

Tetrazole antifungals designed to target fungal lanosterol 14α-demethylase (LDM) appear to be effective against a range of fungal pathogens. In addition, a crystal structure of the catalytic domain of Candida albicans LDM in complex with the tetrazole VT-1161 has been obtained.
KEYWORDS: CYP51, Candida albicans, Candida glabrata, Saccharomyces cerevisiae, VT-1161, antifungal resistance, crystal structure, fungal infections, posaconazole, tetrazole inhibitor
ABSTRACT
Tetrazole antifungals designed to target fungal lanosterol 14α-demethylase (LDM) appear to be effective against a range of fungal pathogens. In addition, a crystal structure of the catalytic domain of Candida albicans LDM in complex with the tetrazole VT-1161 has been obtained. We have addressed concern about artifacts that might arise from crystallizing VT-1161 with truncated recombinant CYP51s and measured the impact on VT-1161 susceptibility of genotypes known to confer azole resistance. A yeast system was used to overexpress recombinant full-length Saccharomyces cerevisiae LDM with a C-terminal hexahistidine tag (ScLDM6×His) for phenotypic analysis and crystallographic studies with VT-1161 or with the widely used triazole drug posaconazole (PCZ). We determined the effect of characterized mutations in LDM on VT-1161 activity and identified drug efflux pumps from fungi, including key fungal pathogens, that efflux VT-1161. The relevance of these yeast-based observations on drug efflux was verified using clinical isolates of C. albicans and Candida glabrata. VT-1161 binding elicits a significant conformational difference between the full-length and truncated enzymes not found when posaconazole is bound. Susceptibility to VT-1161 is reduced by ATP-binding cassette (ABC) and major facilitator superfamily (MFS) drug efflux pumps, the overexpression of LDM, and mutations within the drug binding pocket of LDM that affect interaction with the tertiary alcohol of the drug.
INTRODUCTION
The triazoles used in medicine and agriculture include compounds with relatively broad antifungal specificities, such as voriconazole (VCZ) and posaconazole (PCZ), and prodrugs, such as prothioconazole and isavuconazonium sulfate. However, the use of azoles can be compromised due to drug interactions, by the innate resistance of the Zygomycetes and Aspergillus species to some azoles, and through the acquisition of drug resistance via various mechanisms (1–4). The investigational clinical candidate tetrazoles VT-1161, VT-1129, and VT-1598 developed by ViaMet preferentially target the eburicol or lanosterol 14α-demethylases (LDMs) of fungi rather than their human orthologues (5, 6). Preclinical evidence has been gathered to support the use of VT-1161 against superficial infections, such as onychomycosis caused by tinea (7) and recurrent vulvovaginal candidiasis (8), and for the treatment of disseminated fungal disease, including cryptococcal meningitis (9–11), coccidioidomycosis (12, 13) and mucormycosis (14, 15). VT-1161 and its congener VT-1129 have also been reported to show efficacy against intrinsically azole-resistant clinical isolates of Candida glabrata and Candida krusei (16). The tetrazole VT-1598 is claimed to have efficacy against common yeast and molds, including endemic pathogens (4). VT-1161 has completed phase 2b and VT-1129 phase 1 clinical trials (ViaMet).
Informative X-ray crystal structures of the catalytic domain of trypanosomal and fungal sterol 14α-demethylases in complex with various ligands have been obtained using recombinant enzymes that were truncated of their N-terminal monospanning transmembrane helices in order to aid crystallization. These include Trypanosoma cruzi LDM (TcCyp51-5×His) and Candida albicans lanosterol 14α-demethylase (NtruncCaLDM6×His) in complex with VT-1161 and Aspergillus fumigatus sterol 14α-demethylase (AfCYP51B6×His) in complex with VT-1598 (17, 18). We have obtained crystal structures of full-length Saccharomyces cerevisiae LDM (ScLDM6×His) in complex with its substrate and a wide range of medically relevant azole drugs and azole agrochemicals (3, 19–21). We have now investigated the structure of the full-length protein in complex with VT-1161 to determine the impact of this compound on the ligand binding pocket (LBP) that comprises a substrate entry channel (SEC), a deeply buried heme-containing active site, and a putative product exit channel (PPEC). We have also determined the efficacy of the tetrazole class of antifungals in response to structurally characterized mutations in LDMs and the overexpression of LDMs or drug efflux pumps, all of which are known to confer significant resistance to azole drugs (2). A yeast system (22) was used to overexpress recombinant full-length ScLDM6×His for phenotypic analysis and crystallization in complex with VT-1161 and with the established triazole drug posaconazole (PCZ). The same system was used to determine the effect of characterized mutations in LDM on VT-1161 activity and identify drug efflux pumps from prominent fungal pathogens that efflux VT-1161 (22–24). The relevance of the yeast-based observations on drug efflux was validated using clinical isolates of C. albicans and C. glabrata (23, 25).
RESULTS
Activity of VT-1161 is affected by fungal ATP-binding cassette and major facilitator superfamily efflux pumps.
Agarose diffusion assays, using the glucan synthase inhibitor micafungin (MCF) as an independent control antifungal and the triazole drugs VCZ and PCZ as positive controls for azole-based antifungal activity, showed that VT-1161 and isavuconazole (IVC) inhibited the growth of hypersusceptible S. cerevisiae host strains (strain Y785, ADΔ; strain Y2411, AD2Δ) that are deleted of seven ATP-binding cassette (ABC) multidrug efflux pump genes and the PDR3 transcriptional regulator gene (Fig. 1A).
FIG 1.
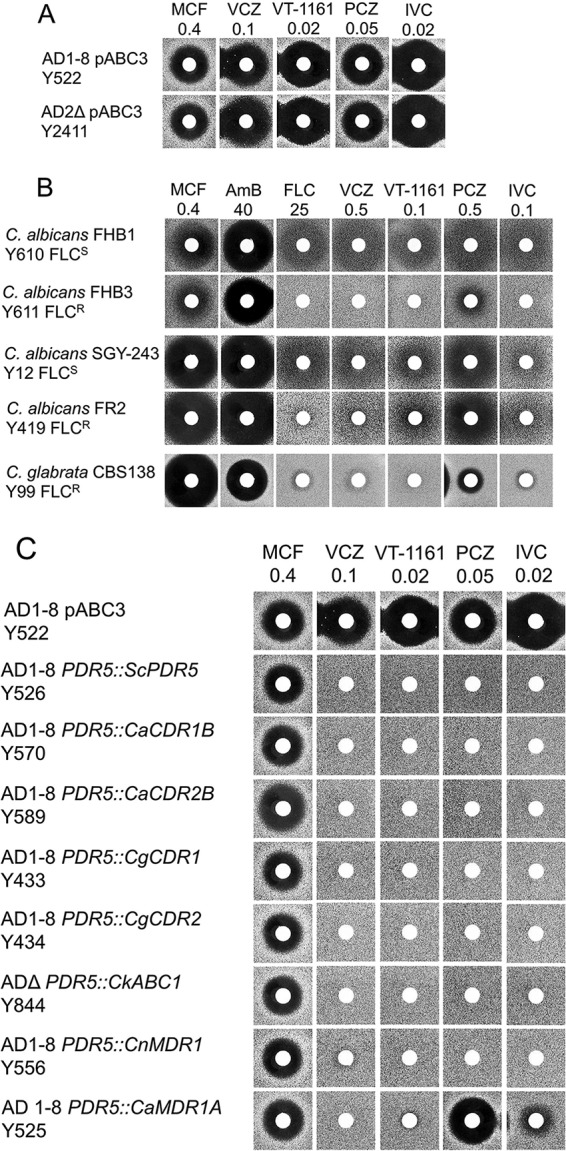
Susceptibilities of yeast constructs and clinical isolates to azole drugs, including VT-1161. (A) Susceptibilities of S. cerevisiae host strains to micafungin and azole drugs. The drugs micafungin (MCF), voriconazole (VCZ), VT-1161, posaconazole (PCZ), and isavuconazole (IVC) were applied to the disks at the indicated nanomolar amounts and agarose diffusion carried out as described in Materials and Methods. (B) Susceptibilities of C. albicans and C. glabrata clinical isolates to MCF and azole drugs. The drugs MCF, amphotericin B (AmB), fluconazole (FLC), VCZ, VT-1161, PCZ, and IVC were applied to disks at the indicated nanomolar amounts and agarose diffusion carried out as described in Materials and Methods. (C) Susceptibilities of S. cerevisiae host strains and strains expressing individual ABC and MFS drug efflux pumps from fungal pathogens to micafungin and azole drugs. The drugs MCF, VCZ, VT-1161, PCZ, and IVC were applied to disks at the indicated nanomolar amounts and agarose diffusion carried out as described in Materials and Methods.
VT-1161 inhibited the growth of azole-sensitive clinical isolates of wild-type C. albicans (Fig. 1B, strains SGY-243 and FHB1) in agarose diffusion assays. These strains and their daughter clinical isolates were susceptible to both MCF and amphotericin B (AMB). On a molar basis, VT-1161 showed activity ∼250-fold greater than that of the gold standard triazole fluconazole (FLC), about 5-fold greater than those of VCZ and PCZ, and activity comparable to that of IVC. Strain FHB3, a daughter clinical isolate of strain FHB1, overexpresses the ABC transporter CaCdr1 (25). Like the triazoles VCZ, PCZ, and IVC, the tetrazole VT-1161 (Fig. 1B) was not effective against strain FHB3. These results suggested both VT-1161 and IVC are substrates of CaCdr1. C. albicans strain FR2, a daughter clinical isolate of strain SGY-243, expresses the major facilitator superfamily (MFS) transporter CaMdr1 at much higher levels than it does CaCdr1 (23). As expected, FR2 showed resistance to FLC and VCZ but not to PCZ. It was also resistant to IVC and was significantly less susceptible to VT-1161 than its parent strain. Thus, IVC appears to be a substrate of CaMdr1, while VT-1161 appears to be a weaker substrate. C. glabrata strain CBS138 is a clinical isolate known to be resistant to azole drugs due to the induction of drug efflux pumps (26). CBS138 was found to be sensitive to MCF and AMB but resistant to VT-1161 and the triazole drugs FLC, VCZ, PCZ, and IVC (Fig. 1B) (26).
The effect of transporter-mediated drug efflux on susceptibility to VT-1161 and IVC was assessed by comparing the susceptibility to azole drugs of control hypersensitive S. cerevisiae host strains with derivative strains that constitutively overexpress individual fungal ABC and MFS efflux pumps from the PDR5 locus (Fig. 1C) (24, 27). VCZ, which is effluxed by both fungal ABC and MFS transporters, was used as a positive control. MCF, which is not transported by these drug efflux pumps, was used as a negative control (26). Figure 1C shows that the strains overexpressing individual ABC drug efflux pumps from S. cerevisiae (ScPdr5), C. albicans (CaCdr1B and CaCdr2A), C. glabrata (CgCdr1 and CgCdr2), C. krusei (CkAbc1), and Cryptococcus neoformans (CnMdr1) were all sensitive to MCF but highly resistant to VCZ, PCZ, IVC, and VT-1161 when exposed to quantities of each drug that inhibited growth of the control parental host strain AD1-8/pABC3. Overexpression of the MFS transporter CaMdr1 in yeast conferred resistance to VCZ, VT-1161, and IVC but not PCZ. This pattern of azole susceptibility of the CaMdr1-overexpressing strain is consistent with the phenotype of the CaMdr1-expressing strain FR2 (Fig. 1C).
The specificity of the drug efflux mechanisms transporting VT-1161 was confirmed using the Cdr1-specific inhibitor RC21v3 and the CaMdr1-specific inhibitor MCC1189 (23, 24). As expected, RC21v3 and MCC1189 had no effect on the growth of the control host strain Y2411 (Fig. 2). Strain Y570, which overexpresses CaCdr1B and confers resistance to FLC and VT-1161, was sensitized to both FLC and VT-1161 by RC21v3 but not MCC1189. Strain Y525, which overexpresses CaMdr1A and thus confers resistance to FLC and VT-1161, was sensitized to FLC and VT-1161 by MCC1189. In this instance, RC21v3 also weakly sensitized strain Y525 to VT-1161. As expected, neither RC21v3 nor MCC1189 sensitized transport by the C. neoformans ABC transporter CneMdr1. These results show VT-1161, like FLC, can be effluxed via CaCdr1 and CaMdr1 (Fig. 2A) and most likely by other fungal ABC and MFS transporters (Fig. 1B).
FIG 2.
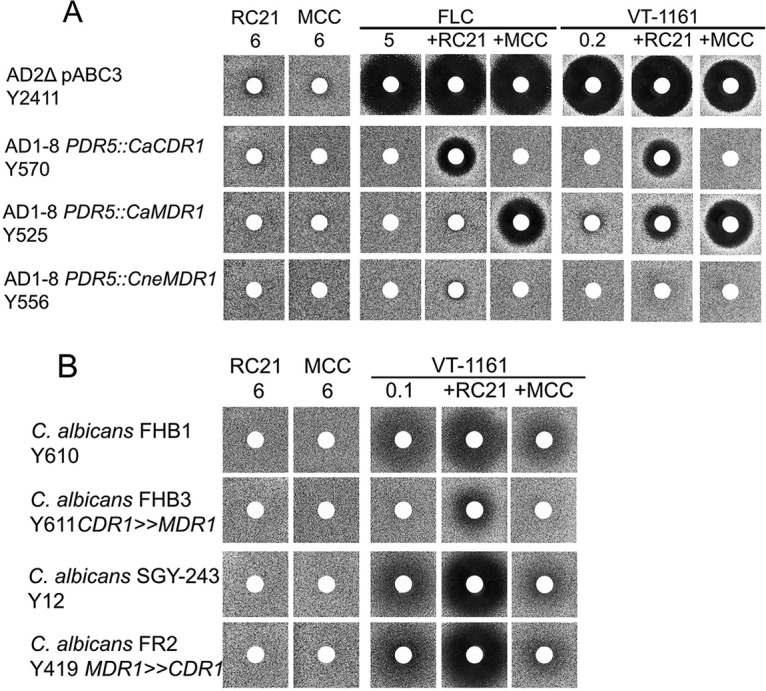
Chemosensitization of VT-1161 efflux via CaCdr1 or CaMdr1 using transporter-specific inhibitors RC23v3 and MCC1189. (A) Chemosensitization of VT-1161 efflux in S. cerevisiae overexpressing CaCdr1 or CaMdr1. The azoles FLC and VT-1161 and the chemosensitizers RC21v3 and MCC1189 were applied to disks at the indicated nanomolar amounts, either individually or in combinations, and agarose diffusion carried out as described in Materials and Methods. (B) Chemosensitization of VT-1161 efflux in C. albicans clinical isolates expressing CaCdr1 or CaMdr1. The agarose diffusion assay was carried out as described for panel A.
In two-dimensional liquid MIC assays, strain Y570 gave MIC80 (80% growth inhibition compared to a no-drug control) values of 30 µM for VT-1161 and 20 µM for RC21v3 (Fig. 3A). Strain Y525 gave MIC80 values of 1.5 µM for VT-1161 and 20 µM for MCC1189 (Fig. 3B). Inhibition of CaCdr1B by RC21v3 in strain Y570 gave fractional inhibitory concentration (FIC) values of 0.025 for VT-1611 and 0.125 for RC21v3, resulting in an FIC index (FICI) of 0.150. Inhibition of CaMdr1A in strain Y525 gave FIC values of 0.0153 for VT-1161 and 0.0625 for MCC1189, resulting in an FICI of 0.0778. These results show strong synergistic chemosensitization of VT-1161 efflux by RC21v3 and MCC1189 for yeast strains constitutively overexpressing CaCdr1B and CaMdr1A, respectively.
FIG 3.
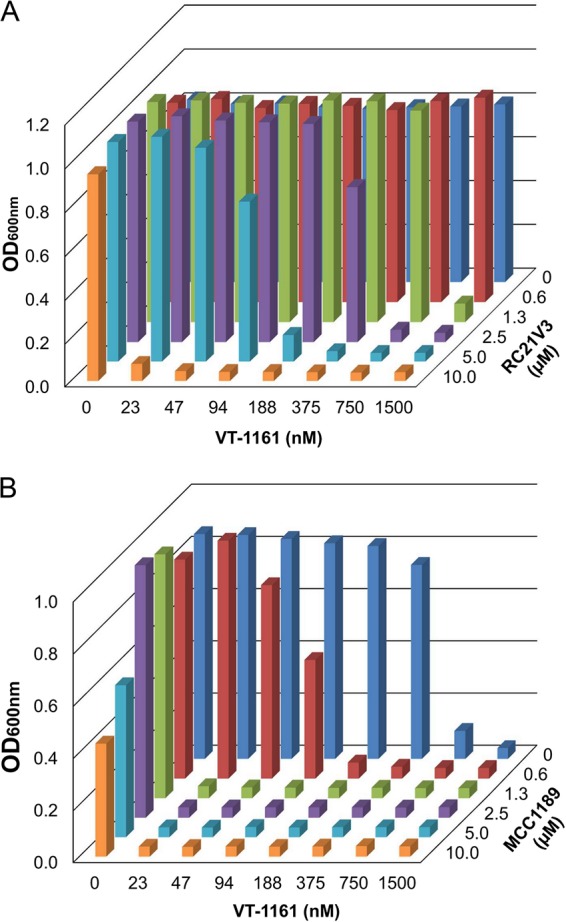
Synergy of drug efflux pump inhibitors on VT-1161 efflux. (A) Effect of RC21v3 on VT-1161 efflux by strain Y570 overexpressing CaCdr1B. (B) Effect of MCC1189 on VT-1161 efflux by strain Y525 overexpressing CaMdr1A. Liquid MIC determinations were carried out as 2-dimensional matrices in microplates incubated for 2 days at 30°C.
The conferral of resistance to VT-1161 due to efflux by CaCdr1 was confirmed by showing that RC21v3 significantly sensitized the C. albicans clinical isolate FHB3 to VT-1161, while MCC1189 did not (Fig. 2B). Surprisingly, MCC1189 did not sensitize the CaMdr1-overexpressing clinical isolate FR2 to VT-1161. This apparent lack of sensitization may be due to VT-1161 binding to the zinc cluster transcription factor Tac1 and activating drug efflux via the native CaCdr1, which can efflux both VT-1161 and MCC1189 (23). The finding that RC21v3 sensitized C. albicans strains SGY-243 and FR2 to VT-1161 (Fig. 2B) supports this interpretation.
Drug target overexpression reduces susceptibility to VT-1161.
LDM overexpression is a mechanism that confers azole resistance in fungi. It occurs due to factors such as the induction of aneuploidy in C. albicans (28), the modification of transcription factors UPC2 in C. albicans (29, 30) and UPC2A in C. glabrata (31), or the duplication of promoter elements in mutant CYP51A LDMs of A. fumigatus (32, 33). The constitutive overexpression of full-length recombinant ScLDM6×His in a S. cerevisiae host deleted of drug efflux pumps provides a convenient and robust experimental model to assess the effect of target overexpression on azole drugs and azole agrochemicals (3, 20, 21). Compared to the control hypersensitive host strain Y522, which expresses wild-type ERG11, constitutive overexpression of ScLDM6×His from the PDR5 locus in strain Y2300 substantially reduced susceptibility to VCZ, PCZ, VT-1161, PCZ, and IVC but not MCF (Fig. 4). These results show that ScLDM6×His is the target for VT-1161 and IVC in strain Y2300. We have shown previously that retention of the native ERG11 gene in strain Y2291 (the parent of Y2300) did not significantly reduce susceptibility to FLC, VCZ, itraconazole (ITC), VT-1161, or PCZ, implying that the recombinant enzyme is expressed at levels severalfold higher than the native enzyme (21). Furthermore, overexpression of full-length recombinant CaLDM (CaLDM6×His) or CgLDM (CgLDM6×His) confers resistance to VT-1161 and other azole drugs, including FLC and PCZ (34).
FIG 4.
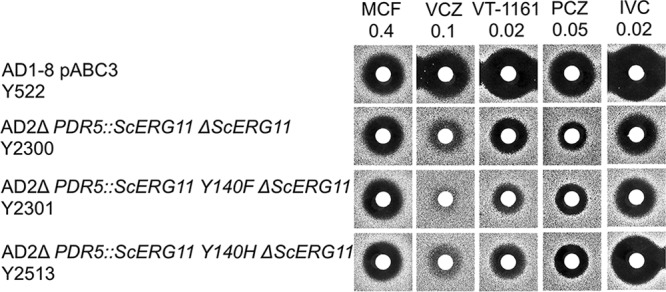
Susceptibilities of S. cerevisiae strains that overexpress recombinant wild-type LDM6×His or LDM6×His Y140F and Y140H to azole drugs. The drugs MCF, VCZ, VT-1161, PCZ, and IVC were applied to disks at the indicated nanomolar amounts and agarose diffusion carried out as described in Materials and Methods.
The ScLDM6×His active-site mutations Y140F and Y140H confer resistance to the short-tailed azoles FLC and VCZ but not long-tailed azoles, such as PCZ and ITC (21). Compared with strain Y2300, which overexpresses wild-type ScLDM, deletion of the native ERG11 gene together with constitutive overexpression of ScLDM6×His Y140F from the PDR5 locus in strain Y2301 conferred susceptibility comparable to those of PCZ and IVC but reduced susceptibility to VCZ and VT-1161 in agarose diffusion assays (Fig. 4). Strain Y2513, which is deleted of the native ScLDM and constitutively overexpresses ScLDM6×His Y140H from the PDR5 locus, showed susceptibility to VCZ and VT-1161 intermediate between strains Y2300 and Y2301. In addition, strain Y2513 had enhanced susceptibility to PCZ and IVC compared with strain Y2300. Thus, the Y140F and Y140H mutations in the binding site of ScLDM6×His appeared to reduce the binding affinities of VCZ and VT-1161. In contrast, the binding of the long-tailed triazoles PCZ and IVC to ScLDM6×His was unaffected by the Y140F mutation and appeared to be slightly stronger due to the Y140H mutation.
Liquid MIC values (Table 1) obtained for yeast overexpressing ScLDM6×His or ScLDM6×His Y140F and Y140H constructs confirmed the agarose diffusion data for VCZ, PCZ, and VT-1161. Overexpression of ScLDM6×His in strain Y2300 reduced susceptibility to FLC, VCZ, ITC, PCZ, and VT-1161. The ScLDM6×His Y140F and Y140H mutations further reduced susceptibility (∼2-fold) to FLC and VCZ but gave comparable or slightly increased susceptibilities to ITC and PCZ. The ScLDM6×His Y140F mutation gave a 2-fold reduction in susceptibility to VT-1161, but the Y140H mutation gave only a 1.6-fold reduction. The Y140F mutation conferred comparable reductions in susceptibility to VT-1161, VCZ, and FLC, but the Y140H mutation conferred a more modest reduction in susceptibility to VT-1161.
TABLE 1.
Liquid MIC values of strains overexpressing wild-type and mutant ScLDM6×His
| Strain | Genotype | MIC80 (nM)a |
||||
|---|---|---|---|---|---|---|
| FLC | VCZ | VT-1161 | ITC | PCZ | ||
| Y2411 | AD2Δ pABC3 | 1,800 ± 50 | 37 ± 0.9 | 29 ± 8 | 91 ± 4 | 160 ± 48 |
| Y2300 | AD2Δ PDR5::ScLDM6×His | 4,200 ± 500 | 93 ± 10 | 75 ± 7 | 180 ± 6 | 423 ± 60 |
| Y2301 | AD2Δ PDR5::ScLDM6×His Y140F ΔERG11 | 8,100 ± 500 | 202 ± 50 | 172 ± 29 | 146 ± 8 | 366 ± 119 |
| Y2513 | AD2Δ PDR5::ScLDM6×His Y140H ΔERG11 | 8,800 ± 1100 | 164 ± 9 | 116 ± 24 | 165 ± 4 | 330 ± 34 |
MIC80 determinations were carried out as described in Materials and Methods using SD medium at pH 6.8. Each determination was carried out at least 3 times using samples in triplicate.
Crystal structures of full-length ScLDM6×His in complex with VT-1161 and PCZ.
ScLDM (ScErg11p, ScCYP51) has provided a surrogate target for the triazole drugs, including FLC, VCZ, itraconazole (ITC), and PCZ. The overexpression of functional full-length recombinant wild-type and mutant versions of ScLDM6×His led to the first full-length structure of a fungal LDM (19) and crystal structures of the wild-type and mutant enzymes in complex with triazole drugs (20, 21, 35), as well as with azole fungicides used in agriculture (3). These structures have given valuable insight into azole susceptibility and resistance.
We have obtained a 2.3-Å-resolution X-ray crystal structure of ScLDM6×His in complex with VT-1161 (PDB identification [ID] 5UL0) and a 2.20-Å-resolution structure in complex with PCZ (PDB ID 6E8Q) (Fig. 5). The statistics for the X-ray data collection and structure refinement are shown in Table S3 in the supplemental material. The structure was determined via molecular replacement using ScLDM6×His in complex with lanosterol (PDB ID 4LXJ) as the template (19). The crystal structures include the complete protein primary sequence from serine 6 (S6) for the ScLDM-VT-1161 complex and from valine 8 (V8) to the first histidine in the C-terminal hexahistidine tag for the ScLDM-PCZ complex. The refined structures reveal the catalytic domain, including the fungus-specific loop, the transmembrane domain, and most of the N-terminal membrane-associated helix. A comparison of the disposition of Cα showed these two structures have the same fold (root mean square deviation [RMSD] = 0.175 Å for 453 atoms). They have the same fold as those obtained for full-length S. cerevisiae (PDB ID 5EQB), C. glabrata (PDB ID 5JLC), and C. albicans (PDB ID 5V5Z) LDM6×His in complex with ITC (36).
FIG 5.
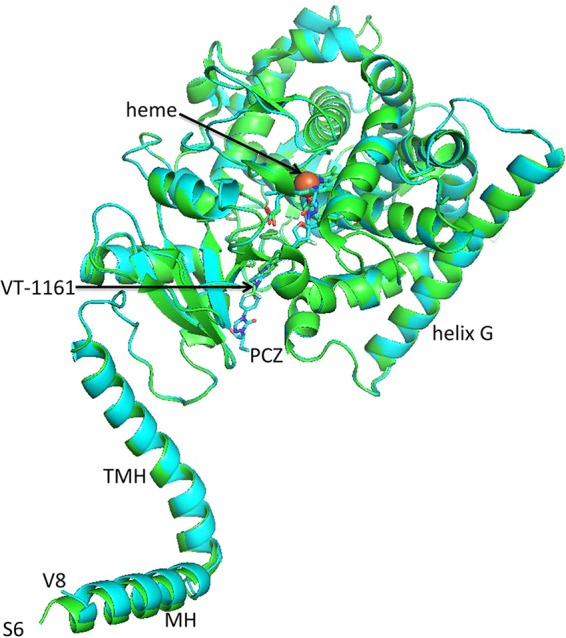
Crystal structure of full-length ScLDM6×His in complex with PCZ and VT-1161. Overlaid crystal structures of ScLDM6×His with PCZ and VT-1161. The cartoon for ScLDM6×His in complex with VT-1161 is shown in green, and the cartoon for ScLDM6×His in complex with PCZ is shown in cyan. The nitrogens of the ligand and heme are shown in blue and the oxygens in red. The iron of the heme is shown in orange. MH, N-terminal membrane-associated helix; TMH, transmembrane helix.
The crystal structure of ScLDM6×His in complex with PCZ shows a 2.1-Å 6th axial (distal) coordination with the heme iron via N-3 of the triazole ring, which projects toward helix I (Fig. 6A), as shown previously for complexes with ITC, FLC, and VCZ (PDB IDs 5EQB, 4WMZ, and 5HS1, respectively) and for the ScLDM6×His Y140F in complex with PCZ (PDB ID 4ZE1) (19–21). The catalytic domain of the ScLDM6×His-PCZ complex has a fold similar (RMSD = 0.569 Å over 415 atoms) to that of the N-terminal-truncated C. albicans LDM (N-truncCaLDM6×His) in complex with PCZ (PDB ID 5FSA) (17). The crystal structures of full-length ScLDM6×His in complex with VT-1161 (Fig. 6B) and N-truncCaLDM6×His in complex with VT-1161 (PDB ID 5TZ1) have similar folds within the enzyme’s catalytic domain (RMSD = 0.579 Å for 400 atoms) but with some significant differences (see below) (37). The N-3 of the tetrazole rings form comparable 2.1-Å to 2.2-Å coordinate bonds with the heme iron. As with wild-type ScLDM6×His or ScLDM6×His Y140F in complex with PCZ or other triazole drugs, the VT-1161 tetrazole ring projects one edge toward the slight kink at the center of helix I (Fig. 6B) and is within 4 Å of G314, G315, and the side chain of T318. The fluorinated phenyl rings of PCZ project deep into the active-site pocket beside helix I and bounded by helix C and the B′-C′ loop similar to that of VCZ (Fig. 5B and C).
FIG 6.

Interactions affecting ligand binding in ScLDM. (A) PCZ lacks a tertiary hydroxyl and has a water-mediated hydrogen bond network in the SEC. The cartoon and stick representation of carbons for ScLDM6×His is shown in green, with key residues within 4 Å of PCZ in magenta. The carbons in heme are shown in salmon and PCZ in yellow. Waters are shown as small red spheres, and the heme iron is a larger orange sphere. Nitrogens are in blue and oxygens are in red. S507 has been deleted to expose the view into the water-containing pocket. Hydrogen bonds are shown as yellow dashed lines. (B) The VT-1161 tertiary alcohol hydroxyl forms a water-mediated hydrogen bond network involving Y140. The cartoon and stick representation of carbons for ScLDM6×His is shown in green, with key residues within 4 Å of VT-1161 shown in magenta. Heme carbons are in salmon and VT-1161 in yellow. The interaction shown between the H381 imidazole and VT-1161 involves a distance measured as 3.5 Å. (C) The N-1 of the PCZ piperazine group forms a water-mediated hydrogen bond network in the SEC. The net indicates the surface of the SEC. The carbons of amino acids that are within 4 Å of PCZ are shown in magenta, while more distant contributors to the SEC are in cyan. The carbons of PCZ are in yellow. RCC, heme ring C propionate carboxyl. RDC, heme ring D propionate carboxyl.
Water-mediated hydrogen bond interactions and the impact of mutations in the ScLDM and CaLDM active sites.
The tertiary alcohol of VT-1161 is one of four substituents on the quaternary carbon that also includes the tetrazole, the difluorinated phenyl ring, and the tail that projects deep into the SEC toward the membrane (Fig. 6B and 7A). The heme ring C propionate carboxyl (RCC) is linked via ionic and hydrogen bonding with the heme bulge (Fig. 6B). The heme ring D propionate carboxyl (RDC) is linked to an internal loop bordering the edge of the active site and the SEC via ionic interaction with R385 and via a hydrogen bond with the Y126 hydroxyl. As in ScLDM6×His complexes with FLC (PBD ID 4WMZ) or VCZ (PDB ID 5HS1), the Y140 hydroxyl forms a hydrogen bond with the heme RCC (20). The tertiary alcohol hydroxyl of VT-1161 in complex with ScLDM6×His or N-truncCaLDM6×His forms a water-mediated hydrogen bond network with the hydroxyl group of structurally aligned ScLDM6×His Y140 and N-truncCaLDM6×His Y132. The water molecule involved (water 743 in the ScLDM6×His-FLC complex) also hydrogen bonds with the heme RDC (20).
FIG 7.
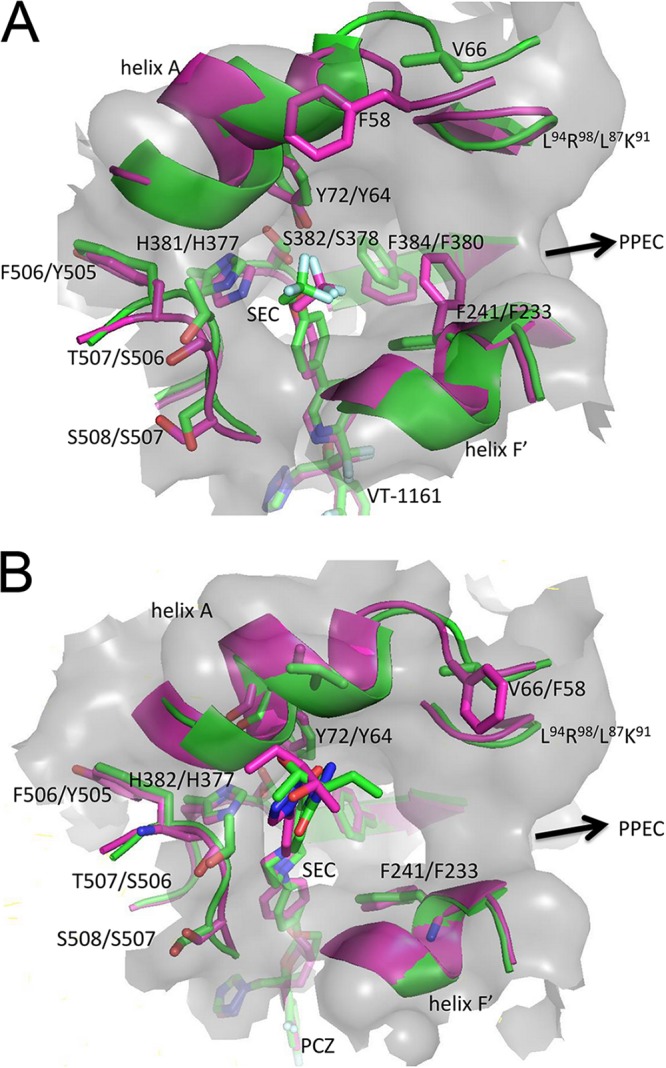
Conformation of structures at the mouth of SEC in full-length ScLDM6×His and N-truncCaLDM6×His in complex with VT-1161 or PCZ. (A) VT-1161. (B) PCZ. A view into the mouth of the SEC is shown. The gray surface of the SEC for ScLDM6×His is shown. The cartoon and stick representation of carbons for ScLDM6×His in complex with VT-1161 or PCZ is shown in green and in magenta for N-truncCaLDM6×His in complex with VT-1161 or PCZ.
The structure of ScLDM6×His in a precatalytic complex with its substrate lanosterol (PDB ID 4LXJ) does not contain a water-mediated hydrogen bonding involving Y140 (19). PCZ (Fig. 6A) and its congener triazole ITC do not contain a tertiary alcohol group. Instead, an oxygen in the dioxolane (ITC) or oxolane (PCZ) group in these drugs fills the space that corresponds to water 743 (21). In the absence of water 743 in the ScLDM6×His-lanosterol, ITC, and PCZ complexes, the Y140 hydroxyl forms a hydrogen bond only with the heme RCC. Within the active site, the other hydrogen bond and ionic contacts found in the ScLDM6×His-VT-1161 complex are maintained in the ScLDM6×His-PCZ complex, apart from the loss of a hydrogen bond with Y126. The hydrogen bond and ionic interactions involving the heme bulge and helix C are maintained in both complexes.
Interactions within the ScLDM LBP.
The crystal structure of ScLDM6×His in complex with VT-1161 shows 20 LBP residues in close proximity (within 4 Å) of the ligand (Fig. 6B and Table 2). These residues are Y72, Y126, L129, T130, I139, Y140, F236, P238, G310, G314, G315, T318, L380, H381, S382, F384, F506+Y, T507+S, S508, and M509 (F506+Y, T507+S, and S508 are the 3 amino acid residues present in the LBP but are >4 Å from VT-1161 in CaLDM6×His; structurally aligned residues ScLDM G315 and CaLDM G308 are 4.0 and 4.1 Å from the VT-1161 tetrazole, respectively, a distance that is not significantly different; +X gives the single-letter code for a structurally aligned but different residue in CaLDM). In comparison, 17 structurally aligned residues in the N-truncCaLDM6×His-VT-1161 complex are within 4 Å of this ligand. Of the residues within 4 Å of VT-1161 in the ScLDM6×His structure, only Y72, F241, H381, S382, F384, F506, T507, and S508 differ significantly in conformation or structure compared with their structurally aligned N-truncCaLDM6×His counterparts Y64, F232, H377, S378, F380, Y505, S506, and S507. These residues are in proximity of the mouth of the SEC or PPEC (Fig. 7A).
TABLE 2.
LDM LBP residues within 4 Å of VT-1161a
| SRS no. or group | ScLDM6×His + VT-1161 | N-truncCaLDM6×His + VT-1161 |
|---|---|---|
| SRS1 | A124AYAHLTTPVFGKGVIYDCP143 | E116AYKHLTTPVFGKGVIYDCP135 |
| SRS4 | I304ANLLIGVLMGGQHTSAA321 | I297ANLLIGILMGGQHTSAS314 |
| SRS5 | H378PLHSLFR385 | M374PLHSIFR381 |
| SRS6 | D505FTSMVTLPTG515 | D504YSSMVVLPTE514 |
| Helix A′ | A69V70, Y72GM74 | A61A62, Y64GQ66 |
| Helix FF′ | F236, P238, F241 | F228, P230, F233 |
| Additional residues in LBP surface |
V66, P76 L95L96, R98, M100, L147, Q150K151, V154, I239, V242, H405, I471 |
F58, P68 L87L88
K90, M92 L139, Q142K143, A146, I231, V234, Y401, I471 |
Amino acid residues in the LBP are shown in italics, and residues within 4 Å of VT-1161 are in bold.
The trifluoroethoxy tail of VT-1161 is located within the SEC. It has slightly different conformations in complexes with ScLDM6×His and N-truncated CaLDM6×His (Fig. 6B and 7A). The oxygen of the VT-1161 trifluoroethoxy group is closer to the collar of the F506, T507, and S508 carbonyls in ScLDM6×His than to the structurally aligned Y505, S506, and S507 carbonyls in N-truncCaLDM6×His. For N-truncCaLDM6×His, a 2.8-Å moderate-strength interaction has been proposed between the oxygen of the trifluoroethoxy group and N-2 of H377 in N-truncCaLDM6×His (17). The structure of ScLDM6×His in complex with VT-1161 shows a distance of 3.6 Å and thus a significantly weaker interaction.
Twenty-three ScLDM6×His LBP residues are within 4 Å of PCZ (A69, V70, Y72, G73, Y126, L128, F134, I139, Y140, F236, P238, G310, V311, G314, T318, L380, H381, S382, F384, F506, T507, S508, M509; Fig. 6A and Table 3). Two N-truncCaLDM6×His LBP residues structurally aligned with the ScLDM residues listed above (in bold) are >4 Å from the PCZ, as follows: CaLDM A62 in the mouth of the SEC is 4.3 Å, and CaLDM I131 in the SRS1 BC-loop is 4.1 Å. The measurement for CaLDM I131 is unlikely to differ significantly from 4 Å. V311 in helix I of the ScLDM active site is structurally aligned to the chemically similar I304 in CaLDM. Three chemically similar residues located at or near the mouth of the SEC in ScLDM6×His (Fig. 7B) differ structurally from their N-truncCaLDM6×His counterpart (ScLDM6×His/N-truncCaLDM6×His; V70/A62, F506/Y505, and T507/S506). In addition, the N-truncCaLDM6×His Q66 residue, which structurally aligns with ScLDM6×His M74 at the mouth of the SEC, adopts a conformation that places its side-chain amide within 4 Å of the PCZ in monomer B but not monomer A. Finally, the 2-hydroxypentan-3-yl-4,5-dihydro-1H-1,2,4-triazol group in the tail of PCZ is rotated 180° between the ScLDM6×His and N-truncCaLDM6×His structures (Fig. 7B). This conformation of the PCZ ligand in N-truncCaLDM6×His has been proposed to give a polar interaction with the carbonyl of A61 in helix A′ at the mouth of the SEC (17). The same interaction was not seen in the ScLDM6×His-PCZ complex.
TABLE 3.
LDM LBP residues within 4 Å of PCZa
| SRS no. or group | ScLDM6×His + PCZ | N-truncCaLDM6×His + PCZ |
|---|---|---|
| SRS1 | A124AYAHLTTPVFGKGVIYDCP143 | E116AYKHLTTPVFGKGVIYDCP135 |
| SRS4 | I304ANLLIGVLMGGQHTSAA321 | I297ANLLIGILMGGQHTSAS314 |
| SRS5 | H378PLHSLFR385 | M374PLHSIFR381 |
| SRS6 | D505FTSMVTLPTG515 | D504YSSMVVLPTE514 |
| Helix A′ | A69V70, Y72GM74 | A61, Y64GQ66 |
| Helix FF′ | F236, P238, F241 | F228, P230, F233 |
| Additional residues in LBP surface |
V66 L95L96, R98, M100, L147, Q150K151, V154, I239, V242, H405, I471 |
F58, P68, L87L88, K90, M92, L139, Q142K143, A146, I231, V234, Y401, I471 |
Amino acid residues in the LBP are shown in italics, and residues within 4 Å of PCZ are in bold.
The presence of PCZ instead of VT-1161 in the SEC allows N-truncCaLDM6×His A61-Q66, F238-F235, and Y506-M509 to adopt a conformation significantly closer to that found in the full-length ScLDM6×His structures. The PCZ ligand has a conformation essentially identical to its congener ITC in crystal structures obtained with full-length ScLDM6×His and full-length CaLDM6×His (36). Like ScLDM6×His in complex with ITC, the PCZ complex has the piperazine ring in a chair conformation (Fig. 6A and C). This allows the piperazine N-1 to contribute to a water-mediated hydrogen bond network that involves 3 waters, the main chain amides of H381 and S382 and the main-chain carbonyls of P379, S382, S508, and M509 (Fig. 6C). Despite adopting a chair conformation, the same stabilization of the piperazine ring was not detected in the N-truncCaLDM6×His-PCZ complex, possibly due to low resolution (2.86 Å).
Compared to ScLDM6×His in complex with VT-1161, the N-truncCaLDM6×His structure differed in protein conformation and gave larger B-factor values between V51, in the loop upstream of helix A, and P68, at the N terminus of helix A in CaLDM6×His (Fig. 8). The loop has polar and nonpolar interactions with the transmembrane helix in full-length ScLDM complexed with several ligands (3, 19, 20) and CaLDM in complex with ITC (PDB ID 5V5Z) (36). Subtle differences between the conformation for the F′ helix and its C-terminal loop (P230–H241) and Y505–M508 in the loop between the β4-1 and β4-2 sheets differentially shape the sides and mouth of the SEC (Fig. 7). For example, F241 in the full-length ScLDM6×His structure and its structural equivalents in the full-length CaLDM6×His (F233) and CgLDM6×His (F242) line the nexus of the SEC and PPEC. In N-truncCaLDM6×His, F233 blocks the entry to the PPEC when VT-1161 but not when PCZ is the ligand (Fig. 7A and B). In contrast, the differences in conformation and B-factors were minimal when the complexes of PCZ with N-truncCaLDM6×His and ScLDM6×His were compared. We conclude that the differences in protein conformation identified in full-length ScLDM6×His or N-truncated CaLDM6×His in complexes with VT-1161 or PCZ are likely to be due to artifacts of the truncation or crystal contacts and not the primary sequence differences between the catalytic domains of these enzymes.
FIG 8.
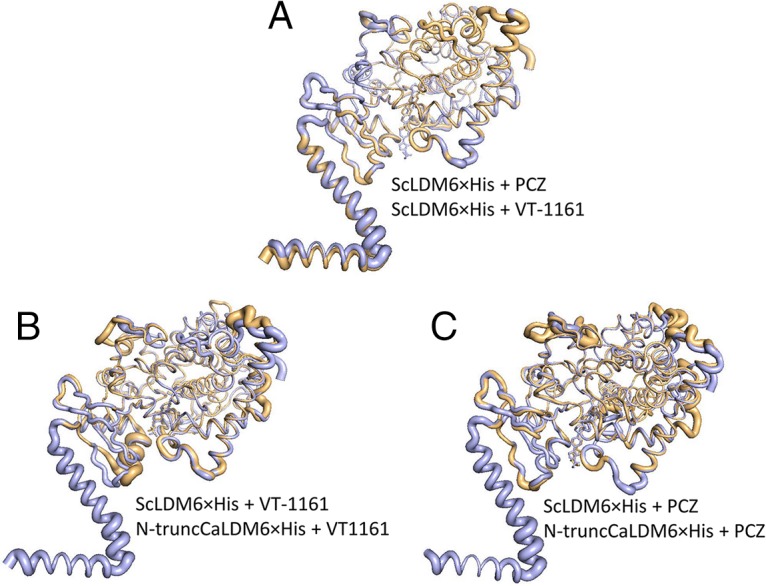
Conformational differences among crystal structures of ScLDM6×His and N-truncCaLDM6×His in complex with VT-1161 and PCZ. (A) ScLDM6×His in complex with PCZ or VT-1161. ScLDM in complex with PCZ is in aquamarine and ScLDM in complex with VT-1161 is in gold. (B) ScLDM6×His in complex with VT-1161 and N-truncCaLDM6×His in complex with VT-1161. ScLDM in complex with VT-1161 is in aquamarine and N-truncCaLDM in complex with VT-1161 is in gold. (C) ScLDM6×His in complex with PCZ and N-truncCaLDM6×His in complex with PCZ. ScLDM in complex with PCZ is in aquamarine and N-truncCaLDM in complex with PCZ is in gold. PyMOL was used to generate and overlay the B-factor putty figures.
Mutations in the drug target affect the efficacy of VT-1161.
The X-ray crystal structures of S. cerevisiae, C. albicans, and C. glabrata LDMs identified ∼50 residues that contribute to the surface of the LBP, i.e., the heme-containing active site, the SEC, and the PPEC. Structural alignment of the primary sequences of LDMs from S. cerevisiae and a range of prominent ascomycete, basidiomycete, and zygomycete fungal pathogens of humans and plants (fungal pathogens of humans [Candida albicans, Candida glabrata, Candida parapsilosis, Candida tropicalis, Ajellomyces capsulatus, Pneumocystis jirovecii, Cryptococcus neoformans, Aspergillus fumigatus, Coccidioides immitis, Rhizopus arrhizus, Rhizopus microsporus, and Mucor circinelloides] and plants [Zymoseptoria tritici and Phakopsora pachyrhizi]) together with the LDMs of human and plant hosts (soybean and wheat), identified 4 conserved LBP residues, i.e., Y126 and F134 (SRS1, B′-C loop), Q150 (helix C), and H317 (helix I) (Fig. 6A, S. cerevisiae numbering). These residues are all located within the catalytic site and are thus likely to have roles in catalysis. H317 is thought to provide protons via the solvent (S) channel using a “reverse” proton delivery pairing with D233 (17, 38). Y126 hydrogen bonds with the heme ring D propionate plus the main-chain amino of F384 when in complex with PCZ and additionally with the main-chain carbonyl of F384 when in complex with VT-1161 or lanosterol (PDB ID 4LXJ) (19). Together with residues F134 and Q150 located deeper in the active site adjacent to helix I, Y126 might help bind and orient the substrate in the catalytic state. For example, the F134 aromatic group could interact with the lanosterol C5-C6 double bond required for it to be an LDM substrate, while the side chain of Q150 could form a hydrogen bond with the C-3 hydroxyl of lanosterol.
Twenty-three LBP residues within 5 Å of ScLDM’s substrate lanosterol in a precatalytic state are conserved in the LDMs of ascomycete and basidiomycete fungal pathogens. These residues (S. cerevisiae numbering) are distributed among 6 separate structures of the fungal enzyme that interact with distinct parts of the substrate, i.e., Y72 and G73 (helix A′, mouth of the SEC); Y126, Y129, T130, I139, and Y140 (SRS1, B′-C′ loop, active site); F236, P238, and F241 (helix F′′, SEC and PPEC); G310, M313, and G314 (SRS4, helix I); L380, H381, S382, L383, and F384 (SRS5, internal loop); and F506, T507, S508, M509, and V510 (SRS6, β4-1 to β4-2 loop). Of this group, only Y126 is conserved in the human, wheat, and soybean hosts. Only 5 of these LBP residues are conserved among ascomycete and basidiomycete pathogens, are not found in humans and plant hosts, and are not subject to known mutations, i.e., Y72, F241, M313, H381, and S382 (S. cerevisiae numbering). One other residue in the ∼50-residue LBP surface, L147 (S. cerevisiae numbering), is conserved among the ascomycete and basidiomycete pathogens and is not found in the host organisms or subject to known mutations. Are these 6 residues required for enzyme function, or do they affect the binding of the triazole and tetrazole drugs?
The first question was addressed by structural alignment with residues in the LDM F1 and F5 isoforms of distantly related zygomycete pathogens. In both isoforms, Y72, M313, and H381 (S. cerevisiae numbering) are substituted with F, L147 is substituted with I, and S382 is substituted with Q/N (F1/F5). Thus, only F241 is conserved among ascomycete, basidiomycete, and zygomycete pathogens (data not shown). We have also demonstrated that alanine mutagenesis of Y72, L147, F241, M313, H381, or S382 had no effect on cell viability during growth reliant on constitutive overexpression of recombinant mutant ScLDM6×His from the PDR5 locus (data not shown). This was achieved by replacing the promoter of the native ScERG11 with a GAL1 promoter in a host strain and testing constructs for their ability to grow on glucose, which repressed the function of the GAL1 promoter. All constructs that constitutively expressed wild-type or mutant ScLDM6×His from the PDR5 locus grew normally in either galactose-containing medium or in glucose-containing medium (Fig. 9A). In an independent experiment, the native ERG11 gene was replaced with a HIS marker in a strain constitutively overexpressing the mutant LDM from the PDR5 locus. All the genetically modified strains grew normally in the presence of glucose (Fig. 9B). Thus, the side chains of Y72, L147, F241, M313, H381, and S382 are not required for LDM function. Agarose diffusion assays using glucose medium showed that the H381A mutation in the ScLDM SEC slightly reduced susceptibility to VT-1161 but not the other azole drugs tested. SEC residues Y72A, F241A, and S382A had no effect on susceptibility to ITC or PCZ but gave increased susceptibility to FLC, VCZ, IVC, and VT-1161 (Fig. 8A and B). In contrast, the L147A and M313A mutations in the ScLDM active site gave increased susceptibility to all the azoles tested. Thus, the binding of FLC, VCZ, IVC, VT-1161, ITC, and PCZ is unlikely to require interactions with the side chains of the mutated residues apart from a weak interaction between VT-1161 and H381.
FIG 9.

Azole susceptibilities of ScLDM6×His alanine mutants. (A) ScLDM6×His mutants with the native ERG11 promoter replaced with the GAL1 promoter. (B) ScLDM6×His mutants with the native ERG11 gene deleted.
The X-ray crystal structure N-truncCaLDM6×His in complex with VT-1161 predicts that an alanine substitution of H377 removes the interaction between the trifluoroethoxyphenyl oxygen of VT-1161 and N-2 of the imidazole ring of CaLDM H377 (H381, S. cerevisiae numbering) (17). The proposed hydrogen bond was expected to significantly (1 to 5 kcal/mol) stabilize the N-truncCaLDM6×His complex with VT-1161. The absence of this hydrogen bond in the structure of ScLDM6×His in complex with VT-1161 is consistent with the slightly reduced susceptibility to VT-1161 of the ScLDM6×His H381A mutant. The susceptibility of zygomycete pathogens to both PCZ and VT-1161, despite having a phenylalanine in place of the histidine, supports the idea that the interaction between VT-1161 and the CaLDM H377 imidazole is of limited importance for drug binding (14, 15, 39).
DISCUSSION
VT-1161 efficacy is reduced by LDM overexpression and drug efflux.
The structurally related compounds VT-1161 and VT-1129 have been proposed as antifungal agents that effectively target the LDMs of several fungal pathogens, ranging from Ascomycetes and Basidiomycetes to Zygomycetes (1–12). These drugs use the weaker affinity for the heme iron conferred by a tetrazole instead of a triazole, together with additional contacts within the LBP, to bind preferentially with their fungal target and minimize interaction with human cytochrome P450s (5–16, 40). We have used a yeast expression system and clinical isolates of the fungal pathogens C. albicans and C. glabrata to demonstrate that the efficacy of this new class of azole drugs can be compromised by mechanisms of antifungal resistance common in these organisms. These mechanisms include target (LDM) overexpression, mutations in the active site of LDM, and overexpression of drug efflux pumps of both the ABC (CaCdr1p) and MFS (CaMdr1p) transporter classes. A better understanding of fungal physiology and the structures of target-inhibitor complexes is needed to overcome these increasingly significant problems.
Crystal structure of VT-1161 in complex with full-length LDM provides a more reliable structural model than with N-truncated LDM.
The crystal structures of full-length ScLDM in complex with VT-1161 and PCZ reported here complement and extend understanding of the mode of action of these drugs previously shown with structures of the LDM catalytic domain alone (17). The ScLDM6×His and N-truncCaLDM6×His crystal structures reveal several comparable interactions between the drugs and the enzyme’s LBP, especially within the active site. This includes the coordination of the VT-1161 tetrazole with the heme iron and the projection of one edge of the tetrazole ring toward the kink in helix I. The position of the azole rings appears to prevent a water molecule found in the ScLDM6×His in complex with lanosterol (PDB ID 4LXJ) but not in complexes with triazoles including PCZ, from associating with helix I. This water, or possibly a hydronium ion, is stabilized via interactions with the main-chain carbonyls of M313 and G314 and the main-chain nitrogens of H317 and T318. As expected, the difluorinated phenyl group locates in the space between helix I and the B′-C′ loop, and we detect a water-mediated hydrogen bond network involving the VT-1161 tertiary alcohol, the heme RCC, and ScLDM6×His Y140. LDM mutations structurally aligned with ScLDM6×His Y140F and Y140H are expected to significantly affect the binding with the tertiary hydroxyl-containing short-tailed azole drugs, as we have previously demonstrated with the S. cerevisiae enzyme (21). The structurally aligned CaLDM6×His Y132F and Y132H (41, 42) and ScLDM6×His Y140F and Y140H mutations (21) each confer significant resistance to the short-tailed triazoles FLC, VCZ, and VT-1161 but not to the long-tailed triazoles ITC and PCZ. The crystal structures presented in this report confirm that the Y140F and Y140H mutations will deleteriously affect the binding of VT-1161 but not PCZ.
ScLDM6×His in complex with VT-1161 has F241 in helix F′ lining the LBP at the nexus of the SEC and the PPEC, while the structurally aligned N-truncCaLDM6×His Y233 residue blocks the putative product exit channel (17). Finally, the mouth of the SEC in N-truncCaLDM6×His in complex with VT-1161 differs significantly from those in N-truncCaLDM6×His in complex with PCZ and with full-length ScLDM6×His in complex with VT-1161 (Fig. 7 and 8) or lanosterol, the long-tailed triazoles PCZ and ITC, and the short-tailed triazoles FLC and VCZ. These structural differences could be due to contacts with aqueous solution, the use of a detergent surrogate for a lipid bilayer, or the impact of crystallization conditions (0.1 M HEPES. 0.2 M NaCl, 10% polyethylene glycol 600 [PEG-600] [pH 7.4] for the truncated enzyme versus 0.1 M glycine, 40% PEG-400 [pH 9.4] for the full-length yeast enzyme). A comparison of crystal structures suggests that the structural difference, which is ameliorated by the use of PCZ as a ligand, is not due to primary sequence differences in the catalytic domains but differing interprotomer crystal contacts enabled by the truncation of CaLDM. Crystal contacts in the asymmetric unit of N-truncCaLDM in complex with VT-116, which involve the main-chain carbonyl of Y53 forming a hydrogen bond with a side-chain nitrogen of W53 in the second LDM protomer, appear to affect structures nearby in the SEC. This crystal contact is not found in the structure of N-terminal-truncated A. fumigatus CYP51B6×His in complex with the VT-1598, a tetrazole with a tail significantly longer than VT-1161 (37). This crystal structure shares the same fold as the ScLDM6×His structures but does not have the distorted loop 1-helix A′ found in the N-truncCaLDM6×His complex with VT-1161 (Fig. 8). The structure-based discovery of novel antifungals that interact effectively with both the active site and structural features within the SEC requires reliable templates, such as the relatively rigid full-length LDM structures, with the caveat that crystal contacts could still cause significant distortions in these structures.
Our structures of ScLDM6×His were crystallized in the space group P1211, with a single ScLDM6×His-ligand complex in the asymmetric unit. The single exception is ScLDM6×His in complex with fluquinconazole (FQZ), which crystallized in space group P1 with two protomers in the asymmetric unit (3). A comparison of the ScLDM in complex with FQZ with the other ScLDM structures shows almost identical protein structures, including the transmembrane and amphipathic helices. All ligand-protein complexes are isomorphous with the original ScLDM6×His-lanosterol complex (PDB ID 4LXJ) that was used for molecular replacement. We conclude from the structures obtained for ScLDM6×His in complex with a variety of ligands (3, 19, 20), as well as those obtained for CaLDM6×His and CgLDM6×His in complex with ITC (36), that the full-length structures are relatively rigid and therefore are likely to provide a better vehicle for drug design than the N-terminal-truncated structures.
Ways to obtain more effective azole drugs.
Chimeric azoles that include favorable features of existing drugs may be a way to generate more effective antifungals. For example, the tail of the azole drugs provides an important site for further drug modification designed to overcome the impact of mutations in LDM. Tetrazole drug candidates with tail lengths intermediate between VT-1161 and ITC or PCZ deserve significant research focus, i.e., such tails should not interact with the readily mutated residues at the mouth of the SEC (35). Three water molecules located in a pocket associated with the SEC provide opportunity for polar interaction (Fig. 6). For example, the conformations of ITC or PCZ allow the piperazine ring N-1 to form a water-mediated hydrogen bond network with main-chain nitrogens and carbonyls of LDM. In addition, the inclusion of a nitrile triple bond at the end of the azole tail, as in IVC and VT-1598 (37), could mimic the double bond in the tail of lanosterol, eburicol, and obtusifoliol. We are currently testing these and other ideas using our yeast expression system and clinical isolates.
Despite displaying significantly different residues in the LBP from those conserved in the Ascomycetes and Basidiomycetes, the Zygomycetes have been reported to be susceptible to VT-1161 (14, 15). This suggests that the zygomycete Y72F, H381F, and S382Q/N LDM substitutions (S. cerevisiae numbering) may provide a sufficiently polar environment, opposite the main-chain carbonyls of the YTS sequence conserved in Zygomycetes instead of the ScLDM F506TS508, to overcome the effects of the active-site Y140F and V311A substitutions in LDM F5 that cause innate resistance to FLC and VCZ but not PCZ, which lacks a tertiary alcohol.
Overcoming drug tolerance and drug resistance.
Drug binding to transcription factors generates a tolerance to azole drugs and gives time for the development of gain-of-function mutations that enable drug efflux-mediated drug resistance (1). It is therefore important to identify inhibitors of fungal LDMs that do not cause overexpression of CYP51 or drug efflux pumps, i.e., inhibitors that do not bind to relevant transcriptional regulators, such as Upc1, Upc2, or Tac1 in C. albicans or Pdr1 in C. glabrata. Preliminary evidence presented for C. albicans strains SGY-243 and FR2 suggests that VT-1161 may induce CaCdr1-mediated drug efflux that is sensitized by the CaCdr1-specific surface-active peptide RC21v3. This suggests a role in tolerance induction via the binding of VT-1161 to C. albicans Tac1. We have developed a system that monitors related interactions in S. cerevisiae by expressing reporters, such as enhanced green fluorescent protein, from the PDR5 locus under the control of the Pdr1 transcriptional regulator. It will be of interest to determine if this system is activated by VT-1161. We have also sought antifungals that are not susceptible to drug efflux by using the yeast overexpression system to screen for bioactives targeting LDM that are not effluxed by fungal ABC or MFS transporters. For example, PCZ is effluxed by CaCdr1 but not by CaMdr1, while VT-1161, IVC, and many other azole drugs are effluxed by both transporters. Unfortunately, the structures of the substrate binding sites of drug efflux pumps are not sufficiently resolved to enable the design of LDM inhibitors that are either suicide substrates of drug efflux or not substrates of these broadly specific systems. There is a need to produce fungicidal LDM inhibitors that act rapidly enough to reduce membrane sterols and produce toxic fecosterols without inducing mutations that protect LDM function, disable Erg3, or enable drug efflux. Finally, while it may be possible to take advantage of contact points identified as conserved in fungal LDMs among the most prominent ascomycete and basidiomycete pathogens and not found in human and plant hosts, it may be more difficult to extend this approach to the Zygomycetes.
MATERIALS AND METHODS
Strains.
The yeast strains used in the present study are shown in Table S1. Yeast strains were grown on YPD medium, which contains 1% (wt/vol) Bacto yeast extract (BD Difco Laboratories, Inc., Franklin Lakes, NJ), 2% (wt/vol) Bacto peptone (BD Difco), and 2% (wt/vol) glucose. Synthetic defined (SD) medium was used for the selection of transformants. It contained 2% (wt/vol) glucose or galactose, 0.67% (wt/vol) yeast nitrogen base without amino acids (BD Difco), 1.8% (wt/vol) agar (Oxoid Ltd., Hampshire, UK), and either 0.77 g/liter uracil drop-out (QBioGene, Irvine, CA) or 0.77 g/liter histidine drop-out (Formedium, Norfolk, UK) complete supplement mixture. Liquid SD medium with 0.79 g/liter complete supplement mixture (Formedium) containing 10 mM morpholineethanesulfonic acid (MES) and 20 mM HEPES buffered with Tris to pH 6.8 was used for MIC80 determinations.
Materials.
Desalted oligonucleotides (Table S2), AMB, FLC, ITC, VCZ, and PCZ were purchased from Sigma-Aldrich Ltd. (St. Louis, MO). IVC was purchased from Bocsci, Inc. (Shirley, NY). VT-1161 was prepared by MicroCombiChem e.K. (Wiesbaden, Germany) using the methods described by Hoekstra et al. (5). MCC1189 (23) was supplied by MicroCombiChem and RC21v3 by the Massey University Centre for Separation Science (Palmerston North, New Zealand) (24, 43). MCF was supplied by Selleck Chemicals (Houston, TX). Colony PCRs were carried out using DNA polymerase (TaKaRa Bio, Inc., Shiga, Japan). All other PCRs were performed using KOD Hot Start DNA polymerase (Novagen, Madison, WI). PCR cleanup and DNA gel extraction were carried out using kits from Qiagen Pty Ltd. (Limburg, The Netherlands). Genomic DNA from yeast was isolated using the Y-DER kit from Thermo Fisher (Waltham, MA). Yeast DNA transformation was carried out using an Alkali-Cation yeast transformation kit from QBioGene (Irvine, CA). DNA transformation cassettes and genes inserted at the S. cerevisiae PDR5 or ERG11 locus were confirmed by DNA sequence analysis performed at the Genetic Analysis Services facility (University of Otago, Dunedin, New Zealand).
Construction of recombinant strains.
The S. cerevisiae AD2Δ host (19, 21, 22) was used to create several strains for this study (Table S1). The AD2Δ mutant is deleted of the URA3 and HIS1 open reading frames (ORFs), 7 pleiotropic drug resistance (PDR) ABC transporters, and the PDR3 transcriptional regulator, but it includes the mutant pdr1-3 transcriptional regulator that drives constitutive expression from the PDR5 locus (22).
The native promoter of ScERG11 was replaced with an inducible GAL1 promoter by using a three-fragment transformation of the AD2Δ host strain. The first fragment containing Dpl200-flanked CaURA3 was amplified from the pDDB57 vector by PCR (44). The second fragment containing 710 bp upstream of the ScGAL1 ORF was amplified from genomic DNA of the ADΔ mutant strain. The third PCR fragment consisted of 183 bp at the 5′ end of the ScERG11 ORF in the yeast ADΔ mutant strain. To enable homologous recombination, 21- to 25-nucleotide primers used for fragment amplification were designed with flanking regions matching neighboring fragments and for the sequence from −599 to −534 of the ScERG11 promoter. Transformants were selected using –URA dropout SD medium. Successful promoter replacement was confirmed by colony PCR using primers directed against flanking regions and by the ability of transformants to grow on galactose-containing but not glucose-containing SD medium. Subsequent deletion of the URA3 marker, leaving a 245-nucleotide scar, was obtained by passage through 5-fluoroorotic acid (5FOA)-containing medium and confirmed by colony PCR.
Alanine mutants of ScLDM6×His and deletion of the native ERG11 locus were obtained essentially as previously described (21) using the oligonucleotides shown in Table S2.
Agarose diffusion assays.
Agarose diffusion assays were carried out as previously described by Keniya et al. (23), with MCF serving as a control drug with an action independent of ergosterol content or biosynthesis.
MIC determination for yeast constructs and for C. albicans clinical isolates.
The MIC80 of a strain was defined as 80% growth inhibition compared to a no-drug control. This approach was used because the triazole drugs are fungistatic rather than fungicidal and can give trailing growth. MIC80s to AMB, FLC, VCZ, VT-1161, ITC, IVC, and PCZ were determined in 96-well microtiter plates using SD buffered to pH 6.8 for S. cerevisiae constructs (23). RPMI 1640 was used according to the CLSI standard for clinical isolates of C. albicans and C. glabrata (45). Cells were seeded at an optical density at 600 nm (OD600) of 0.005 (1.5 × 104 CFU), and the plates were incubated at 30°C with shaking at 200 rpm for 48 h for S. cerevisiae constructs and 24 h for the clinical isolates. Cell growth was assessed by measuring the OD600 using a Synergy 2 multimode plate reader (BioTek Instruments, VT, USA). Each MIC80 was determined using triplicate measurements for pools of 4 clones of each strain, in three separate experiments.
Preparation of crude membranes.
Full-length ScLDM6×His was overexpressed in S. cerevisiae strain MML Y941 grown in multiple 1.0-liter liquid cultures in baffled flasks (19). The cultures were grown in YPD medium to an OD600 of ∼10 at 30°C with shaking at 200 rpm. Harvested yeast cells were broken using a bead-beating protocol, and crude membranes were prepared by differential centrifugation (19). For some experiments, 50-ml cultures were grown and harvested, cells were broken in Eppendorf tubes with glass beads by vortexing, and crude membranes were prepared by differential centrifugation in 1.5-ml tubes. The concentration of crude membrane protein was estimated using the Lowry method (46), with bovine serum albumin (Thermo Fisher) as a standard.
Enzyme purification.
ScLDM6×His was purified by nickel-nitrilotriacetic acid (Ni-NTA) affinity and size-exclusion chromatography (SEC), essentially as described by Monk et al. (19). Recombinant enzymes were extracted from crude membranes using 17 mM decylmaltoside (DM, 10× critical micelle concentration [cmc]) and ultracentrifugation. Samples for crystallography were partially purified by Ni-NTA chromatography using imidazole as an eluent and purified to near homogeneity by SEC using a Superdex 200 10/300 column in the presence of 150 mM NaCl, 6.8 mM DM (4 × cmc), and 50 mM HEPES (pH 8.0) at 8°C. The protein peak was pooled and concentrated to ≥20 mg/ml by centrifugal filtration in the SEC buffer containing 40 µM VT-1161 or PCZ.
X-ray crystallography.
A hanging-drop vapor-diffusion method was used to crystallize ScLDM6×His in complex with VT-1161 and PCZ (19). The reservoir solution contained 30 to 40% polyethylene glycol-400 in 500 mM glycine-NaOH, at a pH range of 8 to 9.5. Crystals were obtained using 4-µl drops composed of 2 µl of the concentrated LDM6×His preparation (5 to 20 mg/ml) together with 2 µl of reservoir solution. Red boat-shaped crystals formed after 1 to 2 weeks of incubation at 18°C and were collected for crystallographic studies. The crystals were flash-cooled in liquid nitrogen prior to data collection. Data sets were collected on the MX1 beamline (ADSC Quantum 210r detector) at the Australian Synchrotron (Melbourne, Australia). During data collection, the crystals were kept frozen under a cryostream at −180°C. Indexing and integration of data were done using iMosflm (47) and scaling with SCALA (48). Phaser-MR (49) from Phenix was used to carry out molecular replacement using ScLDM6×His in complex with lanosterol (PDB ID 4LXJ) as the template (19). Refinement and modeling were performed using phenix.refine (39) and Coot (50), respectively. Waters were added if at least one hydrogen bond was detected (2.5 to 3.3 Å).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a grant from the Health Research Council of New Zealand awarded to B.C.M. The New Zealand Synchrotron group supported travel to the Australian Synchrotron. This research was undertaken in part using the MX1 beamline at the Australian Synchrotron.
We declare no conflicts of interest.
M.V.K. prepared and genetically characterized recombinant yeast strains, determined phenotypes, including agarose diffusion assays and MIC determinations, and edited the manuscript. M.S. and R.K.W. purified enzyme and contributed to crystal trials and structural resolution. D.O.G., H.F.H., and D.C. created yeast mutants and analyzed their phenotypes. J.D.A.T. assisted with structure determination, assisted with building and assessment of structural models, and edited the manuscript. B.C.M. developed and directed the project, obtained funding, interpreted data, and wrote the manuscript.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.02114-18.
REFERENCES
- 1.Monk BC, Goffeau A. 2008. Outwitting multidrug resistance to antifungals. Science 321:367–369. doi: 10.1126/science.1159746. [DOI] [PubMed] [Google Scholar]
- 2.Perlin DS, Rautemaa-Richardson R, Alastruey-Izquierdo A. 2017. The global problem of antifungal resistance: prevalence, mechanisms, and management. Lancet Infect Dis 117:e383–e392. doi: 10.1016/S1473-3099(17)30316-X. [DOI] [PubMed] [Google Scholar]
- 3.Tyndall JD, Sabherwal M, Sagatova AA, Keniya MV, Negroni J, Wilson RK, Woods MA, Tietjen K, Monk BC. 2016. Structural and functional elucidation of yeast lanosterol 14alpha-demethylase in complex with agrochemical antifungals. PLoS One 11:e0167485. doi: 10.1371/journal.pone.0167485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yates CM, Garvey EP, Shaver SR, Schotzinger RJ, Hoekstra WJ. 2017. Design and optimization of highly-selective, broad spectrum fungal CYP51 inhibitors. Bioorg Med Chem Lett 27:3243–3248. doi: 10.1016/j.bmcl.2017.06.037. [DOI] [PubMed] [Google Scholar]
- 5.Hoekstra WJ, Garvey EP, Moore WR, Rafferty SW, Yates CM, Schotzinger RJ. 2014. Design and optimization of highly-selective fungal CYP51 inhibitors. Bioorg Med Chem Lett 24:3455–3458. doi: 10.1016/j.bmcl.2014.05.068. [DOI] [PubMed] [Google Scholar]
- 6.Warrilow AG, Hull CM, Parker JE, Garvey EP, Hoekstra WJ, Moore WR, Schotzinger RJ, Kelly DE, Kelly SL. 2014. The clinical candidate VT-1161 is a highly potent inhibitor of Candida albicans CYP51 but fails to bind the human enzyme. Antimicrob Agents Chemother 58:7121–7127. doi: 10.1128/AAC.03707-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Warrilow AGS, Parker JE, Price CL, Garvey EP, Hoekstra WJ, Schotzinger RJ, Wiederhold NP, Nes WD, Kelly DE, Kelly SL. 2017. The tetrazole VT-1161 is a potent inhibitor of Trichophyton rubrum through its inhibition of T. rubrum CYP51. Antimicrob Agents Chemother 61:e00333-17. doi: 10.1128/AAC.00333-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garvey EP, Hoekstra WJ, Schotzinger RJ, Sobel JD, Lilly EA, Fidel PL. Jr, 2015. Efficacy of the clinical agent VT-1161 against fluconazole-sensitive and -resistant Candida albicans in a murine model of vaginal candidiasis. Antimicrob Agents Chemother 59:5567–5573. doi: 10.1128/AAC.00185-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nielsen K, Vedula P, Smith KD, Meya DB, Garvey EP, Hoekstra WJ, Schotzinger RJ, Boulware DR. 2017. Activity of VT-1129 against Cryptococcus neoformans clinical isolates with high fluconazole MICs. Med Mycol 55:453–456. doi: 10.1093/mmy/myw089. [DOI] [PubMed] [Google Scholar]
- 10.Warrilow AG, Parker JE, Price CL, Nes WD, Garvey EP, Hoekstra WJ, Schotzinger RJ, Kelly DE, Kelly SL. 2016. The investigational drug VT-1129 is a highly potent inhibitor of Cryptococcus species CYP51 but only weakly inhibits the human enzyme. Antimicrob Agents Chemother 60:4530–4538. doi: 10.1128/AAC.00349-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lockhart SR, Fothergill AW, Iqbal N, Bolden CB, Grossman NT, Garvey EP, Brand SR, Hoekstra WJ, Schotzinger RJ, Ottinger E, Patterson TF, Wiederhold NP. 2016. The investigational fungal Cyp51 inhibitor VT-1129 demonstrates potent in vitro activity against Cryptococcus neoformans and Cryptococcus gattii. Antimicrob Agents Chemother 60:2528–2531. doi: 10.1128/AAC.02770-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shubitz LF, Trinh HT, Galgiani JN, Lewis ML, Fothergill AW, Wiederhold NP, Barker BM, Lewis ER, Doyle AL, Hoekstra WJ, Schotzinger RJ, Garvey EP. 2015. Evaluation of VT-1161 for treatment of coccidioidomycosis in murine infection models. Antimicrob Agents Chemother 59:7249–7254. doi: 10.1128/AAC.00593-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shubitz LF, Roy ME, Trinh HT, Hoekstra WJ, Schotzinger RJ, Garvey EP. 2017. Efficacy of the investigational antifungal VT-1161 in treating naturally occurring coccidioidomycosis in dogs. Antimicrob Agents Chemother 61:e00111-17. doi: 10.1128/AAC.00111-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gebremariam T, Alkhazraji S, Lin L, Wiederhold NP, Garvey EP, Hoekstra WJ, Schotzinger RJ, Patterson TF, Filler SG, Ibrahim AS. 2017. Prophylactic treatment with VT-1161 protects immunosuppressed mice from Rhizopus arrhizus var. arrhizus infection. Antimicrob Agents Chemother 61:e00390-17. doi: 10.1128/AAC.00390-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gebremariam T, Wiederhold NP, Fothergill AW, Garvey EP, Hoekstra WJ, Schotzinger RJ, Patterson TF, Filler SG, Ibrahim AS. 2015. VT-1161 protects immunosuppressed mice from Rhizopus arrhizus var. arrhizus infection. Antimicrob Agents Chemother 59:7815–7817. doi: 10.1128/AAC.01437-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schell WA, Jones AM, Garvey EP, Hoekstra WJ, Schotzinger RJ, Alexander BD. 2017. Fungal CYP51 inhibitors VT-1161 and VT-1129 exhibit strong in vitro activity against Candida glabrata and C. krusei isolates clinically resistant to azole and echinocandin antifungal compounds. Antimicrob Agents Chemother 61:e01817-16. doi: 10.1128/AAC.01817-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hargrove TY, Friggeri L, Wawrzak Z, Qi A, Hoekstra WJ, Schotzinger RJ, York JD, Guengerich FP, Lepesheva GI. 2017. Structural analyses of Candida albicans sterol 14alpha-demethylase complexed with azole drugs address the molecular basis of azole-mediated inhibition of fungal sterol biosynthesis. J Biol Chem 292:6728–6743. doi: 10.1074/jbc.M117.778308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoekstra WJ, Hargrove TY, Wawrzak Z, da Gama Jaen Batista D, da Silva CF, Nefertiti AS, Rachakonda G, Schotzinger RJ, Villalta F, Soeiro Mde N, Lepesheva GI. 2016. Clinical candidate VT-1161’s antiparasitic effect in vitro, activity in a murine model of Chagas disease, and structural characterization in complex with the target enzyme CYP51 from Trypanosoma cruzi. Antimicrob Agents Chemother 60:1058–1066. doi: 10.1128/AAC.02287-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Monk BC, Tomasiak TM, Keniya MV, Huschmann FU, Tyndall JDA, O’Connell JD, Cannon RD, McDonald JG, Rodriguez A, Finer-Moore JS, Stroud RM. 2014. Architecture of a single membrane spanning cytochrome P450 suggests constraints that orient the catalytic domain relative to a bilayer. Proc Natl Acad Sci U S A 111:3865–3870. doi: 10.1073/pnas.1324245111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sagatova AA, Keniya MV, Wilson RK, Monk BC, Tyndall JD. 2015. Structural Insights into binding of the antifungal drug fluconazole to Saccharomyces cerevisiae lanosterol 14alpha-demethylase. Antimicrob Agents Chemother 59:4982–4989. doi: 10.1128/AAC.00925-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sagatova AA, Keniya MV, Wilson RK, Sabherwal M, Tyndall JD, Monk BC. 2016. Triazole resistance mediated by mutations of a conserved active site tyrosine in fungal lanosterol 14alpha-demethylase. Sci Rep 6:26213. doi: 10.1038/srep26213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lamping E, Monk BC, Niimi K, Holmes AR, Tsao S, Tanabe K, Niimi M, Uehara Y, Cannon RD. 2007. Characterization of three classes of membrane proteins involved in fungal azole resistance by functional hyperexpression in Saccharomyces cerevisiae. Eukaryot Cell 6:1150–1165. doi: 10.1128/EC.00091-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Keniya MV, Fleischer E, Klinger A, Cannon RD, Monk BC. 2015. Inhibitors of the Candida albicans major facilitator superfamily transporter Mdr1p esponsible for fluconazole resistance. PLoS One 10:e0126350. doi: 10.1371/journal.pone.0126350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Niimi K, Harding DR, Holmes AR, Lamping E, Niimi M, Tyndall JD, Cannon RD, Monk BC. 2012. Specific interactions between the Candida albicans ABC transporter Cdr1p ectodomain and a d-octapeptide derivative inhibitor. Mol Microbiol 85:747–767. doi: 10.1111/j.1365-2958.2012.08140.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.White TC, Marr KA, Bowden RA. 1998. Clinical, cellular, and molecular factors that contribute to antifungal drug resistance. Clin Microbiol Rev 11:382–402. doi: 10.1128/CMR.11.2.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Niimi M, Nagai Y, Niimi K, Wada S, Cannon RD, Uehara Y, Monk BC. 2002. Identification of two proteins induced by exposure of the pathogenic fungus Candida glabrata to fluconazole. J Chromatogr B Analyt Technol Biomed Life Sci 782:245–252. doi: 10.1016/S1570-0232(02)00668-2. [DOI] [PubMed] [Google Scholar]
- 27.Lamping E, Baret PV, Holmes AR, Monk BC, Goffeau A, Cannon RD. 2010. Fungal PDR transporters: phylogeny, topology, motifs and function. Fungal Genet Biol 47:127–142. doi: 10.1016/j.fgb.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berman J. 2016. Ploidy plasticity: a rapid and reversible strategy for adaptation to stress. FEMS Yeast Res 16:fow020. doi: 10.1093/femsyr/fow020. [DOI] [PubMed] [Google Scholar]
- 29.Morschhauser J. 2016. The development of fluconazole resistance in Candida albicans an example of microevolution of a fungal pathogen. J Microbiol 54:192–201. doi: 10.1007/s12275-016-5628-4. [DOI] [PubMed] [Google Scholar]
- 30.Dunkel N, Liu TT, Barker KS, Homayouni R, Morschhauser J, Rogers PD. 2008. A gain-of-function mutation in the transcription factor Upc2p causes upregulation of ergosterol biosynthesis genes and increased fluconazole resistance in a clinical Candida albicans isolate. Eukaryot Cell 7:1180–1190. doi: 10.1128/EC.00103-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Whaley SG, Caudle KE, Vermitsky JP, Chadwick SG, Toner G, Barker KS, Gygax SE, Rogers PD. 2014. UPC2A is required for high-level azole antifungal resistance in Candida glabrata. Antimicrob Agents Chemother 58:4543–4554. doi: 10.1128/AAC.02217-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Snelders E, Huis In ‘t Veld RA, Rijs AJ, Kema GH, Melchers WJ, Verweij PE. 2009. Possible environmental origin of resistance of Aspergillus fumigatus to medical triazoles. Appl Environ Microbiol 75:4053–4057. doi: 10.1128/AEM.00231-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Snelders E, van der Lee HA, Kuijpers J, Rijs AJ, Varga J, Samson RA, Mellado E, Donders AR, Melchers WJ, Verweij PE. 2008. Emergence of azole resistance in Aspergillus fumigatus and spread of a single resistance mechanism. PLoS Med 5:e219. doi: 10.1371/journal.pmed.0050219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Keniya MV, Ruma YN, Tyndall JDA, Monk BC. 2018. Heterologous expression of full-length lanosterol 14alpha-demethylases of prominent fungal pathogens Candida albicans and Candida glabrata provides tools for antifungal discovery. Antimicrob Agents Chemother 62:e01131-18. doi: 10.1128/AAC.01131-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sagatova AA, Keniya MV, Tyndall JDA, Monk BC. 2017. The impact of homologous resistance mutations from pathogenic yeast on Saccharomyces cerevisiae lanosterol 14alpha-demethylase. Antimicrob Agents Chemother 62:e02242-17. doi: 10.1128/AAC.02242-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Keniya MV, Sabherwal M, Wilson RK, Woods MA, Sagatova AA, Tyndall JDA, Monk BC. 2018. Crystal structures of full-length lanosterol 14alpha-demethylases of prominent fungal pathogens Candida albicans and Candida glabrata provide tools for antifungal discovery. Antimicrob Agents Chemother 62:e01134-18. doi: 10.1128/AAC.01134-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hargrove TY, Garvey EP, Hoekstra WJ, Yates CM, Wawrzak Z, Rachakonda G, Villalta F, Lepesheva GI. 2017. Crystal structure of the new investigational drug candidate VT-1598 in complex with Aspergillus fumigatus sterol 14alpha-demethylase provides insights into its broad-spectrum antifungal activity. Antimicrob Agents Chemother 61:e00570-17. doi: 10.1128/AAC.00570-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lepesheva GI, Waterman MR. 2011. Structural basis for conservation in the CYP51 family. Biochim Biophys Acta 1814:88–93. doi: 10.1016/j.bbapap.2010.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Garvey EP, Hoekstra WJ, Moore WR, Schotzinger RJ, Long L, Ghannoum MA. 2015. VT-1161 dosed once daily or once weekly exhibits potent efficacy in treatment of dermatophytosis in a guinea pig model. Antimicrob Agents Chemother 59:1992–1997. doi: 10.1128/AAC.04902-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Flowers SA, Colon B, Whaley SG, Schuler MA, Rogers PD. 2015. Contribution of clinically derived mutations in ERG11 to azole resistance in Candida albicans. Antimicrob Agents Chemother 59:450–460. doi: 10.1128/AAC.03470-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sanglard D, Ischer F, Koymans L, Bille J. 1998. Amino acid substitutions in the cytochrome P-450 lanosterol 14alpha-demethylase (CYP51A1) from azole-resistant Candida albicans clinical isolates contribute to resistance to azole antifungal agents. Antimicrob Agents Chemother 42:241–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hayama K, Ishibashi H, Ishijima SA, Niimi K, Tansho S, Ono Y, Monk BC, Holmes AR, Harding DR, Cannon RD, Abe S. 2012. A d-octapeptide drug efflux pump inhibitor acts synergistically with azoles in a murine oral candidiasis infection model. FEMS Microbiol Lett 328:130–137. doi: 10.1111/j.1574-6968.2011.02490.x. [DOI] [PubMed] [Google Scholar]
- 44.Wilson RB, Davis D, Enloe BM, Mitchell AP. 2000. A recyclable Candida albicans URA3 cassette for PCR product-directed gene disruptions. Yeast 16:65–70. doi: 10.1002/(SICI)1097-0061(20000115)16:1<65::AID-YEA508>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 45.Clinical and Laboratory Standards Institute. 2012. Reference method for broth dilution antifungal susceptibility testing of yeasts; 4th informational supplement. CLSI document M27-S4. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 46.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. 1951. Protein measurement with the Folin phenol reagent. J Biol Chem 193:265–275. [PubMed] [Google Scholar]
- 47.Battye TG, Kontogiannis L, Johnson O, Powell HR, Leslie AG. 2011. iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr D Biol Crystallogr 67:271–281. doi: 10.1107/S0907444910048675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Evans P. 2006. Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr 62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 49.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. 2007. Phaser crystallographic software. J Appl Crystallogr 40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.