

Paracoccidioidomycosis (PCM), caused by Paracoccidioides, is a systemic mycosis with granulomatous character and a restricted therapeutic arsenal. The aim of this work was to search for new alternatives to treat largely neglected tropical mycosis, such as PCM.
KEYWORDS: paracoccidioidomycosis, chorismate synthase, molecular modeling, new antifungal, virtual screening
ABSTRACT
Paracoccidioidomycosis (PCM), caused by Paracoccidioides, is a systemic mycosis with granulomatous character and a restricted therapeutic arsenal. The aim of this work was to search for new alternatives to treat largely neglected tropical mycosis, such as PCM. In this context, the enzymes of the shikimate pathway constitute excellent drug targets for conferring selective toxicity because this pathway is absent in humans but essential for the fungus. In this work, we have used a homology model of the chorismate synthase (EC 4.2.3.5) from Paracoccidioides brasiliensis (PbCS) and performed a combination of virtual screening and molecular dynamics testing to identify new potential inhibitors. The best hit, CP1, successfully adhered to pharmacological criteria (adsorption, distribution, metabolism, excretion, and toxicity) and was therefore used in in vitro experiments. Here we demonstrate that CP1 binds with a dissociation constant of 64 ± 1 μM to recombinant chorismate synthase from P. brasiliensis and inhibits enzymatic activity, with a 50% inhibitory concentration (IC50) of 47 ± 5 μM. As expected, CP1 showed no toxicity in three cell lines. On the other hand, CP1 reduced the fungal burden in lungs from treated mice, similar to itraconazole. In addition, histopathological analysis showed that animals treated with CP1 displayed less lung tissue infiltration, fewer yeast cells, and large areas with preserved architecture. Therefore, CP1 was able to control PCM in mice with a lower inflammatory response and is thus a promising candidate and lead structure for the development of drugs useful in PCM treatment.
INTRODUCTION
Paracoccidioidomycosis (PCM) is a systemic granulomatous disease caused by thermally dimorphic fungi of the genus Paracoccidioides (1, 2). PCM is endemic in Latin America, mainly southern Brazil, and features one of the highest mortality rates among chronic fungal diseases. With an average of 2.7 new cases per 100.000 habitants per year, this mycosis is among the 10 largest causes of death in predominantly chronic diseases and has high mortality rates among systemic mycoses (3, 4). Recent studies have shown that PCM is responsible for approximately 51.2% of deaths caused by systemic mycoses in Brazil (5). In addition, PCM has a high social and economic cost since it may leave important sequelae in individuals during their productive age. Despite these social problems, the prevalence of this mycosis is underestimated since it is based on reported cases or hospitalization and mortality data (5).
PCM presents two principal clinical forms, acute/subacute and chronic. The acute or subacute form, also known as the juvenile form, predominantly affects young adults of both sexes. The infection rapidly evolves and disseminates to multiple organs, with many severe symptoms, such as lymphadenomegaly, hepatosplenomegaly, fever, weight loss, and anorexia (5, 6). This form is responsible for up to 25% of PCM cases. The chronic form represents the majority of PCM cases (74 to 96%), affecting mainly male adults. The chronic form of PCM is characterized by slow progression, frequently with pulmonary involvement, and the mucocutaneous tissue may be affected (6). Therefore, PCM could result in serious sequelae or lead to death if not detected early. Thus, this disease fits into the neglected endemic mycoses and requires epidemiological data and surveillance to fully comprehend the magnitude of the problem (7).
In clinical practice, the following three medications are commonly used for the treatment of PCM: azole derivatives (itraconazole [ITZ]), sulfanilamide compounds (trimethoprim-sulfamethoxazole), and polyenes (amphotericin B [AmB]). Unlike for other pathogenic fungi, there is no solid evidence to indicate that P. brasiliensis develops resistance to antifungal agents (6). However, treatment may range from several months to a year depending on the patient’s condition. Thus, the limitations for the treatment of PCM are linked to few therapeutic options, drug interactions, infusion-related events, nephrotoxicity, and prolonged antifungal treatment (5). The eukaryotic nature shared by fungi and the host is the major challenge for treatment of infectious diseases (8).
A promising strategy for selective antimicrobial therapy has been to exploit the inhibition of unique targets essential to the pathogen but absent in humans (9). In this sense, the shikimate pathway is an attractive target since it is present in bacteria, fungi, plants, and apicomplexan parasites but absent from mammals (10–12). Equally important, this pathway is essential for the survival of these organisms, and therefore, the enzymes are promising targets for drug development (13). Notably, the broad-spectrum herbicide glyphosate, one of the most successful herbicides currently used worldwide, inhibits the sixth enzyme of the pathway, 5-enolpyruvylshikimate 3-phosphate synthase (14).
The enzyme chorismate synthase (EC 4.2.3.5) is the seventh enzyme in the shikimate pathway and is responsible for the synthesis of chorismate, the branch point in the biosynthesis of several important aromatic molecules, including aromatic amino acids, folate, naphthoquinones, and menaquinones. This enzyme catalyzes the conversion of 5-enolpyruvylshikimate-3-phosphate (EPSP) to chorismate in the presence of reduced flavin mononucleotide (FMNH2), which is not consumed in the reaction (15). Although the three-dimensional structure of chorismate synthase (CS) from Paracoccidioides brasiliensis (PbCS) has not been solved yet, its amino acid sequence shares 67.9% identity with that of the homologous enzyme from Saccharomyces cerevisiae (16).
Therefore, the aim of this study was to identify potential inhibitors of CS from Paracoccidioides brasiliensis by in silico methodologies and to validate potential candidates for their usefulness as compounds or lead structures for the development of new antifungals. In this work, we describe the identification and (partial) characterization of a new compound possessing promising antifungal activity against Paracoccidioides species. Most importantly, we demonstrate that this compound significantly reduces the fungal burden in the lungs of infected mice, resulting in diminished tissue damage.
RESULTS
Protein modeling and evaluation.
The high sequence identity of PbCS to the main template from Saccharomyces cerevisiae, 63.9%, enabled homology modeling of the three-dimensional structure (17, 18). The short insertions and deletions observed in the multiple alignments did not affect the active site with regard to substrate or cofactor binding. The Psipred server suggested that the two segments in PbCS did not have the secondary-structure element compatible with the template. Thus, no restriction was added to these segments during the modeling process. The high degree of freedom regarding protein movement in the protomer could compromise the active-site configuration, essentially structured by random segments of structure (loops and turns). For this reason, the protein was modeled as a homotetramer (Fig. 1). Therefore, symmetry restraints were imposed on alpha-carbons of the subunits to generate symmetry and to decrease possible model inconsistencies for virtual screening (VS). Once modeled, only the A subunit was used in the docking studies. The final model of PbCS (Fig. 1) exhibited excellent stereochemical quality, with 91.4% of residues in the most favorable regions and 8.6% in additional allowed regions of the Ramachandram plot.
FIG 1.

(A) Ribbon model of biological unity of PbCS bonded to FMNH2 (red) and EPSP (purple). (B) Homotetramer protein chains. (C) Monomeric structure colored by electrostatic potential, highlighting the sites of FMNH2 and EPSP with electropositive potential (blue). (D) Close view of EPSP binding site. The most important residues in the docking of both CP1 (brown) and EPSP (black), as presented in Table S1, are labeled. FMNH2 is represented by a ball and stick.
Virtual screening.
The application of the validated docking protocols shows that the substrate EPSP overlay almost perfectly to the minimized pose, exhibiting an average root mean square deviation (RMSD) of 0.57 ± 0.13 Å and a mean ranking of −12.7 ± 1.06 kcal mol−1. At the end of the virtual screening, 53 compounds were selected and were used in an additional 5 screening repetitions using AutoDock and 5 screenings using Molegro, totaling 10 repetitions. The compounds Zinc13370291 (CP3), Zinc06445857 {CP1; (1S,2S,3aS,4S,9bR)-1-chloro-6-nitro-2-(2-nitrophenyl)sulfanyl-2,3,3a,4,5,9b-hexahydro-1H-cyclopenta[c]quinoline-4-carboxylic acid}, and Zinc06445743 (CP2) were always ranked among the best 5 in all 10 repetitions, exhibiting mean ΔGbinding values of −14.2 ± 0.13, −13.3 ± 0.17, and −13.1 ± 0.19 kcal mol−1, respectively (Fig. 2). These 3 molecules were subjected to ADMETox (adsorption, distribution, metabolism, excretion, and toxicity) evaluation, which includes Lipinski’s rule of 5 by means of the server FAF-Drugs (19). After passing these criteria, the molecules were purchased for in vitro evaluations. Statistically, when different programs select a common molecule using different search and ranking algorithms, the chance of getting a false-positive result significantly decreases and the chance of the selected molecule exhibiting in vitro or in vivo activity increases.
FIG 2.
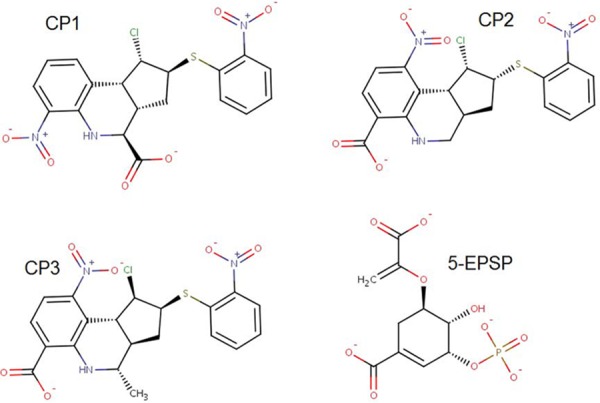
Chemical structures of the 3 molecules selected by virtual screening and the reference ligand EPSP.
MD simulations.
In order to investigate the structural stability and folding of PbCS, as well as to obtain information about the residues involved in stabilizing ligands, molecular dynamics (MD) simulations were carried out for 20 ns with the protein in the apo- and holo-forms. In these simulations, the final model of PbCS with bound FMNH2 and EPSP, or the best pose of potential inhibitors, was minimized and then equilibrated as described in Materials and Methods. The thermodynamic equilibrium was reached when the RMSD of the main chain atoms of the protein as a function of time had stabilized on a constant level for at least 5 ns. The results showed that the evaluated structures reached an equilibrium after 6 ns of simulation (Fig. 3A). The radius of gyration (Rgyr) from the protein-ligand complexes was constant during the simulations for all structures evaluated (Fig. 3B), indicating that the structures were stable and the presence of the ligands did not lead to protein unfolding. Furthermore, the oscillations of Rgyr were lower in the tetrameric structures (biological units) than in the monomeric form, which indicates greater structural stability in this form.
FIG 3.

Analysis of the molecular dynamics trajectory for PbCS structures. Shown are the A-chain in the complex with FMNH2 plus EPSP (pink), tetramer apo (black), tetramer with FMNH2 plus EPSP bound (red), tetramer with bound FMNH2 plus CP 1 (gray), tetramer with bound FMNH2 plus CP2 (green), and tetramer with bound FMNH2 plus CP3 (blue). The RMSD (A) and radius of gyration (B) were obtained from the oscillation of the protein main chain atoms relative to the fully minimized structure.
The protein regions with increased flexibility were evidenced by the root mean square fluctuation (RMSF) of each residue. The RMSF was calculated from Cα atoms from the last 5 ns of the simulation, i.e., during the thermodynamic equilibrium. The residues with greater flexibility were in the N-terminal regions of the 4 chains, a fact that is common to most proteins, and in the case of PbCS, refers to a helix with a high degree of freedom of rotation due to a turn that connects it to the remainder of the protein (see Fig. S2 in the supplemental material). Because of this, the last 10 residues were disregarded in the calculation of RMSD and Rgyr. The RMSF plot also showed that residues making contact with EPSP are located in the most stable regions of the protein with smaller RMSF values (Fig. S2).
Residues 290 to 298 constitute a very flexible loop placed on the surface of the protein. Since there are no intramolecular interactions to stabilize this region, it becomes quite flexible in all protein chains. The pronounced increase in this flexibility, as observed in the protein-inhibitor complexes, is not related to the presence of the ligands because this loop is located in a place far away from the ligand binding site, suggesting that this is solely a random variation in the simulation. The great flexibility of these residues is also reflected by the lack of electron density in homologous regions of the crystallographic templates.
Active site and EPSP docking.
There is some flexibility in the EPSP binding site, which is structured mainly by loop elements. This prompted us to take special care in modeling this region because this site showed substantial flexibility in the templates (20). Thus, we opted for an interactive model, prioritizing the templates in which the flexible area is in contact with EPSP, i.e., in a closed conformation.
In this work, the binding site was delimited from the residues that had a frequency of contact with the EPSP greater than 70% over the course of the simulation (Table S1). This is because each ligand has a specific geometry and composition, making them interact differently with the protein and thus structuring distinct sites.
Ligand CP1.
From the 3 potential inhibitors identified by virtual screening, CP1 was the most promising to present fungicidal activity in vitro. Thus far, no application of this compound has been reported. CP1 binds to PbCS with fewer hydrogen bonds than does EPSP (Fig. S2); however, the salt bridges with Arg45, Arg124, Arg353, and Arg361 are still preserved. Several other interactions occur between CP1 and PbCS, such as hydrogen bonds and van der Waals contacts, which were reflected in a better ranking than for EPSP.
To support our findings from virtual screening and molecular docking simulations, we produced and purified recombinant CS from P. brasiliensis and analyzed the binding of CP1 to the protein. As shown in Fig. 4A, the UV-visible (UV-Vis) absorption spectrum of CP1 changes as a function of the PbCS concentration, allowing for the calculation of a dissociation constant of 64 ± 1 μM from the hyperbolic plot (Fig. 4, inset). Next the influence of CP1 on the enzymatic activity of PbCS was analyzed using an enzyme-coupled assay. As shown in Fig. 4B, CP1 also affects the activity, yielding a 50% inhibitory concentration (IC50) of 47 ± 5 μM, which fits nicely with the observed dissociation constant. Thus, our biochemical results clearly indicate that CP1 specifically binds and inhibits the target enzyme of our screening approach.
FIG 4.

Binding of CP1 to PbCS (A) and the effect of CP1 on PbCS activity (B). (A) CP1 was dissolved in 50 mM MOPS (pH 7.5; ∼20 μM [black spectrum]) and titrated with PbCS (∼443 μM) using 10-μl aliquots (inset). Spectral changes were monitored at 300 to 800 nm. The orange and pink traces represent the intermediate spectra at ∼39 μM PbCS and ∼72 μM PbCS, respectively. The blue trace corresponds to the final spectrum recorded at ∼111 μM PbCS. The inset shows the mean absorption changes at 500 nm (from 3 determinations) as a function of the protein concentration, yielding a dissociation constant of 64 ± 1 μM (the SD at each PbCS concentration is shown by an error bar). (B) The influence of CP1 on the activity of PbCS was tested in an assay involving 80 μM EPSP, 4 μM PbCS, and 20 μM anthranilate synthase at 37°C. By forward coupling, the chorismate synthase reaction and PbCS activity could be studied spectrofluorometrically by following the increase in fluorescence intensity at 390 nm (λex = 340 nm) for 5 min, which correlates with the amount of anthranilate formed during the course of the reaction. The slopes of the initial velocities were determined and plotted as a function of the log of the corresponding CP1 concentration. Using a nonlinear curve fit based on the Hill function, an IC50 of 47 ± 5 μM was determined (rates were determined in triplicates, with the SDs shown as error bars).
In vitro CP1 antifungal activity against Paracoccidioides spp.
CP1 inhibited the growth of Paracoccidioides spp., with MICs of 2 to 16 mg/liter for P. brasiliensis isolates and 16 to 32 mg/liter for P. lutzii isolates (Table 1). The minimal fungicidal concentrations (MFCs) were 4 mg/liter for P. brasiliensis (Pb18) and 32 to 128 mg/liter for P. lutzii isolates (Table 1). All isolates tested showed itraconazole (ITZ) susceptibility (MIC of 1 mg/liter). Although the CP1 MICs were higher than for azole, CP1 showed potent antifungal activity, suggesting a fungicidal effect against several Paracoccidioides isolates (Table 1). However, there was no synergistic action between CP1 and the conventional drugs tested (data not shown).
TABLE 1.
MICs and MFCs of CP1 and itraconazole (ITZ) for Paracoccidioides species isolates
| Isolate | Species | MIC of CP1 (mg/liter) |
MFC of CP1 (mg/liter) |
MIC of ITZ (mg/liter) |
|---|---|---|---|---|
| Pb18 | P. brasiliensis | 2 | 4 | 1 |
| Mg14 | P. brasiliensis | 16 | —a | 1 |
| Pb01 | P. lutzii | 16 | 128 | 1 |
| ROSC | P. lutzii | 32 | 32 | 1 |
| 8334 | P. lutzii | 16 | 64 | 1 |
—, there was no fungal growth on the MFC plate.
CP1 had no cytotoxic effect on human cell lines.
Initially, J774 macrophages were used because these cells are the first to come into contact with pathogens. In addition, 2 human cell lines, HUVEC and HeLa, were also evaluated. As shown in Fig. 5, all cell lines tested showed high viability after exposure to various CP1 concentrations. Even at concentrations of 3× MIC (128 mg/liter), cell viability was higher than 80% for all cell lines tested, and the differences (P > 0.05) were not statistically significant (Fig. 5).
FIG 5.

CP1 in vitro cytotoxicity assessment. Three cell lines were subjected to cytotoxicity by MTS assay. Various CP1 concentrations were tested (0.0 to 128 mg/liter) without showing significant toxicity. The 3 cell lines evaluated showed high viability after 24 h in contact with CP1, even at concentrations of 2× MIC (64 mg/liter). The results are representative of those from 3 independent experiments. Bars indicate SDs.
Morphological alteration in P. brasiliensis caused by CP1 treatment.
CP1 at 2× MIC (64 mg/liter) was able to kill all clinical isolates tested, and this CP1 concentration was chosen for further experiments. Electron micrographs revealed deformations in the P. brasiliensis cells, including depression and rough surfaces (Fig. 6C). Irregularly shaped cells, as well as aggregating cells, were also observed (Fig. 6D). These hallmark changes were seen in approximately 80% of the population after treatment. In the control, the typical morphology of P. brasiliensis cells was observed (Fig. 6A and B).
FIG 6.

Scanning electron microscopy (SEM) of Paracoccidioides brasiliensis (Pb18). Cells of the isolated Pb18 strain were grown at a CP1 concentration of 64 mg/liter. (A and B) Control consisting of P. brasiliensis cells without treatment. (C and D) Cells treated with compound CP1 (64 mg/liter). Control cells feature preserved cell surfaces and multibudding. Note the leakage of cellular content and cell surface squashing and depression (arrows). In addition, aggregation of cells was observed (D). Samples were observed at magnifications of ×3,000 and ×1,000.
Effective in vivo antifungal activity.
Intraperitoneal treatment with CP1 (5 mg/kg of body weight) was able to significantly reduce the fungal burden in the lungs of animals infected with P. brasiliensis. The group treated with ITZ (5 mg/kg) showed a similar efficacy (P > 0.05) (Fig. 7). Both CP1 and ITZ were able to reduce approximately 90% of CFU recovered from the lungs compared to the control.
FIG 7.

Antifungal activity of CP1 in the treatment of PCM. CFU for lung tissue infected with P. brasiliensis are shown. The control group consisted of animals treated with a vehicle. All treatments were performed for 14 days. The experiments were performed in triplicate, and the bars indicate SDs. *, P < 0.05 (statistically significant compared to the control).
The semiquantitative histopathological analysis indicates that the CP1 performance is superior to the treatment with ITZ, considering the tissue damage. The CP1 compound was able to significantly reduce the pulmonary fibrosis and inflammatory infiltrate caused by infection with P. brasiliensis (Fig. 8). Figure 8A shows a significant reduction in pulmonary fibrosis sequelae compared with the control and ITZ groups. The level of inflammation was lower in the lung of animals treated with CP1, presenting a significant reduction compared with the control group (Fig. 8B). Fungal cells were visualized in all treatment groups, with the largest amount of the P. brasiliensis cells in the control group. Otherwise, the animals treated with CP1 showed lower scores in the fungal-cell parameter, corroborating the results of the determination of pulmonary fungal burden by CFU counting (Fig. 8C).
FIG 8.

Histopathological score of three factors indicative of lung tissue impairment of animals infected with Paracoccidioides brasiliensis (Pb18) after 14 days of treatment of ITZ, CP1, and control groups. (A) Pulmonary fibrotic profile. (B) Recruitment of inflammatory infiltrates in the lung. (C) Concentration of fungal cells of the Pb18 in the lung. *, P < 0.05 (statistically significant compared to the control).
The histopathological analysis results are represented in Fig. 9. The group treated with CP1 showed large areas of lung tissue with the preserved structure (Fig. 9C), less inflammatory infiltration (Fig. 9F), and fewer fungal cells (Fig. 9I). ITZ was less effective in reducing tissue damage caused by P. brasiliensis infection, since this group presented poorly preserved lung tissue (Fig. 9B) with large areas containing inflammatory infiltration (Fig. 9E) and fungal cells (Fig. 9H). As expected, the control group presented damage throughout the lung tissue, exhibiting multiple pulmonary foci of epithelioid inflammation with early pulmonary fibrosis (Fig. 9B). Therefore, CP1 treatment was shown to be more efficient in protecting lung tissue against pulmonary sequelae than the conventional treatment with ITZ in an experimental murine PCM model.
FIG 9.

Photomicrographs of the lungs of mice infected with 1 × 106 P. brasiliensis (Pb18) cells following treatment and hematoxylin-eosin staining. Representative images of each treatment group are given. Infected mice and mice given vehicle treatment (1% DMSO and 0.2% Pluronic) served as the controls (A, D, and G). Images for infected mice and mice treated with 5 mg/kg of ITZ (B, E, and H) and 5 mg/kg of CPI (C, F, and I) are shown. (A) The lung structure was not preserved, showing large fibrotic masses (magnification, ×100). (B) The lung structure was severely damaged, showing confluent fibrotic masses (magnification, ×100). (C) Preserved lung tissue with single fibrotic masses (magnification, ×100). (D and E) Lung tissue with high agglomeration inflammatory infiltrate in the parenchyma (blue arrows; magnification, ×400). (F) Moderate inflammatory infiltrate (magnification, ×400). (G and H) Accumulation of a large amount of fungal cells (arrows; magnification, ×400). (I) Scarce presence of fungal cells in the lung tissue (magnification, ×400).
DISCUSSION
PCM constitutes a substantial public health problem with high mortality rates in Latin America. Several antifungal drugs have been shown to be effective in treating different clinical forms of PCM, including amphotericin B (AmB; formulations in deoxycholate, lipid, and liposomal complex), sulfonamide derivatives (trimethoprim-sulfamethoxazole), and azole derivatives (ITZ and voriconazole or posaconazole). However, the main challenge of systemic mycosis is the requirement for long periods of treatment with antifungal medications that present several limitations (21).
Amphotericin B has been a polyene recommended for severe cases of PCM since 1958, mainly in the initial phase (5, 6). However, it should be noted that this drug has many important limitations, such as intravenous administration and problems related to infusion and high nephrotoxicity. Although there are current liposomal forms of amphotericin B, these are costly and still have several adverse effects (22). In the maintenance phase of treatment, trimethoprim-sulfamethoxazole has been frequently used (23, 24). However, therapeutic failure was described for 5% of patients, and the main disadvantage is the need for long-term treatments (over 12 months), in addition to adverse effects such as hypersensitivity reactions, leukopenia, megaloblastic anemia, and thrombocytopenia (24). For the azole derivatives, ITZ is the best option for PCM treatment in patients not hospitalized (5, 6). The new azoles, such as voriconazole and posaconazole, present high costs and lack of clinical studies demonstrating advantages for their use in clinical practice (24). The main limitations of the azoles are adverse reactions, such as abnormal vision, chromatopsia, rash, headache, and an increase in hepatic transaminases (6). Unfortunately, over the last 30 years only one new antifungal class has been discovered (echinocandins), reinforcing the need to use truly innovative antifungal discovery approaches (25).
The rational design and development of new drugs that specifically act on targets of the pathogenic fungi without producing collateral damage to mammalian cells constitute a great pharmacological challenge. In this study, we identified a new potential antifungal compound against Paracoccidioides spp., selected by virtual screening using CS as the target. Our study clearly demonstrates that CP1 binds and inhibits recombinant CS from P. brasiliensis (PbCS). CS is the seventh enzyme of the common shikimate pathway and occurs only in the fungus and not in humans and thus may help to minimize side effects of the compound on human cells. Since CP1 showed promising activity in vitro and in vivo, our study identified CS and the shikimate pathway as a new target for the development of antifungal compounds.
Although the most efficient drug in PCM treatment, amphotericin B, is considered a fungicidal agent (2, 26) its administration is limited by toxicity and occasionally discontinuation of the therapy is required owing to its high nephrotoxicity (27). In contrast to AmB, our in vitro and in vivo study did not indicate any toxicity of CP1 to human cells and thus supports the concept that CP1 has selective activity against fungus, proving to be promising for the new antifungal development.
Although the need for new therapies for PCM (and other diseases caused by pathogenic fungi) is obvious, only a few studies have been reported recently. Recently, 3 thioredoxin reductase inhibitors were selected by virtual screening with antifungal activity against Paracoccidioides spp., with MICs of 8 to 32 mg/liter (28). Furthermore, two compounds (HS1 and HS2) have been identified as homoserine dehydrogenase inhibitors showing antifungal activity (32 to 64 mg/liter) against P. brasiliensis (29). A Brazilian medicinal plant called Schinus terebinthifolius presented inhibitory activity against isolates Pb18 and Pb01, with MICs of 62.5 to 250 mg/liter (30). Finally, the antifungal activity of a synthetic compound derived from chalcones was analyzed. This 6-quinolinyl N-oxide could inhibit the growth of many Paracoccidioides isolates, with a MIC of 5.9 mg/liter against Pb18 (31). In comparison with these recent reports, the MIC determined for CP1 (2 mg/liter) against P. brasiliensis isolate Pb18 indicates superior antifungal activity, encouraging future efforts to target CS.
Yet another aspect of the usability and efficacy of a drug revolves around synergistic and/or antagonistic effects that are related to the drug target(s) and potential interactions with other drugs. Synergistic relationships can occur in series, in parallel, and in combination, while antagonistic interactions involve positive-feedback interactions (32). In the case of CP1, neither synergistic nor antagonistic effects with ITZ or AmB were observed. This may be rationalized by the fact that CP1, ITZ, and AmB target different pathways, i.e., possess entirely different modes of action. Scanning electron microscopy showed a defect in the budding of yeast cells, suggesting that CP1 interfered with the cell cycle. This effect can be explained by the inhibition of the shikimate pathway, which provides essential metabolites such as aromatic amino acids.
CP1 treatment of animals infected by P. brasiliensis was highly effective. This compound was able to reduce pulmonary fibrosis, inflammatory infiltration, and yeast cells at a dosage of 5 mg/kg over 14 days. The efficiency of CP1 in reducing the lung fungal burden was very similar to that of treatment with ITZ, but the host inflammatory response was better controlled in the animals treated with CP1. Similar results were found with treatment with chalcone (compound 4c), which presented pulmonary parenchyma preserved with mild areas of inflammation (31). Although ITZ is an oral drug with good activity against P. brasiliensis, it leads to important adverse reactions (33). During treatment with ITZ, patients suffer from irreversible changes in the hepatobiliary system that indicate hepatotoxicity and, consequently, result in the discontinuation of treatment (34).
Another limitation for treatment with ITZ is the development of fibrotic sequelae that was observed in approximately 60% of patients even after prolonged treatment (35, 36). It is important to note that despite the morbidity and mortality associated with pulmonary fibrosis in patients with PCM, there is no effective therapy for this condition (6). Thus, the ability of CP1 to reduce the fungal burden and inflammatory response in the lungs of mice infected with P. brasiliensis appears to be very promising for controlling pulmonary sequelae in PCM. It is noteworthy that a similar effect was achieved by a combination of antifungal treatment and immunomodulatory components. The immunization with P10 in association with conventional drugs resulted in the absence of granulomas and areas with very few or undetectable yeast cells (37). Similarly, Naranjo and collaborators combined itraconazole-pentoxifylline treatment to reduce lung fibrosis in the murine model of PCM (36), and Puerta-Arias and collaborators were able to reduce the infection and pulmonary fibrosis with a combination of ITZ and monoclonal antibody against neutrophils (38).
Conclusions.
In conclusion, we have successfully demonstrated the identification of a new antifungal lead structure by a combination of virtual screening and molecular dynamics simulations. Furthermore, we have shown that the identified compound, CP1, was bound to the putative target enzyme, CS, and inhibited its activity. This new compound combines a potent antifungal activity in vivo with a significant reduction in lung inflammation that may contribute to the reduction of pulmonary fibrosis, a serious complication frequently occurring in PCM patients. In addition, CP1 could be used as a lead structure to further develop this new class of antifungal compounds, targeting chorismate synthase of the respective pathogenic fungus. Therefore, we are confident that our results provide a promising new direction for the treatment of PCM.
MATERIALS AND METHODS
Sequence analysis and protein modeling.
The amino acid sequence of PbCS (39) (Uniprot number C1GBW5) was evaluated by InterProScan server (40) for identification of structural motifs. This sequence was also used to search for structural templates in two ways, via BLASTp (41) and through structural alignment by means of fold recognition using pGenThreader (42), which also provides the prediction of secondary structure. The crystallographic structures of homotetrameric forms of CS from Saccharomyces cerevisiae (15) (PDB codes 1R53 and 1R52), from Streptococcus pneumoniae (43) (PDB code 1QXO), and from Helicobacter pylori (44) (PDB code 1UM0) were used as the main protein templates. Figure S1 in the supplemental material shows the alignment of the amino acid sequences from templates with PbCS. The structure with PDB code 1UM0 was also used as the template for the cofactor FMNH2, while the structure with PDB code 1QXO was used as the template for the substrate 5-EPSP. The Modeller-v9.11 program (45) generated 1,000 models using a symmetry restraint applied on the alpha-carbons of all four chains. The best five models were selected by the modeler Dope score, where the best one was chosen based on the stereochemical quality evaluated by the program Procheck (CCP4 1994). An additional 1,500 models were generated for correction of eventual residues placed in regions not allowed in the Ramachandram plot, and the best one was chosen according the criterion described above.
Partial charge calculations.
The force field Charmm C35b2-C36a2 (46) was used in the protein molecular dynamics (MD) simulations. The files containing spatial coordinates of the ligands 5-EPSP (zinc4095612), CP1 (zinc6445857), CP2 (zinc6445743), CP3 (zinc13370291), and FMNH2 (zinc31976681) were obtained from the Zinc Database (47), adjusted to its protonation state at pH 7.0, and then submitted to the SwissParam server (48) to generate their force field parameters. The Mülliken charges for each atom of the ligands were calculated using the B3LYP/6-311G* level of theory by means of the program Orca (49).
Molecular dynamics simulations.
The MD simulations were done through the program NAMD2 (50). Initially, the modeled homotetramer structure was solvated in a periodic box with TIP3 water, where the limits were at least from 10 Å away from the outer surface of the protein. A sufficient amount of sodium counterions was added to neutralize the system charges. The MD simulation was carried out in four steps. In the first step, all system atoms were minimized by 60,000 steps of conjugate gradient. In the second step, the atoms of protein and ligands were fixed in space and water and ions were equilibrated for 60 ps. In the third step, all atoms of the system were minimized again for 60,000 steps, where the resulting structure (minimized 3) was used in redocking and virtual screening simulations. In the fourth and final step, the entire system was equilibrated for 20 ns. All simulations were performed at 300 K, 1 atm, and pH 7.0. All other simulation parameters were adjusted according to the protocol previously described (51). The simulations were evaluated in terms of root mean square deviation (RMSD) using the full minimized structure (minimized 3) as the reference. The radius of gyration (Rgyr) of the protein-ligand complexes, the protein residues in contact with inhibitors until a distance of 4.0 Å, the root mean square fluctuation (RMSF) of alpha-carbons of each protein residue, and the number of hydrogen bonds between protein and ligands were extracted from the trajectory file. The simulations were carried out in four nodes of an SGI Altix ICE 8400 LX running parallel and using two processors, Intel Six Core 5680 of 3.33 GHz in each node (48 cores) and 36GB RAM, at CENAPAD/Unicamp, Brazil.
VS simulations.
The docking protocol was established from redocking evaluations of 5-EPSP on the full minimized structure of the protein-cofactor complex by means of two programs, Autodock v.4.2.3 (52) implemented in the Pyrx-0.9 graphical interface (53) and the program Molegro Virtual Docker v.6 (54). The Autodock program uses the default search and rank algorithms and a default box size centered on 5-EPSP. The search and rank protocols for Molegro were Moldock Simplex Evolution and Moldock Score (Grid), respectively. The 10-Å radius of the search sphere was centered on 5-EPSP. Both protocols were validated by five repetitions, providing an average docking RMSD of 0.57 ± 0.130 Å, which was applied in virtual screening (VS). The library used in the VS was buildt through a literature search and by searching in the Binding Database (55) and Brenda enzymes (56) by molecules with known inhibitory activities on CS. For each identified molecule, a new search was made in the Zinc database for molecules with 80% structural similarity and commercial availability (2,555 molecules). The Natural Products library from Zinc (204,400 molecules) was added to the first, totaling 206,955 structures, plus the structure of 5-EPSP, so that at the end of the simulation only the best-ranked substrates were considered.
Compound CP1.
The CP1 compound{(1S,2S,3aS,4S,9bR)-1-chloro-6-nitro-2-(2-nitrophenyl)sulfanyl-2,3,3a,4,5,9b-hexahydro-1H-cyclopenta[c]quinoline-4-carboxylic acid} was purchased from Specs, Netherlands. The stock solution was prepared in dimethyl sulfoxide (DMSO) at a concentration of 100 mg/ml. For the assay, this solution was diluted in culture medium or phosphate-buffered saline (PBS; 0.1 M, pH 7) and complete solubilization was achieved with Pluronic (0.2%; Sigma-Aldrich, St. Louis, MO).
Plasmid construction.
The CS gene of the Pb18 isolate of P. brasiliensis (GenBank accession no. XM_010761971.1; 1,218 bp) was commercially synthesized with the preferred codons of Escherichia coli (Epoch Biolabs, Missouri City, TX) and cloned into a pET21a vector using the NdeI and XhoI restriction sites at the 5′ and 3′ ends, respectively. Thereby, a nucleotide sequence coding for a C-terminal hexahistidine tag was fused to the gene of interest to allow for purification of the recombinant protein by affinity chromatography. Clones containing the correct insert were transformed with E. coli BL21(λDE3) cells (Novagen, San Diego, CA) for gene expression.
Production and purification of recombinant PbCS.
For expression of PbCS, Luria-Bertani (LB) medium containing 100 μg/ml of ampicillin was inoculated with an overnight culture to an optical density at 600 nm (OD600) of 0.1, and cultures were grown to an OD600 of 0.7 at 37°C and 140 rpm. Then expression was induced by adding isopropyl-β-d-1-thiogalactopyranoside (IPTG) to a final concentration of 0.1 mM, and cultures were further incubated at 20°C and 140 rpm overnight (16 h). Cells were harvested by centrifugation (5,500 × g for 15 min) and the resulting cell pellets were stored at −20°C until further use.
To obtain pure PbCS, cell pellets (approximately 15 g of cells [wet weight]) were resuspended in about 40 ml of binding buffer (50 mM NaH2PO4, 150 mM NaCl, 10 mM imidazole [pH 8]) and lysed by sonication (2 × 5 min) using a Labsonic L sonication probe (B. Braun Biotech, Berlin, Germany). The lysate was cleared by centrifugation (38,500 × g for 45 min) and loaded onto a nickel-nitrilotriacetic acid (Ni-NTA) column (5 ml) equilibrated with binding buffer. After extensive washing (10 column volumes) with wash buffer (50 mM NaH2PO4, 150 mM NaCl, 20 mM imidazole [pH 8]) to remove all nonspecifically bound proteins, PbCS was eluted with elution buffer (50 mM NaH2PO4, 150 mM NaCl, 300 mM imidazole [pH 8]) and SDS-PAGE analysis was used to identify the fractions containing PbCS (>98% purity). The latter were pooled and dialyzed against 50 mM C7H15NO4S (MOPS; pH 7.5) overnight before concentrating the protein to about 400 μM using centripreps (30-kDa cutoff, Merck-Millipore, Darmstadt, Germany). The protein was flash frozen using liquid nitrogen and stored at −80°C until further use.
Determination of the dissociation constant of CP1.
To assay the binding of CP1 to PbCS, changes in the UV-Vis absorption spectrum of CP1 were studied as a function of the concentration of CS. To this end, 800 μl of CP1 diluted to a final concentration of 20 μM using 50 mM MOPS (pH 7.5; A439 ∼ 0.2) was transferred to a quartz cuvette and an absorption spectrum was recorded between 300 and 800 nm. Then 10-μl aliquots of PbCS (400 μM) were added to both the measurement and reference cuvettes, and the absorption spectrum was recorded after each addition. To quantify the binding affinity of CP1 to PbCS, absorption changes at 500 nm were plotted as a function of the enzyme concentration in the cuvette, with the standard deviation (SD; from three titrations) represented by error bars. Using a hyperbolic fit, a dissociation constant (Kd) could be determined.
Effect of CP1 on chorismate synthase activity.
To test the inhibitory effect of CP1 on PbCS, a coupled assay involving EPSP, CS, and anthranilate synthase was used (57, 58). This special assay setup allowed forward coupling of the CS reaction, leading to the formation of anthranilate, a stable product that exhibits a fluorescence emission maximum at 390 nm and can thus be detected spectrofluorometrically.
Reaction mixtures containing 3 mM MgSO4, 7.5 mM l-Gln, 22.5 mM (NH4)2SO4, 1 mM dithiothreitol (DTT), 10 μM FMN, 80 μM EPSP, 4 μM PbCS, ∼20 μM anthranilate synthase, and various concentrations of CP1 (0.1 to 500 μM) were prepared in 100 mM potassium phosphate (pH 7.6) and pipetted into a 96-well plate (triplicate determinations). After the addition of 500 μM NADPH, the plate was incubated at 37°C for 60 s before recording the fluorescence emission changes at 390 nm (λex = 340 nm) using a Spectramax Gemini XS microplate spectrofluorometer (Molecular Devices, San Jose, CA) for 5 min (data points every 12 s). Initial velocities were extracted from the first 15 data points, and relative rates were plotted as a function of the log (CP1 micromolar concentration), with the standard deviations shown as error bars (triplicate determinations). Using the program GraphPad Prism 5 (GraphPad Software, Inc., San Diego, CA), the data were fitted applying the nonlinear curve fit “log (inhibitor) versus normalized response − variable slope,” which allowed for the determination of IC50s.
Fungal growth conditions.
In this study, five isolates from Paracoccidioides spp. were used, two from P. brasiliensis (Mg14 and Pb18) and three from P. lutzii (Pb01, 8334, and ROSC). These isolates belong to the Medical Mycology Laboratory Collection of Universidade Estadual de Maringá. Fungal yeast cells were maintained by weekly subcultivation in semisolid Fava Netto culture medium containing 4% glucose at 35°C and were used on the seventh day of culture (59).
Animals.
Six-week-old male BALB/c mice bred under specific-pathogen-free conditions at the animal facilities of the Universidade Estadual de Maringá, Brazil, were used. All the procedures were performed according to the regulations of the institutional Ethical Committee for animal experimentation, State University of Maringá, Brazil (approval no. CEP 053/2014). The animals were handled according to the guideline for the care and use of laboratory animals of CONCEA (67).
MICs.
The antifungal activity assays were performed by the broth microdilution method according to guidelines of the Clinical and Laboratory Standards Institute (CLSI; document M27-A3) (60), with modifications. The CP1 concentration ranged from 0.2 to 128 mg/liter, and only RPMI 1640 medium was used as a negative control. Briefly, yeast cell suspensions (0.5 × 105 to 2.5 × 105 CFU/ml) were prepared in RPMI 1640 and diluted 1:2 at the well plate. The MIC plates were incubated at 35°C for 7 days (61). The minimal fungicidal concentrations (MFCs) were determined by subculture from each well onto brain heart infusion (BHI) agar plates incubated at 35°C for 7 days. The MFC was the lowest drug concentration that showed no growth (30). The synergistic interaction between CP1 and itraconazole or amphotericin B was evaluated by microdilution checkerboard (62).
Cytotoxicity evaluation.
The CP1 cytotoxicity evaluation was performed in HeLa, J774, and HUVEC cells using the CellTiter 96 assay (Promega, Madison, WI) based on the reduction of MTS [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium] (63).
Scanning electron microscopy (SEM).
Yeast (Pb18 isolate) was incubated for 72 h at 35°C with CP1 (64 mg/liter). The cells were prepared as previously described (64). Samples were then observed with a Shimadzu SS-550 Super Scan (Shimadzu, Tokyo, Japan) at magnifications of ×1,000 and ×3,000.
Experimental paracoccidioidomycosis and treatment.
The mice were infected through the intratracheal (i.t.) route (1 × 106 viable yeast cells, by trypan blue). After 48 h of infection, the animals were treated intraperitoneally daily with CP1 (5 mg/kg), ITZ (5 mg/kg), or the control for 14 days. The control group was treated with a vehicle (PBS, DMSO [1%], and Pluronic [0.2%]) used for diluting the CP1 compound. Then the mice were euthanized, and organs were aseptically removed for fungal burden determination (CFU per gram) (65). Briefly, the organs were weighed and macerated in PBS. CFU were recovered in supplemented BHI agar and incubated at 37°C for 7 days or until there was no growth. The CFU count was determined, and the results are presented as log10 of the average value ± standard error. These experiments were repeated three independent times.
Histopathological analysis.
Parts of the organs were fixed in PBS–10% formalin, dehydrated, and embedded in paraffin. Five-micrometer sections were stained with hematoxylin and eosin (H&E), and the severity and size of the tissue lesion were examined by microscopy. Pathological changes were analyzed based on the size, morphology, and cell composition of granulomatous lesions and the presence of fungi and inflammatory infiltrates. Morphometric analysis was performed using a Nikon DXM 1200c digital camera (magnifications of ×50 and ×400) and Nikon NIS Elements AR 2.30 software (66). Histopathological analysis was performed on the lungs of all animals. The following three parameters were analyzed: fibrosis, inflammatory infiltration, and fungal cells. In the semiquantitative analysis, each one of the parameters was assigned a score that ranged from 1 to 3, increasing and depending on the degree of tissue damage. When the quantifier was not observed, the score was assigned as 0. The obtained scores were plotted and subjected to statistical analysis.
Statistical analysis.
Data are expressed as the means ± standard deviations (SDs) from at least three independent experiments. Significant differences among means of the variables before and after treatment with CP1 were identified using Student’s t distribution. The data were analyzed using Prism 6.0 software (GraphPad, San Diego, CA). P values of ≤0.05 were considered statistically significant.
Supplementary Material
ACKNOWLEDGMENTS
We thank the Brazilian agencies CAPES, CNPq, and Fundação Araucária (147/14 and 40/16) for financial support and CENAPAD/SP and LNCC for computational facilities. P.M. expresses his gratitude to the Austrian Science Fund for providing financial support through DK Molecular Enzymology (W901).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.01097-18.
REFERENCES
- 1.da Costa MM, da Silva SHM. 2014. Epidemiology, clinical, and therapeutic aspects of paracoccidioidomycosis. Trop Mycol Curr Trop Med Rep 1:138–144. [Google Scholar]
- 2.Bocca AL, Amaral AC, Teixeira MM, Sato PK, Shikanai-Yasuda MA, Felipe MSS. 2013. Paracoccidioidomycosis: eco-epidemiology, taxonomy and clinical and therapeutic issues. Future Microbiol 8:1177–1191. doi: 10.2217/fmb.13.68. [DOI] [PubMed] [Google Scholar]
- 3.Bellissimo-Rodrigues F, Machado AA, Martinez R. 2011. Paracoccidioidomycosis epidemiological features of a 1,000-cases series from a hyperendemic area on the southeast of Brazil. Am J Trop Med Hyg 85:546–550. doi: 10.4269/ajtmh.2011.11-0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coutinho ZF, Silva D, Lazerda M. 2002. Paracocidioidomycosis mortality in Brazil (1980–1995). Cad Saude Publica 18:1441–1454. doi: 10.1590/S0102-311X2002000500037. [DOI] [PubMed] [Google Scholar]
- 5.Martinez R. 2017. New trends in paracoccidioidomycosis epidemiology. J Fungi (Basel) 3:E1. doi: 10.3390/jof3010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shikanai-Yasuda MA, Mendes RP, Colombo AL, Queiroz-Telles F, Kono ASG, Paniago AMM, Nathan A, Valle ACFD, Bagagli E, Benard G, Ferreira MS, Teixeira MM, Silva-Vergara ML, Pereira RM, Cavalcante RS, Hahn R, Durlacher RR, Khoury Z, Camargo ZP, Moretti ML, Martinez R. 2017. Brazilian guidelines for the clinical management of paracoccidioidomycosis. Rev Soc Bras Med Trop 50:715–740. doi: 10.1590/0037-8682-0230-2017. [DOI] [PubMed] [Google Scholar]
- 7.Queiroz-Telles F, Fahal AH, Falci DR, Caceres DH, Chiller T, Pasqualotto AC. 2017. Neglected endemic mycoses. Lancet Infect Dis 17:e367–e377. doi: 10.1016/S1473-3099(17)30306-7. [DOI] [PubMed] [Google Scholar]
- 8.Haanstra JR, Gerding A, Dolga AM, Sorgdrager FJH, Buist-Homan M, Du Toit F, Faber KN, Holzhütter HG, Szöör B, Matthews KR, Snoep JL, Westerhof HV, Bakker BM. 2017. Targeting pathogen metabolism without collateral damage to the host. Sci Rep 7:40406. doi: 10.1038/srep40406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abadio AK, Kioshima ES, Teixeira MM, Martins NF, Maigret B, Felipe MS. 2011. Comparative genomics allowed the identification of drug targets against human fungal pathogens. BMC Genomics 27:12–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bentley R. 1990. The shikimate pathway—a metabolic tree with many branches. Crit Rev Biochem Mol Biol 25:307–384. doi: 10.3109/10409239009090615. [DOI] [PubMed] [Google Scholar]
- 11.Duke SO, Powles SB. 2008. Glyphosate: a once-in-a-century herbicide. Pest Manag Sci 648:319–325. doi: 10.1002/ps.1518. [DOI] [PubMed] [Google Scholar]
- 12.Herrmann KM, Weaver LM. 1999. The shikimate pathway. Annu Rev Plant Physiol Plant Mol Biol 50:473–503. doi: 10.1146/annurev.arplant.50.1.473. [DOI] [PubMed] [Google Scholar]
- 13.Dias MV, Ely F, Palma MS, de Azevedo WFJ, Basso LA, Santos DS. 2007. Chorismate synthase: an attractive target for drug development against orphan diseases. Curr Drug Targets 8:437–444. doi: 10.2174/138945007780058924. [DOI] [PubMed] [Google Scholar]
- 14.Schönbrunn E, Eschenburg S, Shuttleworth WA, Schloss JV, Amrhein N, Evans JN, Kabsch W. 2001. Interaction of the herbicide glyphosate with its target enzyme 5-enolpyruvylshikimate 3-phosphate synthase in atomic detail. Proc Natl Acad Sci U S A 98:1376–1380. doi: 10.1073/pnas.98.4.1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Macheroux P, Schmid J, Amrhein N, Schaller A. 1999. A unique reaction in a common pathway: mechanism and function of chorismate synthase in the shikimate pathway. Planta 207:325–334. doi: 10.1007/s004250050489. [DOI] [PubMed] [Google Scholar]
- 16.Quevillon-Cheruel S, Leulliot N, Meyer P, Graille M, Bremang M, Blondeau K, Sorel I, Poupon A, Janin J, van Tilbeurgh H. 2004. Crystal structure of the bifunctional chorismate synthase from Saccharomyces cerevisiae. J Biol Chem 279:619–625. doi: 10.1074/jbc.M310380200. [DOI] [PubMed] [Google Scholar]
- 17.Rost B. 1999. Twilight zone of protein sequence alignments. Protein Eng 12:85–94. doi: 10.1093/protein/12.2.85. [DOI] [PubMed] [Google Scholar]
- 18.Yang AS, Honig B. 2000. An integrated approach to the analysis and modeling of protein sequences and structures. III. A comparative study of sequence conservation in protein structural families using multiple structural alignments. J Mol Biol 301:691–711. doi: 10.1006/jmbi.2000.3975. [DOI] [PubMed] [Google Scholar]
- 19.Miteva MA, Violas S, Montes M, Gomez D, Tuffery P, Villoutreix BO. 2006. FAF-Drugs: free ADME/tox filtering of compound collections. Nucleic Acids Res 34:W738–W744. doi: 10.1093/nar/gkl065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tapas S, Kumar A, Dhindwal S, Preeti KP. 2011. Structural analysis of chorismate synthase from Plasmodium falciparum: a novel target for antimalarial drug discovery. Int J Biol Macromol 49:767–777. doi: 10.1016/j.ijbiomac.2011.07.011. [DOI] [PubMed] [Google Scholar]
- 21.Cruz RC, Werneck SMC, Oliveira CS, Santos PC, Soares BM, Santos DA, Cisalpino PS. 21 November 2012. Conditions for determining the minimal inhibitory concentration (MIC) of seven antifungal agents against Paracoccidioides brasiliensis by microdilution: influence of different media, incubation times and temperatures. J Clin Microbiol. doi: 10.1128/JCM.02231-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aguirre JPB, Hamid AMR. 2015. Amphotericin B deoxycholate versus liposomal amphotericin B: effects on kidney function. Cochrane Database Syst Rev 23:CD010481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Borges SR, Silva GM, Chambela MC, Oliveira RV, Costa RL, Wanke B, Valle AC. 2014. Itraconazole vs. trimethoprim-sulfamethoxazole: a comparative cohort study of 200 patients with paracoccidioidomycosis. Med Mycol 52:303–310. doi: 10.1093/mmy/myt012. [DOI] [PubMed] [Google Scholar]
- 24.Shikanai-Yasuda MA, Benard G, Higaki Y, Del Negro GM, Hoo S, Vaccari EH, Gryschek RC, Segurado AA, Barone AA, Andrade DR. 2002. Randomized trial with itraconazole, ketoconazole and sulfadiazine in paracoccidioidomycosis. Med Mycol 40:411–417. doi: 10.1080/mmy.40.4.411.417. [DOI] [PubMed] [Google Scholar]
- 25.Roemer T, Krysan DJ. 2014. Antifungal drug development: challenges, unmet clinical needs, and new approaches. Cold Spring Harb Perspect Med 4: a019703. doi: 10.1101/cshperspect.a019703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Macedo PM, Almeida-Paes R, Freitas DFS, Brito-Santos F, Figueiredo-Carvalho MHG, Soares JCA, Freitas AD, Zancopé-Oliveira RM, do Valle ACF. 2017. Hepatic disease with portal hypertension and acute juvenile paracoccidioidomycosis: a report of two cases and literature review. Mycopathologia 182:915–919. doi: 10.1007/s11046-017-0152-6. [DOI] [PubMed] [Google Scholar]
- 27.Laniado-Laborín R, Cabrales-Vargas MN. 2009. Amphotericin B: side effects and toxicity. Rev Iberoam Micol 26:223–227. doi: 10.1016/j.riam.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 28.Abadio AKR, Kioshima ES, Leroux V, Martins NF, Maigret B, Felipe MSS. 2015. Identification of new antifungal compounds targeting thioredoxin reductase of Paracoccidioides genus. PLoS One 10:e0142926. doi: 10.1371/journal.pone.0142926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Batagin MC, Pimentel AL, Biavatti DC, Basso EA, Kioshima ES, Seixas FAV, Gauze GF. 2017. Targeting homoserine dehydrogenase from Paracoccidioides species against systemic fungal infections. Antimicrob Agents Chemother 61:e00165-17. doi: 10.1128/AAC.00165-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johann S, Sá NP, Lima LA, Cisalpino PS, Cota BB, Alves TMA, Siqueira EP, Zani CL. 2010. Antifungal activity of schinol and a new biphenyl compound isolated from Schinus terebinthifolius against the pathogenic fungus Paracoccidioides brasiliensis. Ann Clin Microbiol Antimicrob 9:30. doi: 10.1186/1476-0711-9-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Sá NP, Cisalpino PS, Tavares LC, Espíndsaola L, Pizzolatti MG, Santos PC, de Paula TP, Rosa CA, Souza DG, Santos DA, Johann S. 2015. Antifungal activity of 6-quinolinyl N-oxide chalcones against Paracoccidioides. J Antimicrob Chemother 70:841–845. doi: 10.1093/jac/dku427. [DOI] [PubMed] [Google Scholar]
- 32.Yin N, Ma W, Pei J, Ouyang Q, Tang C, Lai L. 2014. Synergistic and antagonistic drug combinations depend on network topology. PLoS One 9:e93960. doi: 10.1371/journal.pone.0093960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Queiroz-Telles F, Goldani LZ, Schlamm HT, Goodrich JM, Espinel-Ingroff A, Shikanai-Yasuda MA. 2007. An open-label comparative pilot study of oral voriconazole and itraconazole for long-term treatment of paracoccidioidomycosis. Clin Infect Dis 45:1462–1469. doi: 10.1086/522973. [DOI] [PubMed] [Google Scholar]
- 34.Levorato AD, Moris DV, Cavalcante RS, Sylvestre TF, de Azevedo PZ, de Carvalho LR, Mendes RP. 2018. Evaluation of the hepatobiliary system in patients with paracoccidioidomycosis treated with cotrimoxazole or itraconazole. Med Mycol 56:531–540. doi: 10.1093/mmy/myx080. [DOI] [PubMed] [Google Scholar]
- 35.Tobón AM, Agudelo CA, Osorio ML, Alvarez DL, Arango M, Cano LE, Restrepo A. 2003. Residual pulmonary abnormalities in adult patients with chronic paracoccidioidomycosis: prolonged follow-up after itraconazole therapy. Clin Infect Dis 37:898–904. doi: 10.1086/377538. [DOI] [PubMed] [Google Scholar]
- 36.Naranjo TW, Lopera DE, Diaz-Granados LR, Duque JJ, Restrepo AM, Cano LE. 2011. Combined itraconazole-pentoxifylline treatment promptly reduces lung fibrosis induced by chronic pulmonary paracoccidioidomycosis in mice. Pulm Pharmacol Ther 24:81–91. doi: 10.1016/j.pupt.2010.09.005. [DOI] [PubMed] [Google Scholar]
- 37.Marques AF, Silva MB, Juliano MAP, Munhõz JE, Travassos LR, Taborda CP. 2008. Additive effect of P10 immunization and chemotherapy in anergic mice challenged intratracheally with virulent yeasts of Paracoccidioides brasiliensis. Microb Infect 10:1251–1258. doi: 10.1016/j.micinf.2008.07.027. [DOI] [PubMed] [Google Scholar]
- 38.Puerta-Arias JD, Pino-Tamayo PA, Arango JC, Salazar-Peláez LM, González A. 2018. Itraconazole in combination with neutrophil depletion reduces the expression of genes related to pulmonary fibrosis in an experimental model of paracoccidioidomycosis. Med Mycol 56:579–590. doi: 10.1093/mmy/myx087. [DOI] [PubMed] [Google Scholar]
- 39.Desjardins CA, Champion MD, Holder JW, Muszewska A, Goldberg J, Bailao AM, Brigido MM, Ferreira ME, Garcia AM, Grynberg M, Grynberg M, Gujja S, Heiman DI, Henn MR, Kodira CD, León-Narváez H, Longo LV, Ma LJ, Malavazi I, Matsuo AL, Morais FV, Pereira M, Rodríguez-Brito S, Sakthikumar S, Salem-Izacc SM, Sykes SM, Teixeira MM, Vallejo MC, Walter ME, Yandava C, Young S, Zeng Q, Zucker J, Felipe MS, Goldman GH, Haas BJ, McEwen JG, Nino-Vega G, Puccia R, San-Blas G, Soares CM, Birren BW, Cuomo CA. 2011. Comparative genomic analysis of human fungal pathogens causing paracoccidioidomycosis. PLoS Genet 7:e1002345. doi: 10.1371/journal.pgen.1002345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mulder N, Apweiler R. 2007. InterPro and InterProScan: tools for protein sequence classification and comparison. Methods Mol Biol 396:59–70. doi: 10.1007/978-1-59745-515-2_5. [DOI] [PubMed] [Google Scholar]
- 41.Mount DW. 2007. Using the Basic Local Alignment Search Tool (BLAST). CSH Protoc 2007:pdb.top17. doi: 10.1101/pdb.top17. [DOI] [PubMed] [Google Scholar]
- 42.Lobley A, Sadowski MI, Jones DT. 2009. pGenTHREADER and pDomTHREADER: new methods for improved protein fold recognition and superfamily discrimination. Bioinformatics 25:1761–1767. doi: 10.1093/bioinformatics/btp302. [DOI] [PubMed] [Google Scholar]
- 43.Maclean J, Ali S. 2003. The structure of chorismate synthase reveals a novel flavin binding site fundamental to a unique chemical reaction. Structure 11:1499–1511. doi: 10.1016/j.str.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 44.Ahn HJ, Yoon HJ, Lee B, Suh SW. 2004. Crystal structure of chorismate synthase: a novel FMN-binding protein fold and functional insights. J Mol Biol 336:903–915. doi: 10.1016/j.jmb.2003.12.072. [DOI] [PubMed] [Google Scholar]
- 45.Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen MY, Pieper U, Sali A. 2006. Comparative protein structure modeling using Modeller. Curr Protoc Bioinformatics Chapter5:Unit 5.6. doi: 10.1002/0471250953.bi0506s15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mackerell AD, Feig M, Brooks CL. 2004. Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J Comput Chem 25:1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 47.Irwin JJ, Sterling T, Mysinger MM, Bolstad ES, Coleman RG. 2012. ZINC: a free tool to discover chemistry for biology. J Chem Inf Model 52:1757–1768. doi: 10.1021/ci3001277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zoete V, Cuendet MA, Grosdidier A, Michielin O. 2011. SwissParam: a fast force field generation tool for small organic molecules. J Comput Chem 32:2359–2368. doi: 10.1002/jcc.21816. [DOI] [PubMed] [Google Scholar]
- 49.Neese F. 2012. The ORCA program system. Wiley Interdiscip Rev Comput Mol Sci 2:73–78. doi: 10.1002/wcms.81. [DOI] [Google Scholar]
- 50.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. 2005. Scalable molecular dynamics with NAMD. J Comput Chem 26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Homem DP, Flores R, Tosqui P, Rozada TC, Basso EA, Gasparotto A, Seixas FAV. 2013. Homology modeling of dihydrofolate reductase from T. gondii bonded to antagonists: molecular docking and molecular dynamics simulations. Mol Biosyst 9:1308–1315. doi: 10.1039/c3mb25530a. [DOI] [PubMed] [Google Scholar]
- 52.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. 2009. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem 30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wolf LK. 2009. New software and websites for the chemical enterprise. Chem Eng News 87:32. doi: 10.1021/cen-v087n005.p032. [DOI] [Google Scholar]
- 54.Thomsen R, Christensen MH. 2006. MolDock: a new technique for high-accuracy molecular docking. J Med Chem 49:3315–3321. doi: 10.1021/jm051197e. [DOI] [PubMed] [Google Scholar]
- 55.Gilson MK, Liu T, Baitaluk M, Nicola G, Hwang L, Chong J. 2016. BindingDB in 2015: a public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res 44:D1045–D1053. doi: 10.1093/nar/gkv1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schomburg I, Chang A, Placzek S, Sohngen C, Rother M, Lang M, Munaretto C, Ulas S, Stelzer M, Grote A, Scheer M, Schomburg D. 2013. BRENDA in 2013: integrated reactions, kinetic data, enzyme function data, improved disease classification: new options and contents in BRENDA. Nucleic Acids Res 41:D764–D772. doi: 10.1093/nar/gks1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schaller A, Windhofer V, Amrhein N. 1990. Purification of chorismate synthase from a cell culture of the higher plant Corydalis sempervirens Pers. Arch Biochem Biophys 282:437–442. doi: 10.1016/0003-9861(90)90141-K. [DOI] [PubMed] [Google Scholar]
- 58.Fitzpatrick TB, Killer P, Thomas RM, Jelesarov I, Amrhein N, Macheroux P. 2001. Chorismate synthase from the hyperthermophile Thermotoga maritima combines thermostability and increased rigidity with catalytic and spectral properties similar to mesophilic counterparts. J Biol Chem 276:18052–18059. doi: 10.1074/jbc.M100867200. [DOI] [PubMed] [Google Scholar]
- 59.Fava-Netto C, Vegas VS, Sciannaméa IM, Guarnieri DB. 1969. The polysaccharidic antigen from Paracoccidioides brasiliensis. Study of the time of cultivation necessary for the preparation of the antigen. Rev Inst Med Trop 11:177–181. [PubMed] [Google Scholar]
- 60.Clinical and Laboratory Standards Institute. 2008. Reference method for broth dilution antifungal susceptibility testing of yeast, 3rd ed Approved standard CLSI M27-A3 Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 61.Cruz RC, Werneck SMC, Oliveira CS, Santos PC, Soares BM, Santos DA, Cisalpino PS. 2013. Influence of different media, incubation times, and temperatures for determining the MICs of seven antifungal agents against Paracoccidioides brasiliensis by microdilution. J Clin Microbiol 51:436–443. doi: 10.1128/JCM.02231-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hemaiswarya S, Kruthiventi AK, Doble M. 2008. Synergism between natural products and antibiotics against infectious diseases. Phytomedicine 15:639–652. doi: 10.1016/j.phymed.2008.06.008. [DOI] [PubMed] [Google Scholar]
- 63.Capoci IRG, Bonfim-Mendonça PS, Arita GS, Pereira RRA, Consolaro MEL, Bruschi ML, Negri M, Svidzinski TIE. 2015. Propolis is an efficient fungicide and inhibitor of biofilm production by vaginal Candida albicans. Evid Based Complement Alternat Med 1:1–9. doi: 10.1155/2015/287693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sakita KM, Faria DR, Silva EMD, Tobaldini-Valério FK, Kioshima ES, Svidzinski TI, Bonfim-Mendonça PS. 2017. Healthcare workers’ hands as a vehicle for the transmission of virulent strains of Candida spp. A virulence factor approach. Microb Pathog 113:225–232. doi: 10.1016/j.micpath.2017.10.044. [DOI] [PubMed] [Google Scholar]
- 65.Kioshima ES, Aliperti F, Maricato JT, Mortara RA, Bagagli E, Mariano M, Lopes JD. 2011. A synthetic peptide selectively kills only virulent Paracoccidioides brasiliensis yeasts. Microbes Infect 13:251–260. doi: 10.1016/j.micinf.2010.10.019. [DOI] [PubMed] [Google Scholar]
- 66.Costa TA, Bazan SB, Feriotti C, Araújo EF, Bassi Ê, Loures FV, Calich VL. 2013. Pulmonary paracoccidioidomycosis IL-10 deficiency leads to increased immunity and regressive infection without enhancing tissue pathology. PLoS Negl Trop Dis 7:2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ministério da Ciência, Tecnologia e Inovação, Conselho Nacional de Controle de Experimentação Animal (CONCEA), Brazil. 2016. Guia brasileiro de produção, manutenção ou utilização de animais em atividades de ensino ou pesquisa científica. https://www.mctic.gov.br/mctic/export/sites/institucional/legislacao/Arquivos/Anexo_Res_Normativa_Concea_33_2016.pdf. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.