

Abstract
Understanding the mechanisms underlying autoantibody development will accelerate therapeutic target identification in autoimmune diseases such as Systemic Lupus Erythematosus (SLE)1. Follicular helper T cells (Tfh) have long been implicated in SLE pathogenesis. Yet, a fraction of SLE patients’ autoantibodies are unmutated, supporting that autoreactive B cells also differentiate outside germinal centers (GCs)2. Here, we describe a CXCR5− CXCR3+ PD1hi CD4+ T cell helper population distinct from Tfh and expanded in both SLE blood and the tubulointerstitial areas of patients with Proliferative Lupus Nephritis (PLN). These cells produce IL10 and accumulate mitochondrial ROS (mtROS) as the result of reverse electron transport (RET) fueled by succinate. Furthermore, they provide B cell help, independently of IL21, through IL10 and succinate. Similar cells are generated in vitro upon priming naïve CD4+ T cells with plasmacytoid DCs (pDCs) activated with Oxidized mitochondrial DNA (Ox mtDNA), a distinct class of interferogenic TLR9 ligand3. Targetting this pathway might blunt the initiation and/or perpetuation of extrafollicular humoral responses in SLE.
Activation of pDCs with either chromatin-containing immune complexes (IC)4,5 or neutrophil-derived Ox mtDNA3 leads to type I IFN production. As antigen presenting cells, pDCs also shape adaptive immune responses6,7. Indeed, pDC activation with CpGA induces naïve CD4+ T cells to become regulatory (Tr1)8. Mechanistically, CpGA activates IRF7- but only minimally NFkB-related pathways9, as detected by lower expression of IL6 and CD86 as well as decreased p65 nuclear translocation compared to CpGB (Fig. 1a, b and Supplementary Fig. 1a–c). Ox mtDNA exclusively triggers IFN production. As CpGA, it up-regulates major histocompatibility antigens (HLA), CD83 and CD40 (Fig. 1a, b and Supplementary Fig. 1a–d). It uniquely induces however the IL3 receptor α-chain (CD123), which upon engagement with IL3 promotes pDC survival10 (Fig. 1b and Supplementary Fig. 1e). Activation of pDCs with either CpGA or Ox mtDNA downregulates expression of the chemokine receptors CXCR4 and CXCR3 while increasing CCR7, which promotes migration to secondary lymphoid organs11 (Fig. 1b and Supplementary Fig. 1f).
Figure 1. Ox mtDNA induces a unique pDC phenotype.
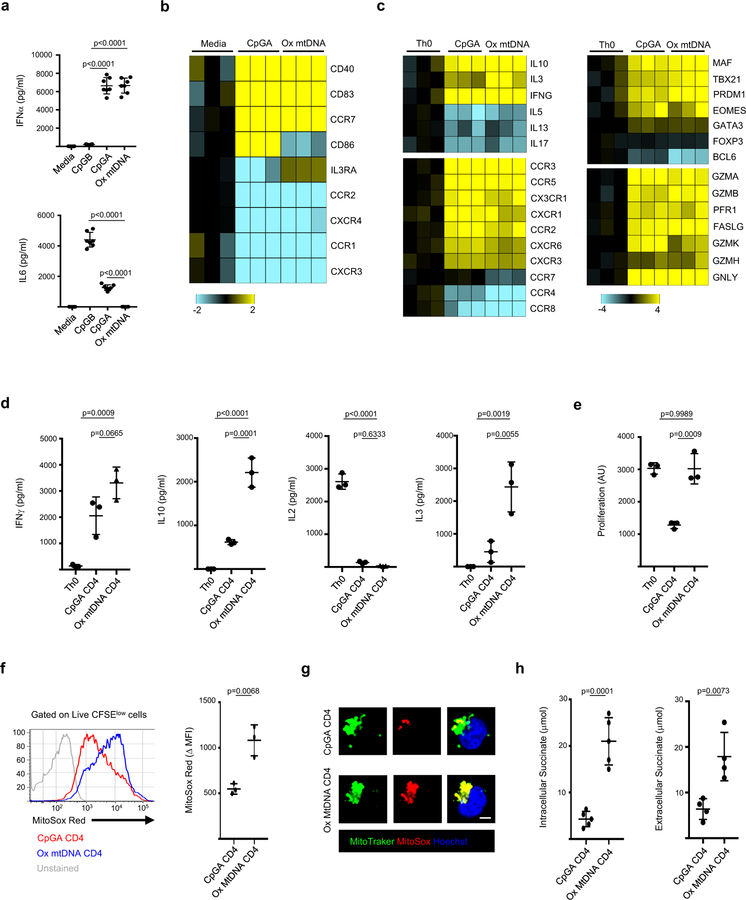
a, Cytokine profile of pDCs activated for 24 h with media, CpGB, CpGA or Ox mtDNA (n=7 independent experiments). b, Gene expression profile of pDCs in response to CpGA or Ox mtDNA (n=3 independent experiments). c, Gene expression profile of Th0, CpA and Ox mtDNA CD4+ T cell (n=3 independent experiments). d, e Cytokine profile (d) and proliferation (e) of Th0, CpGA or Ox mtDNA CD4+ T cells upon reactivation with CD3/CD28 (n=3 independent experiments). f, g MtROS production by CpGA or Ox mtDNA CD4+ T cells was assessed by flow cytometry (f, n=3 independent experiments) or by immunofluorescence microscopy (g, one representative of three independent experiments). Scale bar = 7 μm. h, Intracellular (left) and extracellular (right) succinate levels in CpGA or Ox mtDNA CD4+ T cells (n=5 independent experiments). Shown are mean ± s.e.m.; statistical analysis by nonparametric one-way ANOVA (a-e) and two-tailed nonparametric unpaired t-test at 95% CI (f, h).
To explore the biological outcome of activating pDCs with these two different TLR9 ligands, we co-cultured either type of activated pDCs with naïve CD4+ T cells (hereafter referred to as CpGA or Ox mtDNA CD4+ T cells) using CD3/CD28 activation as a control (hereafter referred to as Th0 cells). Upon sorting and restimulating proliferating (CFSElow) CD4+ T cells (Supplementary Fig. 2a), both CpGA and Ox mtDNA CD4+ T cells expressed similar proinflammatory chemokine receptors and cytotoxic molecules. They also produced high levels of IFNγ and low levels of IL2 (Fig. 1c, d and Supplementary Fig. 2b). Ox mtDNA CD4+ T cells, however, secreted significantly higher levels of IL10 and IL3 (Fig. 1c, d and Supplementary Fig. 2c).
In agreement with the reported Th1 origin of IFNγ+ IL10+ T cells, both CpGA and Ox mtDNA CD4+ T cells expressed the Th1-associated transcription factors Tbet (encoded by TBX21) and Eomes12 as well as the chemokine receptor CXCR313 (Fig. 1c and Supplementary Fig. 2d). Furthermore, knocking down TBX21 substantially decreased their generation (Supplementary Fig. 2e).
CpGA-activated pDCs induce anergic CD4+ T cells8. Accordingly, CpGA CD4+ T cells proliferated poorly upon reactivation (Fig. 1e and Supplementary Fig. 2f). Lack of expression of D-type Cyclins and failure to phosphorylate the retinoblastoma tumor suppressor protein (p-Rb) suggested their arrest in the cell cycle G1 phase14 (Supplementary Fig. 2g). On the contrary, Ox mtDNA CD4+ T cells proliferated vigorously upon reactivation and expressed D-type Cyclins and p-Rb (Fig. 1e and Supplementary Fig. 2f, g).
As reported for proliferating CD4+ T cells15, Ox mtDNA CD4+ T cells produced higher mtROS levels than CpGA CD4+ T cells (Fig. 1f, g and Supplementary Fig. 3a), and treatment with MitoTempo (MT) attenuated their proliferation (Supplementary Fig. 3b, c). Complex I (CI) of the electron-transport chain is the major site for mtROS production during Reverse Electron Transport (RET), an event characterized by mtROS-mediated disintegration of complex I16. Indeed, CI levels were reduced in a mtROS- and mitochondrial protease-dependent manner in Ox mtDNA CD4+ T cells (Supplementary Fig. 3d–f). CI degradation leads to decreased CI-mediated respiration17 (Supplementary Fig. 3g), which can be compensated by higher CII activity (Supplementary Fig. 3h). Indeed, we detected similar Maximal Respiration Rate (MMR) in CpGA and Ox mtDNA CD4+ T cells (Supplementary Fig. 3i). RET is induced upon succinate accumulation18. Accordingly, Ox mtDNA CD4+ T cells produced and secreted higher levels of succinate than CpGA CD4+ T cells (Fig. 1h). As succinate accumulation leads to HIF-1α stabilization19, HIF-1α was up-regulated in Ox mtDNA compared to CpGA CD4 T cells (Supplementary Fig. 4a).
Constitutive activation of mechanistic target of rapamycin (mTOR) in SLE T cells leads to ROS production20. As expected, rapamycin reduced the ability of naïve CD4+ T cells to proliferate and differentiate into IFNγ+ IL10+ producing T cells in response to Ox mtDNA-activated pDC (Supplementary Fig. 4b). Decreased proliferation as well as mtROS and cytokine production also occurred upon restimulation of Ox mtDNA CD4+ T cells in the presence of rapamycin (Supplementary Fig. 4c).
In line with their capacity to produce high IL10 levels, Ox mtDNA CD4+ T cells support IL10-dependent differentiation of naïve B cells into plasmablasts21,22 (Fig. 2a, b). Contrary to Tfh23, however, these cells do not express CXCR5, Bcl-6 or IL21 (Supplementary Fig. 5a, b). Instead, CXCR4 expression suggests a role in extrafollicular B cell responses. Yet, CpGA CD4+ T cells also produce IL10 but are weaker B cell helpers. Because succinate modulates immune functions upon binding to SUCNR124, which is expressed by all major human blood B cell subsets (Fig. 2c), we tested its B cell helper capacity. Indeed, blocking SUCNR1 partially inhibited Ox mtDNA CD4+ T cell-driven B cell activation (Fig. 2d). Importantly, high levels of IgM and IgG were detected in CpGA CD4+ T cell:B cell co-cultures supplemented with succinate (Supplementary Fig. 5c). Finally, succinate synergized with IL10 in promoting Ig secretion by naïve B cells activated in vitro with CD40L (Supplementary Fig. 5d).
Figure 2. Ox mtDNA CD4+ T cells help B cells through IL10 and succinate.

a, Percentage of IgD− CD27+ and CD27+ CD38+ B cells upon co-culture with CpGA or Ox mtDNA CD4+ T cells (n=3 independent experiments). Representative flow cytometry density plots are also shown. b, IgM and IgG levels in the supernatants from CpGA or Ox mtDNA CD4+ T cells and naïve B cell co-cultures (n=6 independent experiments). c, Immunoblot analysis of succinate receptor (SUCNR1) expression by purified human B cells subsets (n=3 independent experiments). d, IgM and IgG levels in the supernatants from Ox mtDNA CD4+ T cell and naïve B cell co-cultures in the presence of anti-SUCRN1 (n=3 independent experiments). e, IgM and IgG levels in the supernatants from co-cultures of Ox mtDNA CD4+ T cells, generated in the presence of isotype control or anti-PD1 antibody, and naïve B cells (n=3 independent experiments). Shown are mean ± s.e.m.; statistical analysis by nonparametric one-way ANOVA (b) and two-tailed nonparametric unpaired t-test at 95% CI (a, d, e).
To identify the signals leading to succinate accumulation in Ox MtDNA CD4+ T cells, we analyzed expression of co-stimulatory (CD28 and ICOS) and co-inhibitory (CTLA4 and PD1) receptors known to modulate T cell metabolism25,26. Ox mtDNA CD4+ T cells expressed higher PD1 levels (Supplementary Fig. 5e), and PD1 ligation was necessary for succinate and mtROS production as well as for acquisition of B cell helper function (Fig. 2e and Supplementary Fig. 5f–h).
As Ox mtDNA is released upon activation of SLE neutrophils3, we next explored whether cells with the phenotype of Ox mtDNA CD4+ T cells were present in SLE blood. Indeed, CXCR3+ PD1hi CD4+ T cells were significantly expanded in the SLE memory (CD45RA−) non-Tfh (CXCR5−) CD4+ T cell compartment (Fig. 3a). To test their capacity to help B cells, we sorted and co-cultured these cells with naïve B cells. CXCR3+ PD1low CD4+ T cells, CXCR3− PD1hi CD4+ T cells and CD45RA− CXCR5+ CD4+ T cells (Tfh) were used as controls (Supplementary Fig. 6a). Among them, CXCR3+ PD1hi CD4+ T cells and Tfh cells were equally effective at inducing naive and memory B cell differentiation into IgG-producing plasmablasts (Fig. 3b, c and Supplementary Fig. 6b, c).
Figure 3. Memory CXCR5− CXCR3+ PD1hi CD4+ T cells represent the blood counterpart of Ox mtDNA CD4+ T cells.

a, Percentage of CXCR3− PD1hi, CXCR3+ PD1hi and CXCR3+ PD1low CD4+ T cells in the blood CD45RA− CXCR5− compartment of healthy controls (n=13), SLE patients (n=27) or JDM patients (n=6). b-c, IgG levels (b, n=12 independent experiments) and CD20/CD38 expression (c, n=4 independent experiments) on naïve B cells co-cultured with CXCR3− PD1hi CD4+ T cells, CXCR3+ PD1hi CD4+ T cells, CXCR3+ PD1low CD4+ T cells or Tfh cells. d, Cytokine profile of sorted CXCR3+ PD1hi CD4+ T cells and Tfh cells (n=18 independent experiments). e-f, Succinate (e, n=12 independent experiments) and mtROS (f, n=5 independent experiments) levels in CXCR3+ PD1hi CD4+ T cells and Tfh cells. g, IgG levels in the supernatants from co-cultures of CXCR3+ PD1hi CD4+ T cells or Tfh cells and naïve B cells (n=8 independent experiments). h, Top: principal component analysis (PCA) of RNA-seq data corresponding to genes differentially expressed between CXCR3+ PD1hi CD4+ T cells and Tfh cells (n=7 independent experiments). Bottom: volcano plot of up-regulated genes in each population (DESeq2, Wald test, adjusted p-value <0.05 and fold change >2). i, Heat map of differentially expressed transcripts in CXCR3+ PD1hi CD4+ T cells and Tfh cells (n=7 independent experiments). j, Top: PCA of ATAC-seq data on peaks differentially accessible between CXCR3+ PD1hi CD4+ T cells (n=4 independent experiments) and Tfh cells (n=2 independent experiments). Bottom: chromatin sites with differential accessibility. Plot indicates the number of opening/closing chromatin peaks in CXCR3+ PD1hi CD4+ T cells when compared to Tfh cells (EdgeR, adjusted p-value < 0.05 and fold change >2). Shown are mean ± s.e.m.; statistical analysis by nonparametric one-way ANOVA (a-c; g) and two-tailed nonparametric unpaired t-test at 95% CI (d-f).
Upon TCR stimulation, SLE blood Tfh cells produced significant amounts of IL21, CXCL13 and IL2 while CXCR3+ PD1hi CD4+ T cells released the highest levels of IL10 but no IL21 or CXCL13 (Fig. 3d and Supplementary Fig. 6d). Similar results were obtained upon contact with naïve or memory B cells (Supplementary Fig. 6e). Furthermore, CXCR3+ PD1hi CD4+ T cells produced the highest levels of IFNγ, IL3, mtROS and succinate (Fig. 3d-f). Neutralization of IL10 during co-culture of naïve B cells with SLE blood CXCR3+ PD1hi CD4+ T cells or Tfh cells inhibited IgG secretion, while Tfh cell function was mainly dependent on IL2127,28. Conversely, succinate receptor blockade, but not IL21 neutralization, decreased IgG secretion in naïve B cell-CXCR3+ PD1hi CD4+ T cell co-cultures (Fig. 3g).
We next compared the transcriptome of SLE blood CXCR3+ PD1hi CD4+ T cells and Tfh cells. Principal component analysis (PCA) revealed 1230 differentially expressed transcripts (log fold change >1.2, False Discovery Rate FDR<0.01) (Fig. 3h). Among them, CXCR3+ PD1hi CD4+ T cells upregulated chemokine receptors such as CCR2, CCR5 and CX3CR1 (Fig. 3i and Supplementary Fig. 6f). In addition, transcripts (GZMA, GZMB, PRF1, GZMH, GNLY, CTSW, FCRL6, S1PR5, SLAMF7 and NKG7) as well as transcription factors linked to cytotoxic programs29 (ZNF683, RUNX3, EOMES and TBX21) were exclusively upregulated in CXCR3+ PD1hi CD4+ T cells (Fig. 3i and Supplementary Fig. 6g). Gene ontology analysis identified additional gene sets involved in cell cycle regulation, including cyclins (CCNE2, CCNB1, CCNB2 and CCNA2), cyclin dependent kinases (CDK1) and elongation factors (E2F1, E2F2) as the top differentially expressed (Supplementary Fig. 6h). Accordingly, p-Rb levels were increased in CXCR3+ PD1hi CD4+T cells compared to Tfh cells (Supplementary Fig. 6i).
The global chromatin landscape30 of both cell types was also markedly different, with 935 differentially accessible chromatin sites (peaks). Indeed, most sites had increased accessibility in CXCR3+ PD1hi CD4+ T cells. Overall, opening peaks were detected for 690 genes, with 107 up-regulated in the CXCR3+ PD1hi CD4+ T cell transcriptome (Fig. 3j) including cytokines (IL3, IL10 and IFNG), transcription factors (TBX21 and RUNX3), pro-inflammatory chemokine receptors (CCR3, CCR5 and CX3CR1) and cytolytic molecules (GZMB) (Supplementary Fig. 6j).
Strikingly, the frequencies of SLE blood CXCR3+ PD1hi CD4+ T cells and Tfh cells were inversely correlated (Supplementary Fig. 7a). On the contrary, CXCR3+ PD1hi CD4+ T cell frequency correlated positively with IgG and IgA levels (Fig. 4a). As previously reported31,32, plasmablasts were expanded in our patients’ blood. A correlative trend between plasmablast and CXCR3+ PD1hi CD4+ T cell frequencies was found, but it did not reach statistical significance (Supplementary Fig. 7b, c) most likely due to plasmablast fragility during cryopreservation. In addition to plasmablasts, age-associated B cells (ABCs) have been reported in SLE33,34. This cell population was expanded in our pediatric patients and its frequency correlated with that of CXCR3+ PD1hi CD4+ T cells (Fig. 4b, c).
Figure 4. IL10+ IFNγ+ ROS+ PD1+ CD4+ T cells accumulate in PLN lesions.
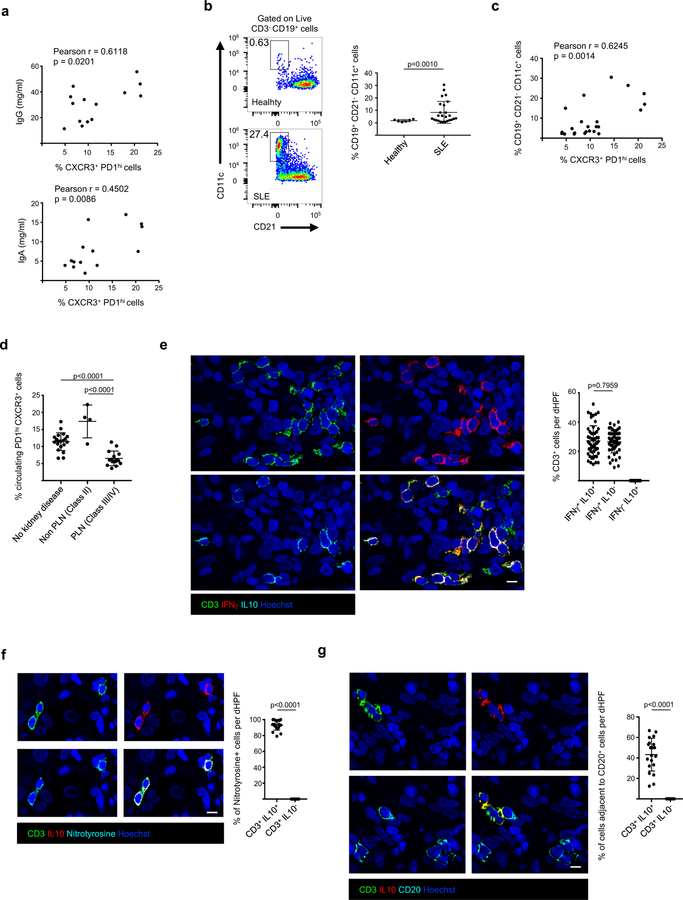
a, Pearson correlation analysis between the frequency of SLE blood CXCR3+ PD1hi CD4+ T cells and serum immunoglobulin IgG and IgA levels (n=14 biologically independent samples). b, Representative flow cytometry density plot (left) and percentage of CD19+CD21−CD11c+ B cells (ABCs) among CD3− CD19+ cells (right) in the blood of healthy donors (n=6) or SLE patients (n=25). c, Pearson correlation analysis between the frequency of blood CXCR3+ PD1hi CD4+ T cells and ABCs. d, Percentage of CXCR3+ PD1hi CD4+ T cells in blood of SLE patients with nephritis Class II (n=4 biologically independent samples), Class III/IV (PLN; n=15 biologically independent samples) or without kidney disease (n=20 biologically independent samples). e, Representative immunofluorescence microscopy of CD3, IFNγ and IL10 staining in the kidney of a class IV LN section. The percentage of CD3+ IFNγ+ IL10+; CD3+ IFNγ+ IL10− and CD3+ IFNγ− IL10+ cells is also shown (n=10 class III/IV PLN samples; 5 dHPF [digital High Power Field] per sample). f, Representative immunofluorescence microscopy of CD3, Nytrotyrosine and IL10 staining in the kidney of a class IV LN section. The percentage of CD3+ IL10+ Nytrotyrosine+ and CD3+ IL10− Nytrotyrosine+ cells is also shown (n=5 class III/IV PLN samples; 4 dHPF per sample). g, Representative immunofluorescence microscopy of CD3, IL10 and CD20 staining in the kidney of a class IV LN section. The percentage of CD3+ IL10+ and CD3+ IL10− cells adjacent to CD20+ B cells is also shown (n=5 class III/IV PLN samples; 4 dHPF per sample). Scale bar = 10 μm. Shown are mean ± s.e.m.; statistical analysis by nonparametric one-way ANOVA (d) and two-tailed nonparametric unpaired t-test at 95% CI (Welch’s correction b; f-g).
Lupus nephritis (LN), which comprises several histological classes, is one of the major drivers of morbidity and mortality. As conventional markers of kidney function are not LN class-specific, we asked if blood CXCR3+ PD1hi CD4+ T cells would represent a LN biomarker. When correlated with clinical and laboratory data, the highest frequency and absolute numbers of blood CXCR3+ PD1hi CD4+ T cells were detected in patients with no LN or with LN class not associated with lymphocytic infiltration in the kidney (Class II; Fig. 4d and Supplementary Fig. 7d). Conversely, the lowest frequency was found in patients with proliferative nephritis (PLN, Classes III and IV), the most severe class of LN in children. This distribution was not driven by immunosuppressive therapy (Supplementary Fig. 7e).
Because lower numbers of blood CXCR3+ PD1hi CD4+ T cells in PLN might reflect their migration into the kidney, tissue sections from SLE patients without LN and with different LN classes were analyzed by immunofluorescence. Different degrees of CD3+ T cell infiltrates were detected in the peritubular areas of a significant fraction (13/17) of PLN sections (Supplementary Fig. 7f, g). Across these PLN sections, 27.1% ± 10.3% (mean ± SEM) of CD3+ T cells co-expressed IFNγ+ and IL10+ (Fig. 4e). While the presence of cytotoxic CD8+ T cells in LN has been reported35, all infiltrating IL10+ CD3+ T cells co-expressed CD4 (Supplementary Fig. 7h). Furthermore, similar to circulating CXCR3+ PD1hi CD4+ T cells, infiltrating IL10+ CD3+ T cells co-expressed PD1 (Supplementary Fig. 7i) and were positive for nitrotyrosine (93% ± 6.1%; mean ± SD; Fig. 4f). Because intrarenal B cells are also a feature of PLN36, we examined the spatial relationship between IL10+ CD3+ T cells and CD20+ B cells. As shown in Fig. 4g, a large fraction (43.2% ± 16%; mean ± SD) of IL10+ CD3+ T cells appeared in close proximity to CD20+ B cells.
Recently, a population of CXCR5− PD1hi CD4+ T cells (Tph) was reported expanded in rheumatoid arthritis (RA). Similar to Tfh, Tph induce B cell differentiation and antibody secretion in an IL21- and CXCL13-dependent manner37. The cells that we describe herein produce neither IL21 nor CXCL13 and exhibit a distinctive transcriptome and chromatin landscape compared to Tfh. Importantly, they help B cells through a unique mechanism involving IL10 and succinate.
New roles for succinate, an intermediate of the tricarboxylic acid (TCA) cycle, outside metabolism have recently emerged, including its synergism with LPS to induce IL1β production by macrophages24. While the effects of succinate on innate immunity are well recognized, this is the first report implicating it in the shaping of adaptive immune responses.
B and T cells are a prominent feature of PLN infiltrates36. Patients with PLN, however, carry the lowest numbers of blood CXCR3+ PD1hi CD4+ T cells. Instead, IFNγ+ IL10+ ROS+ PD1hi CD4+ T cells are found in the peritubular areas in close proximity to B cells. Of note, succinate receptor (SUCNR1) is expressed in a variety of tissues, especially the kidney. Within this organ, the highest receptor density is found in the proximal tubular epithelium. There, triggering of the receptor leads to release of renin from the juxtaglomerular apparatus. Consequently, succinate has been implicated in the pathogenesis of diabetic nephropathy and renovascular hypertension38. Whether T cell-derived succinate directly contributes to kidney damage in PLN, the main class of LN associated with reno-vascular hypertension, deserves further studies.
The novel CD4+ T cell population that we describe, and propose to designate “Th10”, expands the spectrum of B cell-helper T cells and might contribute to SLE pathogenesis and end organ damage in a variety of ways. These cells provide unique mechanistic clues for therapeutic intervention in a disease for which only one new drug has been approved in more than 60 years.
Methods
Patient samples:
This study was approved by the Institutional Review Boards of the University of Texas Southwestern Medical Center, Texas Scottish Rite Hospital for Children, Baylor-Scott&White Health Care Systems, Pathologists Bio-Medical Laboratories and Weill Cornell Medical College. Informed consent was obtained from all patients or their parents/guardians. Blood samples were obtained from patients fulfilling the diagnosis of SLE according to the criteria established by the American College of Rheumatology. Healthy pediatric control samples were obtained via and IRB-approved protocol from children whose parent/guardian completed a questionnaire indicating that the child 1) had no chronic illness and 2) had not been ill, received any vaccinations or suffering from seasonal allergies at the time of blood collection or during the month beforehand. Renal biopsies (FPE) from control (n = 6) or SLE patients displaying class II (n = 4), III (n = 9), class IV (n = 8) Lupus nephritis (LN), as defined by the International Society of Nephrology/Renal Pathology Society revised LN classification criteria and who on review of records fulfilled the American College of Rheumatology revised criteria for the classification of SLE39,40, were obtained from Pathologists Bio-Medical Laboratories (Lewisville, TX). All relevant ethical regulations were followed while conducting this work.
Flow cytometry and cell sorting:
For CXCR3+ PD1hi CD4+ T cell quantification, cryopreserved cells were thawed into warm RPMI/10% FBS, washed once in cold PBS, and stained in PBS/1% BSA with the following antibodies for 45 min: anti-CD4 PE-Cy7 (Clone SK3), anti-CXCR5 AlexaFluor 647 (Clone RF8B2), anti-CD45RA APC-H7 (Clone H100), anti-PD1 Brilliant Violet 421 (Clone EH12.2H7), anti-CD3 V500 (Clone RPA-T4), anti-CXCR3 Brilliant Violet 785 (Clone GO25H7). Antibodies used in additional panels included anti-CD21 FITC (Clone Bu32), anti-CD27 PE (Clone M-T271), anti-IgD PerCP Cy5.5 (Clone IA6–2), anti-CD38 PE-Cy7 (Clone HB7), anti-CD19 AF700 (Clone H1B19), anti-CD11c V450 (Clone B-ly6), anti-CD3 Brilliant Violet 650 (Clone OKT3). Cells were washed in cold PBS, passed through a 70-μm filter, and data acquired on a BD Fortessa, BD Canto II or Cytek Aurora flow cytometer. Data were analysed using FlowJo 10.0.7. For pDC isolation, the total dendritic cell fraction was obtained from healthy buffy coats by magnetic cell sorting with EasySep™ Human pan-DC Enrichment Kit (Stem Cell Technology; cat # 19251) following the manufacturer’s instructions. Highly pure (>99%) pDCs (Lin1- HLADR+ CD11c− CD123+ cells) were then isolated from this fraction by FACS sorting with the following antibodies: anti-Lin1 FITC, anti-HLADR APC-H7 (Clone G46–6), anti-CD11c APC (Clone S-HCL-3) and anti-CD123 PE (Clone 9F5). The “Lin1” cocktail was composed of CD3, CD14, CD16, CD19, CD20, and CD56 antibodies (BD Biosciences; cat # 340546). Fresh peripheral blood naïve CD4 T cells (>99% pure) were isolated using EasySep™ Human Naïve CD4+ T Cell Enrichment Kit (Stem Cell Technology; cat # 19155) following the manufacturer’s instructions. Naïve CD4+ T cells where labeled with 5 μM carboxy-fluorescein diacetate succinimidyl ester (CFSE; Thermo Fisher) following the manufacturer’s instructions. Were described, primed (CFSElow) CD4+ T cells were sorted from pDC/CD4 T cell co-cultures at day 6. 7ADD and anti-CD123 antibody (Clone 9F5) were used to remove dead cells and contaminating pDCs, respectively.
For B cell isolation, CD19+ cells were obtained from healthy buffy coats by magnetic cell sorting with EasySep™ Human B Cell Enrichment Kit (Stem Cell Technology; cat # 19054). Enriched cells were then stained with anti-IgD APC (Clone IgD26), anti-CD27 PE (Clone MT271) and anti-CD19 FITC (Clone H1B19) and then sorted as IgD+CD27−CD19+ cells (naïve), IgD−CD27+CD19+ cells (memory) or IgD+CD27+CD19+ cells (double positive). For blood CD4+ T cell subset sorting, frozen PBMCs from SLE patients were stained with anti-CD4 PE-Cy7, anti-CXCR5 AlexaFluor 647, anti-CD45RA APC-H7, anti-PD1 Brilliant Violet 421, anti-CD3 V500 and anti-CXCR3 Brilliant Violet 785 as described above. Then, CXCR3+ PD1low CD4+ T cells, CXCR3− PD1hi CD4+ T cells, CXCR3+ PD1hi CD4+ T cells populations were sorted from the CD3+ CD4+ CD45RA− CXCR5− cell fraction. Where described, CD3+ CD4+ CD45RA− CXCR5+ CD4+ T cells (Tfh) were also sorted for comparison. Cell sorting was performed on a BD FACSAria Fusion or BD FACSMelody cell sorter using a 100μm nozzle. Sort gates were drawn as depicted in Supplementary Fig. 2a and 6a. Cell purity was routinely >98%. For functional analyses, cells were sorted into cold RPMI/10% FBS. For RNA analyses, sorted cells were lysed in RLT lysis buffer (Qiagen) with 1% β-mercaptoethanol (Sigma).
Ox mtDNA generation and pDC activation:
Ox mtDNA was generated as previously described3. Briefly, healthy neutrophils were pre-incubated with IFNα2β (2000 U/ml; Schering Corp.) for 90 min at 37 C and then extensively washed before incubation with anti-RNP IgG (50 μg/ml) purified from SLE patient sera. Neutrophil supernatants were then collected, centrifuged for 10 min at 1400g and stored at −80 C. PDCs (5×105 cells/well – 96 U bottom plate) were cultured with 40% v/v Ox mtDNA-containing neutrophil supernatants (referred in the text as “Ox mtDNA”) or with 5 μg/ml of either CpGA (ODN-2216; Invivogen) or CpGB (ODN-2006; Invivogen) for 24 h. The volume of Ox mtDNA-containing neutrophil supernatants and the concentration of CpGA were selected based on their capacity to trigger similar levels of IFNα production by pDCs.
PDC analysis:
PDCs were cultured as described in the paragraph above, and cytokine levels in the corresponding supernatants were measured with Flex Set Kit (BD Biosciences). For flow cytometry analysis, cells were stained with anti-CD80 APC (Clone L307), anti-CD86 PE (Clone FUN-1), anti-CD83 FITC (Clone HB15e), anti-CD40 PE (Clone 5C3), anti-HLADR APC-H7 (Clone G46–6), anti-CD123 PE (Clone 9F5), anti-CXCR4 PECy7 (Clone 12G5), anti-CXCR3 Brilliant Violet 785 (Clone GO25H7) or anti-CCR7 APC (Clone 3D12). To assess the effect of IL3 on cell viability, activated pDCs were treated with recombinant human IL3 (50 ng/ml – BD Biosciences), cultured for an additional 24 h and then stained with the annexin V–apoptosis detection kit (BD Biosciences) following the manufacturer’s instructions.
Migration assay:
For Transwell-migration assays, 5 × 104 activated pDCs were applied in 100 μl cRPMI to 6.5-mm diameter Transwell inserts that were separated from the lower chamber by polycarbonate membranes containing 5-μm pores (Costar). The lower compartments were filled with cRPMI and 1 μg/ml CCL19 or CCL21 (R&D Systems). Cells were then allowed to migrate through the bottom of the chamber for 18 h. The number of transmigrated cells relative to input was then measured.
CD4 T cell differentiation, activation and analysis:
12 × 104 freshly sorted allogeneic naïve CD4+ T cells were co-cultured with activated pDCs (DC/T cell ratio of 1:6) in round-bottom 96-wells culture plates for 6 days. Were described, MitoTempo (MT – 50 μM – SantaCruz Biotechnology) Rapamycin (100 nM - SantaCruz Biotechnology) or anti-PD1 antibody (clone EH12.2H7 – 10 μg/ml - Biolegend) were added during the co-culture. As a control, 12 × 104 naïve CD4+ T cells were activated with 2 μl of Dynabeads human T cell activator CD3/CD28 (Thermo Fisher) for 6 days (referred in the text as “Th0”). For intracellular cytokine staining, primed CD4+ T cells were re-stimulated with 50 ng/ml PMA, 2 μg/ml Ionomycin and 1 μl of GolgiPlug (BD Biosciences) for 5 h. Cells were then stained with a combination of anti-IL10 APC (Clone JES3–19F1) and anti-IFNγ PE-Cy7 (Clone B27) antibodies with Cytofix/Cytoperm Fixation and Permeabilization Solution (BD Biosciences) following the manufacturer’s instructions. For re-stimulation experiments, 5 × 104 primed (CFSElow) CD4+ T cells or sorted SLE patient CD4+ T cells were re-stimulated with 10 μg/ml of plate-bound anti-CD3 (OKT3 - Biolegend) and 2 μg/ml of soluble anti-CD28 mAb (Biolegend) for 24 h. Cytokine levels in the corresponding supernatants were measured with Flex Set Kit (BD Biosciences). For Tbet and HIF1α intracellular staining, CD4+ T cells were stained with anti-HIF1α antibody (Clone 546–16; Biolegend) or anti-Tbet antibody (Clone 4B10; Biolegend) with Foxp3 fix/perm buffer set (Biolegend) following the manufacturer’s instructions. Dead cells were excluded from the analysis by labeling with LIVE/DEAD fixable Aqua (Thermo Fisher) prior to fixation/permeabilization.
Proliferation assay:
CD4 T cell proliferation was measured by resazurin reduction, as previously described41. Briefly, primed (CFSElow) CD4+ T cells were seeded at 5 × 105 cells and re-stimulated with 10 μg/ml of plate-bound anti-CD3 (OKT3) and 2 μg/ml of soluble anti-CD28 mAbs. After incubation for 20 h, medium supplemented with 40 μM resazurin was added for an additional 4 hr. Resazurin reduction to resorufin was measured fluorometrically using a SpectraMax M5 (Molecular Devices). Results obtained were expressed in fluorescence arbitrary units (AU). Alternatively, proliferation was assessed by flow cytometry using the Click-IT EdU Flow Cytometry Proliferation Kit, according to the manufacturer’s instructions (Thermo Fisher).
In vitro Tfh cell generation:
Human Tfh cells were generated as previously described42. Briefly, after overnight stimulation of naive CD4+ T cells with Dynabeads human T cell activator CD3/CD28 (Thermo Fisher) in complete RPMI medium, cells were transferred to 96-well plates coated with anti-CD3 and supplemented with 2 μg/ml of soluble anti-CD28 mAb (Biolegend), human recombinant IL-23 (25 ng/ml; Biolegend) and human recombinant TGFβ1 (5 ng/ml; Biolegend). After 4 d cells were collected and used for analysis.
B cell cultures:
For B/T cells co-cultures, naïve or memory B cells were co-cultured with CD4+ T cells (2 × 104 B cells - 2 × 104 CD4 T cells) in the presence of endotoxin-reduced SEB (500 ng/ml; Sigma Aldrich) in cRPMI supplemented with 10% heat-inactivated FBS. Where described, anti-IL10 (10 μg/ml - clone JES3–9D7 - Biolegend LEAF™ purified antibody), anti-SUCNR1/GPR91 (20 μg/ml – Novus Biological), anti- IL21R (10 μg/ml - clone 17A12 - Biolegend LEAF™ purified antibody) or succinate (2 mM – Sigma-Aldrich) were added during the co-cultures. Sodium azide and other preservatives were removed from the antibody preparations by protein desalting with Zeba Spin Desalting Columns (7K MWCO; Thermo Fisher). IgM and IgG concentrations were measured at day 12 in the corresponding supernatants with Flex Set Kit (BD Biosciences). For T cell-independent B cell differentiation, naïve B cells (5 × 104 cells) were co-cultured with irradiated (77Gy) human-CD40L transfected fibroblasts43 (0.5 × 104 cells) in cRPMI supplemented with 10% heat-inactivated FBS. Recombinant human IL10 (Biolegend, 500 ng/ml) and/or succinate (2 mM – Sigma-Aldrich) were added during the co-cultures. IgG and IgM concentrations were measured at day 12 in the corresponding supernatants with Flex Set Kit (BD Biosciences).
Immunofluorescence microscopy (IF):
Cells were settled on poly-L-lysine coated glass coverslips for 20 min at 37C, rinsed with PBS and then fixed with 4% paraformaldehyde for 20 min at room temperature. Cells were permeabilised with 0.05% TritonX-100 in PBS for 5 min at room temperature and then treated with Blocking Buffer (5% goat serum and 1% BSA in PBS) for 30 min at room temperature. Primary (anti-p65 antibody; Abcam, cat # ab16502) and secondary antibody stainings were carried out in Staining Buffer (1% BSA in PBS). Isotype specific anti-mouse or anti-rabbit AlexaFluor-488 or AlexaFluor-568 were used as secondary antibodies. Counterstaining of cell nuclei was performed with Hoechst stains (Molecular Probes). Samples were mounted with ProLong Gold Antifade Reagent (Molecular Probes) and examined with a Leica TCS SP5 confocal laser-scanning microscope equipped with a 63x/1.4 oil objective. ImageJ software (NIH; Bethesda MD) was used for analysis. The percentage of co-localization was calculated from the Manders’ Overlap Coefficient using the ImageJ “Co- localization analysis” plugin (NIH; Bethesda MD).
Quantitative real-time PCR (qPCR):
5 × 104 were lysed with 50 μl of Cell-to-Ct lysis buffer (Thermo Fisher). cDNA was directly synthesized from cell lysates with the Cell-to-Ct Kit (Thermo Fisher). Quantitative real-time PCR was performed with TaqMan Gene Expression Assays (Applied Biosystems) on a 7500 Real-Time PCR System (Applied Biosystems). β-actin was used as housekeeping gene.
SDS-PAGE and Western blot:
Cells were washed in PBS and then lysed in RIPA buffer in the presence of Halt Protease and Phosphate Inhibitor Cocktail (Thermo Fisher). Samples were incubated on ice for 30 min and then centrifuged (13000g for 10 min at 4C). The supernatants containing the protein fraction were collected and stored at −80C until further analysis. Protein concentration was estimated using the BCA kit (Thermo Fisher) following the manufacturer’s instructions. 10–20 µg of proteins were resuspended in 5x Lane Marker Reducing Sample Buffer (Thermo Fisher), boiled for 5 min at 100C and then subjected to electrophoresis with Mini-PROTEAN TGX Precast Gel (BIO-RAD). The proteins were then transferred to PVDF membranes with the TransBlot Turbo System (BIO-RAD), blocked for 1 h 5% nonfat dry milk in TBST (Tris Buffered Saline containing 0.1% Tween20) and incubated overnight at 4C with the primary antibodies. Anti-SUCNR1 (Cat # NBP1–00861; Novusbio), anti-GAPDH (Cat # 2118; Cell Signaling), anti-pRb Ser807/811 (Cat # 9308; Cell Signaling), anti-Cyclin D1 (Cat # 2989; Cell Signaling), anti-Cyclin D2 (Cat # 3741; Cell Signaling), anti-Cyclin D3 (Cat # 2936; Cell Signaling), anti-Nitrotyrosine (Cat # 9691; Cell Signaling), anti-NDUFA9 (Cat # ab110412; Abcam), anti-NDUFA8 (Cat # ab199681; Abcam), anti-SDHA (Cat # ab110412; Abcam), anti-UQCRC2 (Cat # ab110412; Abcam) and anti-ATP5A (Cat # ab110412; Abcam) were used as primary antibodies. After washing in TBST, the membranes were incubated for 1 h at room temperature with Poly HRP-conjugated anti-rabbit or anti-mouse IgG (Thermo Fisher). ECL Plus reagents (Amersham) were used for detection. Digital images were acquired with ChemiDoc MP System (BIO-RAD) and analyzed with Image Lab Software (BIO-RAD).
siRNA knockdown:
Knockdown of TBX21 in primary human naïve CD4+ T cells was done using ACCELL siRNA SMARTpool designed and validated by Dharmacon (Lafayette, CO). A non-targeting siRNA was used as negative control. 12 × 104 freshly sorted allogeneic naïve CD4+ T cells were activated with pDCs (DC/T cell ratio of 1:6) for 24 h before adding 1 μM of ACCELL siRNA. 96 h post-transfection, cells were re-stimulated with 50 ng/ml PMA, 2 μg/ml Ionomycin and GolgiPlug (BD Biosciences) for 5 h and analyzed for intracellular cytokine production as described above. Protein knockdown was validated by flow cytometry.
Microarray analysis of pDCs:
Total RNA was isolated using the RNeasy kit (Qiagen), amplified and then labeled with Illumina TotalPrep RNA amplification kit (Invitrogen). Agilent 2100 Analyzer (Agilent Technologies) was used to assess RNA integrity. Biotinylated complementary RNA (cRNA) was hybridized to Illumina Human-6 Beadchip Array version 2 and scanned on Illumina Beadstation 500. Fluorescent hybridization signals were assessed with Beadstudio software (Illumina), and statistical analysis and hierarchical clustering were performed with GeneSpring 7.3.1 software (Agilent Technologies).
RNA preparation and Sequencing Library Preparation:
Total RNA was isolated from cell lysates using a modified protocol for the RNAqueous™ Micro Total RNA Isolation Kit (Thermo Fisher) including on-column DNase digestion and analyzed for quality using the RNA 6000 Pico Kit (Agilent). For in vitro generated CD4+ T cells, Poly-A enriched NGS library construction was performed using the KAPA mRNA Hyper Prep Kit (KAPA Biosystems) using 500ng of input total RNA according to manufacturer’s protocol using 9 amplification cycles. Individual libraries were quantitated via qPCR using the KAPA Library Quantification Kit, Universal (KAPA Biosystems) and equimolar pooled. Final pooled libraries were sequenced on an Illumina NextSeq 500 with paired-end 75 base read lengths. For ex vivo isolated CD4+ T cells, NGS library construction was performed using the SMART-Seq v4 Ultra Low Input RNA kit (Clontech) using 2ng of input total RNA according to manufacturer’s protocol using 12 amplification cycles to generate cDNA. Sequencing libraries were prepared using the Nextera XT DNA Library Prep Kit (Illumina) using 150pg of cDNA according to manufacturer’s protocol. Individual libraries were quantitated using Qubit dsDNA HS Assay Kit (Thermo Fisher) and equimolar pooled. Final pooled libraries were sequenced on an Illumina NextSeq 500 with paired-end 75 base read lengths.
RNA-seq data processing and analysis:
Quality control of raw reads was performed with FASTQC. Reads were aligned to the reference human genome (GRCh38) using hisat2 after quality and adapter trimming by cutadapt. After sorting bam files by name using samtools, HTSeq-count program was used to quantify total numbers of read counts mapped to genome. The RNA-seq data analysis was perform in R programming language. DESeq2 R package was used for size factor and dispersion estimation calculation and differential gene expression analysis.
ATAC-seq library generation and sequencing:
ATAC-seq was performed as previously described30. 20,000 unfixed nuclei were tagged using Tn5 transposase (Nextera DNA sample prep kit; Illumina) for 30 min at 37°C, and the resulting library fragments were purified using a Qiagen MinElute kit. Libraries were amplified by 10–12 PCR cycles, purified using a Qiagen PCR cleanup kit, and finally sequenced on an Illumina HiSeq 2500 with 75 bp paired-end reads to a minimum depth of 30 million reads per sample. At least two technical replicates were processed per biological sample.
ATAC-seq preprocessing and bioinformatics analysis:
Reads were trimmed using trimmomatic44 and mapped to the GRCh37/hg19 assembly of the human genome using bwa-mem45. Duplicate reads were removed using samtools and technical replicates were merged into a single bam file. Peak calling was performed using MACS2 with “--nomodel --shift 37 --extsize 73 --broad” parameters, and only peaks with q-value < 0.05 were selected. Peaks overlapping blacklisted regions as defined by the ENCODE project were discarded. Consensus peaks present in at least two samples were obtained using the DiffBind R package. Peaks were annotated to the closest transcription start sites in the UCSC hg19 knownGene transcriptome using the ChIPSeeker R package46. Peaks with differential chromatin accessiblility were found using a generalized linear model in EdgeR using the population as covariate. Genome tracks of read coverage per base pair and per million mapped reads were generated using HOMER47 makeUCSCfile with parameters “-res 1 -norm 1e6”, and visualized with the IGV genome browser.
Mitochondrial ROS detection:
Primed (CFSElow) CD4+ T cells were sorted and loaded for 30 min at 370C with MitoSox Red (2.5 μM - Thermo Fisher) and MitoTraker Deep Red (25 nM - Thermo Fisher Cells). Cells were then re-stimulated with 10 μg/ml of plate-bound anti-CD3 (OKT3 - Biolegend) and 2 μg/ml of soluble anti-CD28 (Biolegend) for 1 h. Cells were then washed and subjected to flow cytometry analysis or immunofluorescence microscopy. Alternatively, total PBMCs from healthy donors or SLE patients were loaded for 30 min at 37C with MitoSox Red (2.5 μM - Thermo Fisher). Cells where then stained as described above and analyzed by flow cytometry.
Succinate assay:
Succinate was measured with the Succinate Colorimetric Assay Kit (Abcam) following the manufacturer’s instructions. Briefly, 5 × 104 CD4+ T cells were homogenized with 50 μl of Succinate Assay Buffer for 5 min on ice. 10 μl of lysates were then used for the assay.
Seahorse assays:
Complex I and II-linked mitochondrial respiration was determined using a modified version of a previously described method48. Briefly, XF24 cell culture microplates (Seahorse Bioscience) were coated with Cell-Tak (50uL at 22.4 μg/mL; Corning) and 15 × 104 primed (CFSElow) CD4 T cells were plated in MAS-BSA assay solution (220 mM mannitol, 70 mM sucrose, 10 mM KH2PO4, 5 mM MgCl2, 2mM HEPES, 1 mM EGTA, 0.2% fatty acid free BSA) containing XF Plasma Membrane Permeabilizer (2nM; Seahorse Bioscience) and ADP (4mM; Santa Cruz Biotechnology). Complex I activity was assessed by measuring the oxygen consumption rates (OCR) in response to the complex I substrate pyruvate (10 mM; Sigma Aldrich). Malate (5 mM; Sigma Aldrich) was added togheter with pyruvate to allow oxaloacetate production and condensation with acetyl CoA, allowing normal pyruvate dehydrogenase (PDH) flux48. Complex II (SDH) activity was assessed by measuring the OCR in response to the complex I substrate succinate (10 mM; Sigma Aldrich). Rotenone (2 μM; Sigma Aldrich) was added togheter with succinate to prevent accumulation of oxaloacetate, which is a potent inhibitor of SDH48. For maximal respiration rate (MMR) measurement, the assay was performed in Seahorse medium (DMEM supplemented with 2 mM glutamine, 25 mM glucose and 1 mM pyruvate). MMR was defined as the OCR after the addition of oligomycin (2 μM; Sigma Aldrich) and FCCP (2 μM). All extracellular flux analyses were performed using an XF-24 Extracellular Flux Analyzer (Seahorse Bioscience) as recommended by the manufacturer.
Immunofluorescence of paraffin-embedded kidney sections:
Tissue sections were de-paraffinized in xylene for 10 min, washed with 100% ethanol followed by 95, 80, 70 and 50% ethanol, and then rinsed in distilled water. For antigen retrieval, tissue sections were boiled for 10 min in 10 mM sodium citrate buffer (pH 6.0) containing 0.05% Tween-20. After two washes with distilled water and two washes with PBS, tissue sections were permeabilized for 30 min at room temperature in PBS/0.1% Triton X-100. Sections were blocked for 1 h at room temperature in blocking buffer (PBS/1% BSA/0.5% Fish Gelatin/5% goat serum) and then incubated overnight at 4 °C with the primary antibody diluted in blocking buffer. Anti-CD3 (Clone OKT3; Biolegend), anti-IL10 (Clone A47–25-17; Abcam), anti-IFNγ (Cat # ab25101; Abcam), anti-Nitrotyrosine (Cat # ab42789; Abcam), anti-CD20 (Clone EP459Y; Abcam), anti-CD4 (Clone EPR6855; Abcam), anti-PD1 (Clone EPR4877; Abcam) and anti-Aquaporin1 (Clone B-11; SantaCruz Bio) were used as primary antibodies. Tissue sections were then probed with Alexa Fluor conjugated secondary antibodies, diluted in blocking buffer, at room temperature for 60 min. Counterstaining of cell nuclei in tissue sections was performed with the Hoechst stains (Thermo Fisher). Samples were then mounted with ProLong Gold Antifade Reagent (Thermo Fisher). Digital images were taken using LSM 880 confocal microscope with Airyscan High Resolution Detector (Carl Zeiss Microscopy). ImageJ software (NIH; Bethesda MD) was used for data analysis.
Serum immunoglobulin levels:
Serum levels of IgG and IgA were measured with total human IgG and IgA Flex Set Kits in accordance with the manufacturer’s instruction (BD Biosciences).
Statistical Analysis:
No specific statistical methods were used to predetermine sample size. All results are presented as the mean ± SEM. The significance of the difference between groups was analyzed as described in the figure legends. Pearson’s correlation coefficients with 2-tailed P values were determined in the analysis of correlations. P values less then 0.05 were considered statistically significant. All statistical analyses were performed using GraphPad Prism Software (v7.0e).
Data availability:
Expression array data from pDCs is available at the Gene Expression Omnibus (GEO) GSE93679. Expression array data from in vitro generated CD4+ cells is available at the GEO database under GSE118951. Expression array data from ex vivo isolated CD4+ cells is available at the GEO database under accession GSE109843. ATAC-sequencing data is available at the GEO database under accession GSE110017. Uncropped data for Supplementary Fig. 2g can be accessed in Supplementary Fig. 8a. All other relevant data are available from the corresponding author directly.
Supplementary Material
Acknowledgements
The authors wish to thank Drs. Christine Kusminski and Ruth Gordillo for helping with the metabolic Seahorse experiments. We also thank Drs. Nicole Baldwin and Radu Marches and especially our patients, healthy donors and their parents. This work was supported by grants P50 AR054083–01 and U19 AIO82715 (V.P.), by the Baylor-Scott & White Health Care Research Foundation and the Drukier Institute for Children’s Health at Weill Cornell Medicine.
Research Grant and consulting honorarium from Sanofi-Pasteur (VP).
Footnotes
Competing financial interests
References
- 1.Tsokos GC Systemic lupus erythematosus. The New England journal of medicine 365, 2110–2121 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Tipton CM, et al. Diversity, cellular origin and autoreactivity of antibody-secreting cell population expansions in acute systemic lupus erythematosus. Nat Immunol 16, 755–765 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Caielli S, et al. Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. J Exp Med 213, 697–713 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guiducci C, et al. TLR recognition of self nucleic acids hampers glucocorticoid activity in lupus. Nature 465, 937–941 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Means TK, et al. Human lupus autoantibody-DNA complexes activate DCs through cooperation of CD32 and TLR9. J Clin Invest 115, 407–417 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gilliet M & Liu YJ Human plasmacytoid-derived dendritic cells and the induction of T-regulatory cells. Hum Immunol 63, 1149–1155 (2002). [DOI] [PubMed] [Google Scholar]
- 7.Jego G, et al. Plasmacytoid dendritic cells induce plasma cell differentiation through type I interferon and interleukin 6. Immunity 19, 225–234 (2003). [DOI] [PubMed] [Google Scholar]
- 8.Ito T, et al. Plasmacytoid dendritic cells prime IL-10-producing T regulatory cells by inducible costimulator ligand. J Exp Med 204, 105–115 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gilliet M, Cao W & Liu YJ Plasmacytoid dendritic cells: sensing nucleic acids in viral infection and autoimmune diseases. Nature reviews. Immunology 8, 594–606 (2008). [DOI] [PubMed] [Google Scholar]
- 10.Grouard G, et al. The enigmatic plasmacytoid T cells develop into dendritic cells with interleukin (IL)-3 and CD40-ligand. J Exp Med 185, 1101–1111 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Penna G, Sozzani S & Adorini L Cutting edge: selective usage of chemokine receptors by plasmacytoid dendritic cells. J Immunol 167, 1862–1866 (2001). [DOI] [PubMed] [Google Scholar]
- 12.Glimcher LH & Murphy KM Lineage commitment in the immune system: the T helper lymphocyte grows up. Genes Dev 14, 1693–1711 (2000). [PubMed] [Google Scholar]
- 13.Acosta-Rodriguez EV, et al. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat Immunol 8, 639–646 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Jackson SK, DeLoose A & Gilbert KM The ability of antigen, but not interleukin-2, to promote n-butyrate-induced T helper 1 cell anergy is associated with increased expression and altered association patterns of cyclin-dependent kinase inhibitors. Immunology 106, 486–495 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sena LA, et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 38, 225–236 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nag S, Picard P & Stewart DJ Expression of nitric oxide synthases and nitrotyrosine during blood-brain barrier breakdown and repair after cold injury. Lab Invest 81, 41–49 (2001). [DOI] [PubMed] [Google Scholar]
- 17.Guaras A, et al. The CoQH2/CoQ Ratio Serves as a Sensor of Respiratory Chain Efficiency. Cell Rep 15, 197–209 (2016). [DOI] [PubMed] [Google Scholar]
- 18.Murphy MP How mitochondria produce reactive oxygen species. Biochem J 417, 1–13 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tannahill GM, et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 496, 238–242 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oaks Z, Winans T, Huang N, Banki K & Perl A Activation of the Mechanistic Target of Rapamycin in SLE: Explosion of Evidence in the Last Five Years. Curr Rheumatol Rep 18, 73 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rousset F, et al. Interleukin 10 is a potent growth and differentiation factor for activated human B lymphocytes. Proc Natl Acad Sci U S A 89, 1890–1893 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moore KW, de Waal Malefyt R, Coffman RL & O’Garra A Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol 19, 683–765 (2001). [DOI] [PubMed] [Google Scholar]
- 23.Ueno H, Banchereau J & Vinuesa CG Pathophysiology of T follicular helper cells in humans and mice. Nat Immunol 16, 142–152 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mills E & O’Neill LA Succinate: a metabolic signal in inflammation. Trends Cell Biol 24, 313–320 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Buck MD, O’Sullivan D & Pearce EL T cell metabolism drives immunity. J Exp Med 212, 1345–1360 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Keir ME, Butte MJ, Freeman GJ & Sharpe AH PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol 26, 677–704 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Locci M, et al. Human circulating PD-1+CXCR3-CXCR5+ memory Tfh cells are highly functional and correlate with broadly neutralizing HIV antibody responses. Immunity 39, 758–769 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morita R, et al. Human blood CXCR5(+)CD4(+) T cells are counterparts of T follicular cells and contain specific subsets that differentially support antibody secretion. Immunity 34, 108–121 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Patil VS, et al. Precursors of human CD4(+) cytotoxic T lymphocytes identified by single-cell transcriptome analysis. Sci Immunol 3(2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Buenrostro JD, Giresi PG, Zaba LC, Chang HY & Greenleaf WJ Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10, 1213–1218 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arce E, et al. Increased frequency of pre-germinal center B cells and plasma cell precursors in the blood of children with systemic lupus erythematosus. J Immunol 167, 2361–2369 (2001). [DOI] [PubMed] [Google Scholar]
- 32.Dorner T & Lipsky PE Correlation of circulating CD27high plasma cells and disease activity in systemic lupus erythematosus. Lupus 13, 283–289 (2004). [DOI] [PubMed] [Google Scholar]
- 33.Rubtsov AV, et al. Toll-like receptor 7 (TLR7)-driven accumulation of a novel CD11c(+) B-cell population is important for the development of autoimmunity. Blood 118, 1305–1315 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang S, et al. IL-21 drives expansion and plasma cell differentiation of autoreactive CD11c(hi)T-bet(+) B cells in SLE. Nat Commun 9, 1758 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blanco P, et al. Increase in activated CD8+ T lymphocytes expressing perforin and granzyme B correlates with disease activity in patients with systemic lupus erythematosus. Arthritis Rheum 52, 201–211 (2005). [DOI] [PubMed] [Google Scholar]
- 36.Chang A, et al. In situ B cell-mediated immune responses and tubulointerstitial inflammation in human lupus nephritis. J Immunol 186, 1849–1860 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rao DA, et al. Pathologically expanded peripheral T helper cell subset drives B cells in rheumatoid arthritis. Nature 542, 110–114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peti-Peterdi J High glucose and renin release: the role of succinate and GPR91. Kidney Int 78, 1214–1217 (2010). [DOI] [PubMed] [Google Scholar]
- 39.Weening JJ, et al. The classification of glomerulonephritis in systemic lupus erythematosus revisited. Kidney Int 65, 521–530 (2004). [DOI] [PubMed] [Google Scholar]
- 40.Hochberg MC Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 40, 1725 (1997). [DOI] [PubMed] [Google Scholar]
- 41.Anoopkumar-Dukie S, et al. Resazurin assay of radiation response in cultured cells. Br J Radiol 78, 945–947 (2005). [DOI] [PubMed] [Google Scholar]
- 42.Schmitt N, et al. The cytokine TGF-beta co-opts signaling via STAT3-STAT4 to promote the differentiation of human TFH cells. Nat Immunol 15, 856–865 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Garrone P, et al. Fas ligation induces apoptosis of CD40-activated human B lymphocytes. J Exp Med 182, 1265–1273 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bolger AM, Lohse M & Usadel B Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li H & Durbin R Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yu G, Wang LG & He QY ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31, 2382–2383 (2015). [DOI] [PubMed] [Google Scholar]
- 47.Heinz S, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Salabei JK, Gibb AA & Hill BG Comprehensive measurement of respiratory activity in permeabilized cells using extracellular flux analysis. Nat Protoc 9, 421–438 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Expression array data from pDCs is available at the Gene Expression Omnibus (GEO) GSE93679. Expression array data from in vitro generated CD4+ cells is available at the GEO database under GSE118951. Expression array data from ex vivo isolated CD4+ cells is available at the GEO database under accession GSE109843. ATAC-sequencing data is available at the GEO database under accession GSE110017. Uncropped data for Supplementary Fig. 2g can be accessed in Supplementary Fig. 8a. All other relevant data are available from the corresponding author directly.