

Abstract
Histone deacetylase (HDAC) is overexpressed in multiple cancers including pancreatic cancer (PC). However, the effects of histone deacetylase inhibitor (HDACi) on apoptosis and epithelial-mesenchymal transition (EMT) differ in various cancers. In this study, we aimed to investigate the anti-tumor effects of a novel multitargets HDACi, CUDC-101, combined with gemcitabine in PC cell lines. In vitro, we found that Co-treatment with CUDC-101 and gemcitabine results in greater levels of apoptosis and significantly inhibited cell proliferation on PC cells. In addition, CUDC-101 enhanced gemcitabine-induced apoptosis via inhibited PI3K/Akt/mTOR and Erk pathway activation, as indicated by the phosphorylation status of Akt, 4EBP1, S6 and Erk. We also found that co-treatment with gemcitabine and CUDC-101 not only synergistically suppressed ability of PC cell migration and invasion, but also synergistically inhibited EMT signaling pathway through modulation of cadherin, vimentin and transcription factors Snail, Slug and MMP-9. In vivo, the co-treatment group showed a significant anti-tumor function in the growth of xenograft tumors. Overall, combination of CUDC-101 and gemcitabine significantly increased anti-tumor activities compared with single drug alone, thus supporting a further evaluation of combination treatment for PC. Accordingly, it provides a rationale to investigate the combination of gemcitabine and CUDC-101 as a potential therapeutic strategy for PC.
Keywords: CUDC-101, pancreatic cancer, gemcitabine, HDAC inhibitor, antitumor
Introduction
Pancreatic cancer (PC) is one of the most fatal cancers, with a five-year survival rate for PC was reported at 8%, which was the lowest among many other common types of cancer [1]. Surgery remains the primary treatment modality for PC, however, more than one-half of cases are usually diagnosed at a later stage with metastases. At present, gemcitabine is the main chemotherapy drug and has been demonstrated to show a benefit to patients with PC [2]. Unfortunately, most patients with unrespectable PC either do not respond or respond transiently and modestly to gemcitabine. Ultimately, these patients die because of therapeutic resistance and subsequent metastatic disease [3]. Therefore, there is an urgent need to develop more effective treatments to improve PC patient survival.
At present, HDAC inhibitors (HDACi) have been widely applied in cancer research. Recent study have shown that HDACi displayed anti-tumor activities consist of cell apoptosis-inducing, anti-angiogenesis, double strand breakage of DNA and DNA damage repair [4-7]. CUDC-101 is a synthetic small-molecule, multi-targeted inhibitor that targets HDAC, epidermal growth factor receptor (EGFR) and Her2, and it has an in vitro IC50 of 4.4 nM compared with HDACi vorinostat (SAHA), at 40 nM [8]. Multi-target drugs can improve the efficacy of anti-cancer drugs and reduce their side effects which are an important direction in the development of anti-cancer drugs. CUDC-101 has been shown to exhibit a wide range of anti-tumor activities in a variety of human cancer cells [9-11]. Recently, some HDACis were proven to work synergistically to enhance the anti-cancer activities of conventional chemotherapeutic drugs [12], thus suggesting their potential clinical activity when combined with chemotherapeutic agents.
The epithelial-mesenchymal transition (EMT) is a developmental program that enables stationary epithelial cells to gain the ability to migrate and invade as single cells. Tumor cells reactivate EMT to acquire molecular alterations that enable the partial loss of epithelial features and the partial gain of mesenchymal phenotype [13]. Numerous studies have suggested that EMT is pivotal for the invasion and metastasis of PC and contributes to the early-stage dissemination of cancer cells [14-17]. In addition, previous research has demonstrated that a variety of HDACis could reverse or reduce EMT by up-regulating E-cadherin in different solid tumors, such as hepatocellular carcinoma [18], breast cancer [19,20], esophageal cancer [21] and ovarian tumors [22]. Therefore, controlling the mechanisms of EMT is crucial for the development of new therapeutic strategies for cancer metastasis.
In this study, we determined the combined effects of gemcitabine and CUDC-101 on human PC cells. The results showed that co-treatment could inhibit cell proliferation and EMT progression and induce cell apoptosis. In addition, in vivo studies showed that these two agents combined inhibition of tumor growth and promoted tumor necrosis. These results will provide a more effective prognostic evaluation and a new target for the clinical treatment of PC.
Materials and methods
Cell lines and culture conditions
Human PC cell line PANC-1, MIA PaCa-2were purchased from the Korean Cell Line Bank (KCLB, Seoul, Korea). These cells were maintained in DMEM (Gibco BRL, Grand Island, NY, USA) supplemented with 10% heat inactivated fetal bovine serum (FBS), penicillin (100 U/ml) and streptomycin (100 μg/ml) (Gibco BRL, Grand Island, NY, USA). The cells were incubated in 5% CO2 and 95% humidity at 37°C chamber. The growth medium was changed every 3 days.
Reagents and antibodies
Cell culture medium (DMEM) and fetal bovine serum (FBS) were purchased from Gibco (Grand Island, USA). Gemcitabine (HANSOH PHARMA, China) was prepared in saline and CUDC-101 (MedChem Express, China) was prepared in 100% dimethyl sulfoxide (DMSO), and diluted in culture medium for assays, with 500 nM or 1 μM gemcitabine and 500 nM or 1 μM CUDC-101 alone or together treated.
Antibodies PI3K, AKT, p-AKT, p70S6, p-p70S6, 4E-BP1, p-4E-BP1, p-EGFR, E-cadherin, cleaved caspase-3, Snail, Slug, Cyclin B1 were obtained from Cell Signaling Technology (Danvers, MA, USA). HDAC1, HDAC3, HDAC4, HDAC6, MMP-9, Bax, Bcl-2, ERK1/2, p-ERK1/2 were purchased from Santa Cruz Biotechnoligy Inc. (Santa Cruz, CA). And Vimentin, anti-β-actin, CDK1, acetyl-Histone H3 were purchased from Millipore (Billerica, MA, USA).
Transfection
Cell were transfected with 30 nM siRNA (si-HDAC1) using Lipofectamine 3000 (Invitrogen) according to the manufacturer’s instructions.
Cell viability assay
Cells were plated at 5000 cells per well in complete culturing medium in 96-well tissue culture plates for 24 hours and then incubated with indicated treatments for 48 hours, followed by the addition of 5 mg/ml MTT (Sigma, St. Louis, MO). After incubation for 4 hours, the supernatants were removed. 200 μl of DMSO were added, and the absorbance value (OD) at 570 nm was measured using an ELISA reader system (TECAN-infinite M200 pro, Mannedorf, Switzerland). Growth inhibitory IC50 were calculated using Graphpad Prism 5 software. The combination index (CI) was obtained using commercially available software (Calcusyn; Biosoft, Ferguson, MO).
Cell apoptosis assay
Cells were seeded on 6-well plates and treated with gemcitabine and CUDC-101 for 48 hrs, and then cells were harvested, washed with PBS and stained with Annexin V-FITC/PI at room temperature (RT) for 15 min in the darkness. Samples were analyzed by flow cytometry.
Wound healing assay
PANC-1 and MIA PaCa-2 cells were plated in 6-well plates and allowed to form a confluent monolayer. A wound was introduced by running a P200 pipette tip evenly across the monolayer. Afterwards, received gemcitabine and CUDC-101 diluted in media with 0.5% FBS. Plates were photographed after 0 and 48 h.
Cell invasion and migration assays
PANC-1 and MIA PaCa-2 cells were seeded in Millicell cell culture insert (Millipore) that contained polycarbonate filter membranes with 8-mm diameter pores. For the invasion assay, the lower chambers of matrigel coated transwell insert were used. Tumor cells from the control group were maintained in DMEM supplemented with 0.5% FBS and 1% antibiotics, and the treatment group received 500 nM of CUDC-101 or 500 nM gemcitabine diluted in media with 0.5% FBS. The lower chamber contained DMEM supplemented with 10% FBS and 1% antibiotics. The cells were incubated at 37°C in 5% CO2 humidified incubator. After 48 hours, invasive cells in the lower chamber were fixed, stained with hematoxylin and eosin (H&E) and counted with microscopy.
Tumor cell clonogenic assay
To mimic individual cell development into macroscopic cell clones, the cell clonogenic assay was performed as previously described. Single-cell suspension (500 cells per well) were seeded into 6-well plate and allowed to grow for 24 h. Cells were then treated with different concentrations of drug. After 2 weeks, colonies were fixed in 3.7% paraformaldehyde, stained with 0.1% crystal violet and counted manually.
Carboxyfluorescein diacetate succinimidyl ester (CFSE) and cell proliferation
Cell proliferation is evaluated by the Cell TraceTM CFSE cell proliferation kit according to manufacturer protocol. Briefly, mammospheres were dissociated and incubated with PBS containing 5 μM CFSE/1 × 106 cells and 0.5% FBS, for 15 minutes, at 37°C. The reaction was stopped with cold culture medium and after washing with medium, cells were grown on individual sterile coverslips, after 24 hours, with 500 nM or 1 μM gemcitabine and 500 nM or 1 μM CUDC-101 alone or together treated.
After 48 hours, fixed in 4% paraformaldehyde for 10 minutes, at RT. Cell nuclei were stained with DAPI (C1006, Beyotime, Shanghai, China). Coverslips were mounted with mounting medium (P0126, Beyotime, Shanghai, China) and examined at a Leica SP5II confocal microscope (Leica Microsystems, Mannheim, Germany).
Immunoblot analysis
Total protein was extracted, and the protein concentration was measured by the BCA protein assay kit (Pierce, Rockford, Illinois). Then, 20-40 µg protein from each sample was separated on Bis-Tris polyacrylamide gel through electrophoresis and blotted on PVDF membranes (Bio-Rad, USA). Membranes were blocked in 5% skim-milk for 30 min and primary antibody was added at 4°C overnight, and then the secondary antibody at room RT for 1 hour. Proteins were visualized by using ECL prime western blotting detection reagent (Amersham biosciences, Uppsala, Sweden) and results were analyzed quantitatively using chemiluminescent and fluorescent imaging system.
Immunofluorescence (IF) microscopy
Individual sterile coverslips were placed in the 6-well plate and PC cells were added and incubated for 24 h. Subsequently, the cells were treated with drugs diluted in DMEM media with 0.5% FBS for 48 h. Fixed in 4% paraformaldehyde for 20 min at RT, permeabilized with 0.1% Triton X-100 (CWBIO, China) for 10 min and blocking solution was then added for 30 min. After washing, the primary antibodies were added at 4°C overnight, followed by incubation with a fluorescent-labeled secondary antibody for 1 hour at RT. After washing with PBS, cells were counter stained with 49-6-diamidino-2-phenylindole (DAPI) (C1006, Beyotime, Shanghai, China) and the coverslips were mounted with Antifade Mounting Medium (P0126, Beyotime, Shanghai, China). Finally, the immunofluorescence signals were visualized and recorded by Leica SP5II confocal microscope (Leica Microsystems, Mannheim, Germany).
Immunohistochemical (IHC) analysis
IHC analysis was performed using the DAKO LSAB kit (DAKO, Glostrup, Denmark). Before IHC staining, all sections were deparaffinized, rehydrated and incubated with 3% H2O2 in methanol for 15 min at RT. The antigen was retrieved at 95°C for 20 min by placing the slides in 0.01 M sodium citrate buffer (pH 6.0). The slides were then incubated with primary antibody at 4°C overnight. Subsequent to incubation with a biotinylated secondary antibody (cat. no. PV9000; Origene Technologies, Inc., Beijing, China) at RT for 30 min, the slides were then incubated with a streptavidin-peroxidase complex (cat. no. PV9000; Origene Technologies, Inc., Beijing, China) at RT for 30 min. IHC staining was developed using 3,3’-diaminobenzidine, and Mayer’s hematoxylin (cat. no., ZLI9610; Origene Technologies, Inc.) was used for counterstaining. Mouse IgG (cat. no. PV9000; Origene Technologies, Inc., Beijing, China) was used as an isotype control. In addition, tissue sections were processed omitting the primary antibody as the negative control.
In vivo animal studies
Four-week-old female BALB/c nude mice were s.c. injected into right back with 5 × 106 of PC cells. Treatment was initiated when tumor size reached ~200 mm2, Nude mice were randomly divided into four groups (n = 5, per group) as follows: control group and drug group, CUDC-101 (80 mg/kg), gemcitabine (50 mg/kg) and combination group (gemcitabine 50 mg/kg + CUDC-101 80 mg/kg), by intraperitonally (i.p.) every 3 days for a total of 6 times, the control group was given the same volume of saline. In the xenograft mouse model of PANC-1, the body weight of mice was checked twice a week, and tumor volume was checked every 6 days. The length and width of each tumor were measured, and volume was calculated using above mentioned formula: tumor volume = (length × width2) × 3.1415926/6 [23]. To observe the tumor inhibition rate of the transplanted tumor, and to determine the difference of the effect between the two groups.
Statistical analysis
Significant differences between values obtained in a population of control and treated PC cells were determined using factorial design of two factors and one-way ANOVA. P values less than 0.05 were assigned significance. For the in vivo studies, significant differences in the mean tumor volumes after 6 times of treatment with gemcitabine and/or CUDC-101 were determined using a two-tailed paired t-test.
Results
HDACs were overexpressed in human PC specimens
To determine the clinical relevance of HDAC expression in PC, we first analyzed the expression of HDAC proteins in clinical specimens from the human protein atlas (www.proteinatlas.org). The results showed that HDAC1, HDAC3, HDAC6 and HDAC9 had higher expression in PC tissues compared with normal tissues (Figure 1A). In addition, according to oncomine data (www.oncomine.org), the HDAC1 (P < 0.0001), HDAC2 (P < 0.0001), HDAC8 (P < 0.0001) and HDAC9 (P < 0.001) mRNA levels were higher in PC tissues than in normal tissues (Figure 1B).
Figure 1.

HDAC is upregulated in human PC specimens. A. HDAC1, HDAC3, HDAC6 and HDAC9 expression in normal pancreas tissues. Images were taken from the Human Protein Atlas (htt://www.proteinatlas.org) online database. B. Oncomine data (www.oncomine.org) showing HDAC1, HDAC2, HDAC8, HDAC9 expression in normal vs tumor of pancreas (**P < 0.01, ***P < 0.0001). C. HDAC1, HDAC3 and HDAC4 were highly expressed in PANC-1 and MIA PaCa-2 cells. D. Cell viability of PANC-1 and MIA PaCa-2 cells transfect with si-HDAC1 (P < 0.05). E. Western blot analysis of bax, bcl-2, E-cadherin and Vimentin expressed in si-HDAC1 cells.
The results of western blot also showed that HDACs (HDAC1, HDAC3, and HDAC4) were highly expressed in pancreatic ductal epithelial cancer cell PANC-1 and pancreatic epithelial cancer cell MIA PaCa-2, while they were almost invisible in the human breast epithelial cell MCF-10A which was used as a control here (Figure 1C). Furthermore, to explore the potential role of HDACs in PC progress, HDAC1 gene was knock-downed with si-RNA in PC cells PANC-1 and MIA PaCa-2. The results revealed that HDAC1 knockdown led to a remarkable inhibition in cell proliferation (P < 0.05) (Figure 1D). The results of western blot showed that the knockdown of HDAC1 gene induced apoptosis as determined by increased anti-oncogene Bax and reduced proto-oncogene bcl-2. Importantly, the ratio of Bax/Bcl-2 was significantly increased, which suggests that the Bax/bcl-2 plays an important role in PC cell progression regulated by HDAC1. Moreover, the silencing of HDAC1 significantly increased epithelial marker E-cadherin expression and decreased mesenchymal marker Vimentin in PC cells (Figure 1E). These data indicated that increased expression of HDACs promoted the malignant potential of PC, and HDAC inhibitors are promising compounds for the therapy of PC and warrant further research.
CUDC-101 synergizes with gemcitabine to inhibit the proliferation of human PC cells
To investigate the anti-cancer activity of CUDC-101 and/or gemcitabine, the PC cell lines PANC-1 and MIA PaCa-2 were subjected to a MTT assay. The cell proliferation assay results indicated that an increased concentration of 100-1000 nM CUDC-101 or 100-2000 nM gemcitabine inhibited the proliferation of PANC-1 and MIA PaCa-2 cells (Figure 2A) in a dose-dependent manner. More importantly, the isobologram analysis indicated that the effect of the combined treatment was highly synergistic in PANC-1 (combination index, CI = 0.75) when the concentration of gemcitabine and CUDC-101 were 1 μM and 1 μM, respectively. Similarly, synergistic effects of gemcitabine plus CUDC-101 were also observed in the MIA PaCa-2 line with CI values below 1 (Figure 2B). Additionally, co-treatment of CUDC-101 and gemcitabine showed more effectively inhibitory effect on the PC cells proliferation than the agents alone (Figure 2C).
Figure 2.

Cell proliferation in the PC cell lines following treatment CUDC-101 with/or gemcitabine. A. Cell viability of PANC-1 and MIA PaCa-2 treated with the CUDC-101 or gemcitabine for 48 h was measured by MTT assay. B. The combination index was calculated according to the approach described by Chou and Talalay. CI = 1 indicates an additive effect, CI < 1 a synergistic effect, and CI > 1 an antagonistic effect. C. Combination of CUDC-101 and gemcitabine synergistically inhibited the viability of PC cells compared with single agents (*P < 0.05, ***P < 0.0001). D. CUDC-101 and gemcitabine synergistically inhibited the colony formation of PANC-1 and MIA PaCa-2 cells by the plate colony forming assay. E. CFSE staining in control and treated mammospheres.
To further determine the anti-proliferation effect of combined therapy in PC cells, the colony formation assay was conducted. The results showed that co-treatment significantly inhibited the colony formation of PC cells compared with a single drug alone (Figure 2D). CFSE is a vital dye stable in the cytoplasm for about 7-8 generations, but the intensity of CFSE fluorescence declines due to its progressive halving within daughter cells following each cell division. In our results, the fluorescence intensity of the combination group was significantly higher than that of single drug group. These data indicated that co-treatment of CUDC-101 and gemcitabine inhibited the proliferation of PC cells more effectively compare with the agents alone (Figure 2E).
CUDC-101 increases gemcitabine-induced apoptosis in PC cells
To determine if the synergistic growth inhibition of gemcitabine and CUDC-101 results from apoptosis, flow cytometric analysis was performed in the PANC-1 and MIA PaCa-2 cell lines. We found that treatment with gemcitabine resulted in an approximately 8.4% increase in early and late apoptosis in PANC-1 cells and that treatment with CUDC-101 resulted in a 5.6% increase in early and late apoptosis. Importantly, co-treatment resulted in a significantly increased rate of apoptosis, up to 39.2% (Figure 3A). Similarly, a synergistic apoptosis-inducing effect was found in MIA PaCa-2 cells, for which the co-treatment group had a 57.2% apoptotic rate compared with the gemcitabine group, with 32%, and the CUDC-101 group, with 15.9%.
Figure 3.

Co-treatment with CUDC-101 and gemcitabine synergistically induced apoptosis signaling. A. Annexin V-FITC/PI stained of of PC cells after treatment with CUDC-101 and gemcitabine alone or combination for 48 h. Apoptotic cells were analyzed by flow cytometry. B. Western blot analysis showed CUDC-101 increased inhibition of gemcitabine on HDAC1, HDAC3, HDAC4 and p-EGFR proteins. Cyclin B1/CDK1 protein expression in CUDC-101 group was decreased. Acetylased histone H3 protein expression in CUDC-101 group and co-treatment group. C. Protein levels of p-p53, cl-caspase3 and the ratio of bax/bcl-2 were markedly increased in co-treatment group. Statistical significance was determined by a standard test (*P < 0.05, **P < 0.01).
We have detected the multi-targeted inhibition of CUDC-101 in PC cells. The results of western blot, CUDC-101 reduced protein levels of HDAC1, HDAC3, HDAC6 and p-EGFR. We also found that CUDC-101 treatment alone group and co-treatment group significantly increased the acetylation of histone H3 in PC cells. Cyclin B1/CDK1 is an important regulatory protein that is involved in mitosis. To determine whether cyclin B1/CDK1 is involved in the cell cycle arrest-related effects of CUDC-101 on PC cells, a western blot assay was applied. Here, gemcitabine treatment had no significant effect on cyclin B1/CDK1 expression in PANC-1 and MIA PaCa-2 cells. In contrast, CUDC-101 treatment decreased cyclin B1/CDK1 expression in both PC cell lines. The effects of the combined therapy on cyclin B1/CDK1 expression were similar to those observed for the CUDC-101 treatment alone group, suggesting that cyclin B1/CDK1 does not play an important role in the synergistic effect induced by the drug combination (Figure 3B). The apoptotic pathway include a caspase-dependent and a mitochondrial pathway. We investigated the involvement of the caspase family of proteins and the Bcl-2 family of proteins during the induction of apoptosis in PC cells. Western blot analysis revealed that gemcitabine increased the expression level of p-p53 in two PC cell lines and CUDC-101 increased the ratio of Bax/bcl-2 in PANC-1 cells. Importantly, co-treatment with gemcitabine and CUDC-101 resulted in a marked increase in the ratio of bax/bcl-2 and p-p53 as well as cleaved caspase-3 protein (Figure 3C).
Effects of co-treatment on EMT in PC cells in vitro
To determine the effect on cell migration and invasion, several functional studies were performed. Wound healing assays showed that the migratory ability of two PC cell lines was significantly inhibited in the co-treatment group compared with the single treatment groups (Figure 4A). Furthermore, in a trans-well migration assay and invasion experiments, CUDC-101 combined with gemcitabine significantly inhibited the migration and invasion abilities of PC cells (Figure 4B).
Figure 4.
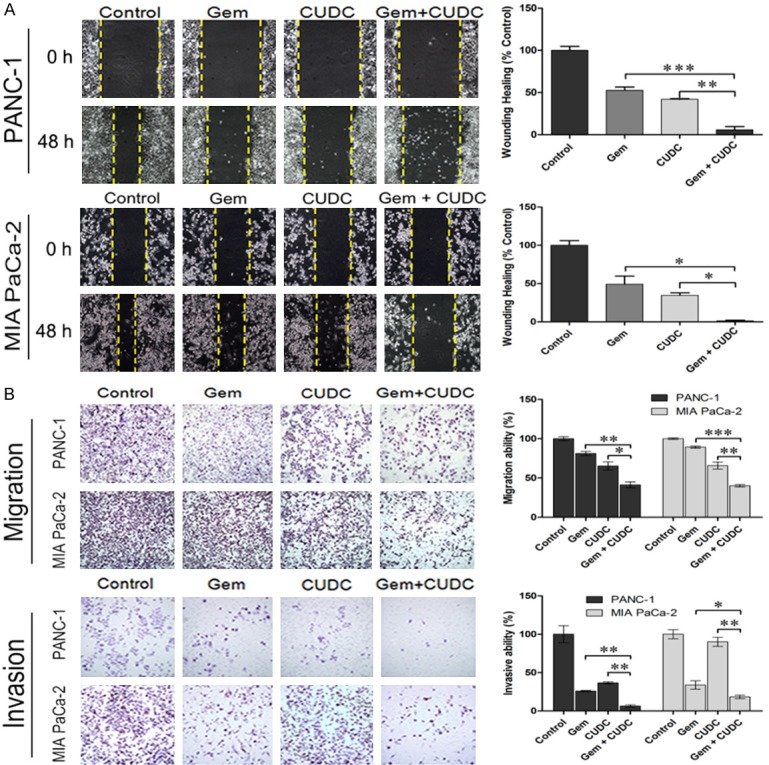
Effects of co-treatment on migration and invasion of PC cells. A. Co-treatment inhibited cell migration of PANC-1 and MIA PaCa-2 cells in the wound-healing migration assay. Photographs were taken at o h and 48 h, after the wound was made. (*P < 0.05, **P < 0.01, ***P < 0.001). B. Migration and invasion assay of PANC-1 and MIA PaCa-2 cells treated with CUDC-101 and/or gemcitabine for 48 h, and the number of migrate and invasive cells were determined using a transwell penetration assay. (*P < 0.05, **P < 0.01, ***P < 0.001).
EMT plays a key role in regulating gene expression and tumor development. Many studies have reported that HDAC inhibitors reverse or attenuate EMT by altering expression levels of E-cadherin and Vimentin. After treatment with gemcitabine and CUDC-101 for 48 h, the expression levels of EMT markers were detected by IF. Our results indicated that gemcitabine with CUDC-101 synergistically suppresses the mesenchymal marker Vimentin, and increases ZO-1 expression in PANC-1 cells (Figure 5A). Similarly, increased E-cadherin protein and decreased Vimentin protein were found in MIA PaCa-2 cells (Figure 5B). Western blot analysis also showed that the epithelial marker E-cadherin was significantly increased, where as the mesenchymal markers Vimentin and MMP-9 were decreased. The transcription factors Snail and Slug were also decreased (Figure 5C).
Figure 5.

Effects of co-treatment on EMT in PC cells. (A) PANC-1 and (B) MIA PaCa-2 cells were treated with the indicated concentration of CUDC-101 and/or gemcitabine for 48 h. Expression of EMT markers (E-cadherin, ZO-1, Vimentin) were detected by IF staining. (C) Western blot analyses was performed for EMT-associated markers (E-cadherin, Vimentin, MMP-9, Snail and Slug).
PI3K/Akt/mTOR and Erk pathways are involved in anti-tumor effects of combined therapy in PC cells
The PI3K/Akt/mTOR and Erk signaling pathways are major pathways activated in PC cells. They play a variety of physiological roles, including the regulation of EMT, cell growth, cell cycle, and cell survival. To confirm whether the anti-tumor activity of CUDC-101 and gemcitabine was regulated by the PI3K/Akt and Erk signaling pathways, we evaluated the activation of proteins in PC cells by western blot. As shown in Figure 6, the western blot analysis revealed that the expression level of PI3K, p-S6, p-4EBP1 were significantly lower in PANC-1 cell with co-treatment when compared with those treated with single treatment (P < 0.05). And, the expression level of p-AKT (P < 0.01) and p-ERK (P < 0.05) were significantly lower with co-treatment compared with gemcitabine alone. In MIA PaCa-2 cell, the expression level of p-AKT (P < 0.01), p-S6 (P < 0.01), p-4EBP1 (P < 0.05) and p-ERK (P < 0.01) were significantly lower with co-treatment compared with gemcitabine alone. However, the levels of PI3K was significantly lower with co-treatment when compared with single treatment (P < 0.05).
Figure 6.
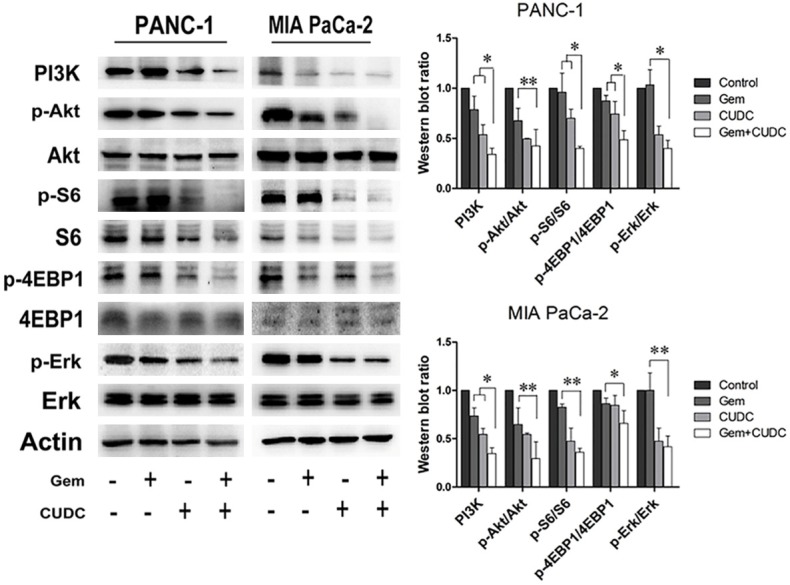
PI3K/Akt/mTOR and Erk pathway involved in anti-tumor activity of combined therapy in PC cells. PANC-1 and MIA PaCa-2 cells were treated with CUDC-101 and/or gemcitabine for 48 h, and western blot analysis showed increased inhibition of the PI3K, p-Akt, p-S6, p-4EBP1 and p-Erk proteins (*P < 0.05, **P < 0.01).
Combined treatment synergistically inhibits tumor growth in PC xenograft models
To better model of the clinical setting, we evaluated whether CUDC-101 combined with gemcitabine showed a synergistic reaction in human PC cell xenograft models. As shown in Figure 7A, the treatment of CUDC-101 and/or gemcitabine at the given concentration had no discernable effect on body weights in mice, compared to the control or individual drugs groups, it showed that the dose had no toxic and side effect on nude mice, however, single agent groups effectively inhibited tumor growth, whereas the co-treatment group exhibited a 60% further tumor regression (P < 0.0001). Additionally, at the end of treatment, the combination treatment had a significantly decreased xenograft tumor size (Figure 7B). Furthermore, IHC staining showed a lower expression of HDAC6 and Ki-67 in the co-treatment group compared to the other groups. An increased expression of E-cadherin was also observed in the co-treatment group, but the mesenchymal marker Vimentin was decreased, consistent with the in vitro findings (Figure 7C).
Figure 7.
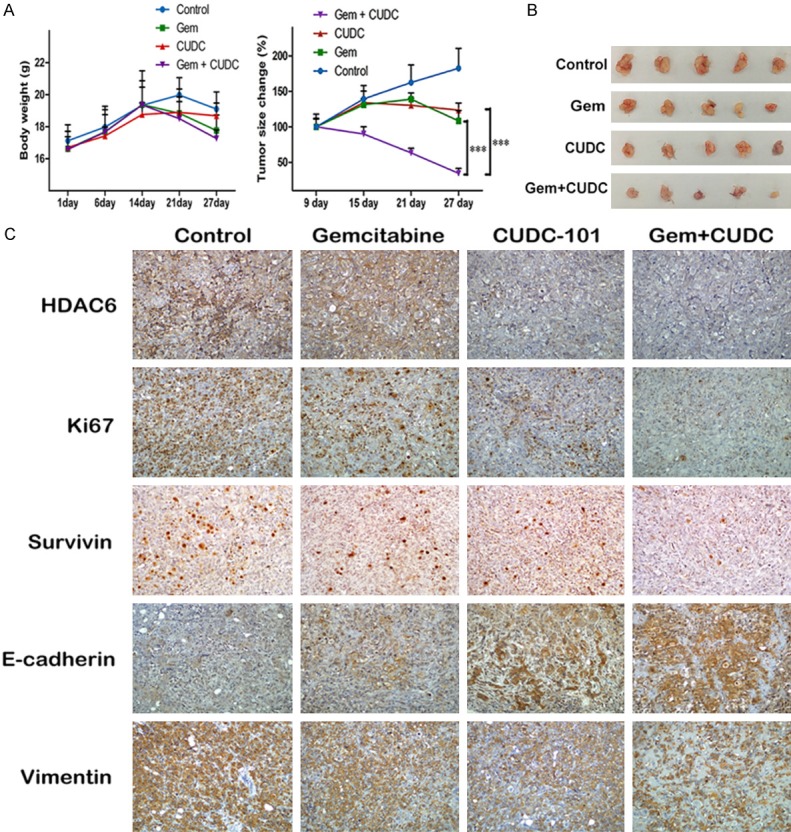
CUDC-101 and gemcitabine synergistically suppressed the growth of PANC-1 cells in vivo. A. The mice were sacrificed 27 days after drug treatment initiation, and the tumors were removed. The body weights and tumor volumes were measured every 6 days during the treatment. B. Image of tumor size. C. Immunohistochemistry (IHC) was utilized for detecting proteins of HDAC6, Ki-67, Survivin, E-cadherin and Vimentin from xenograft tumor model.
Discussion
The average survival time of PC patients is still short due to the rapid recurrence and metastasis. Gemcitabine is the standard chemotherapy regimen for treating advanced PC [24]. However, the efficiency of gemcitabine in the clinic is less than 20% of treated patients due to the low response to gemcitabine [25]. With an improved understanding of the genetic changes that are involved in cancer initiation and progression, researchers can develop many targeted anti-cancer therapies.
Previous studies have reported that HDACs are overexpressed in many tumors including PC, and that inhibiting HDACs could induce tumor cell differentiation and apoptosis, inhibit metastasis and reverse or reduce EMT in different solid tumors [26-28]. About the HDAC family, there’re at least eighteen members as we known. Among them [29], HDAC1 plays an important role in the tumors, the cellular and biochemical functions of HDAC1 are relatively well characterized. High expression of HDAC1 in tumor cells can significantly increase the proliferation, and enhance the migration and invasion [30]. In this study, when HDAC1 gene was knock-downed, cell proliferation was decreased in MTT assay and induced apoptosis and down-regulated EMT pathways. These results proved that HDAC1 was involved in cell proliferation, apoptosis and metastasis. Recently, researchers have found that some HDACis improved the sensitivity of cancer cells to chemotherapeutic drugs [31-33]. As a hybrid molecular targeted agent, CUDC-101 can avoid interacting with drugs during combined applications and can reduce adverse drug reactions and resistance. To date, there are no reports on the combination of traditional chemotherapy drugs with CUDC-101. In this study, for the first time, we found that co-treatment with CUDC-101 and gemcitabine synergistically exerted anti-tumor activity in PC cells in vitro and in vivo.
Apoptosis is a major mechanism for eliminating cancer cells [34] and mainly includes the death receptor signaling pathway and the mitochondrial apoptosis pathway. Bcl-2 and Bax protein are important regulatory factors for changing the permeability of the mitochondrial membrane potential [35]. A loss of mitochondrial membrane potential could release pro-apoptotic factors from the mitochondrial membrane space, leading to caspase activation. Caspase3 is the final executor of apoptosis. When the pro-caspase-3 is fragmented into active cleaved caspase-3, it can specifically cut DNA, leading to the inactivation of PARP and DNA-dependent protein kinase (DNA-PK) and promoting chromatin condensation and nuclease activation-induced apoptosis [36]. In this study, statistical analysis showed that the ratio of pro-apoptotic proteins Bax and anti-apoptotic protein Bcl-2 were significantly increased. Moreover, cleaved caspase-3 was up-regulated by the combined therapy. These data indicated that co-treatment inhibits cell proliferation and induces apoptosis via the mitochondrial pathway.
The PI3K/Akt/mTOR and Erk signaling pathways play important roles in the regulation of cell survival, invasion and metastasis [37-39]. Akt can activate mTOR, resulting in increased protein synthesis via its effectors 4EBP1 and S6K [40]. Moreover, MAPKs are a family of kinases that regulate cell proliferation, cell cycle arrest, differentiation and metastasis. Lisa Z et al. reported that CUDC-101 is effective in anaplastic thyroid cancer cell lines with gene mutations in both the MAPK and PI3K/Akt pathways [11]. So we conceived that CUDC-101 inhibited the proliferation and induced apoptosis of PC cells might be associated with the regulation of PI3K/mTOR and Erk signaling pathways. In this study, we showed that the expression levels of PI3K/mTOR and Erk signaling pathways were not significantly altered in PC cells with gemcitabine therapy. However, CUDC-101 combined with gemcitabine resulted in significant downregulation of the PI3K/mTOR and Erk pathway on PC cells, as indicated by effectively reducing the phosphorylation of Akt, S6, 4EBP1 and Erk in PC cells. These data suggested that CUDC-101 exerted its synergistic effect in gemcitabine-treated PC may occur via inactivation of the PI3K/mTOR and Erk signaling pathways.
Many studies have reported that HDAC inhibitors reverse or attenuate EMT through the up-regulation of E-cadherin in different solid tumors. Piao J et al. reported that the HDAC inhibitor TSA with BEZ235 synergistically suppressed EMT progression in lung cancer cells [41]. Wang J et al. reported that CUDC-101 also efficiently inhibited the proliferation of MET-overexpressing non-small cell lung cancer and as well as migration, invasion and EMT [42]. In the wound healing and trans-well experiments, we showed that gemcitabine combined with CUDC-101 significantly inhibited the migration and invasion of PC cells. Furthermore, western blot and IF staining showed that epithelial markers such as E-cadherin and ZO-1 were significantly up-regulated in the combined drug group, whereas the mesenchymal marker Vimentin and MMP-9 expression were significantly down-regulated compared with the control groups. Similarly, in vivo experiments showed that CUDC-101 combined with gemcitabine could up-regulate E-cadherin and down-regulate Vimentin in a nude mouse xenograft model. Taken together, these results suggest that the CUDC-101 and gemcitabine combination can effectively suppress the malignant transformation of PC by driving the cancer cells toward an epithelial state. The mechanisms driving EMT warrant further elucidation.
Although tumor cell dissemination and EMT begins early in progression [17], clinically relevant distant metastasis is a late event [43] and EMT may be more important for therapy resistance than metastatic spread [44]. In recent years, many research found that Erk signaling pathway may be involved in the drug resistance process of tumor cells, and its mechanism may be related to the regulation of drug-resistance genes and protein expression. Use of an Erk1/2 inhibitor can overcome acquired resistance to both BRAF and MEK1/2 inhibitors in melanoma, breast and colon cancer cell lines [45,46]. Fryer RA et al. have reported that Erk activity underlies sensitivity to gemcitabine and that addition of an agent that reduces this activity, such as lenalidomide, enhances gemcitabine efficacy [47]. Snail is a transcriptional factor that represses multiple other factors, and its overexpression in a trigger of EMT. Tomono T et al. have reported that snail-induced EMT enhances multidrug resistance in non small cell lung cancer cells [48]. Our findings show that gemcitabine did not effectively inhibit the Erk1/2 phosphorylation, but co-treatment of cells with CUDC-101 effectively inhibited the phosphorylation of Erk1/2. Moreover, the transcription factors Snail was also decreased. These results indicated that CUDC-101 and gemcitabine combination can effectively control the mechanism of gemcitabine-resistance, and inhibited EMT in PC maybe via Erk/Snail signaling pathway.
In conclusion, we demonstrated that CUDC-101 potentiated the antitumor effects of gemcitabine by inhibiting cell proliferation and driving the cancer cells toward an epithelial state. We conceived that CUDC-101 combing with gemcitabine not only induce apoptosis via PI3K/mTOR signaling, but also reduce sensitivity to gemcitabine in PC via Erk signaling. These results may provide a potential rational basis for a combination strategy for chemotherapy treatment of PC.
Additionally, the mechanisms driving EMT warrant further elucidation. In the next study, we will detect weather CUDC-101 could reduce the drug resistance of gemcitabine resistant cell line of PC, by regulating EMT pathway.
Acknowledgements
This study was supported by grants from National Natural Science Funds of China (No. 81460399, 31760313), The Funds of Changbai Mountain Scholar Project and Key Laboratory of the Science and Technology Department of Jilin Province (No. 20170622007JC).
Disclosure of conflict of interest
None.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 2.Lee HW, Chung MJ, Kang H, Choi H, Choi YJ, Lee KJ, Lee SW, Han SH, Kim JS, Song SY. Gemcitabine-induced hemolytic uremic syndrome in pancreatic cancer: a case report and review of the literature. Gut Liver. 2014;8:109–112. doi: 10.5009/gnl.2014.8.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ryan DP, Hong TS, Bardeesy N. Pancreatic adenocarcinoma. N Engl J Med. 2014;371:1039–1049. doi: 10.1056/NEJMra1404198. [DOI] [PubMed] [Google Scholar]
- 4.Sanaei M, Kavoosi F, Roustazadeh A, Shahsavani H. In vitro effect of the histone deacetylase inhibitor valproic acid on viability and apoptosis of the PLC/PRF5 human hepatocellular carcinoma cell line. Asian Pac J Cancer Prev. 2018;19:2507–2510. doi: 10.22034/APJCP.2018.19.9.2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Qian DZ, Kato Y, Shabbeer S, Wei Y, Verheul HM, Salumbides B, Sanni T, Atadja P, Pili R. Targeting tumor angiogenesis with histone deacetylase inhibitors: the hydroxamic acid derivative LBH589. Clin Cancer Res. 2006;12:634–642. doi: 10.1158/1078-0432.CCR-05-1132. [DOI] [PubMed] [Google Scholar]
- 6.Ruscetti M, Dadashian EL, Guo W, Quach B, Mulholland DJ, Park JW, Tran LM, Kobayashi N, Bianchi-Frias D, Xing Y, Nelson PS, Wu H. HDAC inhibition impedes epithelial-mesenchymal plasticity and suppresses metastatic, castration-resistant prostate cancer. Oncogene. 2016;35:3781–3795. doi: 10.1038/onc.2015.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gaymes TJ, Padua RA, Pla M, Orr S, Omidvar N, Chomienne C, Mufti GJ, Rassool FV. Histone deacetylase inhibitors (HDI) cause DNA damage in leukemia cells: a mechanism for leukemia-specific HDI-dependent apoptosis? Mol Cancer Res. 2006;4:563–573. doi: 10.1158/1541-7786.MCR-06-0111. [DOI] [PubMed] [Google Scholar]
- 8.Cai X, Zhai HX, Wang J, Forrester J, Qu H, Yin L, Lai CJ, Bao R, Qian C. Discovery of 7-(4-(3-ethynylphenylamino)-7-methoxyquinazolin-6-yloxy)-N-hydroxyheptanamide (CUDc-101) as a potent multi-acting HDAC, EGFR, and HER2 inhibitor for the treatment of cancer. J Med Chem. 2010;53:2000–2009. doi: 10.1021/jm901453q. [DOI] [PubMed] [Google Scholar]
- 9.Lai CJ, Bao R, Tao X, Wang J, Atoyan R, Qu H, Wang DG, Yin L, Samson M, Forrester J, Zifcak B, Xu GX, DellaRocca S, Zhai HX, Cai X, Munger WE, Keegan M, Pepicelli CV, Qian C. CUDC-101, a multitargeted inhibitor of histone deacetylase, epidermal growth factor receptor, and human epidermal growth factor receptor 2, exerts potent anticancer activity. Cancer Res. 2010;70:3647–3656. doi: 10.1158/0008-5472.CAN-09-3360. [DOI] [PubMed] [Google Scholar]
- 10.Zhang L, Boufraqech M, Lake R, Kebebew E. Carfilzomib potentiates CUDC-101-induced apoptosis in anaplastic thyroid cancer. Oncotarget. 2016;7:16517–16528. doi: 10.18632/oncotarget.7760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang L, Zhang Y, Mehta A, Boufraqech M, Davis S, Wang J, Tian Z, Yu Z, Boxer MB, Kiefer JA, Copland JA, Smallridge RC, Li Z, Shen M, Kebebew E. Dual inhibition of HDAC and EGFR signaling with CUDC-101 induces potent suppression of tumor growth and metastasis in anaplastic thyroid cancer. Oncotarget. 2015;6:9073–9085. doi: 10.18632/oncotarget.3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Won HR, Ryu HW, Shin DH, Yeon SK, Lee DH, Kwon SH. A452, an HDAC6-selective inhibitor, synergistically enhances the anticancer activity of chemotherapeutic agents in colorectal cancer cells. Mol Carcinog. 2018;57:1383–1395. doi: 10.1002/mc.22852. [DOI] [PubMed] [Google Scholar]
- 13.Yeung KT, Yang J. Epithelial-mesenchymal transition in tumor metastasis. Mol Oncol. 2017;11:28–39. doi: 10.1002/1878-0261.12017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hotz B, Arndt M, Dullat S, Bhargava S, Buhr HJ, Hotz HG. Epithelial to mesenchymal transition: expression of the regulators snail, slug, and twist in pancreatic cancer. Clin Cancer Res. 2007;13:4769–4776. doi: 10.1158/1078-0432.CCR-06-2926. [DOI] [PubMed] [Google Scholar]
- 15.Arumugam T, Ramachandran V, Fournier KF, Wang H, Marquis L, Abbruzzese JL, Gallick GE, Logsdon CD, McConkey DJ, Choi W. Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res. 2009;69:5820–5828. doi: 10.1158/0008-5472.CAN-08-2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Taube JH, Herschkowitz JI, Komurov K, Zhou AY, Gupta S, Yang J, Hartwell K, Onder TT, Gupta PB, Evans KW, Hollier BG, Ram PT, Lander ES, Rosen JM, Weinberg RA, Mani SA. Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc Natl Acad Sci U S A. 2010;107:15449–15454. doi: 10.1073/pnas.1004900107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, Reichert M, Beatty GL, Rustgi AK, Vonderheide RH, Leach SD, Stanger BZ. EMT and dissemination precede pancreatic tumor formation. Cell. 2012;148:349–361. doi: 10.1016/j.cell.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.DI Fazio P, Montalbano R, Quint K, Alinger B, Kemmerling R, Kiesslich T, Ocker M, Neureiter D. The pan-deacetylase inhibitor panobinostat modulates the expression of epithelial-mesenchymal transition markers in hepatocellular carcinoma models. Oncol Lett. 2013;5:127–134. doi: 10.3892/ol.2012.951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shah P, Gau Y, Sabnis G. Histone deacetylase inhibitor entinostat reverses epithelial to mesenchymal transition of breast cancer cells by reversing the repression of E-cadherin. Breast Cancer Res Treat. 2014;143:99–111. doi: 10.1007/s10549-013-2784-7. [DOI] [PubMed] [Google Scholar]
- 20.Srivastava RK, Kurzrock R, Shankar S. MS-275 sensitizes TRAIL-resistant breast cancer cells, inhibits angiogenesis and metastasis, and reverses epithelial-mesenchymal transition in vivo. Mol Cancer Ther. 2010;9:3254–3266. doi: 10.1158/1535-7163.MCT-10-0582. [DOI] [PubMed] [Google Scholar]
- 21.Taylor MD, Liu Y, Nagji AS, Theodosakis N, Jones DR. Combined proteasome and histone deacetylase inhibition attenuates epithelial-mesenchymal transition through E-cadherin in esophageal cancer cells. J Thorac Cardiovasc Surg. 2010;139:1224–32. 1232.e1. doi: 10.1016/j.jtcvs.2010.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meng F, Sun G, Zhong M, Yu Y, Brewer MA. Anticancer efficacy of cisplatin and trichostatin A or 5-aza-2’-deoxycytidine on ovarian cancer. Br J Cancer. 2013;108:579–586. doi: 10.1038/bjc.2013.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu J, Chen M, Chen N, Ma A, Zhu C, Zhao R, Jiang M, Zhou J, Ye L, Fu H, Zhang X. Glycyrrhetinic acid induces G1phase cell cycle arrest in human nonsmall cell lung cancer cells through endoplasmic reticulum stress pathway. Int J Oncol. 2015;46:981–988. doi: 10.3892/ijo.2015.2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heinemann V, Boeck S, Hinke A, Labianca R, Louvet C. Meta-analysis of randomized trials: evaluation of benefit from gemcitabine-based combination chemotherapy applied in advanced pancreatic cancer. BMC Cancer. 2008;8:82. doi: 10.1186/1471-2407-8-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tamburrino A, Piro G, Carbone C, Tortora G, Melisi D. Mechanisms of resistance to chemotherapeutic and anti-angiogenic drugs as novel targets for pancreatic cancer therapy. Front Pharmacol. 2013;4:56. doi: 10.3389/fphar.2013.00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seo J, Min SK, Park HR, Kim DH, Kwon MJ, Kim LS, Ju YS. Expression of histone deacetylases HDAC1, HDAC2, HDAC3, and HDAC6 in invasive ductal carcinomas of the breast. J Breast Cancer. 2014;17:323–331. doi: 10.4048/jbc.2014.17.4.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ma Y, Yue Y, Pan M, Sun J, Chu J, Lin X, Xu W, Feng L, Chen Y, Chen D, Shin VY, Wang X, Jin H. Histone deacetylase 3 inhibits new tumor suppressor gene DTWD1 in gastric cancer. Am J Cancer Res. 2015;5:663–673. [PMC free article] [PubMed] [Google Scholar]
- 28.Ischenko I, Petrenko O, Hayman MJ. A MEK/PI3K/HDAC inhibitor combination therapy for KRAS mutant pancreatic cancer cells. Oncotarget. 2015;6:15814–15827. doi: 10.18632/oncotarget.4538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Halkidou K, Gaughan L, Cook S, Leung HY, Neal DE, Robson CN. Upregulation and nuclear recruitment of HDAC1 in hormone refractory prostate cancer. Prostate. 2004;59:177–189. doi: 10.1002/pros.20022. [DOI] [PubMed] [Google Scholar]
- 30.Mottamal M, Zheng S, Huang TL, Wang G. Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules. 2015;20:3898–3941. doi: 10.3390/molecules20033898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hanke NT, Garland LL, Baker AF. Carfilzomib combined with suberanilohydroxamic acid (SAHA) synergistically promotes endoplasmic reticulum stress in non-small cell lung cancer cell lines. J Cancer Res Clin Oncol. 2016;142:549–560. doi: 10.1007/s00432-015-2047-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen JH, Zheng YL, Xu CQ, Gu LZ, Ding ZL, Qin L, Wang Y, Fu R, Wan YF, Hu CP. Valproic acid (VPA) enhances cisplatin sensitivity of non-small cell lung cancer cells via HDAC2 mediated down regulation of ABCA1. Biol Chem. 2017;398:785–792. doi: 10.1515/hsz-2016-0307. [DOI] [PubMed] [Google Scholar]
- 33.Gallagher SJ, Gunatilake D, Beaumont KA, Sharp DM, Tiffen JC, Heinemann A, Weninger W, Haass NK, Wilmott JS, Madore J, Ferguson PM, Rizos H, Hersey P. HDAC inhibitors restore BRAF-inhibitor sensitivity by altering PI3K and survival signalling in a subset of melanoma. Int J Cancer. 2018;142:1926–1937. doi: 10.1002/ijc.31199. [DOI] [PubMed] [Google Scholar]
- 34.Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626–629. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- 35.Lin HH, Chen JH, Huang CC, Wang CJ. Apoptotic effect of 3,4-dihydroxybenzoic acid on human gastric carcinoma cells involving JNK/p38 MAPK signaling activation. Int J Cancer. 2007;120:2306–2316. doi: 10.1002/ijc.22571. [DOI] [PubMed] [Google Scholar]
- 36.El-Khattouti A, Selimovic D, Haikel Y, Hassan M. Crosstalk between apoptosis and autophagy: molecular mechanisms and therapeutic strategies in cancer. J Cell Death. 2013;6:37–55. doi: 10.4137/JCD.S11034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9:550–562. doi: 10.1038/nrc2664. [DOI] [PubMed] [Google Scholar]
- 38.Thoreen CC, Chantranupong L, Keys HR, Wang T, Gray NS, Sabatini DM. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature. 2012;485:109–113. doi: 10.1038/nature11083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yue X, Li M, Chen D, Xu Z, Sun S. UNBS5162 induces growth inhibition and apoptosis via inhibiting PI3K/AKT/mTOR pathway in triple negative breast cancer MDA-MB-231 cells. Exp Ther Med. 2018;16:3921–3928. doi: 10.3892/etm.2018.6675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hsieh AC, Liu Y, Edlind MP, Ingolia NT, Janes MR, Sher A, Shi EY, Stumpf CR, Christensen C, Bonham MJ, Wang S, Ren P, Martin M, Jessen K, Feldman ME, Weissman JS, Shokat KM, Rommel C, Ruggero D. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature. 2012;485:55–61. doi: 10.1038/nature10912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Piao J, Chen L, Quan T, Li L, Quan C, Piao Y, Jin T, Lin Z. Superior efficacy of co-treatment with the dual PI3K/mTOR inhibitor BEZ235 and histone deacetylase inhibitor trichostatin A against NSCLC. Oncotarget. 2016;7:60169–60180. doi: 10.18632/oncotarget.11109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang J, Pursell NW, Samson ME, Atoyan R, Ma AW, Selmi A, Xu W, Cai X, Voi M, Savagner P, Lai CJ. Potential advantages of CUDC-101, a multitargeted HDAC, EGFR, and HER2 inhibitor, in treating drug resistance and preventing cancer cell migration and invasion. Mol Cancer Ther. 2013;12:925–936. doi: 10.1158/1535-7163.MCT-12-1045. [DOI] [PubMed] [Google Scholar]
- 43.Yachida S, Jones S, Bozic I, Antal T, Leary R, Fu B, Kamiyama M, Hruban RH, Eshleman JR, Nowak MA, Velculescu VE, Kinzler KW, Vogelstein B, Iacobuzio-Donahue CA. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010;467:1114–1117. doi: 10.1038/nature09515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, Wu CC, LeBleu VS, Kalluri R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. 2015;527:525–530. doi: 10.1038/nature16064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morris EJ, Jha S, Restaino CR, Dayananth P, Zhu H, Cooper A, Carr D, Deng Y, Jin W, Black S, Long B, Liu J, Dinunzio E, Windsor W, Zhang R, Zhao S, Angagaw MH, Pinheiro EM, Desai J, Xiao L, Shipps G, Hruza A, Wang J, Kelly J, Paliwal S, Gao X, Babu BS, Zhu L, Daublain P, Zhang L, Lutterbach BA, Pelletier MR, Philippar U, Siliphaivanh P, Witter D, Kirschmeier P, Bishop WR, Hicklin D, Gilliland DG, Jayaraman L, Zawel L, Fawell S, Samatar AA. Discovery of a novel ERK inhibitor with activity in models of acquired resistance to BRAF and MEK inhibitors. Cancer Discov. 2013;3:742–750. doi: 10.1158/2159-8290.CD-13-0070. [DOI] [PubMed] [Google Scholar]
- 46.Hatzivassiliou G, Liu B, O’Brien C, Spoerke JM, Hoeflich KP, Haverty PM, Soriano R, Forrest WF, Heldens S, Chen H, Toy K, Ha C, Zhou W, Song K, Friedman LS, Amler LC, Hampton GM, Moffat J, Belvin M, Lackner MR. ERK inhibition overcomes acquired resistance to MEK inhibitors. Mol Cancer Ther. 2012;11:1143–1154. doi: 10.1158/1535-7163.MCT-11-1010. [DOI] [PubMed] [Google Scholar]
- 47.Fryer RA, Barlett B, Galustian C, Dalgleish AG. Mechanisms underlying gemcitabine resistance in pancreatic cancer and sensitisation by the iMiD lenalidomide. Anticancer Res. 2011;31:3747–3756. [PubMed] [Google Scholar]
- 48.Tomono T, Yano K, Ogihara T. Snail-Induced Epithelial-to-Mesenchymal Transition Enhances P-gp-Mediated Multidrug Resistance in HCC827 Cells. J Pharm Sci. 2017;106:2642–2649. doi: 10.1016/j.xphs.2017.03.011. [DOI] [PubMed] [Google Scholar]