

Abstract
Background/Aims
Indoxyl sulfate (IS) is a uremic toxin and an important causative factor in the progression of chronic kidney disease. Recently, paricalcitol (19-nor-1,25-dihydroxyvitamin D2) was shown to exhibit protective effects in kidney injury. Here, we investigated the effects of paricalcitol treatment on IS-induced renal tubular injury.
Methods
The fluorescent dye 2ʹ,7ʹ-dichlorofluorescein diacetate was used to measure intracellular reactive oxygen species (ROS) following IS administration in human renal proximal tubular epithelial (HK-2) cells. The effects of IS on cell viability were determined using MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assays and levels of apoptosis-related proteins (Bcl-2-associated protein X [Bax] and B-cell lymphoma 2 [Bcl-2]), nuclear factor-κB (NF-κB) p65, and phosphorylation of mitogen-activated protein kinase (MAPK) and protein kinase B (Akt) were determined by semiquantitative immunoblotting. The promoter activity of NF-κB was measured by luciferase assays and apoptosis was determined by f low cytometry of cells stained with f luorescein isothiocyanate-conjugated Annexin V protein.
Results
IS treatment increased ROS production, decreased cell viability and induced apoptosis in HK-2 cells. IS treatment increased the expression of apoptosis-related protein Bax, decreased Bcl-2 expression, and activated phosphorylation of MAPK, NF-κB p65, and Akt. In contrast, paricalcitol treatment decreased Bax expression, increased Bcl-2 expression, and inhibited phosphorylation of MAPK, NF-κB p65, and Akt in HK-2 cells. NF-κB promoter activity was increased following IS, administration and was counteracted by pretreatment with paricalcitol. Additionally, flow cytometry analysis revealed that IS-induced apoptosis was attenuated by paricalcitol treatment, which resulted in decreased numbers of fluorescein isothiocyanate-conjugated Annexin V positive cells.
Conclusions
Treatment with paricalcitol inhibited IS-induced apoptosis by regulating MAPK, NF-κB, and Akt signaling pathway in HK-2 cells.
Keywords: Apoptosis, Proximal tubular epithelial cell, Indican, Paricalcitol, Signal transduction
INTRODUCTION
The accumulation of indoxyl sulfate (IS), a protein-bound uremic toxin, accelerates the progression of chronic kidney disease and renal failure involving progressive tubulointerstitial fibrosis and glomerular sclerosis [1-3]. IS is an end metabolite of tryptophan in dietary proteins. Tryptophan is cleaved to indole by tryptophanase present in intestinal bacteria, such as Escherichia coli. The indole is transported to the liver through the portal vein, where it is oxidized to indoxyl by cytochrome p450 2E1 and sulfated by sulfotransferases 1A1, which is normally excreted in urine mainly via direct secretion by organic anion transporters 1, 3, and 4 in proximal tubular cells [2,4-6]. However, under conditions of reduced renal function, IS is not excreted in the urine and accumulates in the serum due to its reduced renal clearance [7]. Accumulated IS is then incorporated into the basolateral membrane of renal proximal tubular cells by mediating organic anion transporter 1 and 3 [6,8].
IS stimulates renal expression of fibrotic genes such as transforming growth factor-β1 and a-smooth muscle actin, and inflammatory genes, including interleukin-6, interleukin-1β, tumor necrosis factor-a, and monocyte chemotactic protein-1 [9-11]. Importantly, IS induces intracellular production of reactive oxygen species (ROS) through nicotinamide adenine dinucleotide phosphate oxidase and extracellular O2− formation [12]. IS-induced ROS production activates nuclear factor-κB (NF-κB), c-Jun N-terminal domain kinase (JNK), signal transducer and activator of transcription 3, and p53, resulting in increased oxidative stress in tubular epithelia cells [13,14].
ROS production in renal tubular epithelia cells induces cell death via both apoptosis and necrosis dependent upon concentration and exposure time. High IS concentrations induce cell death in renal proximal tubular cells through the apoptosis pathway [15,16], and IS induces oxidative stress, promoting free-radical production and disturbing antioxidant enzymes, such as superoxide dismutase [10,17,18].
Paricalcitol is a synthetically manufactured analog of calcitriol, the biologically active form of vitamin D. Paricalcitol includes modifications to the D2 side chain and the A (19-nor-1,25(OH)2D2) ring, enabling binding to the vitamin D receptor [19] and resulting in attenuation of renal fibrosis [20,21]. The therapeutic effects of paricalcitol treatment may be attributed to its anti-inflammatory and anti-fibrotic effects in experimentally induced kidney diseases [22-24]. Additionally, decreased vitamin D concentration in renal-disease patients leads to increased cardiovascular mortality, as well as renal disease progression [25]. In the study, we determined the capability of paricalcitol to exert beneficial effects on IS-induced kidney tubule-cell injury, as well as the underlying molecular mechanisms associated with its anti-apoptotic effects.
METHODS
Cell culture and reagents
Human renal proximal tubular epithelial cells (HK-2, ATCC, Manassas, VA, USA) were cultured and passaged every 3 to 4 days in 100-mm dishes containing Dulbecco’s modified Eagle’s medium-F-12 medium (Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin (Sigma-Aldrich). Cells were incubated in a humidified atmosphere of 5% CO2 and 95% air at 37°C for 24 hours and sub-cultured at 70% to 80% confluence. For experiments, HK-2 cells were plated onto 60-mm dishes in medium containing 10% fetal bovine serum for 24 hours, and cells were then switched to Dulbecco’s modified Eagle’s medium F12 serum-free media for 24 hours. Cells were harvested at the end of treatment for further analysis. IS was obtained from Sigma-Aldrich (Steinheim, Germany), and paricalcitol was obtained from Abbott Laboratories (North Chicago, IL, USA). PD98059 (a mitogen-activated protein kinase [MAPK]/extracellular signal-related kinase [Erk] inhibitor), SP600125 (a specific Jnk inhibitor), and SB203580 (a p38 MAPK inhibitor) were obtained from Calbiochem (San Diego, CA, USA). Ly294 (a phosphoinositide 3-kinase/protein kinase B [Akt] inhibitor) and N-acetyl-L-cysteine were obtained from Sigma-Aldrich (Steinheim). Bay 11-7082 was obtained from Biomol (Plymouth Meeting, PA, USA).
MTT assay
Viability of HK-2 cells was determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. HK-2 cells were sub-cultured in 96-well plates at an initial density of 5 × 104 cells/mL. Cells were incubated with fresh medium containing 0, 0.3, 0.5, 1, or 2 mM IS for 24 hours. At the end of the experimental period, MTT (Sigma-Aldrich) was added into each well to a final concentration of 0.5 mg/mL and subsequently incubated for 4 hours at 37°C. Supernatants were removed by aspiration, and dimethyl sulfoxide was added to solubilize the precipitated dyes. Absorbance was measured at 570 nm, and cell viability was expressed as the fraction of surviving cells relative to untreated controls.
Intracellular ROS levels
HK-2 cells were cultured in 48-well plates until reaching 60 % to 70 % confluence. Cells were incubated with fresh medium containing 0, 0.3, 0.5, 1, or 2 mM IS for 24 hours. At the end of the experimental period, cells were preloaded with 10 μM 2ʹ,-7ʹ-dichlorofluorescein diacetate (DCF-DA, Molecular Probes, Eugene, OR, USA) for 30 minutes at 37°C. Fluorescence intensity was analyzed using a fluorescence reader (Fluoroskan Ascent FL, Lab systems, Helsinki, Finland) with a 485-nm excitation and 538-nm emission filter. Additional HK-2 cells were cultured on a 6-well plate for DCF-DA staining. Cells were incubated with 1 mM IS for 24 hours and washed twice with Hank’s balanced salt solution, then incubated with Hank’s balanced salt solution (without phenol red) containing DCF-DA for 30 minutes at 37°C in the dark. Images were obtained with a fluorescence microscope (Nikon, Tokyo, Japan).
Nuclear extracts preparation
For nuclear extracts, cells were lysed using NE-PER nuclear-extraction reagent (Pierce Biotechnology, Rockford, IL, USA) according to manufacturer’s protocol as described previously [23].
Western blot analysis
Western blots were performed as described previously [23]. Cell lysates were prepared with RIPA (radioimmunoprecipitation assay) buffer (150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 50 mM tris-HCl, pH 7.5, 2 mM EDTA, 5 mM NaF) containing protease inhibitors. Antibodies, including anti-Erk, anti-phosphorylated Erk (p-Erk), anti-B-cell lymphoma 2 (Bcl-2), anti-Bcl-2-associated protein X (Bax), anti-NF-κB p65 (Cell Signaling Technology, Beverly, MA,USA), anti-Jnk, anti-phosphorylated Jnk (p-Jnk), anti-IκBa (Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-phosphorylated p38 MAPK (p-p38 MAPK, New England Biolabs, Ipswich, MA, USA), histone H3 (Cell Signaling Technology), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH, Sigma-Aldrich), were diluted in blocking buffer and incubated with the blots overnight at 4°C. The bound antibodies were detected with a 1:2,500 dilution of horseradish peroxidase-conjugated secondary antibody according to the instructions provided with the enhanced chemiluminescence kit (Amersham, Franklin Lakes, NJ, USA).
NF-κB-promotor activity
The transcriptional regulation of NF-κB was examined by transient transfection of a NF-κB-promoter luciferase-reporter construct (pGL3-NF-κB). HK-2 cells (5 × 105) were seeded and grown until they reached 60% to 70% confluence, followed by transfection of pGL3-NF-κB wild-type and pGL3-empty into the cells using FuGENE HD reagent (Promega, Durham, NC, USA) according to manufacturer protocol. The pRL-null plasmid encoding Renilla luciferase was included in all samples to monitor transfection efficiency. At 24 hours post-transfection, the levels of firefly and Renilla luciferase activity were measured sequentially from a single sample using the Dual-Glo Luciferase Assay system (Promega). Firefly luciferase activity was normalized to Renilla activity and the relative amount of luciferase activity in the untreated cells.
Flow cytometry
An Annexin V FLUOS staining kit (Sigma-Aldrich) was used to measure the level of Annexin V binding according to manufacturer instructions. Briefly, after treatment with 0, 0.5, or 1 mM IS for 24 hours, HK-2 cells were harvested and washed twice with pre-cooled phosphate-buffered saline and resuspended in a binding buffer containing Annexin V. After incubation in the dark for 15 minutes, cells were analyzed by flow cytometry (Becton-Dickinson, San Jose, CA, USA). Several controls were used to optimize the instrument settings and determine the gating for the Windows-based platform.
Statistical analysis
Results are presented as the mean ± standard error of the mean of three individual experiments. Differences were analyzed by analysis of variance with post hoc comparison. Statistical significance of differences was accepted at the level of p < 0.05.
RESULTS
ROS production and cytotoxicity
IS treatment (0, 0.3, 0.5, 1, and 2 mM) for 24 hours decreased HK-2 cell viability in a concentration-dependent manner as determined by MTT assay (Fig. 1A). IS-induced decrease in cell viability, which was attenuated by paricalcitol pretreatment (Fig. 1B). ROS formation was detected using the ROS-sensitive fluorescent dye DCF-DA in HK-2 cells. The level of intracellular ROS increased progressively after incubation with 0 to 2 mM IS for 24 hours, whereas pretreatment with paricalcitol attenuated IS-induced ROS production (Fig. 1C-1G).
Figure 1.
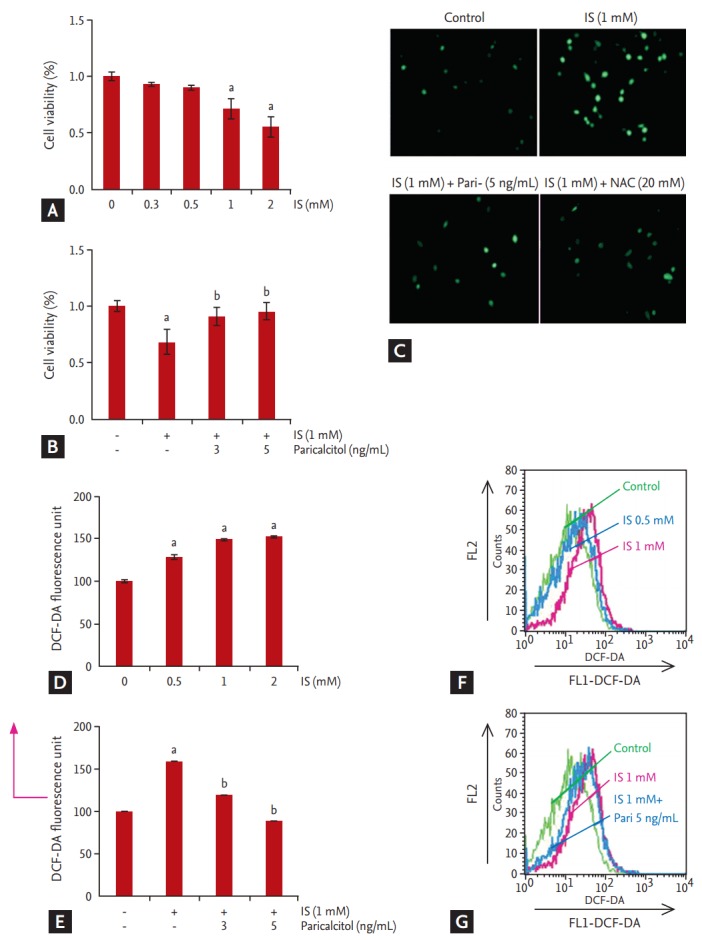
Effects of paricalcitol on cell viability and reactive oxidative stress formation in indoxyl sulfate (IS)-treated human renal proximal tubular epithelial (HK-2) cells. HK-2 cells were treated with IS at various concentrations, and after 24 hours, cell viability was assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. (A) IS exposure indicated dose-dependent decreases in cell viability, (B) which was attenuated by paricalcitol treatment. (C-G) IS caused a dose-dependent increase in 2ʹ,7ʹ-dichlorof luorescein diacetate f luorescence following a 24 hours incubation, whereas treatment with paricalcitol attenuated increased reactive oxidative stress production. Results are presented as the mean ± standard error of the mean of three individual experiments. DCF-DA, 2ʹ,-7ʹ-dichlorof luorescein diacetate; FL1, f luorescent light 1; FL2, fluorescent light 2. ap < 0.05 vs. control, bp < 0.05 vs. IS-treated HK-2 cells.
Cell apoptosis
The expression of pro-apoptotic Bax and anti-apoptotic Bcl-2 was measured by immunoblotting. Incubation with 1 mM IS for 3, 6, 12, or 24 hours increased the ratio of Bax/Bcl-2 expression (Fig. 2A). Additionally, IS-induced increase in the Bax/Bcl-2 ratio in HK-2 cells, which was attenuated by pretreatment with 5 ng/mL paricalcitol for 1 hour (Fig. 2B). Furthermore, HK-2 cells treated with IS exhibited increased Annexin V positive staining, which was attenuated by pretreatment with 5 ng/mL paricalcitol (Fig. 2C and 2D).
Figure 2.
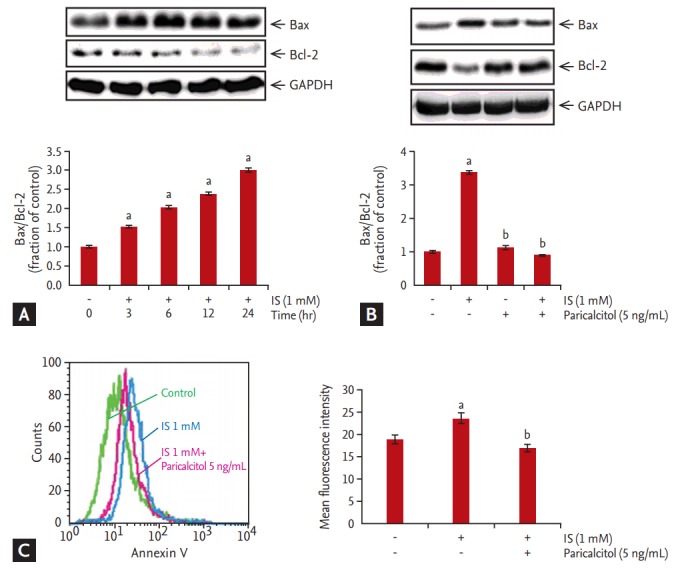
Effect of paricalcitol treatment on expression of pro-/anti-apoptotic Bcl-2-associated protein X (Bax)/B-cell lymphoma 2 (Bcl-2) in indoxyl sulfate (IS)-treated human renal proximal tubular epithelial (HK-2) cells. (A) Incubation with IS for 3, 6, 12, or 24 hours increased the ratio of Bax/Bcl-2 expression. (B) The increased Bax/Bcl-2 ratio was counteracted by treatment with paricalcitol (5 ng/mL). (C) HK-2 cells treated with 1 mM IS for 24 hours exhibited a progressive increase in Annexin V positive staining, which was counteracted by paricalcitol treatment. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. ap < 0.05 vs. control, bp < 0.05 vs. IS-treated HK-2 cells.
MAPK and Akt expression
The expression of p-Erk, p-Jnk, and p-p38 was increased after incubation of HK-2 cells with 1 mM IS for 0.5, 1, 2, or 4 hours. Akt expression was also increased in HK-2 cells incubated with 1 mM IS for 2, 4, 8, and 12 hours. MAPK phosphorylation was peaked at 2 hours after IS treatment (Fig. 3A), whereas p-Akt levels peaked at 8 hours after IS treatment (Fig. 3B). In contrast, total Erk, Jnk, p38, and Akt expression was not affected by IS treatment. To investigate the effects of paricalcitol pretreatment on phosphorylation of MAPK and Akt, HK-2 cells were co-treated with IS and paricalcitol. IS-induced increases in MAPK phosphorylation and p-Akt levels in HK-2 cells, which was attenuated by pretreatment with 5 ng/mL paricalcitol for 1 hour (Fig. 4).
Figure 3.

Effects of indoxyl sulfate treatment on extracellular signal-related kinase 1/2 (Erk1/2), c-Jun N-terminal domain kinase (Jnk), p38, and Akt phosphorylation. (A) Levels of phospho-Erk, phospho-Jnk, phospho-p38, phosphorylated mitogen-activated protein kinase, and (B) phospho-Akt increased significantly following treatment with 1 mM indoxyl sulfate. GAPDH, glyceraldehyde-3-phosphate dehydrogenase; IS, indoxyl sulfate. ap < 0.05 vs. control.
Figure 4.

Effects of paricalcitol treatment on mitogen-activated protein kinase- and Akt-signaling pathway activation in indoxyl sulfate-treated human renal proximal tubular epithelial (HK-2) cells. (A) Levels of phospho-extracellular signal-related kinase 1/2 (phospho-Erk1/2), phospho-c-Jun N-terminal domain kinase (phospho-Jnk), phospho-p38, and (B) phospho-Akt in HK-2 cells incubated with indoxyl sulfate after pretreatment with paricalcitol. Increased levels of phospho-Erk1/2, phospho-Jnk, phospho-p38, and phospho-Akt were attenuated by paricalcitol treatment. GAPDH, glyceraldehyde-3-phosphate dehydrogenase; IS, indoxyl sulfate. ap < 0.05 vs. control, bp < 0.05 vs. indoxyl sulfate-treated HK-2 cells.
NF-κB expression
We then examined changes in NF-κB p65-subunit levels in nuclear extracts of HK-2 cells incubated with 1 mM IS. The expression of p65 subunits began to increase after a 1 hour incubation with IS, whereas total cytoplasmic IκBa expression began to decrease at 1 hour, continued to decrease at 2 hours, then remained stationary for at least 4 hours (Fig. 5A).
Figure 5.

Effect of paricalcitol treatment on nuclear factor-κB (NF-κB) p65 subunit in indoxyl sulfate-treated human renal proximal tubular epithelial (HK-2) cells. (A) NF-κB p65-subunit levels in nuclear extracts of HK-2 cells incubated with indoxyl sulfate. (B-D) Treatment with the inhibitors PD98059, SB203580, SP600125, Bay 11-7082, Ly294, or paricalcitol attenuated nuclear NF-κB p65-subunit levels. Cells were transiently transfected with 1 μg pGL3-NF-κB-reporter construct and incubated with varying concentrations of indoxyl sulfate for 24 hours. (E) HK-2 cells transfected with pGL3-NF-κB showed dose-dependent increases in NF-κB-promoter activity following indoxyl sulfate treatment. (F) Pretreatment of pGL3-NF-κB transfected HK-2 cells with 5 ng/mL paricalcitol exhibited attenuated indoxyl sulfate-induced increases in NF-κB-promoter activity. GAPDH, glyceraldehyde-3-phosphate dehydrogenase; IS, indoxyl sulfate. ap < 0.05 vs. control, bp < 0.05 vs. indoxyl sulfate-treated HK-2 cells.
The effects of specific chemical inhibitors, such as those toward Erk (PD98059), Jnk (SP600125), and p38 (SB203580), are shown in Fig. 5B. Treatment of HK-2 cells with PD98059, SP600125, or SB203580 attenuated IS-induced levels of nuclear NF-κB p65 subunit. As shown in Fig. 5C, Bay 11-7082 (NF-κB inhibitor) and Ly294 (Akt inhibitor) noticeably reduced levels of nuclear NF-κB p65 subunit. These findings suggested that MAPK and Akt activation are involved in IS-induced NF-κB nuclear activation. The IS-induced activation of NF-κB p65 subunit in nuclear extracts of HK-2 cells was also attenuated by pretreatment with 5 ng/mL paricalcitol for 1 hour (Fig. 5D).
HK-2 cells transfected with pGL3-NF-κB showed ~2-fold increases in NF-κB-promoter activity after treatment with 1 mM IS (Fig. 5E). However, as shown in Fig. 5F, HK-2 cells treated with 5 ng/mL paricalcitol before adding IS showed decreases in NF-κB-promoter activity.
DISCUSSION
IS induces nephrotoxic kidney injury by increased production of ROS and induction of inflammatory and fibrotic gene expression [26,27]. In this study, we showed that treatment of proximal tubular HK-2 cells with IS enhanced ROS production and led to dose-dependent cell death. Accordingly, IS treatment led to phosphorylation of MAPK and Akt and subsequent initiation of their signaling pathway, resulting in NF-κB nuclear translocation. However, following paricalcitol treatment, we observed reduced MAPK and Akt phosphorylation and decreased NF-κB activation.
Although some studies demonstrated that altered IS levels promote antioxidant functions under normal physiological conditions, including those catalyzed by superoxide dismutase [28-30], high concentration of IS induced ROS production. Miyamoto et al. [31] reported that high serum IS levels could function as a promoter of pro-oxidant activity due to enhanced oxidative stress, while it might also function as a promoter of antioxidant activity under normal physiological conditions. Our study showed that high IS concentrations induced ROS production and possibly initiated apoptosis in HK-2 cells. We also observed that IS-induced oxidative stress enhanced the phosphorylation of Erk1/2-, Jnk-, p38-, and Akt-signaling pathways. Other studies reported a role for p38- and MAPK-signaling pathways in apoptosis, as well as ROS-induced apoptosis in kidney epithelial cells [32,33]. Here, we showed that paricalcitol pretreatment attenuated IS-induced increases in p-Erk1/2, p-Jnk, p-p38, and p-Akt levels. Furthermore, treatment with the inhibitors PD98059, SB203580, SP600125, and Ly294002 attenuated nuclear levels of NF-κB p65 subunit. These findings suggest the involvement of MAPK- and Akt-signal pathway in NF-κB translocation in HK-2 cells. Activation of NF-κB pathways promote initiation of cell death signal and pro-inflammatory responses [34]. Here, we observed that IS treatment increased nuclear translocation of the NF-κB p65 subunit and increased NF-κB-promoter activity in HK-2 cells, whereas paricalcitol pretreatment attenuated this activity. Additionally, paricalcitol treatment rescued IS-treated apoptotic HK-2 cells from NF-κB-dependent cell death through inactivation of the MAPK and Akt pathways.
Apoptosis describes programmed cell death initiated by specific signaling pathways. ROS is capable of damaging kidney tubular epithelial cells and promoting formation of kidney disease through the activation of apoptosis pathways, such as the mitochondria-mediated intrinsic cell-death pathway. Mitochondria-mediated apoptosis is regulated by Bcl-2 family proteins, which can be classified into two groups: pro-apoptotic proteins; such as Bax, Bad, and Bid; and anti-apoptotic proteins, such as Bcl-2 and Bcl-XL. Bax resides in the cytosol under normal circumstances; however, ROS-mediated Bax activation initiates apoptosis by altering mitochondrial membrane permeability [35].
Our results indicated that treatment of HK-2 cells with IS significantly upregulated Bax expression and decreased Bcl-2 expression. We also showed that IS-treated HK-2 cells exhibited significant progressive increases in Annexin V positive staining, indicating induction of apoptosis. We were subsequently able to modulate Bax and Bcl-2 expression by pretreatment with paricalcitol. These findings suggest that pretreatment of HK-2 cells with paricalcitol contributes to the inhibition of IS-induced apoptosis, and paricalcitol prevents initiation of mitochondria-mediated apoptosis triggered by IS through regulation of Bcl-2 and Bax.
In summary, IS induces apoptosis in renal tubular epithelial cells via excessive intracellular ROS and subsequent activation of the NF-κB-signaling pathway through activation of the MAPK and Akt pathways. Paricalcitol treatment attenuates IS-induced apoptosis by regulating MAPK, Akt, and NF-κB signaling pathway in HK-2 cells.
KEY MESSAGE
1. Indoxyl sulfate induces human proximal tubular cell apoptosis by a production of reactive oxygen species.
2. Paricalcitol inhibits indoxyl sulfate-induced apoptosis in human renal proximal tubular epithelial (HK-2) cells.
3. Inhibition effect by paricalcitol of indoxyl sulfate-induced apoptosis is regulate by mitogen-activated protein kinase, nuclear factor-κB, and Akt signaling pathway in HK-2 cells.
Acknowledgments
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2014R1A1A2008333), by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and future Planning (2016R1A2B4007870), by the Pioneer Research Center Program through the National Research Foundation of Korea funded by the Ministry of Science, ICT &Future Planning (2014M3C1A3053036), by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health and Welfare, Republic of Korea (grant number: HI14C2084), and by the Bio & Medical Technology Development Program of the NRF funded by the Korean government, MSIP (2017M3A9E8023001).
Footnotes
No potential conflict of interest relevant to this article was reported.
REFERENCES
- 1.Mutsaers HA, Stribos EG, Glorieux G, Vanholder R, Olinga P. Chronic kidney disease and fibrosis: the role of uremic retention solutes. Front Med (Lausanne) 2015;2:60. doi: 10.3389/fmed.2015.00060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ellis RJ, Small DM, Vesey DA, et al. Indoxyl sulphate and kidney disease: causes, consequences and interventions. Nephrology (Carlton) 2016;21:170–177. doi: 10.1111/nep.12580. [DOI] [PubMed] [Google Scholar]
- 3.Vanholder R, Schepers E, Pletinck A, Nagler EV, Glorieux G. The uremic toxicity of indoxyl sulfate and p-cresyl sulfate: a systematic review. J Am Soc Nephrol. 2014;25:1897–1907. doi: 10.1681/ASN.2013101062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Niwa T. Uremic toxicity of indoxyl sulfate. Nagoya J Med Sci. 2010;72:1–11. [PMC free article] [PubMed] [Google Scholar]
- 5.Ito S, Yoshida M. Protein-bound uremic toxins: new culprits of cardiovascular events in chronic kidney disease patients. Toxins (Basel) 2014;6:665–678. doi: 10.3390/toxins6020665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Enomoto A, Takeda M, Tojo A, et al. Role of organic anion transporters in the tubular transport of indoxyl sulfate and the induction of its nephrotoxicity. J Am Soc Nephrol. 2002;13:1711–1720. doi: 10.1097/01.asn.0000022017.96399.b2. [DOI] [PubMed] [Google Scholar]
- 7.Niwa T, Ise M. Indoxyl sulfate, a circulating uremic toxin, stimulates the progression of glomerular sclerosis. J Lab Clin Med. 1994;124:96–104. [PubMed] [Google Scholar]
- 8.Taki K, Nakamura S, Miglinas M, Enomoto A, Niwa T. Accumulation of indoxyl sulfate in OAT1/3-positive tubular cells in kidneys of patients with chronic renal failure. J Ren Nutr. 2006;16:199–203. doi: 10.1053/j.jrn.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 9.Shimizu H, Bolati D, Adijiang A, et al. NF-κB plays an important role in indoxyl sulfate-induced cellular senescence, fibrotic gene expression, and inhibition of proliferation in proximal tubular cells. Am J Physiol Cell Physiol. 2011;301:C1201–C1212. doi: 10.1152/ajpcell.00471.2010. [DOI] [PubMed] [Google Scholar]
- 10.Vaziri ND, Oveisi F, Ding Y. Role of increased oxygen free radical activity in the pathogenesis of uremic hypertension. Kidney Int. 1998;53:1748–1754. doi: 10.1046/j.1523-1755.1998.00947.x. [DOI] [PubMed] [Google Scholar]
- 11.Jofre R, Rodriguez-Benitez P, Lopez-Gomez JM, Perez-Garcia R. Inflammatory syndrome in patients on hemodialysis. J Am Soc Nephrol. 2006;17:S274–S280. doi: 10.1681/ASN.2006080926. [DOI] [PubMed] [Google Scholar]
- 12.Gelasco AK, Raymond JR. Indoxyl sulfate induces complex redox alterations in mesangial cells. Am J Physiol Renal Physiol. 2006;290:F1551–F1558. doi: 10.1152/ajprenal.00281.2004. [DOI] [PubMed] [Google Scholar]
- 13.Shimizu H, Bolati D, Adijiang A, et al. Senescence and dysfunction of proximal tubular cells are associated with activated p53 expression by indoxyl sulfate. Am J Physiol Cell Physiol. 2010;299:C1110–C1117. doi: 10.1152/ajpcell.00217.2010. [DOI] [PubMed] [Google Scholar]
- 14.Ito S, Osaka M, Higuchi Y, Nishijima F, Ishii H, Yoshida M. Indoxyl sulfate induces leukocyte-endothelial interactions through up-regulation of E-selectin. J Biol Chem. 2010;285:38869–38875. doi: 10.1074/jbc.M110.166686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang WJ, Chang CH, Sun MF, Hsu SF, Weng CS. DPP-4 inhibitor attenuates toxic effects of indoxyl sulfate on kidney tubular cells. PLoS One. 2014;9:e93447. doi: 10.1371/journal.pone.0093447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim SH, Yu MA, Ryu ES, Jang YH, Kang DH. Indoxyl sulfate-induced epithelial-to-mesenchymal transition and apoptosis of renal tubular cells as novel mechanisms of progression of renal disease. Lab Invest. 2012;92:488–498. doi: 10.1038/labinvest.2011.194. [DOI] [PubMed] [Google Scholar]
- 17.Himmelfarb J, Stenvinkel P, Ikizler TA, Hakim RM. The elephant in uremia: oxidant stress as a unifying concept of cardiovascular disease in uremia. Kidney Int. 2002;62:1524–1538. doi: 10.1046/j.1523-1755.2002.00600.x. [DOI] [PubMed] [Google Scholar]
- 18.Vaziri ND, Dicus M, Ho ND, Boroujerdi-Rad L, Sindhu RK. Oxidative stress and dysregulation of superoxide dismutase and NADPH oxidase in renal insufficiency. Kidney Int. 2003;63:179–185. doi: 10.1046/j.1523-1755.2003.00702.x. [DOI] [PubMed] [Google Scholar]
- 19.Tan X, Wen X, Liu Y. Paricalcitol inhibits renal inflammation by promoting vitamin D receptor-mediated sequestration of NF-kappaB signaling. J Am Soc Nephrol. 2008;19:1741–1752. doi: 10.1681/ASN.2007060666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nolan KA, Brennan EP, Scholz CC, et al. Paricalcitol protects against TGF-β1-induced fibrotic responses in hypoxia and stabilises HIF-a in renal epithelia. Exp Cell Res. 2015;330:371–381. doi: 10.1016/j.yexcr.2014.07.034. [DOI] [PubMed] [Google Scholar]
- 21.He W, Kang YS, Dai C, Liu Y. Blockade of Wnt/β-catenin signaling by paricalcitol ameliorates proteinuria and kidney injury. J Am Soc Nephrol. 2011;22:90–103. doi: 10.1681/ASN.2009121236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park JW, Bae EH, Kim IJ, et al. Paricalcitol attenuates cyclosporine-induced kidney injury in rats. Kidney Int. 2010;77:1076–1085. doi: 10.1038/ki.2010.69. [DOI] [PubMed] [Google Scholar]
- 23.Kim CS, Joo SY, Lee KE, et al. Paricalcitol attenuates 4-hydroxy-2-hexenal-induced inflammation and epithelialmesenchymal transition in human renal proximal tubular epithelial cells. PLoS One. 2013;8:e63186. doi: 10.1371/journal.pone.0063186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim CS, Kim SW. Vitamin D and chronic kidney disease. Korean J Intern Med. 2014;29:416–427. doi: 10.3904/kjim.2014.29.4.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakagawa Y, Koizumi M, Fukagawa M. Current topics on vitamin D: vitamin D and chronic kidney disease. Clin Calcium. 2015;25:413–423. [PubMed] [Google Scholar]
- 26.Shimizu H, Yisireyili M, Nishijima F, Niwa T. Stat3 contributes to indoxyl sulfate-induced inflammatory and fibrotic gene expression and cellular senescence. Am J Nephrol. 2012;36:184–189. doi: 10.1159/000341515. [DOI] [PubMed] [Google Scholar]
- 27.Shimizu H, Yisireyili M, Higashiyama Y, Nishijima F, Niwa T. Indoxyl sulfate upregulates renal expression of ICAM-1 via production of ROS and activation of NF-κB and p53 in proximal tubular cells. Life Sci. 2013;92:143–148. doi: 10.1016/j.lfs.2012.11.012. [DOI] [PubMed] [Google Scholar]
- 28.Cuzzocrea S, Riley DP, Caputi AP, Salvemini D. Antioxidant therapy: a new pharmacological approach in shock, inflammation, and ischemia/reperfusion injury. Pharmacol Rev. 2001;53:135–159. [PubMed] [Google Scholar]
- 29.Harrison D, Griendling KK, Landmesser U, Hornig B, Drexler H. Role of oxidative stress in atherosclerosis. Am J Cardiol. 2003;91:7A–11A. doi: 10.1016/s0002-9149(02)03144-2. [DOI] [PubMed] [Google Scholar]
- 30.Lassègue B, Griendling KK. Reactive oxygen species in hypertension; an update. Am J Hypertens. 2004;17:852–860. doi: 10.1016/j.amjhyper.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 31.Miyamoto Y, Iwao Y, Tasaki Y, et al. The uremic solute indoxyl sulfate acts as an antioxidant against superoxide anion radicals under normal-physiological conditions. FEBS Lett. 2010;584:2816–2820. doi: 10.1016/j.febslet.2010.04.046. [DOI] [PubMed] [Google Scholar]
- 32.Guo C, Yuan H, He Z. Melamine causes apoptosis of rat kidney epithelial cell line (NRK-52e cells) via excessive intracellular ROS (reactive oxygen species) and the activation of p38 MAPK pathway. Cell Biol Int. 2012;36:383–389. doi: 10.1042/CBI20110504. [DOI] [PubMed] [Google Scholar]
- 33.Lu Y, Yuan H, Deng S, et al. Arsanilic acid causes apoptosis and oxidative stress in rat kidney epithelial cells (NRK-52e cells) by the activation of the caspase-9 and -3 signaling pathway. Drug Chem Toxicol. 2014;37:55–62. doi: 10.3109/01480545.2013.806532. [DOI] [PubMed] [Google Scholar]
- 34.Skaug B, Jiang X, Chen ZJ. The role of ubiquitin in NF-kappaB regulatory pathways. Annu Rev Biochem. 2009;78:769–796. doi: 10.1146/annurev.biochem.78.070907.102750. [DOI] [PubMed] [Google Scholar]
- 35.Wei MC, Zong WX, Cheng EH, et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]