include a variety of regimens including tobramycin for inhalation, with or without ciprofloxacin, and/ or intravenous antibiotics. Infants with respiratory symptoms should be treated using regimens guided by clinical condition, respiratory culture and susceptibility results.
Anti-pseudomonal prophylaxis in infants has only been studied in one small retrospective study of inhaled gentamicin.122 There was a significantly increased risk of acquisition of Pseudomonas after treatment was discontinued (after a mean of 42 months), whereas none of the infants who continued treatment for a mean of 79 months were found to be colonized with Pseudomonas. The CF Foundation Pulmonary Guidelines Committee in a review of chronic pulmonary therapies in older patients found good evidence and substantial net benefit of chronic anti-pseudomonal treatment in patients with moderate to severe lung disease88 and found a fair level of evidence and moderate net benefit for use of tobramycin for inhalation to reduce exacerbations in patients over 6 years of age who are asymptomatic or with mild lung disease and with Pseudomonas persistently present in cultures of the airways. With the exception of the studies listed above for which complete results are unavailable, no primary prospective studies were found that addressed the use of this medication as a chronic therapy in patients with CF under 2 years of age. We recommend that infants who remain persistently infected with Pseudomonas aeruginosa after two attempts at eradication be treated chronically with alternate month tobramycin solution for inhalation.
Recommendations # 32 through 34 are in Table I.
Surveillance for Pulmonary Disease
Pulse Oximetry.
Pulse oximetry is a widely-available, noninvasive tool to monitor adequacy of oxygenation. Two cross-sectional studies have looked at the use of pulse oximetry as a modality to detect lung disease in infants. One found that patients with failure to thrive (FTT) and pulmonary symptoms at the time of diagnosis had lower oxygen saturation than those presenting with either meconium ileus or FTT alone.123 Another study found that pulse oximetry in asymptomatic infants with CF was not different than in control subjects, but infants with CF and young children with mild airway inflammation such as rhinitis, cough and pharyngeal erythema developed oxygen desaturation with sleep.124 Pulse oximetry is recommended in the infant with acute respiratory symptoms and clinicians may choose to use it routinely as a supportive method to detect lung disease. Possible clinical interventions for patients with low oxygen saturation are shown in Figure 5, which is based on expert opinion and experience.
Recommendations # 35 and 36 are in Table I.
Chest X-Rays.
Chest x-rays may detect early abnormalities, and changes in standardized scores can be seen as early as 1 year of age.79 In one small study, the presence of consolidation on chest x-ray in children <6 years of age was shown to be correlated with peak flow measurements and survival.125 A baseline chest x-ray should be obtained within the first 3 to 6 months and once again within the first 2 years of life. If subtle findings such as hyperinflation, peribronchial cuffing or streakiness are seen, decisions regarding further assessment and treatment may be aided by the presence of other objective measures of disease such as physical examination, pulse oximetry, respiratory cultures or nutritional variables. Abnormalities such as atelectasis or infiltrate should be treated or evaluated further (Figure 5). Chest x-rays should also be obtained in infants with a change in respiratory status that does not respond to basic interventions. Guidelines on standards of care developed by other organizations, such as the United Kingdom CF Trust126 and the European Consensus Conference,127 recommend obtaining chest x-rays annually.
Recommendation # 37 is in Table I.
Chest Computed Tomography (CT).
Studies evaluating controlled breathing CT scans during infancy have demonstrated thickened airway walls, bronchial dilatation/bronchiectasis, as well as air trapping.128–130 In one cross-sectional study, infants with CF who were clinically stable (without acute respiratory symptoms for at least 3 weeks before the evaluation) had thicker airway walls, narrower lumens and more air trapping on chest CT scans than infants without CF.128 Another also demonstrated thicker airway walls as well as an increased airway lumen diameter in young CF subjects compared with controls.129 In a different study, controlled breathing CT scans of the chest performed before and after pulmonary exacerbations revealed significant improvement in modified Brody scores especially in bronchial dilatation/bronchiectasis and hyperinflation subscores.130 In addition, regional inflammation identified via bronchialveolar lavage correlated with areas of worse disease identified on the CT scan. More abnormalities were identified by ultrafast high resolution CT (HRCT) scans than by chest x-rays in another study, although only 8 of 36 participants were less than two years of age.131 However, HRCT may not be as sensitive to functional changes of early lung disease as a test of regional ventilation heterogeneity, the multiple breath washout (MBW) lung clearance index.132 Although not widely employed, several studies have shown the utility of measuring ventilation heterogeneity in CF patients with MBW techniques. Two issues combine to make routine use of chest CT scans problematic. First, for best quality chest CT scans, infants require sedation. Second, although scanners and techniques are being developed that expose patients to lower levels of radiation, the exposure is still greater than a chest x-ray. In their 2007 review, Brenner and Hall133 focused on the increasing number of CT scans being performed and the consequent cancer risks ‘‘particularly in children.’’ They suggested that age at exposure to diagnostic radiation matters, with much higher rates of death due to cancer in people exposed in childhood. Patients with CF likely will undergo radiologic procedures every year of their life, and their cumulative lifetime exposure to radiation will be increased if routine use of CT scans begins in infancy. There is insufficient evidence to recommend use of chest CT scans for routine surveillance, however this imaging modality may be helpful in infants with symptoms or signs of lung disease who fail to respond to basic interventions (Figure 5).
Recommendations # 38 and 39 are in Table I.
Infant Pulmonary Function Tests.
Pulmonary function testing to detect airways obstruction is part of routine CF care in older patients, both for routine monitoring and as a measure of response to therapy. Two cross-sectional studies compared the raised volume rapid thoracoabdominal compression (RVRTC) technique with conventional rapid thoracic compression where flows are assessed from tidal breathing. Both studies reported that the RVRTC technique was more sensitive.134,135 Reduced forced expiratory flows and volumes have been demonstrated in infants with CF utilizing the RVRTC technique shortly after diagnosis and at follow-up 5 to 10 months later136 even in the absence of symptoms137 and in infants diagnosed through NBS.80 These abnormalities have been shown to persist through the preschool years.138 A cross-sectional study found the RVRTC technique to be equivalent to the multiple breath washout technique for early detection of airways obstruction.139 We found 3 older studies that used whole body plethysmography to detect changes in lung function in infants with CF.140–142 Widespread use of infant pulmonary function tests has been limited by the need for sedation, expertise in performing the technique and the need for specialized equipment. There is insufficient evidence to recommend for or against routine monitoring of infant lung function. However, sites capable of performing infant pulmonary function testing may elect to monitor lung function testing routinely, and testing may be of use in patients who are symptomatic (Figure 5). As noted above, the MBW technique for determining lung clearance index132 shows promise to detect early changes in lung function, but at present it remains a research tool.
Recommendation # 40 is in Table I.
Other Chronic Pulmonary Interventions
A variety of pulmonary interventions have been shown to be associated with significantly improved pulmonary outcomes in older children and adults with CF. Despite strong evidence in older patients,88 there is insufficient evidence in infants to recommend the routine use of dornase alfa (recombinant human DNase), 7% hypertonic saline, or chronic azithromycin. Clinical trials evaluating safety and efficacy of some of these interventions in infants and toddlers are planned. We did not specifically search for studies of ibuprofen, chronic use of β-adrenergic agonists, cromolyn, or leukotriene modifiers in CF infants. Extrapolating from data in older patients with CF, inhaled corticosteroids should not be used unless airways reactivity has been demonstrated.
Dornase Alfa (Recombinant Human DNase).
A Cochrane review concluded that therapy with dornase alfa over a 1-month and 6-month period of time was associated with improved lung function in patients with CF.143 However, none of the eligible trials in this review included children younger than 5 years of age. The CF Foundation evidence-based guidelines on chronic pulmonary medications concluded that for patients over 6 years of age, there was good evidence and a substantial net benefit of dornase alfa in patients with moderate to severe lung disease; the evidence was judged to be fair and the benefit moderate for asymptomatic patients or those with mild lung disease.88 An open label, randomized, placebo-controlled crossover pilot study compared the use of dornase alfa with normal saline in stable infants with CF.144 There was a mean increase in flows referenced to functional residual capacity and a trend towards improved overnight desaturation index associated with use of dornase alfa.
7% Hypertonic Saline.
The CF Foundation Pulmonary Guidelines Committee found a fair level of evidence and a moderate net benefit for use of chronic inhaled 7% hypertonic saline in patients over 6 years of age.88 Two before-after pilot studies, one identified after the completion of the evidence review, assessed single doses of 7% hypertonic saline in infants who had been pre-treated with albuterol and both found it to be safe and well-tolerated.145,146
Chronic Azithromycin.
The CF Foundation Pulmonary Guidelines Committee found a fair level of evidence and substantial net benefit for use of azithromycin in patients over 6 years of age who were chronically colonized with Pseudomonas.88 No primary studies were found that addressed the use of this medication as a chronic therapy in patients with CF under two years of age, although the drug is approved for acute use in infants 6 months of age and older.
Inhaled corticosteroids.
The CF Foundation Pulmonary Guidelines recommended against the use of inhaled corticosteroids to improve lung function and reduce exacerbations in patients over 6 years of age, however this review excluded studies addressing patients with asthma.88 No primary studies were found that addressed the use of inhaled corticosteroids as a chronic therapy in patients with CF under two years of age. Inhaled corticosteroids should not be used unless airways reactivity has been demonstrated.
Recommendations # 41 through 44 are in Table I.
Future Research
Our knowledge of the best ways to care for infants diagnosed in the newborn period is hampered by a lack of good evidence in very young patients with CF. Well-designed clinical trials are the best way to close this information gap. It is easier to design clinical trials based on nutritional than respiratory outcomes: Infants grow rapidly in the first few years of life, anthropometric measures are more standardized than measures of lung health in this age group and growth measurements are included in the CF Foundation Patient Registry. As more states adopt CF NBS, a review of the baseline data from the CF Foundation Patient Registry would be a critical first step to better understand the current status of growth in the first two years of life among US infants diagnosed with CF through NBS.
We set out to develop guidelines for management of infants with CF based on the best available evidence, but much of what is recommended is based on expert consensus due to lack of evidence. Health outcomes studies could be used to evaluate the optimal implementation of the recommendations and help us to learn what needs to be changed. Optimal therapeutic interventions could be evaluated using controlled clinical trial study design. Nutritional issues to be considered for clinical trials might include routine use of high-calorie formulas rather than use as an intervention for weight loss or lack of adequate weight gain, studies to answer questions related to essential fatty acid supplementation in infants, and studies of salt replacement therapy, to name a few.
Outcome measures for use in infants need further development. Anthropometric measures have been standardized and can be used in studies of nutritional interventions, but better measures of lung health are needed. All studies in infants should include development assessments and quality of life measures that are specific to patients in this age range.
Conclusion
Newborn screening offers the opportunity to significantly improve the health of individuals with CF, enabling pre-symptomatic monitoring and treatment when needed to prevent or delay nutritional and pulmonary decline. Although infants with CF may appear well, subtle changes occur early in life. Rigorous and careful monitoring and timely medical intervention will permit the best possible outcome for individuals diagnosed with CF in infancy.■
Glossary
- AAP
American Academy of Pediatrics
- BMI
Body mass index
- CF
Cystic fibrosis
- CFA
Coefficient of fat absorption
- CFTR
Cystic fibrosis transmembrane conductance regulator
- CT
Computed tomography
- DHA
Docosahexaenoic acid
- DRI
Dietary Reference Intake
- FE
Fecal elastase
- FTT
Failure to thrive
- MCT
Medium chain triglyceride
- NBS
Newborn screening
- OP
Oropharyngeal
- P&PD
Percussion and postural drainage
- PCP
Primary care provider
- PERT
Pancreatic enzyme replacement therapy
- PFT
Pulmonary function test
- PI
Pancreatic insufficiency
- PS
Pancreatic sufficiency
- RTC
Rapid thoracic compression
- RVRTC
Raised volume rapid thoracoabdominal compression
- RSV
Respiratory syncytial virus
- USPSTF
United States Preventive Services Task Force
Contributor Information
Drucy Borowitz, From the Women and Children’s Hospital of Buffalo, State University of New York at Buffalo, NY.
Karen A. Robinson, Johns Hopkins School of Medicine, Baltimore, MD.
Margaret Rosenfeld, University of Washington, Seattle WA,.
Stephanie D. Davis, University of North Carolina, Chapel Hill, NC.
Kathryn A. Sabadosa, Dartmouth College, Hanover, NH.
Stephanie L. Spear, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH.
Suzanne H. Michel, Children’s Hospital of Philadelphia, Philadelphia, PA.
Richard B. Parad, Brigham and Women’s Hospital, Boston, MA.
Terry B. White, Cystic Fibrosis Foundation, Bethesda, MD.
Philip M. Farrell, University of Wisconsin School of Medicine and Public Health, Madison, WI.
Bruce C. Marshall, Cystic Fibrosis Foundation, Bethesda, MD.
Frank J. Accurso, Children’s Hospital, Denver, CO.
References
- 1.Robinson KA, Saldanha I, McKoy N. Management of infants with cystic fibrosis: A summary of the evidence for the Cystic Fibrosis Foundation Working Group on Care of Infants with Cystic Fibrosis. J Pediatr (Insert proper citation for the submitted methodology paper in this supplement). [DOI] [PubMed] [Google Scholar]
- 2.Sawaya GF, Guirguis-Blake J, LeFevre M, Harris R, Petitti D. Update on the methods of the US Preventive Services Task Force: estimating certainty and magnitude of net benefit. Ann Intern Med 2007;147: 871–5. [DOI] [PubMed] [Google Scholar]
- 3.Giglio L, Candusso M, D’Orazio C, Mastella G, Faraguna D. Failure to thrive: the earliest feature of cystic fibrosis in infants diagnosed by neonatal screening. Acta Paediatr 1997;86:1162–5. [DOI] [PubMed] [Google Scholar]
- 4.Accurso FJ, Sontag MK, Wagener JS. Complications associated with symptomatic diagnosis in infants with cystic fibrosis. J Pediatr 2005; 147(suppl):S37–41. [DOI] [PubMed] [Google Scholar]
- 5.Jedlicka-Kohler I, Gotz M, Eichler I. Parents’ recollection of the initial communication of the diagnosis of cystic fibrosis. Pediatrics 1996;97: 204–9. [PubMed] [Google Scholar]
- 6.Tluczek A, Koscik RL, Modaff P, Pfeil D, Rock MJ, Farrell PM, et al. Newborn screening for cystic fibrosis: parents’ preferences regarding counseling at the time of infants’ sweat test. J Genet Couns 2006;15:277–91. [DOI] [PubMed] [Google Scholar]
- 7.Sims EJ, Mugford M, Clark A, Aitken D, McCormick J, Mehta G, et al. Economic implications of newborn screening for cystic fibrosis: a cost of illness retrospective cohort study. Lancet 2007;369:1187–95. [DOI] [PubMed] [Google Scholar]
- 8.Stallings VA, Stark LJ, Robinson KA, Feranchak AP, Quinton H. Evidence-based practice recommendations for nutrition-related management of children and adults with cystic fibrosis and pancreatic insufficiency: results of a systematic review. J Am Diet Assoc 2008;108:832–9. [DOI] [PubMed] [Google Scholar]
- 9.Konstan MW, Butler SM, Wohl ME, Stoddard M, Matousek R, Wagener JS, et al. Growth and nutritional indexes in early life predict pulmonary function in cystic fibrosis. J Pediatr 2003;142:624–30. [DOI] [PubMed] [Google Scholar]
- 10.Feranchak AP, Sontag MK, Wagener JS, Hammond KB, Accurso FJ, Sokol RJ. Prospective, long-term study of fat-soluble vitamin status in children with cystic fibrosis identified by newborn screen. J Pediatr 1999;135:601–10. [DOI] [PubMed] [Google Scholar]
- 11.Shepherd RW, Holt TL, Greer R, Cleghorn GJ, Thomas BJ. Total body potassium in cystic fibrosis. J Pediatr Gastroenterol Nutr 1989;9:200–5. [DOI] [PubMed] [Google Scholar]
- 12.Borowitz D Update on the evaluation of pancreatic exocrine status in cystic fibrosis. Curr Opin Pulm Med 2005;11:524–7. [DOI] [PubMed] [Google Scholar]
- 13.Bronstein MN, Sokol RJ, Abman SH, Chatfield BA, Hammond KB, Hambridge KM, et al. Pancreatic insufficiency, growth, and nutrition in infants identified by newborn screening as having cystic fibrosis. J Pediatr 1992;120:533–40. [DOI] [PubMed] [Google Scholar]
- 14.Walkowiak J, Sands D, Nowakowska A, Piotrowski R, Zybert K, Herzig KH, et al. Early decline of pancreatic function in cystic fibrosis patients with class 1 or 2 CFTR mutations. J Pediatr Gastroenterol Nutr 2005;40:199–201. [DOI] [PubMed] [Google Scholar]
- 15.Gaskin K, Waters D, Dorney S, Gruca M, O’Halloran M, Wilcken B. Assessment of pancreatic function in screened infants with cystic fibrosis. Pediatr Pulmonol Suppl 1991;7:69–71. [DOI] [PubMed] [Google Scholar]
- 16.Bellentani S, Grisendi A, Rinaldi M, Bertolani P, Costa G, Agostini M, et al. BT-Paba test in the diagnosis of pancreatic exocrine insufficiency in cystic fibrosis: urinary and serum determinations compared. Eur J Pediatr 1984;143:145–8. [DOI] [PubMed] [Google Scholar]
- 17.Greer R, Shepherd R, Cleghorn G, Bowling FG, Holt T. Evaluation of growth and changes in body composition following neonatal diagnosis of cystic fibrosis. J Pediatr Gastroenterol Nutr 1991;13:52–8. [DOI] [PubMed] [Google Scholar]
- 18.Durie PR, Largman C, Brodrick JW, Johnson JH, Gaskin KJ, Forstner GG, et al. Plasma immunoreactive pancreatic cationic trypsinogen in cystic fibrosis: a sensitive indicator of exocrine pancreatic dysfunction. Pediatr Res 1981;15:1351–5. [DOI] [PubMed] [Google Scholar]
- 19.Gillard BK, Cox KL, Pollack PA, Geffner ME. Cystic fibrosis serum pancreatic amylase: useful discriminator of exocrine function. Am J Dis Child 1984;138:577–80. [DOI] [PubMed] [Google Scholar]
- 20.Watkins JB, Schoeller DA, Klein PD, Ott DG, Newcomer AD, Hofmann AF. 13C-trioctanoin: a nonradioactive breath test to detect fat malabsorption. J Lab Clin Med 1977;90:422–30. [PubMed] [Google Scholar]
- 21.Colombo C, Maiavacca R, Ronchi M, Consalvo E, Amoretti M, Giunta A. The steatocrit: a simple method for monitoring fat malabsorption in patients with cystic fibrosis. J Pediatr Gastroenterol Nutr 1987;6:926–30. [PubMed] [Google Scholar]
- 22.Remtulla MA, Durie PR, Goldberg DM. Stool chymotrypsin activity measured by a spectrophotometric procedure to identify pancreatic disease in infants. Clin Biochem 1986;19:341–7. [DOI] [PubMed] [Google Scholar]
- 23.Beharry S, Ellis L, Corey M, Marcon M, Durie P. How useful is fecal pancreatic elastase 1 as a marker of exocrine pancreatic disease? J Pediatr 2002;141:84–90. [DOI] [PubMed] [Google Scholar]
- 24.Nissler K, Katte IV, Huebner A, Henker J. Pancreatic elastase 1 in feces of preterm and term infants. J Pediatr Gastroenterol Nutr 2001;33:28–31. [DOI] [PubMed] [Google Scholar]
- 25.Kori M, Maayan-Metzger A, Shamir R, Sirota L, Dinari G. Faecal elastase 1 levels in premature and full-term infants. Arch Dis Child Fetal Neonatal Ed 2003;88:F106–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Castellani C, Cuppens H, Macek M Jr., Cassiman JJ, Kerem E, Durie P, et al. Consensus on the use and interpretation of cystic fibrosis mutation analysis in clinical practice. J Cyst Fibros 2008;7:179–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Durie PR, Newth CJ, Forstner GG, Gall DG. Malabsorption of medium-chain triglycerides in infants with cystic fibrosis: correction with pancreatic enzyme supplement. J Pediatr 1980;96:862–4. [DOI] [PubMed] [Google Scholar]
- 28.McClean P, Harding M, Coward WA, Green MR, Weaver LT. Measurement of fat digestion in early life using a stable isotope breath test. Arch Dis Child 1993;69:366–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Caliari S, Benini L, Sembenini C, Gregori B, Carnielli V, Vantini I. Medium-chain triglyceride absorption in patients with pancreatic insufficiency. Scand J Gastroenterol 1996;31:90–4. [DOI] [PubMed] [Google Scholar]
- 30.Borowitz DS, Grand RJ, Durie PR. Use of pancreatic enzyme supplements for patients with cystic fibrosis in the context of fibrosing colonopathy: consensus committee. J Pediatr 1995;127:681–4. [DOI] [PubMed] [Google Scholar]
- 31.Smyth RL, van Velzen D, Smyth AR, Lloyd DA, Heaf DP. Strictures of ascending colon in cystic fibrosis and high-strength pancreatic enzymes. Lancet 1994;343:85–6. [DOI] [PubMed] [Google Scholar]
- 32.Schwarzenberg SJ, Wielinski CL, Shamieh I, Carpenter BL, Jessurun J, Weisdorf SA, et al. Cystic fibrosis-associated colitis and fibrosing colonopathy. J Pediatr 1995;127:565–70. [DOI] [PubMed] [Google Scholar]
- 33.FitzSimmons SC, Burkhart GA, Borowitz D, Grand RJ, Hammerstrom T, Durie PR, et al. High-dose pancreatic-enzyme supplements and fibrosing colonopathy in children with cystic fibrosis. N Engl J Med 1997;336:1283–9. [DOI] [PubMed] [Google Scholar]
- 34.Hendeles L, Dorf A, Stecenko A, Weinberger M. Treatment failure after substitution of generic pancrelipase capsules: correlation with in vitro lipase activity. JAMA 1990;263:2459–61. [PubMed] [Google Scholar]
- 35.Borowitz D, Baker RD, Stallings V. Consensus report on nutrition for pediatric patients with cystic fibrosis. J Pediatr Gastroenterol Nutr 2002;35:246–59. [DOI] [PubMed] [Google Scholar]
- 36.Sinaasappel M, Stern M, Littlewood J, Wolfe S, Steinkamp G, Heijerman HG, et al. Nutrition in patients with cystic fibrosis: a European Consensus. J Cyst Fibros 2002;1:51–75. [DOI] [PubMed] [Google Scholar]
- 37.Taylor C, Willson NB, Littlewood J, Morton A, Watson H, Wolfe S, et al. Nutritional management of cystic fibrosis 2002. Cystic Fibrosis Trust; Web site. Available at: http://www.cftrust.org.uk/aboutcf/publications/consensusdoc/C_3500Nutritional_Management.pdf. Accessed January 20, 2009. [Google Scholar]
- 38.Holliday KE, Allen JR, Waters DL, Gruca MA, Thompson SM, Gaskin KJ. Growth of human milk-fed and formula-fed infants with cystic fibrosis. J Pediatr 1991;118:77–8. [DOI] [PubMed] [Google Scholar]
- 39.Parker EM, O’Sullivan BP, Shea JC, Regan MM, Freedman SD. Survey of breast-feeding practices and outcomes in the cystic fibrosis population. Pediatr Pulmonol 2004;37:362–7. [DOI] [PubMed] [Google Scholar]
- 40.Colombo C, Costantini D, Zazzeron L, Faelli N, Russo MC, Ghisleni D, et al. Benefits of breastfeeding in cystic fibrosis: a single-centre follow-up survey. Acta Paediatr 2007;96:1228–32. [DOI] [PubMed] [Google Scholar]
- 41.Sokol RJ, Reardon MC, Accurso FJ, Stall C, Narkewicz M, Abman SH, et al. Fat-soluble-vitamin status during the first year of life in infants with cystic fibrosis identified by screening of newborns. Am J Clin Nutr 1989;50:1064–71. [DOI] [PubMed] [Google Scholar]
- 42.Ellis L, Kalnins D, Corey M, Brennan J, Pencharz P, Durie P. Do infants with cystic fibrosis need a protein hydrolysate formula? A prospective, randomized comparitive trial. J Pediatr 1998;132:270–6. [DOI] [PubMed] [Google Scholar]
- 43.Farrell PM, Mischler EH, Sondel SA, Palta M. Predigested formula for infants with cystic fibrosis. J Am Diet Assoc 1987;87:1353–6. [PubMed] [Google Scholar]
- 44.Canciani M, Mastella G. Absorption of a new semielemental diet in infants with cystic fibrosis. J Pediatr Gastroenterol Nutr 1985;4:735–40. [DOI] [PubMed] [Google Scholar]
- 45.Kleinman R, editor. Pediatric Nutrition Handbook 5th ed. Elk Grove Village, IL: American Academy of Pediatrics; 2004. [Google Scholar]
- 46.Piazza-Waggoner C, Driscoll KA, Gilman DK, Powers SW. A comparison using parent report and direct observation of mealtime behaviors in young children with cystic fibrosis: implications for practical and empirically based behavioral assessment in routine clinical care. Child Health Care 2008;37:38–48. [Google Scholar]
- 47.Stark LJ, Jelalian E, Powers SW, Mulvihill MM, Opipari LC, Bowen A, et al. Parent and child mealtime behavior in families of children with cystic fibrosis. J Pediatr 2000;136:195–200. [DOI] [PubMed] [Google Scholar]
- 48.Duff AJ, Wolfe SP, Dickson C, Conway SP, Brownlee KG. Feeding behavior problems in children with cystic fibrosis in the UK: prevalence and comparison with healthy controls. J Pediatr Gastroenterol Nutr 2003;36:443–7. [DOI] [PubMed] [Google Scholar]
- 49.Powers SW, Patton SR, Byars KC, Mitchell MJ, Jelalian E, Mulvihill MM, et al. Caloric intake and eating behavior in infants and toddlers with cystic fibrosis. Pediatrics 2002;109:E75–5. [DOI] [PubMed] [Google Scholar]
- 50.Sanders MR, Turner KM, Wall CR, Waugh LM, Tully LA. Mealtime behavior and parent-child interaction: a comparison of children with cystic fibrosis, children with feeding problems, and nonclinic controls. J Pediatr Psychol 1997;22:881–900. [DOI] [PubMed] [Google Scholar]
- 51.Stark LJ, Opipari LC, Jelalian E, Powers SW, Janicke DM, Mulvihill MM, et al. Child behavior and parent management strategies at mealtimes in families with a school-age child with cystic fibrosis. Health Psychol 2005;24:274–80. [DOI] [PubMed] [Google Scholar]
- 52.Simmons RJ, Goldberg S, Washington J, Fischer-Fay A, Maclusky I. Infant-mother attachment and nutrition in children with cystic fibrosis. J Dev Behav Pediatr 1995;16:183–6. [PubMed] [Google Scholar]
- 53.Birch LL. Development of food acceptance patterns. Dev Psychol 1990; 26:515–9. [Google Scholar]
- 54.Carruth BR, Ziegler PJ, Gordon A, Barr SI. Prevalence of picky eaters among infants and toddlers and their caregivers’ decisions about offering a new food. J Am Diet Assoc 2004;104(1 Suppl. 1):s57–64. [DOI] [PubMed] [Google Scholar]
- 55.Stark LJ, Jelalian E, Mulvihill MM, Powers SW, Bowen AM, Spieth LE, et al. Eating in preschool children with cystic fibrosis and healthy peers: behavioral analysis. Pediatrics 1995;95:210–5. [PubMed] [Google Scholar]
- 56.Singer LT, Nofer JA, Benson-Szekely LJ, Brooks LJ. Behavioral assessment and management of food refusal in children with cystic fibrosis. J Dev Behav Pediatr 1991;12:115–20. [PMC free article] [PubMed] [Google Scholar]
- 57.Powers SW, Byars KC, Mitchell MJ, Patton SR, Schindler T, Zeller MH. A randomized pilot study of behavioral treatment to increase calorie intake in toddlers with cystic fibrosis. Child Health Care 2003;32:297–311. [Google Scholar]
- 58.Powers SW, Jones JS, Ferguson KS, Piazza-Waggoner C, Daines C, Acton JD. Randomized clinical trial of behavioral and nutrition treatment to improve energy intake and growth in toddlers and preschoolers with cystic fibrosis. Pediatrics 2005;116:1442–50. [DOI] [PubMed] [Google Scholar]
- 59.Powers SW, Piazza-Waggoner C, Jones JS, Ferguson KS, Daines C, Acton JD. Examining clinical trial results with single-subject analysis: an example involving behavioral and nutrition treatment for young children with cystic fibrosis. J Pediatr Psychol 2006;31:574–81. [DOI] [PubMed] [Google Scholar]
- 60.Rayner RJ, Tyrrell JC, Hiller EJ, Marenah C, Neugebauer MA, Vernon SA, et al. Night blindness and conjunctival xerosis caused by vitamin A deficiency in patients with cystic fibrosis. Arch Dis Child 1989;64:1151–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sitrin MD, Lieberman F, Jensen WE, Noronha A, Milburn C, Addington W. Vitamin E deficiency and neurologic disease in adults with cystic fibrosis. Ann Intern Med 1987;107:51–4. [DOI] [PubMed] [Google Scholar]
- 62.Marcus MS, Sondel SA, Farrell PM, Laxova A, Carey PM, Langhough R, et al. Nutritional status of infants with cystic fibrosis associated with early diagnosis and intervention. Am J Clin Nutr 1991;54:578–85. [DOI] [PubMed] [Google Scholar]
- 63.Koscik RL, Farrell PM, Kosorok MR, Zaremba KM, Laxova A, Lai HC, et al. Cognitive function of children with cystic fibrosis: deleterious effect of early malnutrition. Pediatrics 2004;113:1549–58. [DOI] [PubMed] [Google Scholar]
- 64.Wilson DC, Rashid M, Durie PR, Tsang A, Kalnins D, Andrew M, et al. Treatment of vitamin K deficiency in cystic fibrosis: effectiveness of a daily fat-soluble vitamin combination. J Pediatr 2001;138:851–5. [DOI] [PubMed] [Google Scholar]
- 65.Krebs NF, Westcott JE, Arnold TD, Kluger BM, Accurso FJ, Miller LV, et al. Abnormalities in zinc homeostasis in young infants with cystic fibrosis. Pediatr Res 2000;48:256–61. [DOI] [PubMed] [Google Scholar]
- 66.Easley D, Krebs N, Jefferson M, Miller L, Erskine J, Accurso F, et al. Effect of pancreatic enzymes on zinc absorption in cystic fibrosis. J Pediatr Gastroenterol Nutr 1998;26:136–9. [DOI] [PubMed] [Google Scholar]
- 67.Arvanitakis SN, Lobeck CC. Metabolic alkalosis and salt depletion in cystic fibrosis. J Pediatr 1973;82:535–6. [DOI] [PubMed] [Google Scholar]
- 68.Laughlin JJ, Brady MS, Eigen H. Changing feeding trends as a cause of electrolyte depletion in infants with cystic fibrosis. Pediatrics 1981;68: 203–7. [PubMed] [Google Scholar]
- 69.Ozcelik U, Goeman A, Niper N, Coskun T, Yilmaz E, Ozgue M. Sodium chloride deficiency in cystic fibrosis patients. Eur J Pediatr 1994;153: 829–31. [DOI] [PubMed] [Google Scholar]
- 70.Food and Nutrition Board, Institute of Medicine. Dietary Reference Intakes for Water, Potassium, Sodium, Chloride, and Sulfate Washington, DC: The National Academies Press; 2004. [Google Scholar]
- 71.Coates A, Crofton PM, Marshall T. Prospective audit of salt supplementation in CF infants [abstract]. Pediatr Pulmonol Suppl 2005;28: 345–6. [Google Scholar]
- 72.Adair SM, Bowen WH, Burt BA, Kumar JV, Levy SM, Pendrys DG, et al. Recommendations for Using Fluoride to Prevent and Control Dental Caries in the United States 2001. Centers for Disease Control and Prevention; Web site. Available at: www.cdc.gov/mmwrhtml/rr5014a1.htm. Accessed January 20, 2009. [Google Scholar]
- 73.Cantin AM, White TB, Cross CE, Forman HJ, Sokol RJ, Borowitz D. Antioxidants in cystic fibrosis: conclusions from the CF Antioxidant Workshop. Free Radic Biol Med 2007;42:15–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Steinkamp G, Demmelmair H, Ruhl-Bagheri I, von der Hardt H, Koletzko B. Energy supplements rich in linoleic acid improve body weight and essential fatty acid status of cystic fibrosis patients. J Pediatr Gastroenterol Nutr 2000;31:418–23. [DOI] [PubMed] [Google Scholar]
- 75.van Egmond AW, Kosorok MR, Koscik R, Laxova A, Farrell PM. Effect of linoleic acid intake on growth of infants with cystic fibrosis. Am J Clin Nutr 1996;63:746–52. [DOI] [PubMed] [Google Scholar]
- 76.Hooper L, Thompson RL, Harrison RA, Summerbell CD, Moore H, Worthington HV, et al. Omega 3 fatty acids for prevention and treatment of cardiovascular disease. Cochrane Database of Systematic Reviews 2004;2004:CD003177.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Use of formula fortified with DHA in infants with cystic fibrosis 2009. National Institutes of Health ClinicalTrials.gov; Web site. Available at:http://clinicaltrials.gov/ct/show/NCT00530244?order=1. Accessed January 20, 2009. [Google Scholar]
- 78.Farrell PM, Li Z, Kosorok MR, Laxova A, Green CG, Collins J, et al. Longitudinal evaluation of bronchopulmonary disease in children with cystic fibrosis. Pediatr Pulmonol 2003;36:230–40. [DOI] [PubMed] [Google Scholar]
- 79.Armstrong DS, Grimwood K, Carzino R, Carlin JB, Olinsky A, Phelan PD. Lower respiratory infection and inflammation in infants with newly diagnosed cystic fibrosis. Br Med J 1995;310:1571–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Linnane BM, Hall GL, Nolan G, Brennan S, Stick SM, Sly PD, et al. Lung function in infants with cystic fibrosis diagnosed by newborn screening. Am J Respir Crit Care Med 2008;178:1238–44. [DOI] [PubMed] [Google Scholar]
- 81.Khan TZ, Wagener JS, Bost T, Martinez J, Accurso FJ, Riches DW. Early pulmonary inflammation in infants with cystic fibrosis. Am J Respir Crit Care Med 1995;151:1075–82. [DOI] [PubMed] [Google Scholar]
- 82.Gilljam H, Stenlund C, Ericsson-Hollsing A, Strandvik B. Passive smoking in cystic fibrosis. Respir Med 1990;84:289–91. [DOI] [PubMed] [Google Scholar]
- 83.Campbell PW 3rd, Parker RA, Roberts BT, Krishnamani MR, Phillips JA 3rd. Association of poor clinical status and heavy exposure to tobacco smoke in patients with cystic fibrosis who are homozygous for the F508 deletion. J Pediatr 1992;120:261–4. [DOI] [PubMed] [Google Scholar]
- 84.Flume PA, Robinson KA, O’Sullivan BP, Finder JD, Vender RL, Willey-Courand D- B, et al. Cystic fibrosis pulmonary guidelines: airway clearance therapies. Respir Care 2009;54:522–37. [PubMed] [Google Scholar]
- 85.McCool FD, Rosen MJ. Nonpharmacologic airway clearance therapies: ACCP evidence-based clinical practice guidelines. Chest 2006;129(1 Suppl):250S–9S. [DOI] [PubMed] [Google Scholar]
- 86.Hardy KA, Wolfson MR, Schidlow DV, Shaffer TH. Mechanics and energetics of breathing in newly diagnosed infants with cystic fibrosis: effect of combined bronchodilator and chest physical therapy. Pediatr Pulmonol 1989;6:103–8. [DOI] [PubMed] [Google Scholar]
- 87.Maayan C, Bar-Yishay E, Yaacobi T, Marcus Y, Katznelson D, Yahav Y, et al. Immediate effect of various treatments on lung function in infants with cystic fibrosis. Respiration 1989;55:144–51. [DOI] [PubMed] [Google Scholar]
- 88.Flume PA, O’Sullivan BP, Robinson KA, Goss CH, Mogayzel PJ Jr., Willey-Courand DB, et al. Cystic fibrosis pulmonary guidelines: chronic medications for maintenance of lung health. Am J Respir Crit Care Med 2007;176:957–69. [DOI] [PubMed] [Google Scholar]
- 89.Phillips GE, Pike SE, Rosenthal M, Bush A. Holding the baby: head downwards positioning for physiotherapy does not cause gastro-oesophageal reflux. Eur Respir J 1998;12:954–7. [DOI] [PubMed] [Google Scholar]
- 90.Button BM, Heine RG, Catto-Smith AG, Phelan PD, Olinsky A. Postural drainage and gastro-oesophageal reflux in infants with cystic fibrosis. Arch Dis Child 1997;76:148–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Button BM, Heine RG, Catto-Smith AG, Olinsky A, Phelan PD, Ditchfield MR, et al. Chest physiotherapy in infants with cystic fibrosis: to tip or not? A five-year study. Pediatr Pulmonol 2003;35:208–13. [DOI] [PubMed] [Google Scholar]
- 92.Button BM, Heine RG, Catto-Smith AG, Phelan PD, Olinsky A. Chest physiotherapy, gastro-oesophageal reflux, and arousal in infants with cystic fibrosis. Arch Dis Child 2004;89:435–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nixon GM, Armstrong DS, Carzino R, Carlin JB, Olinsky A, Robertson CF, et al. Clinical outcome after early Pseudomonas aeruginosa infection in cystic fibrosis. J Pediatr 2001;138:699–704. [DOI] [PubMed] [Google Scholar]
- 94.Kosorok MR, Jalaluddin M, Farrell PM, Shen G, Colby CE, Laxova A, et al. Comprehensive analysis of risk factors for acquisition of Pseudomonas aeruginosa in young children with cystic fibrosis. Pediatr Pulmonol 1998;26:81–8. [DOI] [PubMed] [Google Scholar]
- 95.Saiman L, Siegel J. Infection control recommendations for patients with cystic fibrosis: microbiology, important pathogens, and infection control practices to prevent patient-to-patient transmission. Infect Control Hosp Epidemiol 2003;24(5 Suppl):S6–52. [DOI] [PubMed] [Google Scholar]
- 96.Healthcare Infection Control Practices Advisory Committee. Guideline for environmental infection control in health-care facilities 2003. Centers for Disease Control and Prevention; Web site. Available at: www.cdc.gov/ncidod/dhqp/pdf/guidelines/Enviro_guide_03.pdf. Accessed January 20, 2009. [Google Scholar]
- 97.American Academy of Pediatrics Committee on Infectious Diseases. Recommended childhood and adolescent immunization schedules– United States, 2009. Pediatrics 2009;123:189–90. [DOI] [PubMed] [Google Scholar]
- 98.Abman SH, Ogle JW, Butler-Simon N, Rumack CM, Accurso FJ. Role of respiratory syncytial virus in early hospitalizations for respiratory distress of young infants with cystic fibrosis. J Pediatr 1988;113: 826–30. [DOI] [PubMed] [Google Scholar]
- 99.American Academy of Pediatrics Committee on Infectious Diseases. Revised indications for the use of palivizumab and respiratory syncytial virus immune globulin intravenous for the prevention of respiratory syncytial virus infections. Pediatrics 2003;112:1442–6. [PubMed] [Google Scholar]
- 100.Giebels K, Marcotte JE, Podoba J, Rousseau C, Denis MH, Fauvel V, et al. Prophylaxis against respiratory syncytial virus in young children with cystic fibrosis. Pediatr Pulmonol 2008;43:169–74. [DOI] [PubMed] [Google Scholar]
- 101.Speer ME, Fernandes CJ, Boron M, Groothuis JR. Use of Palivizumab for prevention of hospitalization as a result of respiratory syncytial virus in infants with cystic fibrosis. Pediatr Infect Dis J 2008;27:559–61. [DOI] [PubMed] [Google Scholar]
- 102.Treggiari MM, Rosenfeld M, Retsch-Bogart G, Gibson R, Ramsey B. Approach to eradication of initial Pseudomonas aeruginosa infection in children with cystic fibrosis. Pediatr Pulmonol 2007;42:751–6. [DOI] [PubMed] [Google Scholar]
- 103.Kosorok MR, Zeng L, West SE, Rock MJ, Splaingard ML, Laxova A, et al. Acceleration of lung disease in children with cystic fibrosis after Pseudomonas aeruginosa acquisition. Pediatr Pulmonol 2001;32:277–87. [DOI] [PubMed] [Google Scholar]
- 104.Emerson J, Rosenfeld M, McNamara S, Ramsey B, Gibson RL. Pseudomonas aeruginosa and other predictors of mortality and morbidity in young children with cystic fibrosis. Pediatr Pulmonol 2002;34:91–100. [DOI] [PubMed] [Google Scholar]
- 105.Abman SH, Ogle JW, Harbeck RJ, Butler-Simon N, Hammond KB, Accurso FJ. Early bacteriologic, immunologic, and clinical courses of young infants with cystic fibrosis identified by neonatal screening. J Pediatr 1991;119:211–7. [DOI] [PubMed] [Google Scholar]
- 106.Armstrong DS, Grimwood K, Carlin JB, Carzino R, Olinsky A, Phelan PD. Bronchoalveolar lavage or oropharyngeal cultures to identify lower respiratory pathogens in infants with cystic fibrosis. Pediatr Pulmonol 1996;21:267–75. [DOI] [PubMed] [Google Scholar]
- 107.Rosenfeld M, Emerson J, Accurso F, Armstrong D, Castile R, Grimwood K, et al. Diagnostic accuracy of oropharyngeal cultures in infants and young children with cystic fibrosis. Pediatr Pulmonol 1999; 28:321–8. [DOI] [PubMed] [Google Scholar]
- 108.Cystic Fibrosis Foundation. Cystic Fibrosis Foundation Patient Registry, 2007 Annual Data Report to the Center Directors, Bethesda, Maryland, 2008. Cystic Fibrosis Foundation; 2007. [Google Scholar]
- 109.Littlewood J, Bevan A, Connett G, Conway S, Dodd M, Govan J, et al. Antibiotic treatment for cystic fibrosis 2002. Cystic Fibrosis Trust; Web site. Available at: http://www.cftrust.org.uk/aboutcf/publications/consensusdoc/C_3200Antibiotic_Treatment.pdf. Accessed January 20, 2009. [Google Scholar]
- 110.Smyth A, Walters S. Prophylactic anti-staphylococcal antibiotics for cystic fibrosis. Cochrane Database of Systematic Reviews 2003;3: CD001912. [DOI] [PubMed] [Google Scholar]
- 111.Harrison CJ, Marks MI, Welch DF, Sharma BB, Baker D, Dice J. A multicenter comparison of related pharmacologic features of cephalexin and dicloxacillin given for two months to young children with cystic fibrosis. Pediatr Pharmacol 1985;5:7–16. [PubMed] [Google Scholar]
- 112.Beardsmore CS, Thompson JR, Williams A, McArdle EK, Gregory GA, Weaver LT, et al. Pulmonary function in infants with cystic fibrosis: the effect of antibiotic treatment. Arch Dis Child 1994;71:133–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Stutman HR, Lieberman JM, Nussbaum E, Marks MI. Antibiotic prophylaxis in infants and young children with cystic fibrosis: a randomized controlled trial. J Pediatr 2002;140:299–305. [DOI] [PubMed] [Google Scholar]
- 114.Weaver LT, Green MR, Nicholson K, Mills J, Heeley ME, Kuzemko JA, et al. Prognosis in cystic fibrosis treated with continuous flucloxacillin from the neonatal period. Arch Dis Child 1994;70:84–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wright GL, Harper J. Fusidic acid and lincomycin therapy in staphylococcal infections in cystic fibrosis. Lancet 1970;1:9–14. [DOI] [PubMed] [Google Scholar]
- 116.Hudson VL, Wielinski CL, Regelmann WE. Prognostic implications of initial oropharyngeal bacterial flora in patients with cystic fibrosis diagnosed before the age of two years. J Pediatr 1993;122:854–60. [DOI] [PubMed] [Google Scholar]
- 117.Aebi C, Bracher R, Liechti-Gallati S, Tschappeler H, Rudeberg A, Kraemer R. The age at onset of chronic Pseudomonas aeruginosa colonization in cystic fibrosis–prognostic significance. Eur J Pediatr 1995; 154(9 Suppl. 4):S69–73. [DOI] [PubMed] [Google Scholar]
- 118.Ratjen F, Munck A, Kho P. Short and long-term efficacy of inhaled tobramycin in early P. aeruginosa infection: the ELITE study. Pediatr Pulm 2008;(Suppl 31):319. [Google Scholar]
- 119.Gibson RL, Emerson J, McNamara S, Burns JL, Rosenfeld M, Yunker A, et al. Significant microbiological effect of inhaled tobramycin in young children with cystic fibrosis. Am J Respir Crit Care Med 2003;167:841–9. [DOI] [PubMed] [Google Scholar]
- 120.Ratjen F, Doring G, Nikolaizik WH. Effect of inhaled tobramycin on early Pseudomonas aeruginosa colonisation in patients with cystic fibrosis. Lancet 2001;358:983–4. [DOI] [PubMed] [Google Scholar]
- 121.Frederiksen B, Koch C, Hoiby N. Antibiotic treatment of initial colonization with Pseudomonas aeruginosa postpones chronic infection and prevents deterioration of pulmonary function in cystic fibrosis. Pediatr Pulmonol 1997;23:330–5. [DOI] [PubMed] [Google Scholar]
- 122.Heinzl B, Eber E, Oberwaldner B, Haas G, Zach MS. Effects of inhaled gentamicin prophylaxis on acquisition of Pseudomonas aeruginosa in children with cystic fibrosis: a pilot study. Pediatr Pulmonol 2002;33: 32–7. [DOI] [PubMed] [Google Scholar]
- 123.Tepper RS, Hiatt P, Eigen H, Scott P, Grosfeld J, Cohen M. Infants with cystic fibrosis: pulmonary function at diagnosis. Pediatr Pulmonol 1988;5:15–8. [DOI] [PubMed] [Google Scholar]
- 124.Villa MP, Pagani J, Lucidi V, Palamides S, Ronchetti R. Nocturnal oximetry in infants with cystic fibrosis. Arch Dis Child 2001;84:50–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Mukhopadhyay S, Kirby ML, Duncan AW, Carswell F. Early focal abnormalities on chest radiographs and respiratory prognosis in children with cystic fibrosis. Br J Radiol 1996;69:122–5. [DOI] [PubMed] [Google Scholar]
- 126.Littlewood J, Balfour-Lynn I, Barnes R, Bilton D, Dinwiddie R, Doull I, et al. Standards for the clinical care of children and adults with cystic fibrosis in the United Kingdom 2001. Cystic Fibrosis Trust; Web site. Available at: http://www.cftrust.org.uk/aboutcf/publications/consensusdoc/C_3000Standards_of_Care.pdf. Accessed January 20, 2009. [Google Scholar]
- 127.Kerem E, Conway S, Elborn S, Heijerman H. Standards of care for patients with cystic fibrosis: a European consensus. J Cyst Fibros 2005;4:7–26. [DOI] [PubMed] [Google Scholar]
- 128.Martinez TM, Llapur CJ, Williams TH, Coates C, Gunderman R, Cohen MD, et al. High-resolution computed tomography imaging of airway disease in infants with cystic fibrosis. Am J Respir Crit Care Med 2005;172:1133–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Long FR, Williams RS, Castile RG. Structural airway abnormalities in infants and young children with cystic fibrosis. J Pediatr 2004;144:154–61. [DOI] [PubMed] [Google Scholar]
- 130.Davis SD, Fordham LA, Brody AS, Noah TL, Retsch-Bogart GZ, Qaqish BF, et al. Computed tomography reflects lower airway inflammation and tracks changes in early cystic fibrosis. Am J Respir Crit Care Med 2007;175:943–50. [DOI] [PubMed] [Google Scholar]
- 131.Lynch DA, Brasch RC, Hardy KA, Webb WR. Pediatric pulmonary disease: assessment with high-resolution ultrafast CT. Radiology 1990;176:243–8. [DOI] [PubMed] [Google Scholar]
- 132.Gustafsson PM, De Jong PA, Tiddens HA, Lindblad A. Multiple-breath inert gas washout and spirometry versus structural lung disease in cystic fibrosis. Thorax 2008;63:129–34. [DOI] [PubMed] [Google Scholar]
- 133.Brenner DJ, Hall EJ. Computed tomography: an increasing source of radiation exposure. N Engl J Med 2007;357:2277–84. [DOI] [PubMed] [Google Scholar]
- 134.Turner DJ, Lanteri CJ, LeSouef PN, Sly PD. Improved detection of abnormal respiratory function using forced expiration from raised lung volume in infants with cystic fibrosis. Eur Respir J 1994;7:1995–9. [PubMed] [Google Scholar]
- 135.Ranganathan SC, Bush A, Dezateux C, Carr SB, Hoo AF, Lum S, et al. Relative ability of full and partial forced expiratory maneuvers to idetify diminished airway function in infants with cystic fibrosis. Am J Respir Crit Care Med 2002;166:1350–7. [DOI] [PubMed] [Google Scholar]
- 136.Ranganathan SC, Stocks J, Dezateux C, Bush A, Wade A, Carr S, et al. The evolution of airway function in early childhood following clinical diagnosis of cystic fibrosis. Am J Respir Crit Care Med 2004;169:928–33. [DOI] [PubMed] [Google Scholar]
- 137.Ranganathan SC, Dezateux C, Bush A, Carr SB, Castle RA, Madge S, et al. Airway function in infants newly diagnosed with cystic fibrosis. Lancet 2001;358:1964–5. [DOI] [PubMed] [Google Scholar]
- 138.Kozlowska WJ, Bush A, Wade A, Aurora P, Carr SB, Castle RA, et al. Lung function from infancy to the preschool years after clinical diagnosis of cystic fibrosis. Am J Respir Crit Care Med 2008;178:42–9. [DOI] [PubMed] [Google Scholar]
- 139.Lum S, Gustafsson P, Ljungberg H, Hulskamp G, Bush A, Carr SB, et al. Early detection of cystic fibrosis lung disease: multiple-breath washout versus raised volume tests. Thorax 2007;62:341–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kraemer R Early detection of lung function abnormalities in infants with cystic fibrosis. J R Soc Med 1989;82(Suppl 16):21–5. [PMC free article] [PubMed] [Google Scholar]
- 141.Beardsmore CS, Bar-Yishay E, Maayan C, Yahav Y, Katznelson D, Godfrey S. Lung function in infants with cystic fibrosis. Thorax 1988; 43:545–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Godfrey S, Mearns M, Howlett G. Serial lung function studies in cystic fibrosis in the first 5 years of life. Arch Dis Child 1978;53:83–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Jones AP, Wallis CE, Kearney CE. Recombinant human deoxyribonuclease for cystic fibrosis. Cochrane Database Syst Rev 2003:CD001127. [DOI] [PubMed] [Google Scholar]
- 144.Berge MT, Wiel E, Tiddens HA, Merkus PJ, Hop WC, de Jongste JC. DNase in stable cystic fibrosis infants: a pilot study. J Cyst Fibros 2003;2:183–8. [DOI] [PubMed] [Google Scholar]
- 145.Dellon EP, Donaldson SH, Johnson R, Davis SD. Safety and tolerability of inhaled hypertonic saline in young children with cystic fibrosis. Pediatr Pulmonol 2008;43:1100–6. [DOI] [PubMed] [Google Scholar]
- 146.Subbarao P, Balkovec S, Solomon M, Ratjen F. Pilot study of safety and tolerability of inhaled hypertonic saline in infants with cystic fibrosis. Pediatr Pulmonol 2007;42:471–6. [DOI] [PubMed] [Google Scholar]