

Summary
Correct localization of Rab GTPases in cells is critical for proper function in membrane trafficking, yet the mechanisms that target Rabs to specific subcellular compartments remain controversial. Guanine nucleotide exchange factors (GEFs) activate and consequently stabilize Rab substrates on membranes, thus implicating GEFs as the primary determinants of Rab localization. A competing hypothesis is that the Rab C-terminal hypervariable domain (HVD) serves as a subcellular targeting signal. In this study we present a unifying mechanism in which the HVD controls targeting of certain Rabs by mediating interaction with their GEFs. We demonstrate that the TRAPP complexes, two related GEFs that use the same catalytic site to activate distinct Rabs, distinguish between Ypt1 (Rab1) and Ypt31/32 (Rab11) via their divergent HVDs. Remarkably, we find that HVD length gates Rab access to the TRAPPII complex by constraining the distance between the nucleotide-binding domain and the membrane surface.
eTOC blurb
Thomas et al. show that certain Rabs are recognized by their guanine nucleotide exchange factors in part through the Rab C-terminal hypervariable domain (HVD). This suggests a model in which the HVD contributes to Rab localization by mediating Rab activation at target organelles.
Graphical Abstract
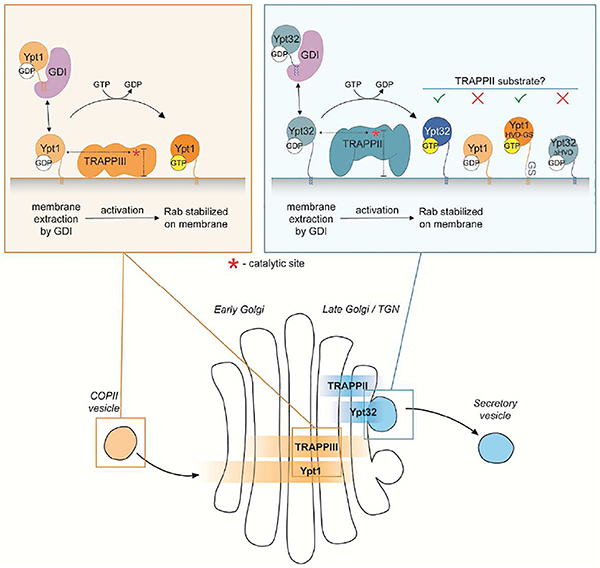
Introduction
In eukaryotic cells Rab family GTPases coordinate virtually every step of membrane transport. More than 60 different Rabs have been identified in mammals and the budding yeast model possesses 11 distinct Rab GTPases (Hutagalung and Novick, 2011). Rabs are critical determinants of organelle identity and membrane organization, with each distinct Rab displaying a characteristic subcellular localization (Pfeffer, 2012). Correct and specific subcellular targeting is essential for proper Rab function, yet the mechanisms controlling Rab localization remain controversial.
Rabs function as molecular switches, cycling between an inactive GDP-bound state and an active GTP-bound state in which they recruit effector proteins to mediate trafficking events (Stenmark, 2009). Rabs are modified at their C-terminus with prenyl groups that function as membrane anchors. Inactive Rabs interact reversibly with a GDP dissociation inhibitor (GDI), which acts as a chaperone to protect the hydrophobic prenyl groups from the cytosol (Goody et al., 2005). The GDI delivers inactive Rabs to organelles, where the Rab binds by inserting its prenylated C-terminus into the membrane. GDI displacement factors (GDFs) have been proposed to catalyze the release of the Rab from GDI during membrane delivery (Sivars et al., 2003). Once at the membrane, the inactive Rab can be re-extracted by a GDI unless it is activated by its guanine nucleotide exchange factor (GEF). GDP exchange for GTP prevents interaction with the GDI and thus stabilizes the Rab on the membrane (Wu et al., 2010). Active Rabs recruit effectors and remain membrane associated until inactivated by a GTPase activating protein (GAP), at which point the Rab can be extracted by the GDI for another cycle of transport.
The mechanisms controlling Rab localization are complex and involve input from multiple factors including GEFs, GAPs, effectors, and membrane lipids. Though the GDI is responsible for Rab membrane delivery, the same GDI chaperones many Rabs and therefore cannot confer specificity. Similarly, only one protein with GDF activity has been identified to date (Sivars et al., 2003); therefore GDFs are unlikely to specify targeting of most Rabs. In contrast, the majority of GEFs display substrate specificity, allowing for precise Rab targeting. Most Rabs become mislocalized in the absence of their GEFs, indicating that GEF activity is required for proper localization (Cabrera and Ungermann, 2013; Gerondopoulos et al., 2012; Morozova et al., 2006; Thomas and Fromme, 2016; Thomas et al., 2018). Moreover, several studies have shown that anchoring GEFs to mitochondria causes a corresponding relocalization of the Rab substrate, suggesting that GEF activity is sufficient for Rab targeting (Blümer et al., 2013; Gerondopoulos et al., 2012). These findings have led to a model in which GEFs are the primary regulators of Rab localization, as they activate and stabilize Rabs on membranes. In this model, the GDI indiscriminately delivers inactive Rabs to organelles, and the Rab will be re-extracted unless activated by its GEF.
Rab proteins consist of a conserved nucleotide-binding domain (NBD) that is connected to the prenylated C-terminus by a flexible linker (Itzen and Goody, 2011). This linker, termed the hypervariable domain (HVD) (Li et al., 2014), has a high degree of sequence divergence and also varies in length, spanning from 27 to greater than 40 residues. Early studies proposed that the HVD functions as a Rab subcellular targeting signal (Brennwald and Novick, 1993; Chavrier et al., 1991; Stenmark et al., 1994). These studies used Rab chimeras to demonstrate that swapping the HVD of distinct Rabs correspondingly swapped Rab localization. However, complementation assays have shown that the sequence of the HVD is not essential, suggesting that HVD identity is not required for Rab function (Beranger et al., 1994; Brennwald and Novick, 1993; Dunn et al., 1993). Moreover, subsequent studies tested a more comprehensive group of Rabs, and found that the HVD might not function as a general Rab targeting signal (Ali et al., 2004; Li et al., 2014). Therefore, it remains unclear as to whether the HVD plays an important role in Rab targeting.
In yeast the Rab GTPases Ypt1 (Rab1) and Ypt31/32 (Rab11) regulate trafficking in the secretory, endocytic, and autophagic pathways (Lipatova et al., 2015). These Rabs are activated by the related multi-subunit TRAPP complexes, TRAPPII and TRAPPIII. Previous studies indicate that TRAPPIII is a specific Ypt1 GEF, yet whether TRAPPII specifically activates Ypt31/32 or promiscuously acts on both Ypt31/32 and Ypt1 has remained unresolved (Morozova et al., 2006; Pinar et al., 2015; Thomas and Fromme, 2016; Riedel et al., 2018). Furthermore, TRAPPII and TRAPPIII use the same active site residues to activate these distinct Rab substrates, indicating the existence of an unknown mechanism for Rab substrate specificity.
Using physiological GEF reconstitution assays, we now demonstrate that TRAPPII is a specific Ypt31/32 GEF when recruited to membranes by its regulatory GTPase Arf1. Using HVD chimeras and mutants, we find that the Rab HVD mediates TRAPP substrate specificity both in vitro and in vivo. As GEFs largely determine the localization of their substrates, our findings support a role for the HVD in Rab intracellular targeting by mediating GEF-Rab interactions. Remarkably, the length of the HVD is a critical determinant of the TRAPPII-Rab interaction, as Ypt1 can be converted into a TRAPPII substrate simply by lengthening its HVD. Therefore we propose that TRAPPII uses a steric gating mechanism to counterselect against Ypt1.
Results
Recruitment of TRAPPII to membranes by Arf1 enforces substrate specificity
Budding yeast possess two related TRAPP complexes that coordinate Rab activation in the secretory pathway. Both complexes share an identical catalytic core made up of seven subunits. The TRAPPIII complex is distinguished by the addition of one distinct subunit whereas TRAPPII contains four additional subunits (Figure 1A). Multiple lines of evidence indicate that TRAPPIII is a specific Ypt1 GEF (Lynch-Day et al., 2010; Tan et al., 2013; Thomas et al., 2018). There is less of a consensus regarding the substrate of TRAPPII. Genetic evidence indicates that the essential role of TRAPPII is to catalyze nucleotide exchange for Ypt31/32 (Pinar et al., 2015; Sciorra et al., 2005; Thomas and Fromme, 2016). Ypt31/32 activation is nearly abolished in a trs130Δ33 TRAPPII mutant, whereas Ypt1 remains activated (Morozova et al., 2006; Thomas and Fromme, 2016). Moreover, TRAPPII is recruited to Golgi compartments directly preceding Ypt31/32, yet coincides with a decline in Ypt1 activation. These findings are consistent with TRAPPII and TRAPPIII being specific, non-redundant GEFs for Ypt31/32 and Ypt1, respectively. However, TRAPPII has been found to activate both Ypt31/32 and Ypt1 in GEF activity assays in vitro (Cai et al., 2008; Morozova et al., 2006; Pinar et al., 2015; Thomas and Fromme, 2016). We therefore sought biochemical conditions that faithfully reconstituted the specificity of TRAPP complexes observed in cells.
Figure 1. TRAPPII and TRAPPIII are specific GEFs for Ypt31/32 and Ypt1, respectively.

(A) Subunit composition of TRAPPII and TRAPPIII complexes. (B) Left: Prenylated Rab/GDI substrates and Arf1 used to recruit TRAPPII to membranes. Right: TRAPPII and rTRAPPIII were purified from yeast or E. coli using the indicated affinity tags. (C) Left: Representative traces showing activation of Rab substrates by TRAPPII in the presence of Ni2+ TGN liposomes. mock = buffer only control reaction. Right: Rates of TRAPPII-mediated nucleotide exchange calculated from the traces at left. Error bars represent 95% CIs for n ≥ 3 reactions. (D) Left: Representative traces showing activation of Rab substrates by TRAPPII in the presence of TGN liposomes (without Ni2+) and Arf1-GTP. Right: Rates of TRAPPII-mediated nucleotide exchange calculated from the traces at left. Error bars represent 95% CIs for n ≥ 3 reactions. n.d., not detectable (the Rab was not activated by the GEF and exponential functions could not be fit to experimental curves). (E) Left: Representative traces showing activation of Rab substrates by TRAPPIII in the presence of TGN liposomes. Right: Rates of TRAPPIII-mediated nucleotide exchange calculated from the traces at left. Error bars represent 95% CIs for n ≥ 3 reactions. (F) Schematic depicting the setup for the GEF-Rab Interaction Test (GRab-IT). (G) Left: TRAPPII localizes largely to Sec7-labeled late Golgi compartments (top) and representative images showing TRAPPII recruitment to mitochondria by nucleotide-free Ypt32, but not Ypt1 (bottom). Right: Quantification of TRAPPII recruitment to mitochondria. Error bars represent 95% CIs for n > 40 cells. See Figure S1 for images of TRAPPII with nucleotide-free Ypt6 and Sec4. (H) Left: TRAPPIII localizes to late Golgi compartments (top) and representative images showing TRAPPIII recruitment to mitochondria by nucleotide-free Ypt1, but not Ypt32 (bottom). Right: Quantification of TRAPPIII recruitment to mitochondria. Error bars represent 95% CIs for n > 40 cells. Scale bars, 2 μm. White arrowheads denote colocalization of TRAPP complexes and Sec7 at late Golgi compartments.
See also Figures S1 and S2.
To closely recapitulate conditions in vivo, we prepared prenylated Rab/GDI substrates by enzymatic synthesis as well as liposome membranes with a lipid composition approximating that of the trans-Golgi network (TGN) (Figure 1B and Table S1; Thomas and Fromme, 2016). We also prepared separate TGN liposomes containing 5% Ni2+-conjugated lipids to anchor His-tagged proteins on membranes. Serendipitously, we observed that the presence of Ni2+ enabled TRAPPII to directly interact with TGN membranes in the absence of any other recruiter and enhanced membrane affinity of TRAPPIII (Figure S1A and B). Ni2+ in membranes has also been shown to increase binding and activity for Arf-GEFs (Gustafson and Fromme, 2017). Our TAP-purified TRAPP complexes lack His-tags; therefore this effect appears to result from the positive charge of the Ni2+ ion, which may mimic some unknown factor in vivo. In GEF reconstitution experiments using prenylated Rab substrates and Ni2+ TGN membranes, TRAPPII robustly activated Ypt32 and, to a lesser extent, Ypt1 (Figure 1C), consistent without our previous findings (Thomas and Fromme, 2016).
In the absence of Ni2+, additional factors are required for TRAPPII to stably associate with membranes. Activated Arf1 recruits TRAPPII to Golgi compartments and enables TRAPPII to bind TGN liposomes lacking Ni2+ (Figure S1C; Thomas and Fromme, 2016). Surprisingly, when recruited by Arf1, TRAPPII no longer catalyzed nucleotide exchange on Ypt1 yet maintained full activity towards Ypt32 (Figure 1D). Thus, in an assay more closely replicating conditions in vivo, TRAPPII displays strong substrate specificity for Ypt31/32 over Ypt1. Furthermore, these data suggest that Arf1 recruitment of TRAPPII to the TGN in cells ensures TRAPPII acts on Ypt31/32 but not Ypt1.
In contrast to TRAPPII, TRAPPIII can associate directly with TGN membranes in the absence of other recruiters (Figure S1B), and we confirmed that TRAPPIII activated Ypt1, but not Ypt32, in the presence of TGN liposomes (Figure 1E; Thomas et al., 2018). Taken together these data indicate that, under physiological conditions, the two TRAPP complexes display distinct Rab substrate specificities.
The Ypt31/32 hypervariable domain (HVD) is required for substrate recognition by TRAPPII
To determine the basis for TRAPP Rab substrate specificity we developed a modified “anchor-away” assay to quantitatively measure GEF-substrate interactions in cells, called the GEF-Rab Interaction Test (GRab-IT; Figure 1F). mRFPmars-tagged Rabs were anchored to mitochondria by replacing their prenylated cysteines with the transmembrane domain of the mitochondrial outer membrane protein Fis1. These Rab “baits” were rendered nucleotide-free by introducing the following mutations: Ypt1(D124N) or Ypt31/32(D129N). These nucleotide-free Rab baits mimic the transition state of the nucleotide exchange reaction and are therefore high-affinity ligands of their GEFs. Rab baits were expressed in cells with fluorescently tagged endogenous TRAPPII (Trs130-mNeonGreen [mNG]) or TRAPPIII (Trs85-mNG). As TRAPP complexes localize largely to late Golgi/TGN compartments (Figure 1G and H), recruitment to the morphologically distinct mitochondria was easily visualized. The extent of colocalization between TRAPP complexes and Rab bait constructs was quantified as a readout of GEF-substrate interaction.
Consistent with our physiological GEF reconstitution assays, TRAPPII was recruited to mitochondria by nucleotide-free Ypt31/32, but not Ypt1 (Figure 1G and S1D). In contrast, TRAPPIII accumulated at mitochondria with nucleotide-free Ypt1, but not Ypt31/32 (Figure 1H and S1D). GEF recruitment in GRab-IT assays indicates a GEF-substrate interaction, as TRAPP complexes only recognized nucleotide-free or GDP-locked Rab substrates, but not wild-type or GTP-locked constructs (Figure S1E and F). TRAPP recruitment was also specific, as TRAPPII and TRAPPIII did not recognize nucleotide-free Ypt6 (Rab6) or Sec4 (Rab8) as substrates (Figure S1G and H). To extend the GRab-IT assay for analysis of other Rab-GEF pairs, we tested recruitment of Sec2 by its substrate Sec4. Indeed, a Sec4 bait construct specifically recruited Sec2 to mitochondria in a nucleotide-dependent manner (Figure S2), indicating that GRab-IT can be used as a tool to characterize other GEF-substrate interactions.
Ypt1 and Ypt31/32 are activated by an identical catalytic site in the TRAPP core (Cai et al., 2008; Thomas and Fromme, 2016), suggesting that complex-specific subunits mediate substrate specificity. The Ypt1 and Ypt31 NBDs share 48% sequence identity yet the Rabs have highly divergent HVDs (19% sequence identity), raising the possibility that TRAPP complexes recognize their substrates using the Rab HVD. To test this hypothesis, we generated HVD chimeras to use in GRab-IT and GEF reconstitution assays. The Ypt1 and Ypt31/32 HVDs diverge in both sequence and length, with the Ypt31/32 HVD being significantly longer than that of Ypt1. To generate HVD chimeras, we therefore swapped the C-terminal 38 residues of Ypt31 with the C-terminal 26 residues of Ypt1 (Figure 2A and Table S2).
Figure 2. The Ypt31/32 HVD provides specificity for TRAPPII-mediated activation.

(A) Schematic depicting Rab HVD chimeras used to test the role of the Rab HVD in TRAPP-mediated nucleotide exchange. (B) Left: Representative images showing recruitment of TRAPPII by nucleotide-free Ypt1HVD31, but not Ypt1. Right: Quantification of TRAPPII recruitment to mitochondria. Error bars represent 95% CIs for n > 40 cells. (C) Left: Representative images showing recruitment of TRAPPII by nucleotide-free Ypt31 but not Ypt31HVD1. Right: Quantification of TRAPPII recruitment to mitochondria. Error bars represent 95% CIs for n > 40 cells. (D) Coomassie-stained SDS-PAGE gel showing prenylated Rab/GDI substrates used to test the role of the HVD in TRAPP complex substrate specificity. (E) Left: Representative traces showing activation of Ypt1 versus Ypt1HVD31 by TRAPPII in the presence of TGN membranes and Arf1-GTP. mock = buffer only control reaction. Right: Rates of TRAPPII-mediated nucleotide exchange calculated from traces at left. Error bars represent 95% CIs for n = 3 reactions. n.d., not detectable (the Rab was not activated by the GEF and exponential functions could not be fit to experimental curves). (F) Left: Representative traces showing activation of Ypt31 versus Ypt31HVD1 by TRAPPII in the presence of TGN membranes and Arf1-GTP. Right: Rates of TRAPPII-mediated nucleotide exchange calculated from traces at left. Error bars represent 95% CIs for n = 3 reactions. Scale bars, 2 μm.
Whereas TRAPPII did not recognize nucleotide-free Ypt1 as a substrate, substitution of the Ypt31 HVD enabled a Ypt1HVD31 chimera to recruit TRAPPII to mitochondria (Figure 2B). In contrast, TRAPPII no longer recognizes Ypt31HVD1 as a substrate (Figure 2C). We also generated Ypt1HVD31 and Ypt31HVD1 chimeras as prenylated Rab/GDI complexes to test as TRAPPII substrates in GEF reconstitution assays (Figure 2D). Consistent with GRab-IT experiments, TRAPPII robustly activated Ypt1HVD31 but not Ypt1 (Figure 2E). In contrast, TRAPPII was not able to catalyze nucleotide exchange for Ypt31HVD1 (Figure 2F). Together these results indicate that the HVD of Ypt31/32 is required for activation by TRAPPII.
The Ypt1 HVD is important for TRAPPIII-mediated nucleotide exchange
We next used HVD chimeras to determine whether TRAPPIII is similarly dependent on the HVD of its substrate. In GRab-IT assays, Ypt1 and Ypt1HVD31 recruited TRAPPIII similarly (Figure 3A). TRAPPIII accumulated at mitochondria in cells expressing nucleotide-free Ypt31HVD1 (Figure 3B), albeit at lower levels compared to the baits containing the Ypt1 NBD. GRab-IT is an endpoint assay, measuring GEF-substrate interactions following stable expression of Rab baits. We therefore used GEF reconstitution assays to measure nucleotide exchange in real time, allowing for more subtle effects to be measured. In the presence of TGN membranes, TRAPPIII activated Ypt1 nearly twice as fast as the Ypt1HVD31 chimera (Figure 3C). TRAPPIII did not activate Ypt31 or Ypt31HVD1 under these conditions (Figure 3C), suggesting that the Ypt1 HVD does not provide enough binding energy to enable TRAPPIII to activate Ypt31. Nevertheless, the slower rate of Ypt1HVD31 activation and GRab-IT recruitment of TRAPPIII by Ypt31HVD1 indicate that the Ypt1 HVD plays a significant role in recognition by TRAPPIII.
Figure 3. The Ypt1 HVD contributes to activation by TRAPPIII.

(A) Left: Representative images showing recruitment of TRAPPIII to mitochondria by nucleotide-free Ypt1 as well as Ypt1HVD31. Right: Quantification of TRAPPIII recruitment to mitochondria. Error bars represent 95% CIs for n > 40 cells. (B) Left: Representative images showing recruitment of TRAPPIII by nucleotide-free Ypt31HVD1 but not Ypt31. Right: Quantification of TRAPPIII recruitment to mitochondria. Error bars represent 95% CIs for n > 40 cells. (C) Left: Representative traces showing activation of Ypt1, Ypt1HVD31, and Ypt31HVD1 by TRAPPIII in the presence of TGN membranes. mock = buffer only control reaction. Right: Rates of TRAPPIII-mediated nucleotide exchange calculated from the traces at left. Error bars represent 95% CIs for n = 3 reactions. n.d., not detectable (the Rab was not activated by the GEF and exponential functions could not be fit to experimental curves). (D) Schematic depicting Ypt6 HVD chimeras used to test whether the Ypt31/32 or Ypt1 HVD is sufficient for recognition by TRAPP complexes. (E) Left: Representative images showing a lack of TRAPP recruitment by nucleotide-free Ypt6HVD31 or Ypt6HVD1. Right: Quantification of TRAPP recruitment to mitochondria. Error bars represent 95% CIs for n > 40 cells. Scale bars, 2 μm.
Our data suggest that TRAPP complexes interact with specific Rab HVDs to facilitate nucleotide exchange. We therefore asked whether the Ypt31/32 or Ypt1 HVDs are sufficient for recognition by TRAPPII or TRAPPIII, respectively. We generated additional Rab chimeras by swapping the Ypt31 or Ypt1 HVD onto the Ypt6 NBD, a distinct Golgi Rab that is not a TRAPP substrate (Figure 3D and Table S2). Nucleotide-free Ypt6HVD31 and Ypt6HVD1 were not able to recruit TRAPPII or TRAPPIII, respectively (Figure 3E). This suggests that the HVD is not sufficient for substrate recognition and that TRAPP complexes engage substrates through a bipartite interaction. In this model, the shared catalytic core binds the conserved Rab NBD, and additional contacts are made with the divergent HVD to confer full substrate specificity.
The Ypt31/32 HVD is required for activation by TRAPPII in vivo
Our GEF reconstitution and GRab-IT experiments indicate that the Rab HVD is an important mediator of TRAPP complex substrate specificity. We therefore examined the functional relevance of GEF-HVD interactions in vivo. We found that Ypt32HVD1 did not complement a ypt31Δypt32Δ mutant (Figure 4A), consistent with our observation that this chimera is not recognized or activated by TRAPPII (Figure 2C and F). Both Ypt32 and Ypt32HVD1 were expressed at similar levels (Figure S3A), indicating that the lack of viability was not due to lower expression of Ypt32HVD1.
Figure 4. The Ypt31/32 HVD is required for activation by TRAPPII in vivo.

(A) Ypt32HVD1 does not complement a ypt31Δypt32Δ mutant in a wild-type background. (B) Left: Localization of mNG-tagged Ypt32 versus Ypt32HVD1 relative to the late Golgi marker Sec7-DsRed. Right: Quantification of Golgi (active) versus cytosolic/ER-localized (inactive) Rab from the images at left. Error bars represent 95% CIs for n ≥ 22 Golgi compartments. (C) Time-lapse imaging series (top) and normalized fluorescence (bottom) for Ypt32 versus Ypt32HVD1 and Sec7 at a single Golgi compartment. Scale bar, 2 μm. White arrowheads denote colocalization of Rabs and Sec7 at late Golgi compartments.
See also Figure S3.
The correct localization of fluorescently tagged Rabs is a readout of their activation by GEFs in cells, as inactive Rabs remain cytosolic (GDI-bound) or at ER membranes (Ali et al., 2004; Blümer et al., 2013; Cabrera and Ungermann, 2013; Gerondopoulos et al., 2012; Stenmark et al., 1994). mNG-Ypt32 is functional and localizes to TGN compartments and polarized secretory vesicles (Figure 4B and S3B). In contrast, mNG-Ypt32HVD1 was largely cytosolic or at ER membranes, indicating that this chimera is not efficiently activated in cells.
We observed small amounts of mNG-Ypt32HVD1 at the Golgi; therefore we performed time-lapse imaging to test whether this chimera is weakly activated by TRAPP complexes. As yeast Golgi compartments undergo cisternal maturation (Losev et al., 2006), we expect a GEF to arrive at the Golgi coincident with or slightly before its substrate. In agreement with previous studies, we found that wild-type mNG-Ypt32 accumulated at the TGN after Sec7, correlating with activation by TRAPPII (Figure 4C and 5D; Kim et al., 2016; McDonold and Fromme, 2014; Thomas and Fromme, 2016). In contrast, mNG-Ypt32HVD1 arrived at the Golgi before Sec7, coinciding with peak TRAPPIII recruitment (Thomas et al., 2018), suggesting that TRAPPIII weakly activates Ypt32HVD1 in cells. Taken together, these results indicate that the Ypt31/32 HVD is required for activation by TRAPPII in vivo.
Figure 5. The HVD provides TRAPP complex substrate specificity in vivo.

(A) Ypt1HVD32 was tested for its ability to complement a ypt1Δ mutant in a trs85Δ (TRAPPIII mutant) background. (B) Left: Localization of mNG-tagged Ypt1 versus Ypt1HVD32 relative to the late Golgi marker Sec7-DsRed. Right: Colocalization analysis of each Rab construct with Sec7. Error bars represent 95% CIs for n > 40 cells. (C) Time-lapse imaging series (top) and normalized fluorescence (bottom) for Ypt1 versus Ypt1HVD32 and Sec7 at a single Golgi compartment. (D) Quantification of peak-to-peak times for wild-type Rabs, HVD chimeras, and TRAPP complexes (Thomas and Fromme, 2016; Thomas et al., 2018) at late Golgi compartments. t = 0 is set to peak Sec7 recruitment. Error bars represent 95% CIs for n ≥ 19 series. (E) Left: Localization of mNG-tagged Ypt1 versus Ypt1HVD32 relative to Sec7-DsRed in a trs85Δ mutant. Right: Quantification of Golgi (active) versus cytosolic/ER-localized (inactive) Rab from the images at left. Error bars represent 95% CIs for n ≥ 25 Golgi compartments. Scale bars, 2 μm. White arrowheads denote colocalization of Rabs and Sec7 at late Golgi compartments.
See also Figure S4.
The Rab HVD dictates substrate specificity in vivo
We next used the reciprocal mNG-Ypt1HVD32 chimera to further test the importance of the HVD for Rab activation in cells. mNG-Ypt1 and mNG-Ypt1HVD32 are expressed at similar levels, and both complement a ypt1Δ mutant in a wild-type background (Figure S4A and B). This is consistent with our observation that TRAPPIII can recognize and activate Ypt1HVD32 (Figure 3A and C), as well as previous studies that found that the Ypt1 HVD is dispensable for cell viability (Brennwald and Novick, 1993). However, in a trs85Δ TRAPPIII mutant background, mNG-Ypt1HVD32 was nearly non-functional (Figure 5A), suggesting that when TRAPPIII activity is compromised the Ypt1 HVD is required for nucleotide exchange.
In wild-type cells, mNG-Ypt1 localizes to the Golgi, overlapping significantly with Sec7 (Figure 5B; Kim et al., 2016; Sclafani et al., 2010). Consistent with its ability to support cell viability, mNG-Ypt1HVD32 was largely activated at Sec7-labeled late-Golgi compartments. mNG-Ypt1HVD32 overlapped significantly more with Sec7 than did mNGYpt1 (Figure 5B), suggesting that the chimera remains at Golgi compartments later than wild-type Ypt1. To test this, we performed time-lapse imaging experiments. Consistent with previous studies, mNG-Ypt1 levels peaked approximately 20 seconds upstream of Sec7, correlating with TRAPPIII-mediated nucleotide exchange (Figure 5C and D; McDonold and Fromme, 2014; Thomas et al., 2018). mNG-Ypt1HVD32 also accumulated upstream of Sec7, but recruitment of the chimera peaked significantly later. Though both mNG-Ypt1 and mNG-Ypt1HVD32 began to accumulate at the same time, mNG-Ypt1 levels declined at the TGN while mNG-Ypt1HVD32 levels continued to rise. Given our previous observations, this suggests that both Rab constructs are initially activated by TRAPPIII at the medial/late Golgi, but that mNG-Ypt1HVD32 continues to be activated by TRAPPII at the TGN.
To further test whether Ypt1HVD32 is activated by both TRAPPIII and TRAPPII, we observed Rab localization in a trs85Δ TRAPPIII mutant. As reported previously (Thomas et al., 2018), mNG-Ypt1 was largely mislocalized to the cytosol and ER in trs85Δ cells (Figure 5E). In contrast, mNG-Ypt1HVD32 remained activated at Golgi compartments, suggesting that TRAPPII catalyzes exchange for Ypt1HVD32. Interestingly, Ypt1HVD32 was efficiently activated and yet not able to support growth in trs85Δ cells. This underscores the importance of proper localization for Rab function: Ypt1HVD32 likely does not support cell viability because it is activated ectopically at the TGN by TRAPPII.
The Ypt1 GAP Gyp1 is recruited by Ypt32 to inactivate Ypt1, resulting in a decline of Ypt1 levels at the TGN (Rivera-Molina and Novick, 2009). We were therefore surprised that Ypt1HVD32 persisted at the TGN, as we expected that Gyp1 should remove any aberrantly activated Rab. This led use to consider whether the Rab HVD might also control GAP substrate specificity. Alternatively Gyp1 activity may be insufficient to counteract nucleotide exchange by both TRAPPIII and TRAPPII. To discern between these two possibilities, we overexpressed Gyp1 in cells and observed mNG-Rab localization. Gyp1 overexpression mislocalized Ypt1 to the cytosol and ER but had no effect on Ypt32, consistent with a specific role for Gyp1 as a GAP for Ypt1 (Figure S4C). Gyp1 overexpression mislocalized Ypt1HVD32 to a similar extent as Ypt1, suggesting that Gyp1 recognizes this chimera as a substrate. This suggests that the Rab HVD is not important for GAP specificity and Ypt1HVD32 remains activated at the TGN due to the combined activity of TRAPPIII and TRAPPII.
Ypt31/32 HVD sequence is important for recognition by TRAPPII
Our combined in vitro and in vivo experiments indicate that the Rab HVD controls TRAPP substrate specificity. In all experiments, the Ypt31/32 HVD was essential for activation by TRAPPII. We therefore asked whether the primary sequence of the Ypt31/32 HVD is important for recognition by TRAPPII, as would be expected for a direct GEF-HVD interaction. Both Rabs possess conserved C-terminal cysteines and a C-terminal interacting motif (CIM) that facilitates interaction with the Rab escort protein (REP) during prenylation (Figure 6A) (Rak et al., 2004). The Ypt31/32 HVD contains conserved residues flanking the CIM as well as a stretch of basic residues upstream of the prenylated cysteines. To test if these motifs are important for interaction with TRAPPII, we made alanine substitution and charge reversal mutations in the Ypt32 HVD.
Figure 6. Conserved residues of the Ypt31/32 HVD are important for TRAPPII-mediated nucleotide exchange.

(A) WebLogo residue conservation of the Ypt1 and Ypt31/32 HVDs. Alanine substitution and charge-reversal mutants are indicated in red; blue boxes designate the CIM. (B) The indicated Ypt32 HVD mutants were tested for their ability to complement a ypt31Δypt32Δ mutant in the trs130Δ33 (TRAPPII mutant) background. Cells were grown at 30°C. (C) Left: Localization of mNG-tagged wild-type Ypt32 versus the indicated HVD mutants relative to Sec7-DsRed. Right: Quantification of Golgi (active) versus cytosolic/ER-localized (inactive) Rab from the images at left. Error bars represent 95% CIs for n ≥ 26 Golgi compartments. Line trace analysis could not be performed for the 205ISL>AAA CIM mutant which lacked any Golgi-localized Rab. See Figure S5 for representative images of Ypt32 194GSN>AAA and 208TPAP>AAAA. (D) Left: Representative images showing recruitment of TRAPPII to mitochondria by Ypt32 with a wild-type HVD versus the indicated HVD mutants. Right: Quantification of TRAPPII recruitment to mitochondria. Error bars represent 95% CIs for n > 40 cells. See Figure S5 for representative images of Ypt32 194GSN>AAA and 208TPAP>AAAA. (E) Coomassie-stained SDS-PAGE gel showing prenylated Ypt32 HVD mutant substrates used in this study. (F) Left: Representative traces showing activation of wild-type Ypt32 versus the indicated HVD mutants by TRAPPII in the presence of TGN membranes and Arf1-GTP. Right: Rates of TRAPPII-mediated Ypt32 activation calculated from traces at left. Error bars represent 95% CIs for n ≥ 3 reactions. Scale bars, 2 μm. White arrowheads denote colocalization of Rabs and Sec7 at late Golgi compartments.
See also Figure S5.
All HVD mutants were expressed at similar levels as wild-type Ypt32 (Figure S5A) and complemented a ypt31Δypt32Δ mutant (Figure S5B). Only the 205ISL>AAA CIM mutant which prevents prenylation was lethal. We and others have found that trs130Δ33 TRAPPII mutants are viable at the permissive temperature, yet Ypt31/32 activation is nearly abolished (Morozova et al., 2006; Thomas and Fromme, 2016), suggesting that low levels of Rab activation are sufficient for cell growth. Therefore, we repeated YPT31/32 complementation tests in trs130Δ33 cells. In this sensitized background, we observed growth defects in the 202GPT>AAA mutant and, to a lesser extent, the 214KKKK>DDDD mutant, which was exacerbated at a higher temperature (Figure 6B and S5C). The 208TPAP>AAAA mutant supported cell viability similarly to wild-type Ypt32, as did 194GSN>AAA, which is mutated in a non-conserved region of the Ypt32 HVD.
We visualized the localization of mNG-Ypt32 mutants as a measurement of Rab activation in cells. Consistent with complementation tests, the 202GPT>AAA and 214KKKK>DDDD mutants were partially mislocalized (Figure 6C and S5D). Inactive mNG-Ypt32 214KKKK>DDDD appeared to localize completely to the cytosol and not ER membranes, raising the possibility that this mutant is impaired in membrane association. A polybasic motif in the Rab35 HVD is important for localizing to the plasma membrane (Li et al., 2014), so the 214KKKK>DDDD may prevent association with anionic late Golgi membranes. The 205ISL>AAA CIM mutant which prevents prenylation was completely cytosolic.
We used GRab-IT to test whether the Ypt32 HVD mutants affect GEF-substrate interaction. This assay allows us to test mutants that interfere with prenylation or membrane association by replacing the prenyl-mediated attachment with a transmembrane domain. Consistent with the results described above, we found that 202GPT>AAA and 214KKKK>DDDD reduced TRAPPII recruitment to mitochondria (Figure 6D and S5E). Intriguingly, the 205ISL>AAA CIM mutant nearly abolished recruitment of TRAPPII; however this result could not be corroborated using other assays due to its effect on prenylation. It is possible that the CIM residues serve a dual purpose in prenylation and TRAPPII-substrate recognition.
Finally, we used GEF reconstitution assays to directly test the importance of HVD sequence in TRAPPII-mediated activation. We prepared prenylated Rab substrates and performed membrane-binding assays to ensure that the mutations did not affect membrane association (Figure 6E and S5F). The 202GPT>AAA and 194GSN>AAA mutants bound membranes similarly to wild-type Ypt32. In contrast, Ypt32 214KKKK>DDDD did not associate with membranes, suggesting that it is not efficiently prenylated or that the charge reversal mutation impairs membrane binding. Ypt32 214KKKK>DDDD was not activated by TRAPPII in GEF reconstitution assays (our unpublished data), consistent with membrane delivery being a prerequisite for nucleotide exchange. The 194GSN>AAA mutant was activated identically to the wild-type (Figure 6F). Consistent with our in vivo experiments, TRAPPII activated Ypt32 202GPT>AAA approximately half as fast as wild-type. Overall, the importance of conserved residues in the Ypt31/32 HVD support a model in which TRAPPII interacts directly with the HVD to facilitate nucleotide exchange.
HVD length determines TRAPPII substrate accessibility
The Ypt1 and Ypt31/32 HVDs diverge not only in sequence and but also in length, with the Ypt31/32 HVD being 12 residues longer than that of Ypt1. The Rab HVD functions in part as a flexible linker, connecting the Rab NBD to its prenylated anchor on the membrane. Therefore, HVD length may play an important role in orienting the Rab NBD for nucleotide exchange. As the TRAPPII complex is significantly larger than TRAPPIII, we wondered whether a longer HVD linker might be required for the Rab NBD to reach the TRAPPII active site (Figure S6A). To test this idea, we generated GRab-IT constructs in which we deleted 12 residues from a non-conserved region of the Ypt32 HVD. These constructs were unable to recruit TRAPPII to mitochondria, but replacement of the missing residues with a 12-residue glycine/serine (GS) linker restored TRAPPII recruitment (Figure 7A and S6B, Ypt32Δ186–197 and Δ189–200). In contrast, truncation of a conserved region of the HVD was not rescued by a GS linker (Figure 7A and S6B, Ypt32Δ201–212). Together, these results indicate that both length and sequence of the HVD are important for recognition of Ypt31/32 by TRAPPII.
Figure 7. HVD length determines substrate accessibility.

(A) Left: Schematic depicting Ypt32 HVD truncations and Gly-Ser rescue constructs, residues likely to be important for recognition by TRAPPII are highlighted in red. Right: Quantification of TRAPPII recruitment to mitochondria by the constructs indicated. Error bars represent 95% CIs for n > 40 cells. See Figure S6B for representative images. (B) Schematic depicting Rab constructs used to test whether HVD length mediates TRAPP complex substrate specificity. (C) Left: Representative images showing TRAPPII recruitment to mitochondria by nucleotide-free Ypt1HVD-GS, but not Ypt1. Right: Quantification of TRAPPII recruitment to mitochondria. Error bars represent 95% CIs for n > 40 cells. (D) Left: Representative traces showing activation of Ypt1 versus Ypt1HVD-GS by TRAPPII in the presence of TGN membranes and Arf1-GTP. mock = buffer only control reaction. Right: Rates of TRAPPII-mediated nucleotide exchange calculated from the traces at left. Error bars represent 95% CIs for n = 3 reactions. n.d., not detectable (the Rab was not activated by the GEF and exponential functions could not be fit to experimental curves). (E) Left: Localization of mNG-tagged Ypt1 versus Ypt1HVD-GS relative to the late Golgi marker Sec7-DsRed. Right: Colocalization analysis of each Rab construct with Sec7. Error bars represent 95% CIs for n > 40 cells. (F) Left: Localization of mNG-tagged Ypt1 versus Ypt1HVD-GS relative to Sec7-DsRed in trs85Δ (TRAPPIII mutant) cells. Right: Quantification of Golgi (active) versus cytosolic/ER-localized (inactive) Rab from the images at left. Error bars represent 95% CIs for n ≥ 25 Golgi compartments. (G) Quantification of TRAPPII recruitment to mitochondria by Ypt32, Ypt1, and Ypt1 baits with increasing Gly-Ser linker lengths at the C-terminus of the HVD. Error bars represent 95% CIs for n > 40 cells. (H) Model for Rab substrate length discrimination by the TRAPPII complex, using negative-stain reconstructions of the TRAPP complexes (Tan et al. 2013; Yip et al. 2010). A red asterisk denotes the approximate position of the active site (Cai et al., 2008). In this model the TRAPPII-specific subunits lift the active site (red asterisk) off the membrane an additional 2 nm relative to TRAPPIII. This additional distance prevents the Ypt1 NBD from reaching the TRAPPII active site due to its shorter HVD. The Ypt31/32 HVD is longer, enabling its NBD to reach the TRAPPII active site. The Ypt31/32 NBD is more difficult for the TRAPP core to activate, and its activation requires the extra binding energy derived from direct interactions between TRAPPII-specific subunits and the Ypt31/32 HVD. These interactions are not made with TRAPPIII, so TRAPPIII can activate Ypt1 but not Ypt31/32. Scale bars, 2 μm. White arrowheads denote colocalization of Rabs and Sec7 at late Golgi compartments.
See also Figures S6 and S7.
We then generated a construct in which we extended the Ypt1 HVD with a 12-residue GS linker (Figure 7B and Table S2). In GRab-IT assays, Ypt1HVD-GS recruited TRAPPII, indicating that a longer HVD facilitates recognition by TRAPPII (Figure 7C). Similarly, extending the Ypt1 HVD enabled TRAPPII-mediated activation in the GEF reconstitution assay (Figure 7D and S6C), further indicating that a longer HVD is required to reach from the membrane to the TRAPPII active site. Considering that the sequence of the HVD was important for TRAPPII recognition of Ypt31/32, we were initially surprised that TRAPPII activation of Ypt1HVD-GS did not require the specific HVD sequence of Ypt31/32. As discussed further below, we attribute these findings to the fact that the Ypt1 NBD is much more readily activated by the TRAPP core than the Ypt31/32 NBD (Wang et al., 2000; Cai et al., 2008; Thomas and Fromme, 2016).
We next visualized mNG-Ypt1HVD-GS in cells. Similar to mNG-Ypt1HVD32, mNGYpt1HVD-GS was activated at late Golgi compartments, and overlapped more with Sec7 than did Ypt1 (Figure 7E). This indicates that mNG-Ypt1HVD-GS remains at the TGN later, likely due to activation by TRAPPII. Furthermore, mNG-Ypt1HVD-GS remained partially activated in trs85Δ TRAPPIII mutant cells (Figure 7F), suggesting that this construct is activated by TRAPPII. Taken together, these data indicate that extending the Ypt1 HVD enables activation by the TRAPPII complex. This raises the possibility that TRAPPII counterselects against Ypt1 using a steric gating mechanism in which the HVD functions as a molecular ruler to mediate substrate specificity, as the endogenous Ypt1 HVD is too short to reach the TRAPPII active site. To determine the minimal HVD length required for TRAPPII to recognize Ypt1, we titrated the number of amino acids added to the Ypt1 HVD in the GRab-IT assay (Table S2). We observed that longer length correlated with an increase in the detected interaction, with a pronounced increase in the interaction occurring at an apparent threshold between 6 and 9 additional amino acids and appearing to plateau above 12 amino acids (Figure 7G).
We next tested whether HVD length is important for TRAPPIII-mediated activation. In GRab-IT assays, Ypt1 and Ypt1HVD-GS recruited TRAPPIII to a similar extent (Figure S7A). TRAPPIII activated Ypt1 and Ypt1HVD-GS nearly identically in GEF reconstitution experiments (Figure S7B), suggesting that TRAPPIII can accommodate a substrate with a longer HVD. However, mNG-Ypt1HVD-GS was not able to complement a ypt1Δ mutant in trs85Δ cells (Figure S7C), indicating that HVD length becomes more important in the absence of robust GEF activity. As with Ypt1HVD32, it is likely that TRAPPII activates Ypt1HVD-GS ectopically at the TGN in trs85Δ cells. Finally, we tested whether HVD length is important for GAP substrate recognition. Consistent with our observation that Gyp1 acts on Ypt1HVD32, Gyp1 overexpression resulted in inactivation of Ypt1HVD-GS (Figure S7D).
Elongating the Ypt1 HVD had no effect on activation by wild-type TRAPPIII. As we hypothesize that TRAPPII substrate accessibility is controlled by HVD length, we wondered whether truncation of the Ypt1 HVD might impose steric constraints during activation by TRAPPIII. We made two Ypt1 HVD truncation mutants by either removing the C-terminal 13 residues of Ypt1 (Ypt1Δ13) or 12 upstream residues in a less conserved region of the HVD (Ypt1Δ181–192). Both HVD truncations significantly impaired recognition of Ypt1 by TRAPPIII (Figure S7E), indicating that HVD length is important for TRAPPIII substrate accessibility. If loss of TRAPPIII recruitment was solely due to HVD length, we would expect that both truncations would similarly impair TRAPPIII recruitment. However Ypt1Δ181–192 had a more severe effect, suggesting that TRAPPIII might interact with specific residues in the Ypt1 HVD to mediate substrate recognition.
A model for substrate selection by steric gating
Previous studies have determined negative stain EM reconstructions of the TRAPPII and TRAPPIII complexes (Tan et al., 2013; Yip et al., 2010). Additionally, structural and biochemical data have defined a catalytic site in the TRAPP core that is used by both Ypt1 and Ypt31/32 (Cai et al., 2008; Thomas and Fromme, 2016). When TRAPP structures are modeled on membranes using predicted binding interfaces (Kim et al., 2005), the TRAPPII catalytic site is significantly farther, about 2 nm, from the membrane surface than that of TRAPPIII (Figure 7H).
In the length-titration experiments (Figure 7G), addition of nine residues, corresponding to ~3 nm assuming ~0.4 nm/residue for a Gly-Ser linker (Van Rosmalen et al., 2017), was sufficient to surpass an apparent length threshold for interaction with TRAPPII. As the HVD links the NBD to the membrane surface, our data indicate the distance of the NBD from the membrane surface is a key determinant for substrate recognition by TRAPPII. We therefore propose a steric gating mechanism in which Rabs with short HVDs are excluded from productive interaction with TRAPPII because the NBD cannot reach the active site. In organisms ranging from yeast to humans, Rab11 homologs have significantly longer HVDs compared to Rab1 homologs of the same species, suggesting that this mechanism of substrate selection may be evolutionarily conserved.
Discussion
Correct Rab localization is essential for proper function, yet the mechanisms that mediate Rab subcellular targeting are not fully understood. Our findings support the hypothesis that Rab localization is controlled largely by GEFs, which activate and consequently stabilize their substrates on specific organelle membrane surfaces. Previous studies have reported that the HVD functions as a subcellular targeting signal for certain Rabs, presumably by interacting with other proteins and/or membrane lipids (Brennwald and Novick, 1993; Chavrier et al., 1991; Stenmark et al., 1994). Some Rabs have been shown to be stabilized by their effectors, for example, TIP47 and RILP play key roles in localizing Rab9 and Rab7 through interactions with their HVDs (Aivazian et al., 2006; Li et al., 2014). We have now determined (using data summarized in Table S3) that the TRAPP GEF complexes distinguish between their distinct Rab substrates using the HVD. Remarkably, the length of the HVD is a critical determinant of substrate recognition by TRAPPII.
Early studies aimed at determining how Rabs localize focused on the highly divergent HVD as a Rab-specific “signature”. These studies made Rab chimeras and reported that swapping HVDs correspondingly switched Rab subcellular localization (Brennwald and Novick, 1993; Chavrier et al., 1991; Stenmark et al., 1994). However, many Rab HVD chimeras were viable, indicating that the HVD might not be essential for Rab function (Beranger et al., 1994; Brennwald and Novick, 1993; Dunn et al., 1993). We found that the Ypt1 HVD plays a significant, but nonessential, role in mediating recognition by TRAPPIII and consequently Ypt1 localization. Moreover, Ypt1 is largely mislocalized in trs85Δ cells, yet these mutants grow normally and show minimal trafficking defects (Sacher et al., 2000; Thomas et al., 2018). This indicates that, at least for some Rabs, low levels of activity are sufficient for viability and a fraction of correctly targeted Rab can support cell growth.
Subsequent studies tested a more comprehensive group of Rab chimeras and mutants and reported that the HVD does not function as a universal targeting signal (Ali et al., 2004; Li et al., 2014). Ali et al. generated HVD chimeras using Rabs that localize to diverse organelles and found that the HVD was not sufficient for Rab localization. They instead proposed that domains overlapping the switch regions mediate targeting. Indeed, our results indicate that the Ypt1 and Ypt31/32 HVDs alone are insufficient for recognition by TRAPP complexes. Our data indicate that TRAPP complexes recognize their substrates through a bipartite interaction, with the Rab NBD activated by the TRAPP core and additional specifying contacts made with the HVD. Crystal structures are available for multiple Rab-GEF pairs, yet GEF-HVD interactions have not been previously reported (Cai et al., 2008; Delprato and Lambright, 2007; Dong et al., 2007; Kiontke et al., 2017). This is likely due to the fact that the highly mobile HVD is not resolved in structures or was truncated to facilitate protein crystallization.
Multiple studies have proposed that GEFs control Rab localization by activating and thereby stabilizing their substrates on membranes. In loss-of-function GEF mutants Rabs become mislocalized from their target compartments, indicating that GEF activity is a primary determinant of Rab localization (Cabrera and Ungermann, 2013; Morozova et al., 2006; Thomas and Fromme, 2016; Thomas et al., 2018). Relocalization of GEFs to mitochondria induced a corresponding relocalization of substrate Rabs (Blümer et al., 2013; Gerondopoulos et al., 2012). Rab relocalization in these cases was rapid and specific, suggesting Rabs are indiscriminately delivered to most or all cellular membranes by GDI but can be re-extracted unless they first are activated by their GEF. Therefore, in many cases the correct localization of GEFs is critical for proper Rab targeting, explaining why most RabGEFs tend to be multi-domain proteins regulated by multiple protein-protein and protein-membrane interactions. In the case of TRAPPII, we previously identified Arf1-binding and the anionic lipid content of the TGN as primary drivers of TRAPPII localization and regulation (Thomas and Fromme, 2016), thus ensuring specifically localized activation of Ypt31/32. We have now found that recruitment by Arf1 enforces TRAPPII substrate specificity, implying that Arf1 orients TRAPPII on the membrane to mediate steric gating.
As Ypt31/32 has a longer HVD and the TRAPPIII active site is likely closer to the membrane, if length was the only consideration then TRAPPIII should be able to activate Ypt31/32. However, TRAPPIII does not recognize or activate wild-type Ypt31/32. It appears that intrinsic differences in the NBD make it more difficult for the TRAPP active site to activate Ypt31/32 compared to Ypt1, because TRAPPII, TRAPPIII, and the TRAPP core are all capable of robust activation of soluble Ypt1 (Sacher et al., 2001; Thomas et al., 2018). In contrast, membranes are required for TRAPPII-mediated Ypt31/32 activation (Thomas and Fromme, 2016), presumably to concentrate GEF and substrate at the membrane surface or to relieve autoinhibition. Our results indicate that both TRAPPII and TRAPPIII interact physically with the Rab HVD, although the HVD interaction appears to be more important for TRAPPII than for TRAPPIII. This GEF-HVD interaction likely provides additional binding energy during catalysis, thus lowering the transition state energy of the nucleotide exchange reaction and enabling TRAPPII to activate Ypt31/32. These contacts do not occur between TRAPPIII and Ypt31/32, thus TRAPPIII is incapable of activating Ypt31/32.
Ectopic activation of the Ypt1 NBD at the TGN by TRAPPII had deleterious effects on cell viability (Figure 5A and E). Similarly, ectopic activation of Ypt1 on secretory vesicles by Sec2 caused trafficking and growth defects (Yuan et al., 2017). These findings underscore the importance of proper localization for Rab function, and reinforce the role of Rabs as determinants of organelle identity.
In summary, we have determined how the large TRAPPII regulatory subunits convert the substrate specificity of the catalytic TRAPP GEF core from Ypt1 to Ypt31/32. We propose a steric gating mechanism that combines direct recognition of the Ypt31/32 HVD together with steric exclusion of the shorter Ypt1 HVD. As the HVD is proposed to be a targeting signal for several other Rabs, it is possible that the HVD mediates substrate specificity for other Rab-GEF pairs.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, J. Christopher Fromme (jcf14@cornell.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All yeast strains and plasmids were generated using standard techniques and are described in the Key Resources Table. To purify endogenous TRAPPII a yeast strain expressing Trs130 with a C-terminal tandem affinity purification (TAP) tag was purchased from Dharmacon. All other yeast strains were derived from the haploid S. cerevisiae strains SEY6210 (MATα) and SEY6210.1 (MATa) with genotype ura3–52 his3-Δ200 leu2–3,112 lys2–801 trp1-Δ901 suc2-Δ9 (Robinson et al., 1988). Yeast were grown in YPD (10 g/L yeast extract, 20 g/L peptone, 20 g/L glucose) or synthetic dropout (SD; 6.7 g/L yeast nitrogen base, 20 g/L glucose, 0.7 g/L amino acid dropout mix) media at 30°C and analyzed in log-phase (OD600 of ~0.5).
For most strains, genetic manipulation was carried out using homologous recombination of PCR-amplified cassettes (Longtine et al., 1998). Endogenous TRS85, TRS130, and SEC2 were tagged with mNeonGreen by amplifying the mNeonGreen::HIS3 cassette from pFA6a-mNeonGreen::HIS3 using primers with homology to the C-termini (without the stop codon) and downstream regions of the genes. TRS85, YPT1, YPT31, and YPT32 were deleted by amplifying the kanMX or HIS3 cassettes from pFA6a-kanMX6 or pFA6a-His3MX6 using primers with homology upstream and downstream of the genes. To generate a trs130Δ33 strain, the HIS3 cassette from pFA6a-His3MX6 was amplified using primers with homology downstream of TRS130 as well as 33 residues upstream of the stop codon. PCR-amplified cassettes were transformed into yeast and clones that underwent homologous recombination were selected by growth on -Histidine media (HIS3 cassette) or media containing G418 (kanMX cassette). ypt1Δ and ypt31Δypt32Δ clones were maintained with pRS416 vectors containing YPT1 or YPT31, respectively, with their endogenous promoters and terminators. SEC7 was tagged at the C-terminus with 6×DsRed using a Sec7–6×DsRed::URA3 integration plasmid (Losev et al., 2006). All clones were screened for correct insertion of cassettes using colony PCR with gene-specific primers flanking the cassette. Strains with integrated fluorescent tags were additionally screened by microscopy.
To visualize Rab localization, cells were transformed with pRS415 containing mNeonGreen-tagged Rabs with the YPT1 promoter and terminator. Though tagged Rabs were introduced as an extra copy, all mNeonGreen tagged Rabs were confirmed to be functional using complementation assays. For GRab-IT, cells were transformed with pRS415 or pRS416 containing mRFPmars-Rab-Fis1 baits with the high copy TDH3 promoter as well as the CYC1 terminator. For Gyp1 overexpression, cells were transformed with pRS414 containing GYP1 with the high copy TDH3 promoter as well as the CYC1 terminator. For complementation (plasmid shuffling) assays, cells were transformed with pRS415 containing non-tagged YPT32 with its endogenous promoter and terminator, or mNeonGreen-tagged YPT1 with its endogenous promoter and terminator. All yeast were freshly transformed with plasmids for each experiment.
METHOD DETAILS
Antibodies and inhibitors
The anti-TAP tag rabbit polyclonal antibody used to detect Trs130-TAP and Trs85-TAP in liposome flotation assays was purchased from Thermo Fisher Scientific and used at a 1:1,000 dilution. The anti-mNeonGreen mouse monoclonal antibody used to assess expression of tagged Rab constructs was purchased from Chromotek and used at a 1:1,000 dilution. The anti–glucose-6-phosphate dehydrogenase (G6PDH) rabbit polyclonal antibody was purchased from Sigma-Aldrich (SAB2100871) and was used at a 1:30,000 dilution. Rabbit and Mouse IgG HRP linked secondaries were purchased from Sigma-Aldrich (NA934 and NA931) and used at a 1:4,000 dilution.
To displace TRAPPII or Sec2 from Golgi compartments or secretory vesicles during GRab-IT assays in Figures S1 and S2, 20 μM of the Sec7 inhibitor 6-methyl-5-nitro-2-(trifluoro-methyl)-4H-chromen-4-one (MNTC; MolPort) was added to growth media 10 min prior to imaging.
Protein purification
All recombinant proteins were purified using Rosetta™ 2 cells (Novagen) with the exception of myristoylated Arf1, which was purified from BL21 (DE3) cells. Recombinant TRAPPIII (rTRAPPIII) was purified by co-expressing the following two plasmids in Rosetta™ 2 cells: a pCOLADuet-1 vector containing the six TRAPP core subunits (TRS33, TRS31, TRS23, TRS20, BET3, and BET5; pLT21) and a pETDuet-1 vector containing the TRAPPIII-specific gene TRS85 with a C-terminal tandem affinity purification (TAP) tag (pLT92). The TAP tag used in this study consists of a calmodulin-binding peptide separated from a protein-A tag by a tobacco etch virus (TEV) protease site. Cells were grown in 4–8 L terrific broth (TB; 12 g/L tryptone, 24 g/L yeast extract, 4 mL/L glycerol, 12.5 g/L K2HPO4, and 2.3 g/L KH2PO4) at 37°C to an OD600 of ~3.0, then protein expression was induced overnight at 18°C with 300 μM IPTG. Cells were harvested by centrifugation (3,000 g, 10 min, 4°C) and lysed by sonication (3 rounds of 20 1-second pulses at 100% power) in CHAPS lysis buffer (20 mM HEPES pH 7.4, 300 mM NaCl, 5% glycerol, 1% CHAPS detergent, 2 mM MgCl2, 1 mM DTT, and 1 mM PMSF). Lysed cells were then then centrifuged (12,500 g, 20 min, 4°C) to remove cell debris and the supernatant was incubated with Sepharose 6B (Sigma-Aldrich) for 30 min at 4°C to remove any proteins that bind non-specifically to Sepharose. The cleared lysate was then incubated with IgG Sepharose (GE Healthcare) for 2–3 h at 4°C to isolate protein A-tagged rTRAPPIII. The resin was washed 3× with IPP300 buffer (25 mM Tris-HCl pH 8.0, 300 mM NaCl, 5% glycerol, 0.1% CHAPS, and 1 mM DTT) followed by 1× with TEV cleavage buffer (IPP300 buffer with 0.5 mM EDTA). The washed resin was resuspended in TEV cleavage buffer and incubated overnight at 4°C with TEV protease to cleave the protein A tag and elute rTRAPPIII. Eluted protein was diluted four-fold in calmodulin binding buffer (25 mM Tris-HCl pH 8.0, 300 mM NaCl, 5% glycerol, 0.1% CHAPS, 1 mM magnesium acetate, 1 mM imidazole, 2 mM CaCl2, and 1 mM DTT) and incubated with Calmodulin Sepharose (GE Healthcare) for 1–2 h at 4°C. The resin was washed 3× with calmodulin binding buffer and rTRAPPIII was eluted using calmodulin elution buffer (25 mM Tris-HCl pH 8.0, 300 mM NaCl, 5% glycerol, 0.1% CHAPS, 1 mM magnesium acetate, 1 mM imidazole, 20 mM EGTA, and 1 mM DTT). rTRAPPIII was further purified by gel filtration chromatography using a Superdex 200 Increase 10/300 GL column (GE Healthcare) equilibrated with calmodulin elution buffer. Fractions were analyzed by SDS-PAGE with BioSafe Coomassie (Bio-Rad Laboratories), and subunit stoichiometry was determined using ImageJ. Fractions containing stoichiometric complexes were pooled and flash frozen in N2(l), then used in GEF reconstitution and membrane-binding assays. rTRAPPIII has activity identical to that of endogenous TRAPPIII purified from yeast (Thomas et al. 2018).
Endogenous TRAPPII was purified from 12–24 L yeast expressing Trs130-TAP (CFY1904). Yeast were grown in YPD at 30°C to log phase, then harvested by centrifugation, washed, and resuspended in CHAPS lysis buffer containing 50 mM NaF, 0.1 mM Na3VO4, and 1× protease inhibitor cocktail (Roche). Resuspended cells were flash frozen dropwise in N2(l) then lysed while frozen (10 2-min cycles) using a freezer mill (SPEX SamplePrep). TRAPPII was purified from the lysate using the same protocol as for rTRAPPIII, but without the final gel filtration step. Each TRAPPII preparation contained approximately stoichiometric amounts of each subunit, with 2 copies of Bet3.
Rab escort protein (REP) was purified from a pET28 vector containing MRS6 with a N-terminal His6 tag (pLT35). Rab geranylgeranyl transferase (Rab GGTase) was purified using a pCDFDuet-1 vector containing BET4 as well as BET2 with an N-terminal His6 tag (pLT41). Cells from 1–8 L of TB were lysed in Ni2+ lysis buffer (40 mM Tris-HCl pH 8.0, 300 mM NaCl, 10% glycerol, 10 mM imidazole, 2 mM MgCl2, 5 mM β-mercaptoethanol [BME], and 1 mM PMSF), and His6-tagged proteins were isolated using Ni-NTA resin (QIAGEN). The resin was washed 4× with Ni2+ lysis buffer, and His6-tagged proteins were eluted using Ni2+ elution buffer (Ni2+ lysis buffer containing 250 mM imidazole). REP and GGTase were further purified by gel filtration with prenylation buffer (20 mM HEPES pH 7.4, 150 mM NaCl, 2 mM MgCl2 and 1 mM DTT).
Full length Rabs and Gdi1 were expressed with N-terminal GST tags using a pGEX-6P vector. Cells from 1–8 L of TB were lysed in lysis buffer (1× PBS, 2 mM MgCl2, 5 mM BME, and 1 mM PMSF) and GST-tagged proteins were affinity purified using glutathione resin (G-Biosciences). The resin was washed in lysis buffer followed by PreScission cleavage buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 2 mM MgCl2, and 1 mM DTT), and the GST tag was cleaved by incubating overnight at 4°C with PreScission (3C) protease. GST tag cleavage eluted the protein from the resin while the GST tag and PreScission remained bound, so purified Rabs and Gdi1 were obtained from the supernatant after pelleting the resin. In most cases GST-tagged proteins were pure enough to be used straight from the resin, but if necessary the eluted protein was further purified by gel filtration with prenylation buffer to remove degradation products.
Myristoylated Arf1 was prepared by co-expressing full length Arf1 with the N-myristoyl transferase Nmt1 in BL21 (DE3) cells in the presence of myristate (Sigma-Aldrich). 1–2 L of cells were grown in Luria-Bertani (LB) broth to an OD600 of ~0.6, then protein expression was induced overnight at 18°C with 1 mM IPTG. Cells were lysed in lysis buffer (20 mM Tris-HCl pH 8.0, 100 mM NaCl, 1 mM MgCl2, 10 mM BME, and 1 mM PMSF) and the lysate was incubated with DEAE Sephacel (GE Healthcare) to remove contaminating proteins. The cleared lysate was incubated with Phenyl resin (Tosoh Bioscience) in high salt buffer (20 mM Tris-HCl pH 8.0, 3 M NaCl, 1 mM MgCl2, 1 mM DTT) to isolate hydrophobic myristoylated Arf1. Proteins were eluted with low salt buffer (20 mM Tris-HCl pH 8.0, 100 mM NaCl, 1 mM MgCl2, 1 mM DTT) and further purified by gel filtration with prenylation buffer.
Preparation of prenylated Rab/GDI substrates
Prenylated Rab/GDI complexes were prepared using an enzymatic synthesis procedure as previously described (Thomas and Fromme 2016). Purified Rab GTPases were labeled with fluorescent mantGDP (Biolog Life Science Institute) using EDTA-based exchange to measure nucleotide exchange during GEF reconstitution assays. For EDTA-based nucleotide exchange, purified Rabs were incubated with a five-fold molar excess of mantGDP and 4mM EDTA for 30 minutes at 30°C. The exchange reaction was stopped by the addition of MgCl2 to a final concentration of 6mM, and the mantGDP-labeled Rab was then buffer exchanged into prenylation buffer. Labeled Rab was incubated with purified Gdi1, His6-REP, and His6-GGTase in a 10:10:1:1 molar ratio with six-fold molar excess geranylgeranyl pyrophosphate (Cayman Chemical) in prenylation buffer at 37°C for 1 h. Imidazole was added to a final concentration of 10 mM to prevent non-specific binding to Ni-NTA resin. The prenylation reaction was then incubated with a 1/10th volume of Ni-NTA resin at 4°C for 1 h to remove His6-tagged REP and GGTase. The resin was pelleted, and the supernatant containing prenylated Rab/GDI complexes was further purified by gel filtration with prenylation buffer. Fractions were analyzed by SDS-PAGE with BioSafe Coomassie, and those containing stoichiometric complexes were pooled and used as substrates in GEF reconstitution assays. Rabs were tested for membrane binding using liposome pelleting assays (see below) to ensure efficient prenylation. Additionally, prenylated Rab/GDI complexes could not be activated in the absence of membranes, indicating that delivery of prenylated Rabs to membranes is a prerequisite for nucleotide exchange.
Liposome preparation
TRAPPII and TRAPPIII localize largely to late Golgi/TGN compartments (Figure 1G and H); therefore we used liposome membranes with a lipid composition approximating that of the TGN (Klemm et al., 2009). Individual lipids were combined in the molar ratios described in Table S1, vacuum-dried in pear shaped flasks, and rehydrated overnight at 37°C in HK buffer (20 mM HEPES pH 7.4, and 150 mM KOAc). Lipids were extruded using a mini-extruder (Avanti Polar Lipids, Inc) with 400-nm filters (Whatman) for pelleting assays or 100-nm filters for GEF reconstitution and flotation assays. Lipids were extruded using 19 passes through the filter, and stored at 4°C. Liposomes were used within 1–2 weeks of extrusion, but generally remain stable without aggregation for at least one month. In previous studies we used liposomes containing 5% Ni2+-DOGS to anchor His-tagged proteins at the membrane surface. We found that the presence of 5% Ni2+-DOGS increased TRAPP complex membrane affinity, and this effect has been reported for other GEFs (Gustafson and Fromme, 2017). GEF reconstitution assays in Figure 1C were performed using TGN liposomes with 5% Ni2+-DOGS in order to recapitulate previously reported results. Otherwise, all GEF assays were performed using TGN liposomes without Ni2+-DOGS in order to rule out the possibility of any artefacts caused by the presence of Ni2+.
Simplified Phosphatidylcholine (PC) liposomes were used in membrane flotation assays to demonstrate the preference of TRAPP complexes for anionic TGN membranes. All liposomes contain 1% DiR near-infrared dye in order to quantify lipid recovery in membrane-binding assays. The lipid compositions of all liposomes used in this study are listed in Table S1.
GEF reconstitution assays
For GEF reconstitution assays with rTRAPPIII, the following components were mixed sequentially in HKM buffer (HK buffer with 2 mM MgCl2 and 1 mM DTT) at 30°C: 333 μM TGN liposomes, 200 μM non-fluorescent GTP, and 250 nM mantGDP-labeled Rab. The reaction was incubated for 2 min to allow the prenylated Rab to begin partitioning into the membrane. Exchange was initiated by the addition of 20 nM rTRAPPIII, and mantGDP fluorescence (365 nm excitation, 440 nm emission) was measured using a fluorometer (Photon Technology International) for 20–25 min to obtain the exchange trace. Release of mantGDP from the Rab results in diminished fluorescence. Assays with TRAPPII and Ni2+ TGN membranes were carried out identically but using Ni2+ TGN liposomes and 13 nM TRAPPII.
For assays using Arf1-GTP to recruit TRAPPII to membranes, Arf1 was pre-activated on TGN liposomes. Myristoylated Arf1 (final concentration of 1.5 μM in GEF assays) was mixed with TGN liposomes and GTP in HKM buffer, then EDTA was added to a final concentration of 2.5 mM for 15 minutes at 30°C to load Arf1 with GTP. MgCl2 was then added to a final concentration of 3.5 mM to stop nucleotide exchange, and proteoliposomes were stored on ice until use in GEF assays. For GEF assays, Arf1-GTP proteoliposomes were incubated at 30°C for 5 min for temperature equilibration before addition of 250 nM mantGDP-labeled Rab followed 2 min later by 13 nM TRAPPII. Arf1-GTP proteoliposomes were used within 2–3 h, but remained stable on ice for at least 6 h.
All assays were performed with n ≥ 3 reactions for each condition. For all assays separate “mock” reactions were carried out by adding buffer in lieu of TRAPP to control for any intrinsic exchange or photobleaching. To determine nucleotide exchange rates, fluorescent traces were fit to a single exponential curve with an additional linear drift term using GraphPad Prism software. Rate constants were then divided by GEF concentration to calculate the exchange rate. For some reactions, the Rab substrate was only minimally activated by the GEF and exponential functions could not be fit to experimental curves. For these reactions nucleotide exchange is reported as “n.d.” (not detectable).
Liposome pelleting assays
Quantitative pelleting assays were performed to test whether Rabs were efficiently prenylated during the preparation of Rab/GDI substrates. 1 μg of Rab in a Rab/GDI complex was mixed with 500 μM TGN liposomes in HKM buffer. 400 μM GDP or GMP-PNP, a non-hydrolyzable GTP analog, was added as well as EDTA to a final concentration of 4 mM. The reaction was incubated at 30°C for 30 minutes to allow EDTA-mediated nucleotide exchange. Exchange was stopped with the addition of MgCl2 to a final concentration of 6 mM. The reactions were centrifuged (15,000 g, 10 min, 4°C) to separate pelleted liposomes from unbound protein in the supernatant. Membrane pellet and supernatant samples were run on SDS-PAGE, stained with BioSafe Coomassie, and visualized on a Odyssey Imager (Li-COR). The amount of membrane-associated Rab was quantified by dividing the amount of Rab in the pellet by the total amount of Rab in the reaction. Reactions lacking liposomes were run in parallel to control for background pelleting. As expected for most Rab/GDI substrates, the majority of the Rab associated with membranes in a nucleotide-dependent manner. All pelleting assays were performed with n ≥ 4 reactions.
Liposome flotation assays
Liposome flotation was used to test association of TRAPPII and TRAPPIII with membranes of different compositions. 1 μg of TRAPP was incubated with 250 μM liposomes in HKM buffer for 20 minutes at 30°C. In separate assays, 4 μg myristoylated Arf1 was pre-activated on membranes using EDTA-based exchange prior to the addition of TRAPPII. Liposomes with any membrane-associated protein were separated from unbound protein using discontinuous sucrose gradient flotation as previously described (Richardson and Fromme 2015). Briefly, membrane-binding reactions were diluted with HKM buffer containing 2.5 M sucrose to a final concentration of 1 M sucrose. The mixture was transferred to 7 × 20 mm Polycarbonate centrifuge tubes (Beckman Coulter), overlayed with HKM containing 0.75 M sucrose followed by HKM without sucrose, and centrifuged (390,000 g, 20 min, 20°C). Following centrifugation, liposomes with bound protein were collected from the top layer of the sucrose gradient. Membrane-bound TRAPP was detected by immunoblotting the recovered liposome fraction with the anti-TAP tag antibody (for Trs130- or Trs85-TAP). Myristoylated Arf1 was visualized using BioSafe Coomassie stain.
Microscopy
Cells were grown overnight in synthetic dropout media at 30°C and imaged in log-phase (OD600 of ~0.5) at room temperature. Each image shown is a single 0.4 μm focal plane. Time-lapse series were generated by imaging a single focal plane every 2 seconds for 2–4 min. Images were processed using ImageJ, adjusting only the minimum/maximum brightness levels for clarity with identical leveling between all images within a figure panel.
Time-lapse series were acquired using a CSU-X spinning-disk confocal system (Intelligent Imaging Innovations) with a DMI6000 B microscope (Leica Microsystems), 100× 1.46 NA oil immersion objective, and a QuantEM electron-multiplying CCD camera (Photometrics). Series were acquired and analyzed using Slidebook 6 software (Intelligent Imaging Innovations). All other images were captured using a DeltaVision Elite system (GE Healthcare) with a 100× 1.35 oil immersion objective and a CoolSNAP HQ2 camera (Photometrics). Images were acquired using softWoRx software (GE Healthcare) and deconvolved (conservative setting; six cycles) to remove out of focus light.
GEF-Rab Interaction Test (GRab-IT)
GRab-IT assays were used to measure GEF-substrate interactions in vivo. Rab genes were cloned into a centromeric LEU2 vector with a high copy TDH3 (GPD) promoter. Rab baits were tagged at their N-termini with a fluorescent mRFPmars tag and rendered nucleotide-free by introducing the following point mutations: Ypt1(D124N), Ypt31/32(D129N), Ypt6(D127N), and Sec4(D136N). Nucleotide-free Rab mutants were designed to mimic the transition state during nucleotide exchange and are therefore bound tightly by their GEFs. Rab baits were targeted to mitochondria by replacing the Rab C-terminal prenylated cysteines with the transmembrane domain of Fis1 (residues 129–155). Rab bait plasmids were transformed into yeast with C-terminal mNG tags on endogenous Trs85 (TRAPPIII), Trs130 (TRAPPII), or Sec2. Given the distinct morphology of mitochondria in yeast, recruitment of TRAPP complexes or Sec2 from Golgi compartments or polarized secretory vesicles could be easily visualized. The GRab-IT setup identifies GEF-substrate interactions, as GEF recruitment is nucleotide dependent (see Figures S1 and S2). Furthermore, GEF recruitment is specific as TRAPPIII, TRAPPII, and Sec2 were only recruited by Ypt1, Ypt31/32, and Sec4, respectively, and not other Rab baits.
Complementation tests
Plasmid shuffling assays were performed to test whether Rab HVD mutants and chimeras are functional in vivo. ypt1Δ or ypt31Δypt32Δ null mutants were maintained by a copy of YPT1 or YPT31 on a centromeric URA3 plasmid. Shuffling strains were transformed with centromeric LEU2 plasmids containing Rab genes under their endogenous promoters. Transformed yeast were plated on -Leu synthetic dropout media to allow for cells with functional constructs to lose the URA3 maintenance plasmid. After 2–3 days of growth transformed cells were resuspended in yeast nitrogen base, diluted to an OD600 of 0.5, then serially diluted onto -Leu and synthetic complete media with 3.9 mM 5-fluoroorotic acid (5-FOA; US Biological). Cells were grown at 30°C unless otherwise noted for 2–3 days prior to imaging.
Rab HVD conservation analysis
A position-specific iterative (PSI)-BLAST was used to identify homologs of Ypt1 and Ypt31. Amino acid sequences with > 95% identity were removed to limit redundancy. Sequences for 142 Ypt1 homologs and 181 Ypt31 homologs were aligned using MUltiple Sequence Comparison by Log-Expectation (MUSCLE). Residue conservation is depicted using WebLogo, with increased letter height corresponding to increased conservation.
TRAPP complex negative stain model
Negative stain reconstructions are shown with positions of relevant subunits (Tan et al. 2013; Yip et al. 2010). A red asterisk denotes the approximate position of the active site (Cai et al., 2008). The membrane binding surface for TRAPPII is predicted based on the fact that the TRAPPII-specific subunits (Trs130, Trs120, Trs65, Tca17) are required for membrane binding (Thomas and Fromme, 2016), and accounting for the 2-fold symmetry of the complex. TRAPPIII is positioned on the membrane in an orientation that corresponds to the orientation of the shared subunits in TRAPPII and is therefore more speculative. However, given the shape of the TRAPPIII complex, the precise orientation of its membrane-binding surface is less important for drawing conclusions from the model. The TRAPPII-specific subunits lift the active site off the membrane an additional 2 nm relative to TRAPPIII, restricting access to GTPases with shorter HVDs.
QUANTIFICATION AND STATISTICAL ANALYSIS
Image analysis
Manders analysis was used to quantify recruitment of mNG-tagged GEFs to mitochondria in GRab-IT assays. Images were cropped to contain 3–7 cells and analyzed with ImageJ using the JACoP plugin. Images were thresholded to only quantify signal at Golgi compartments and mitochondria to avoid any cytoplasmic background. Manders analysis was also used to quantify overlap of mNG-tagged Rabs and Sec7-DsRed at late Golgi/TGN compartments. In all figures the average Manders overlap coefficient (MOC) from n ≥ 40 cells is shown for each condition.
Line-trace analysis was used to quantify activation of Rab chimeras and mutants, as activated Ypt1 and Ypt31/32 localize to Golgi compartments and polarized secretory vesicles. Using ImageJ, line traces were chosen to pass through the brightest Golgi puncta as well as the cytosol/ER and image background. For each trace, the image background value was subtracted from that of the Golgi and cytosol/ER. Rab activation was quantified by dividing background-subtracted Golgi fluorescence by that of the cytosol/ER. In all figures the average Golgi/cytosol fluorescence from n ≥ 20 cells is shown for each condition.
To measure peak-to-peak recruitment times in Figure 5D, time-lapse imaging series were analyzed using Slidebook 6 software. For each pair of proteins, Golgi compartments were chosen that remained distinct from other puncta and in the focal plane throughout the entire lifetime of the compartment. The fluorescent intensity of each protein over time was tracked for isolated Golgi compartments. Fluorescent intensity was normalized between 0.0 and 1.0, and peak fluorescence was defined as the time point when fluorescence = 1.0. To define a timeline of GEF and Rab recruitment to the Golgi, each mNG tagged protein was analyzed pairwise with Sec7-DsRed. For protein, the average peak-to-peak time versus Sec7 was determined from n ≥ 19 time-lapse series.
Statistical analysis
All statistical tests were performed using GraphPad Prism software. For Figures 1G and H, 3E, 6, 7G, S1E and F, S2B, S5, and S7E significance was determined using a one-way ANOVA with Tukey’s test for multiple comparisons. For all other figures, significance was determined using an unpaired two-tailed t test with Welch’s correction. In all figures error bars represent 95% confidence intervals. For all figures, ns indicates not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Supplementary Material
Highlights.
Development of GRab-IT, an assay for investigating GEF-GTPase interactions in vivo
Reconstitution of TRAPP complex activity with physiological substrate specificity
The Rab GTPase hypervariable domain determines GEF substrate specificity
Hypervariable domain length acts as a molecular ruler to control substrate selection
Acknowledgements
We thank the laboratories of J. Baskin, A. Bretscher, S. Emr, B. Glick, R. Kahn, K. Reinisch, and C. Ungermann for generously sharing strains, plasmids, equipment, reagents, and advice. We thank J. Baskin, S. Emr, G. Hollopeter, A. Joiner, and B. Richardson for feedback on the manuscript. B. Richardson also provided assistance with the HVD conservation analysis. This work was funded by National Institutes of Health grant R01GM116942 to J.C.F.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of Interests
The authors declare no competing interests.
References
- Aivazian D, Serrano RL, and Pfeffer S (2006). TIP47 is a key effector for Rab9 localization. J. Cell Biol 173, 917–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali BR, Wasmeier C, Lamoreux L, Strom M, and Seabra MC (2004). Multiple regions contribute to membrane targeting of Rab GTPases. J. Cell Biol 117, 6401–6412. [DOI] [PubMed] [Google Scholar]
- Bahler J, Wu JQ, Longtine MS, Shah NG, McKenzie A 3rd, Steever AB, Wach A, Philippsen P, and Pringle JR (1998). Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast 14, 943–951. [DOI] [PubMed] [Google Scholar]
- Beranger F, Paterson H, Powers S, de Gunzburg J, and Hancock JF (1994). The effector domain of Rab6, plus a highly hydrophobic C terminus, is required for Golgi apparatus localization. Mol. Cell. Biol 141, 744–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blümer J, Rey J, Dehmelt L, Maze T, Wu YW, Bastiaens P, Goody RS, and Itzen A (2013). RabGEFs are a major determinant for specific Rab membrane targeting. J. Cell Biol 200, 287–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennwald P, and Novick P (1993). Interactions of three domains distinguishing the Ras-related GTP-binding proteins Ypt1 and Sec4. Nature 362, 560–563. [DOI] [PubMed] [Google Scholar]
- Cabrera M, and Ungermann C (2013). Guanine nucleotide exchange factors (GEFs) have a critical but not exclusive role in organelle localization of rab GTPases. J. Biol. Chem 288, 28704–28712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y, Chin HF, Lazarova D, Menon S, Fu C, Cai H, Sclafani A, Rodgers DW, De La Cruz EM, Ferro-Novick S, and Reinisch KM (2008). The structural basis for activation of the Rab Ypt1p by the TRAPP membrane-tethering complexes. Cell 133, 1202–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavrier P, Gorvel JP, Stelzer E, Simons K, Gruenberg J, and Zerial M (1991). Hypervariable C-terminal domain of rab proteins acts as a targeting signal. Nature 353, 769–772. [DOI] [PubMed] [Google Scholar]
- Crooks G, Hon G, Chandonia J, and Brenner S (2004). WebLogo: a sequence logo generator. Genome Res 14, 1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delprato A, and Lambright DG (2007). Structural basis for Rab GTPase activation by VPS9 domain exchange factors. Nat. Struct. Mol. Biol 14, 406–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong G, Medkova M, Novick P, and Reinisch KM (2007). A Catalytic Coiled Coil: Structural Insights into the Activation of the Rab GTPase Sec4p by Sec2p. Mol. Cell 25, 455–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn B, Stearns T, and Botstein D (1993). Specificity domains distinguish the Ras-related GTPases Ypt1 and Sec4. Nature 362, 563–565. [DOI] [PubMed] [Google Scholar]
- Duronio RJ, Jackson-Machelski E, Heuckeroth RO, Olins PO, Devine CS, Yonemoto W, Slice LW, Taylor SS, and Gordon JI (1990). Protein N-myristoylation in Escherichia coli: reconstitution of a eukaryotic protein modification in bacteria. Proc. Natl. Acad. Sci. USA 87, 1506–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC (2004). MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32, 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerondopoulos A, Langemeyer L, Liang JR, Linford A, and Barr FA (2012). BLOC-3 mutated in Hermansky-Pudlak syndrome is a Rab32/38 guanine nucleotide exchange factor. Curr. Biol 22, 2135–2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goody RS, Rak A, and Alexandrov K (2005). The structural and mechanistic basis for recycling of Rab proteins between membrane compartments. Cell. Mol. Life Sci 62, 1657–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafson MA, and Fromme JC (2017). Regulation of Arf activation occurs via distinct mechanisms at early and late Golgi compartments. Mol. Biol. Cell 28, 3660–3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutagalung AH, and Novick PJ (2011). Role of Rab GTPases in Membrane Traffic and Cell Physiology. Physiol. Rev 91, 119–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itzen A, and Goody RS (2011). GTPases involved in vesicular trafficking: Structures and mechanisms. Semin. Cell Dev. Biol 22, 48–56. [DOI] [PubMed] [Google Scholar]
- Kim JJ, Lipatova Z, Majumdar U, and Segev N (2016). Regulation of Golgi Cisternal Progression by Ypt/Rab GTPases. Dev. Cell 36, 440–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YG, Sohn EJ, Seo J, Lee KJ, Lee HS, Hwang I, Whiteway M, Sacher M, and Oh BH (2005). Crystal structure of bet3 reveals a novel mechanism for Golgi localization of tethering factor TRAPP. Nat. Struct. Mol. Biol 12, 38–45. [DOI] [PubMed] [Google Scholar]
- Kiontke S, Langemeyer L, Kuhlee A, Schuback S, Raunser S, Ungermann C, and Kümmel D (2017). Architecture and mechanism of the late endosomal Rab7-like Ypt7 guanine nucleotide exchange factor complex Mon1-Ccz1. Nat. Commun 8, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemm RW, Ejsing CS, Surma MA, Kaiser HJ, Gerl MJ, Sampaio JL, de Robillard Q, Ferguson C, Proszynski TJ, Shevchenko A, and Simons K (2009). Segregation of sphingolipids and sterols during formation of secretory vesicles at the trans-Golgi network. J. Cell Biol 185, 601–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Yi L, Zhao L, Itzen A, Goody RS, and Wu YW (2014). The role of the hypervariable C-terminal domain in Rab GTPases membrane targeting. Proc Natl Acad Sci USA 111, 2572–2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipatova Z, Hain AU, Nazarko VY, and Segev N (2015). Ypt/Rab GTPases: Principles learned from yeast. Crit. Rev. Biochem. Mol. Biol 9238, 203–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A 3rd, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, and Pringle JR (1998). Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14, 953–961. [DOI] [PubMed] [Google Scholar]
- Losev E, Reinke CA, Jellen J, Strongin DE, Bevis BJ, and Glick BS (2006). Golgi maturation visualized in living yeast. Nature 441, 1002–1006. [DOI] [PubMed] [Google Scholar]
- Lynch-Day MA, Bhandari D, Menon S, Huang J, Cai H, Bartholomew CR, Brumell JH, Ferro-Novick S, and Klionsky DJ (2010). Trs85 directs a Ypt1 GEF, TRAPPIII, to the phagophore to promote autophagy. Proc Natl Acad Sci USA 107, 7811–7816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonold CM, and Fromme JC (2014). Four GTPases Differentially Regulate the Sec7 Arf-GEF to Direct Traffic at the trans-Golgi Network. Dev. Cell 30, 759–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morozova N, Liang Y, Tokarev AA, Chen SH, Cox R, Andrejic J, Lipatova Z, Sciorra VA, Emr SD, and Segev N (2006). TRAPPII subunits are required for the specificity switch of a Ypt-Rab GEF. Nat. Cell Biol 8, 1263–1269. [DOI] [PubMed] [Google Scholar]
- Pfeffer SR (2012). Rab GTPase regulation of membrane identity. Curr. Opin. Cell Biol 25, 414–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinar M, Arst HN, Pantazopoulou A, Tagua VG, de Los Ríos V, Rodríguez-Salarichs J, Díaz JF, and Peñalva MA (2015). TRAPPII regulates exocytic Golgi exit by mediating nucleotide exchange on the Ypt31 ortholog RabERAB11. Proc. Natl. Acad. Sci. USA 112, 4346–4351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rak A, Pylypenko O, Niculae A, Pyatkov K, Goody RS, and Alexandrov K (2004). Structure of the Rab7:REP-1 complex: Insights into the mechanism of Rab prenylation and choroideremia disease. Cell 117, 749–760. [DOI] [PubMed] [Google Scholar]
- Richardson BC, and Fromme JC (2015). Biochemical methods for studying kinetic regulation of Arf1 activation by Sec7. Methods Cell Biol 130, 101–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedel F, Galindo A, Muschalik N, Munro S (2018) The two TRAPP complexes of metazoans have distinct roles and act on different Rab GTPases. J. Cell. Biol 217, 601–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera-Molina FE, and Novick PJ (2009). A Rab GAP cascade defines the boundary between two Rab GTPases on the secretory pathway. Proc. Natl. Acad. Sci. USA 106, 14408–14413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JS, Klionsky DJ, Banta LM, and Emr SD (1988). Protein sorting in Saccharomyces cerevisiae: isolation of mutants defective in the delivery and processing of multiple vacuolar hydrolases. Mol. Cell Biol 8, 4936–4948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rosmalen M, Krom M, and Merkx M (2017). Tuning the Flexibility of Glycine-Serine Linkers to Allow Rational Design of Multidomain Proteins. Biochemistry 56, 6565–6574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacher M, Barrowman J, Schieltz D, Yates JR, and Ferro-Novick S (2000). Identification and characterization of five new subunits of TRAPP. Eur. J. Cell Biol 80, 71–80. [DOI] [PubMed] [Google Scholar]
- Sacher M, Barrowman J, Wang W, Horecka J, Zhang Y, Pypaert M, and Ferro-Novick S (2001). TRAPPI implicated in the specificity of tethering in ER-to-Golgi transport. Mol. Cell 7, 433–442. [DOI] [PubMed] [Google Scholar]
- Sciorra VA, Audhya A, Parsons AB, Segev N, Boone C, and Emr SD (2005). Synthetic genetic array analysis of the PtdIns 4-kinase Pik1p identifies components in a Golgi-specific Ypt31/rab-GTPase signaling pathway. Mol. Biol. Cell 16, 776–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sclafani A, Chen S, Rivera-Molina F, Reinisch K, Novick P, and Ferro-Novick S (2010). Establishing a role for the GTPase Ypt1p at the late Golgi. Traffic 11, 520–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski RS, and Hieter P (1989). A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122, 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivars Ulf, Alvazian D, and Pfeffer SR (2003). Yip3 catalyses the dissociation of endosomal Rab–GDI complexes. Nature 425, 846–851. [DOI] [PubMed] [Google Scholar]
- Stenmark H (2009). Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol 10, 513–525. [DOI] [PubMed] [Google Scholar]
- Stenmark H, Valencia A, Martinez O, Ullrich O, Goud B, and Zerial M (1994). Distinct structural elements of rab5 define its functional specificity. EMBO J 13, 575–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan D, Cai Y, Wang J, Zhang J, Menon S, Chou HT, Ferro-Novick S, Reinisch KM, and Walz T (2013). The EM structure of the TRAPPIII complex leads to the identification of a requirement for COPII vesicles on the macroautophagy pathway. Proc. Natl. Acad. Sci. USA 110, 19432–19437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas LL, and Fromme JC (2016). GTPase cross talk regulates TRAPPII activation of Rab11 homologues during vesicle biogenesis. J. Cell Biol 215, 499–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas LL, Joiner AMN, and Fromme JC (2018). The TRAPPIII complex activates the GTPase Ypt1 (Rab1) in the secretory pathway. J. Cell Biol 217, 283–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Sacher M, and Ferro-Novick S (2000). TRAPP stimulates guanine nucleotide exchange on Ypt1p. J. Cell Biol 151, 289–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss O, Holden J, Rulka C, and Kahn RA (1989). Nucleotide binding and cofactor activities of purified bovine brain and bacterially expressed ADP-ribosylation factor. J. Biol. Chem 264, 21066–21072. [PubMed] [Google Scholar]
- Wu YW, Oesterlin LK, Tan KT, Waldmann H, Alexandrov K, and Goody RS (2010). Membrane targeting mechanism of Rab GTPases elucidated by semisynthetic protein probes. Nat. Chem. Biol 6, 534–540. [DOI] [PubMed] [Google Scholar]
- Yip CK, Berscheminski J, and Walz T (2010). Molecular architecture of the TRAPPII complex and implications for vesicle tethering. Nat. Struct. Mol. Biol 17, 1298–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H, Davis S, Ferro-Novick S, and Novick P (2017). Rewiring a Rab regulatory network reveals a possible inhibitory role for the vesicle tether, Uso1. Proc. Natl. Acad. Sci. USA 114, 8637–8645. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.