

Abstract
Cyclin-dependent kinase 4 (CDK4) and CDK6 together with D-type cyclins (D1, D2 and D3) to promote cell cycle entry and progression through G1 by inactivating retinoblastoma protein (RB) by inhibiting an INK4 family of CDK inhibitors (CDKN2A/B). Selective cyclin-dependent kinase inhibitors are game changers in the clinical management of hormone receptor-positive/HER2-negative advanced breast cancers. There are currently three CDK4/6 inhibitors that have been approved by the US Food and Drug administration: palbociclib, ribociclib, and abemaciclib. Although, the bulk of the data supporting the use of selective CDK4/6 inhibitors is currently in breast cancer patients with other tumor types are expected to benefit as well from this class of agents, which can counter proliferative signaling pathways and arrest cell cycle in early G1 phase. Areas of active interest include identifying predictive biomarkers for CDK4/6 inhibitors, deciding whether to continue CDK4/6 inhibitor after disease progression, creating novel treatment combinations and expanding the use of CDK4/6 inhibitors beyond hormone receptor-positive/HER2-negative advanced breast cancer. Right now, CCND1 amplification and CDKN2A/B loss have not sorted out biomarkers useful for the purpose. One of the most important clinical questions is how to use a CDK4/6 inhibitor with other targeted therapies. Here we provide a rationale that oncologists can use to sequence the CDK4/6 inhibitors along with the PI3K-AKT-mTOR pathway-specific inhibitor(s); future data will better guide this approach. In this review we have tried to (a) describe the specific cellular signals initiated following alterations in the cell cycle pathway genes and the PI3K pathway genes, (b) interrogate how these alterations/co-alterations influence the action of PI3K and cell cycle pathway-targeted drugs in different clinical trials and (c) understand the role of co-alterations towards the development of cell cycle inhibitors induced drug-resistance in ER+ breast cancers.
Keywords: ER+ breast cancer, PI3K/AKT/mTOR pathway, CYCLIN D1, CDK4/6, retinoblastoma protein (RB)
Introduction
Approximately 75% of breast cancer (BC) patients express ER and/PR biomarkers, and these are indicative of steroid hormone dependency [1]. This hormone dependency in hormone receptor-positive (ER+) breast cancer is exploited therapeutically in three major ways, 1) selective ER modulators (SERMs; tamoxifen), 2) selective ER down-regulators (SERDs; fulvestrant) and 3) blocking estrogen biosynthesis (aromatase inhibitors; AIs). Endocrine therapy remains the therapeutic backbone for the treatment of ER+ cancers. However, anti-estrogen therapies are initially frequently effective, 50% of ER+ patients develop resistance to hormonal manipulation within their lifetime, ultimately leading to therapeutic failure. Two emerging mechanisms of endocrine resistance include activation of growth signaling pathways such as the phosphatidylinositol 3-kinase (PI3K) pathway and more recently, the decoupling of cell cycle control from ER-signaling, via deregulation of the CYCLIN D dependent kinase (CDK-4/6-INK4-RB) pathway besides HER2 amplification [2,3].
Literature references have shown that activation of the PI3K-AKT-mTOR signaling pathway promotes resistance to endocrine-based therapies [4,5]. The PI3K-AKT-mTOR pathway has been the focus of attention in ER+ breast cancer for the last few years. Everolimus (mTOR inhibitor) in combination with aromatase inhibitor was approved in ER+/HER2-negative advanced breast cancers [6]. Pan-PI3K inhibitor in combination with hormonal therapy (HR) showed a modest response in advanced hormone receptor-positive, HER2-negative breast cancer but was associated with significant toxicities [7]. Most recently, p110α specific PI3K inhibitors with better toxicity profile have been developed and are being studied in clinical trials in patients with HR-positive, HER2-negative advanced breast cancer [8].
Cyclin-dependent kinase 4 (CDK4) and CDK6 together with D-type cyclins (D1, D2 and D3) to promote cell cycle entry and progression through G1 by inactivating retinoblastoma protein (RB) by inhibiting an INK4 family of CDK inhibitors (CDKN2A/B). Recent data demonstrated that CDK4 induces cell cycle progression also plays a key role in hormone-independent cell growth [9]. Aberrant activation of CDK leads to dysregulated cell cycle progression from G1 to S phase and is a common feature of many cancers, including BC. Recently, some cell cycle proteins particularly those of CYCLIN D1-CDK4/6-RB network has been shown to exert oncogenic roles in a variety of cancers including BC, suggesting that their therapeutic exploitation may improve outcome in BC patients [9,10]. Published data (PALOMA-1) showed that the addition of palbociclib (an oral small molecule inhibitor for CDK4/6) to letrozole significantly improved progression-free survival (PFS) in women with HR-positive, HER2-negative advanced BC who had not received systemic treatment for advanced disease [11]. Based on this trial, FDA granted accelerated approval to palbociclib and letrozole for treatment of postmenopausal women with HR-positive, HER2-negative metastatic BC who had not received prior endocrine-based therapy. Ribociclib was the second CDK4/6 inhibitor which recently got FDA approval as a first-line treatment for HR-positive, HER2-negative advanced BC in combination with an aromatase inhibitor (AI) in postmenopausal women based on MONALESSA-2 trial [12]. Most recently, abemaciclib (another CDK4/6 inhibitor) in combination with AI has been approved as a first-line treatment in postmenopausal women with ER+/HER2-negative advanced or metastatic breast cancers [13].
Multiple fundamental studies have led to CDK4/6 inhibitors such as palbociclib, ribociclib, and abemaciclib becoming the standard of care for women with metastatic HR-positive/HER2-negative BC. Genetic analyses reveal that these agents are necessary but not sufficient for an effective treatment strategy for some ER+/HER2-negative metastatic breast cancer patients. At the 11th European Breast Cancer Conference in Barcelona, Spain (June 2018), Prof, Nick Turner, a consultant medical oncologist with The Royal Marsden Hospital NHS Trust, explained how HR-positive tumors resist treatment with CDK4/6 inhibitors and how that resistance can be overcome. The PI3K/mTOR pathway is critical in ER-positive breast cancer. In the PALOMA-3 trial of palbociclib, PIK3CA mutations were associated with approximately 5 folds increased the risk of development of resistance of this inhibitor within several months. Prof. Turner mentioned that PI3 kinase inhibitors had demonstrated synergy in several studies with CDK4/6 inhibitors. Remarkably, a recent disclosure of the results of the SOLAR 1 study from Novartis at ESMO, October 2018 showed that BYL719 (PI3K alpha-selective inhibitor, alpelisib) is highly effective in patients’ tumors with ER+/PIK3CA mutations who were already exposed to CDK4/6 inhibitor along with fulvestrant. This article describes a rationale that oncologists can use to sequence the CDK4/6 inhibitors along with the PI3K-AKT-mTOR pathway-specific inhibitor(s); future data will better guide this approach.
Estrogen receptor pathway in breast cancer
Estrogen receptor (ER) is a group of nuclear receptor family. Estrogen receptor (ER) alpha is composed of NH2-terminal domain, two transcriptional activation function domains (AF1, AF2), DNA binding domain, and C-terminal binding domain [14]. The NH2-terminal encodes both ligand-dependent and ligand-independent activation function. AF1 is activated by phosphorylation. AF-2 is integral to ligand binding domain, and its activation requires ligand binding domain to bind to E2. The DNA binding domain contains two zinc finger structures which play an important role in receptor dimerization and the binding of the receptor to specific DNA sequences [14-16]. The C-terminal domain, upon ligand binding causes receptor dimerization, nuclear translocation, and transactivation of target gene expression. The membrane-bound ERs cross-talk with other signaling pathways like IGF-1R, EGFR, FGFR and HER2 which are involved in cell proliferation, survival, differentiation, and apoptosis [17]. The membrane-bound ER can cause activation of mitogen-activated protein kinase (MAPK) signaling, activation of cyclic adenosine monophosphate (cAMP) and activation of the PI3K pathway [18]. Another mechanism by which ER modulates gene expression through protein-protein interactions for example activation protein-1 (AP-1) and nuclear factor k-β (NF-kβ) [19]. Phosphorylation of estrogen receptor can cause ligand-independent ER activation through other signaling pathways including regulation of general phosphorylation state such as protein kinase A (PKA), protein kinase C (PKC), extracellular signals like peptide growth factors, cytokines, cell-cycle regulators [15]. Several cytokines, growth factors, and other pathways phosphorylate ER at a key position in the AF-1 domain and cause ligand-independent activation of ER. For example, ERK 1/2 pathway phosphorylates at serine 104 and serine 106 in the AF-1 domain and causes ligand-independent activation of ER [20]. AKT mediated phosphorylation of ER at serine 167 increases ER α binding to DNA and interaction with co-activator SRC3 in the presence of E2 [20]. Serine 167 is also phosphorylated by p90 ribosomal S 6 kinase (RSK), a kinase downstream of the MEK-ERK pathway [21].
Tamoxifen in ER-positive breast cancer
Tamoxifen is a selective ER modulator which when binds to AF2 causes a conformational change in AF2, releasing the suppression of AF1 and causes the repression of AF2 transcriptional activity [22]. A meta-analysis from EBCTCG showed that adjuvant tamoxifen for 5 years reduced the recurrence of annual BC death by 31% [23]. International breast-cancer-study-group (IBCSG) trial showed that premenopausal women with HR-positive, node-positive BC who received adjuvant tamoxifen for 5 years, after receiving chemotherapy had disease-free survival of 75% as compared to 62% in patients who did not receive adjuvant tamoxifen (HR: 0.59) [24]. ATLAS trial showed that continuing adjuvant tamoxifen for up to 10 years further decreases the cumulative risk of BC during 5-14 years to 21.4% versus 25.1% for women who stopped adjuvant tamoxifen after five years. There was 2.8% decrease in BC mortality during years 5-14 in women who continued adjuvant tamoxifen for 10 years versus who stopped tamoxifen after five years (breast cancer mortality during years 5-14 was 12.2% for women allocated to continue versus 15.0% for controls) [25]. NSABP trial showed that adjuvant tamoxifen in node-negative, HR-positive BC increases disease-free survival to 83% versus 77% in patients who did not receive adjuvant treatment [26]. At 10 years follow-up, patients who received five years of adjuvant treatment with tamoxifen, disease-free survival was 69% as compared to 57% in women who did not receive adjuvant treatment with tamoxifen [27]. Overall, tamoxifen is an effective treatment in the adjuvant setting to decrease the risk of recurrent BC, especially in premenopausal women. Data from the Arimidex study group showed that in postmenopausal women with advanced BC, tamoxifen was associated with increased risk of thromboembolic events and vaginal bleeding as compared to anastrozole [28]. Therefore, aromatase inhibitors (AI) are usually the treatment of choice in postmenopausal women with HR-positive breast cancer in adjuvant and advanced settings.
Aromatase inhibitor (AI) in ER-positive breast cancer
In postmenopausal women, estrogen is no longer produced by ovarian tissue and is predominantly synthesized from non-glandular sources via the aromatase enzyme. Aromatase can be found in several tissues including subcutaneous fat, liver, and muscle; the enzyme has also been isolated in breast cancer cells [Bland KI, Copeland EM. The Breast E-Book: Comprehensive Management of Benign and Malignant Diseases. Elsevier; 2009]. Aromatase inhibitors emerged as an important class of drugs in the early 2000s which significantly improved disease outcome in HR-positive BC, especially in postmenopausal women. AIs block the conversion of adrenal androgens to estrogen in non-ovarian tissue by targeting aromatase enzyme which is the key source of estrogen synthesis in postmenopausal women. Three different AIs are active in clinical use (steroidal and non-steroidal) namely anastrozole (Arimidex), letrozole (Femara) and exemestane (Aromasin).
ATAC trial demonstrated that adjuvant treatment with anastrozole in postmenopausal women with early-stage BC was associated with superior disease-free survival at 4 years (DFS: 86.9% in anastrozole group) as compared with tamoxifen (DFS: 84.5% in tamoxifen group) [29]. A ten-year analysis of ATAC trial continued to show superior DFS, time to recurrence and time to distant metastasis in anastrozole group as compared to tamoxifen [30]. Nabholtz JM et al. showed that anastrozole was associated with improved time to progression (TTP) as compared with tamoxifen when used in the first-line setting in postmenopausal women with HR-positive advanced BC. Median time to progression was 11.1 months in anastrozole group versus 5.6 months in the tamoxifen group [31]. Interestingly, the survival benefits from aromatase inhibitor versus tamoxifen as first-line treatment in postmenopausal women with advanced or metastatic HR-positive, HER2-negative BC is less clear [31,32]. A Cochrane review showed 10% overall survival benefit with AI compared with other non-AI therapies [HR: 0.90, 95% CI: 0.84-0.74] [33]. ESR1 mutant clones evolve in ER+ advanced BC during AI treatment, with ESR1 mutations resulting in constitutively active ER and its downstream signaling activity [34-36]. ESR1 mutations have been recently recognized as one of the mechanisms of endocrine resistance and thought to evolve secondary to hormonal treatment. ESR1 mutations are rarely found in primary BC and have been found in high prevalence in metastatic BC treated with an AI. These mutations occur in the hotspot domain of ligand binding domain (LBD) and cause constitutive activation of the ER pathway and resistance to AI [36]. Jeselsohn R et al. showed that 11 out of 76 patients (11%) with metastatic BC had ESR1 mutation in ligand binding domain (LBD). Nine of these mutations were substitution mutations affecting Y537 and D538 in LBD. None of the 58 primary tumors had the ESR1 mutation [35].
Fulvestrant in advanced breast cancer
Fulvestrant binds to ER monomers and inhibits receptor dimerization, activating function 1 (AF1), and AF2 are rendered inactive, translocation of the receptor to the nucleus is reduced, and degradation of the estrogen receptor is accelerated [37]. Ellis MJ et al. compared fulvestrant 500 mg intramuscularly on day 0, 14, 28 and then every 28 days with anastrozole 1 mg daily in postmenopausal women with HR-positive, HER2-negative metastatic BC. Median overall survival (OS) was 54.1 months in fulvestrant group versus 48.4 months in the anastrozole group [37]. FALCON, a phase 3 randomized, double-blind trial compared fulvestrant 500 mg intramuscularly on day 0, 14, 28 for the first cycle and then every 28 days with anastrozole 1 mg daily in HR-positive, HER2-negative locally advanced breast cancer who had not received prior endocrine therapy. Median PFS was 16.6 month in fulvestrant group versus 13.8 months in anastrozole group [38].
Fribbens C et al. assessed baseline plasma samples of patients (via circulating tumor DNA by digital PCR) enrolled in SOFEA and PALOMA-3 trials for the ESR1 mutation [36]. The SOFEA study randomized patients with advanced HR-positive BC who had progressed on a non-steroidal aromatase inhibitor (NSI), in the adjuvant or metastatic setting, to fulvestrant plus anastrozole, fulvestrant plus placebo, or exemestane [36]. In SOFEA study, ESR1 mutations were detected in 39.1% of patients (63/161). Patients with an ESR1 mutation had improved PFS with fulvestrant containing treatment (N = 45) compared to exemestane alone (N = 18), (HR: 0.52; 95% CI, 0.30 to 0.92; P = .02), whereas patients with wild-type (WT) ESR1 had similar PFS on either treatment (HR: 1.07; 95% CI, 0.68 to 1.67; P = .77).
In PALOMA 3 (Palbociclib Combined with fulvestrant in HR-positive HER2-negative metastatic BC after endocrine failure) trial, patients who had prior endocrine treatment (tamoxifen or an AI) were randomized to receive fulvestrant plus palbociclib, a CDK4/6 inhibitor, or fulvestrant plus placebo [39]. ESR1 mutations were found in the 25.3% of patients (91 of 360), of whom 28.6% (26 of 91) were polyclonal, with mutations associated with acquired resistance to prior AI. Fulvestrant plus palbociclib improved PFS compared with fulvestrant plus placebo in both ESR1 mutant (9.4 months vs. 3.6 months, HR: 0.43; 95% CI, 0.25 to 0.74; P = .002) and ESR1 wild-type patients (9.5 vs. 5.4 months HR: 0.49; 95% CI, 0.35 to 0.70; P < .001). Although median PFS seemed to be slightly worse in the ESR1 mutated patients treated with fulvestrant alone (3.6 months 95% CI, 2.0-5.5) compared to ESR1 wildtype (5.4 months 95% CI, 3.5-7.4), this was not statistically significant. One interesting finding was that ESR1 mutations were almost exclusively found in patients with prior AI exposure with or without tamoxifen and were rare in patients with prior tamoxifen exposure only (28.9% [90 of 311] vs. 2.0% [one of 49], respectively; P = .001) [33,40,41]. Hamilton et al. studied AZD9496, a new oral selective estrogen receptor degrader in ER+, HER2-negative advanced BC in a phase I trial. Total of 45 patients was enrolled; one patient showed partial response and 10 patients (22.2%) showed stable disease at 6 months or longer. The main adverse effects were diarrhea, nausea, upper abdominal pain [42].
In a recent San Antonio breast cancer meeting (2017) Dickler M et al. presented their phase I data with GDC-0927. GDC-0927, an oral selective estrogen receptor degrader (SERD) showed promising activity in postmenopausal women with ER+, HER2-negative metastatic breast cancer who had progressed on prior endocrine treatment and cytotoxic chemotherapy. Nine out of 12 patients who had baseline FES-PET [18F-fluoroestradiol] avid disease showed complete or near complete (> 90%) suppression of FES uptake to background levels, including patients with ESR1 mutations. Evidence of reduced ER levels and Ki67 staining was observed in on-treatment biopsies. Clinical benefit rate (number of patients who remain on study for 24 weeks or greater) was seen in 5 of 12 (CBR = 41.6%). One patient with ESR1 mutation (D538G) remained on study for over 490 days. GDC-0927 had a favorable toxicity profile as almost all adverse effects were grade 1-2. The most common treatment-related adverse effects were nausea (54%, n = 7), diarrhea (46%, n = 6), elevated aspartate aminotransferase (39%, n = 5) and anemia, constipation, (each 31%, n = 4) [43].
Resistance to hormonal therapy in hormone receptor-positive breast cancer
Endocrine treatment in HR-positive metastatic BC is associated with only 30% objective regression, and about another 20% of patients experience prolonged stable disease [18]. There are several mechanisms that have been identified as the cause of endocrine resistance including aberrations at the level of ER, over-expression of ER co-activators, down-regulation of co-repressors, hyperactivation of the PI3K-AKT-mTOR pathway, upregulation of positive regulators of cell cycle causing cell proliferation, downregulation of negative regulators of cell cycle which block the antiproliferative effects of endocrine therapy [18,44,45]. Here, we discuss few mechanisms of endocrine resistance which have been studied extensively and have therapeutic agents already in clinical use or in clinical trials for the treatment of ER-positive BC.
Role of PI3K/AKT/mTOR pathway in endocrine resistance and targeting PI3K/AKT/mTOR pathway
The PI3K/AKT/mTOR pathway is frequently upregulated in estrogen-deprived BC cells. The downstream effectors of the PI3K pathway, AKT-mTOR axis impacts on tumor cell growth, survival, motility, and metabolism (Figure 2A). AKT is an important downstream target of the PI3K pathway that regulates cell survival, proliferation, growth, apoptosis, and glycogen metabolism [46,47]. Increased AKT kinase activity is detected in 20% to 55% of cancers [47]. mTOR is a downstream effector of AKT. mTOR comprises two different structurally similar but functionally different complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). The mTORC1 is the target of rapamycin and rapamycin analogs (rapalogues), such as everolimus. Rapalogues exert their effect mainly on mTORC1, and the incomplete inhibition can lead to feedback loops causing paradoxical activation of AKT to orchestrate proliferative effects via downstream targets. mTOR’s downstream substrate S6 kinase can phosphorylate and activate functional domain of ER leading to the ligand-independent receptor activation [48-52]. The enzymatic activity of PI3K is antagonized by the phosphatase and tensin homolog (PTEN), a protein that catalyzes the dephosphorylation of PIP3, and by inositol polyphosphate-4-phosphatase type II (INPP4B), a protein that catalyzes the dephosphorylation of PIP2 [53].
Figure 2.
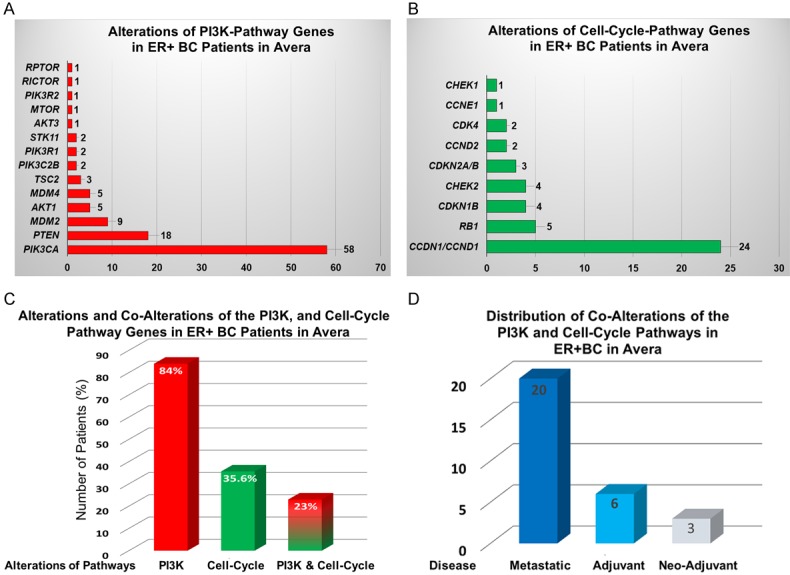
Number of alterations in 14 PI3K-AKT-mTOR signaling genes and 9 cell-cycle pathway genes in patients with ER+ breast cancer at Avera: Alterations in genes in patients with breast cancers enrolled at Avera Cancer Institute with ER+ disease were evaluated for the study. We identified alterations in 14 PI3K-AKT-mTOR pathway genes and 9 cell-cycle pathway genes in 129 patients with ER+ breast cancer at Avera. Alterations of genes were determined from the reports of Foundation Medicine (for DNA sequencing). Genes of the PI3K pathway is altered in 109 patients out of 129 ER+/PR+BC patients (84%; n = 129). A number of alterations of the PI3K pathway showed that the highest alterations were observed in the PIK3CA gene (A). Genes of the cell-cycle pathway are altered in 46 patients out of 129 ER+/PR+BC patients (35.6%; n = 129). A number of alterations of the cell-cycle pathway showed that the highest alterations were observed in the CCND1 gene (B). Percentage of alterations and co-alterations of the PI3K, and cell-cycle pathway genes in ER+BC patients showed 23% of co-alterations (C). Distribution of co-alterations of the PI3K and cell-cycle pathways in ER+BC in Avera showed that co-alterations are predominantly present in patients with metastatic diseases (D).
The study that led to the FDA approval of everolimus was the BOLERO-2-trial. BOLERO-2 compared everolimus (10 mg/day) plus exemestane (25 mg/day) to exemestane alone in 724 postmenopausal women with advanced HR-positive, HER2-negative breast cancer, whose cancer had either progressed or recurred while receiving non-steroidal aromatase inhibitor [6]. The primary endpoint after a median follow-up of 18 months indicated that a co-inhibition of the compensatory pathway via everolimus led to a doubling of PFS (local assessment: 7.8 vs. 3.2 months, HR 0.45, 95% CI 0.38-0.54, log-rank p < 0.0001; central assessment: 11.0 vs. 4.1 months, HR 0.38, 95% CI 0.31 to 0.48, log-rank p < 0.0001) [54]. The biomarker studies from the BOLERO-2 trial showed a greater benefit from everolimus in patients with minimal genetic alterations in PIK3CA/PTEN/CCND1 or FGFR1/2 genes combined, which represented 76% of the next-generation sequencing population. Patients with a single alteration in one of these pathways had a median PFS of 214 days with everolimus, compared to 77 days with placebo (HR: 0.26) [55]. Components of the PI3K pathway are frequently altered in human cancers [56]. Mutations in the gene that constitute the phosphatidylinositol 3-kinase (PI3K) pathway occur in > 70% of breast cancers [45]. The somatic mutations in the components of PI3K pathway include mutations and/or amplification of the genes encoding PI3K catalytic subunits p110α (PI3KCA), p110β (PIK3CB), PIK3R1 which encodes the PI3K regulatory subunit p85α and other genes encoding the PI3K associated modulators (namely PTEN, AKT, MTOR, RICTOR, INPP4B and STK11) [45]. PIK3CA mutations are the most common genetic alterations of this pathway and are seen in 36% of ER+ breast cancers [45,56], Cancer Genome Atlas (TCGA) and the Catalogue of Somatic Mutations in Cancer (COSMIC) databases. These mutations frequently occur in the helical domain (hotspots E545K and E542K) or in the kinase domain (hotspot H1047R) of the PIK3CA-encoded p110α [49,56]. The PIK3R1, the p85α regulatory subunit which inhibits the catalytic activity of p110α (Figure 1B). The p85α also binds to PTEN, preventing PTEN ubiquitination and increasing its protein stability [57]. The increase in PTEN protein expression and stability induced by WT p85α was also inhibited by co-expression of E160 PIK3R1 mutant [58]. Functional mutants in PIK3R1 cannot bind p110α and PTEN, leading to upregulation of PI3K signaling [58]. Very recently, it has been reported by others that a high proportion of tumors harbors multiple mutations, especially PIK3CA plus PIK3R1 mutations [59]. Several preclinical studies showed that PI3K mutant cancers are sensitive to PI3K inhibitors and show synergy with endocrine therapies to decrease tumor progression (Figure 1A) [4,5]. This concept was first studied in the clinical setting by Dr. Baselga et al. in the BELLE-2 trial. BELLE-2 was a phase III double-blind, placebo-controlled randomized trial which included patients with advanced HR-positive, HER2-negative BC whose cancer had progressed on or after AI. The patients were randomly assigned to receive buparlisib (BKM120, a pan PI3K class inhibitor) combined with fulvestrant versus placebo and fulvestrant. Overall median PFS was modestly improved in the buparlisib group versus the placebo group (6.9 months [95% CI 6.8-7.8] vs. 5.0 months [4.0-5.2]; HR: 0.78 [95% CI 0.67-0.89], P = 0.00021). However, buparlisib was poorly tolerated with side-effects including hyperglycemia, diarrhea, stomatitis, and rash, increase in alanine transaminase and aspartate transaminase and frequent mood disorders, including three cases of suicidal ideation in the buparlisib group [7,60]. Exploratory analysis of BELLE-2 trial showed that patients with PIK3CA mutation detected in circulating tumor DNA derived benefit from buparlisib (median PFS 7.0 months vs. 3.2 months in the placebo group, stratified HR: 0.58) whereas patients without PIK3CA mutations did not drive any benefit from buparlisib (median 6.8 months vs. 6.8 months for the placebo group, stratified HR: 1.02). This exploratory analysis strongly suggests that PIK3CA is a potential target, although more tolerable PI3K inhibitors are needed to exploit this potential. PI3K has several catalytic subunits, and the toxicity from buparlisib is partly a consequence of non-selective inhibition of all four isoforms of class 1 PI3Ks (α, β, γ and d). More selective inhibition of the mutant PIK3CA α-subunit has the potential to open the therapeutic window and improve efficacy through more potent inhibition of PI3K in tumor cells [61].
Figure 1.
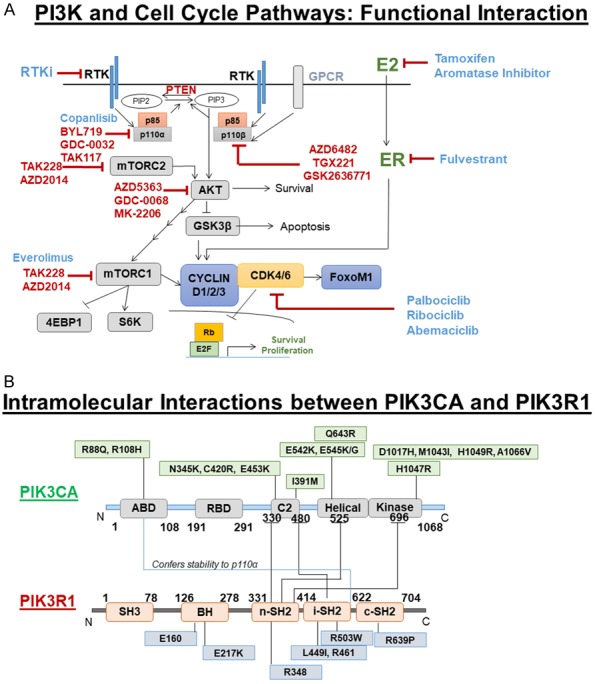
A. The Mechanistic relationship between PI3K pathway activation and cell-cycle in tumor cells: PI3K signaling engages AKT, which drives mTORC1 CAP-dependent mRNA translation encoding CYCLIN D1/D2/D3. CYCLIN D1/D2/D3 bind CDK4/6 to promote RB phosphorylation, which depresses E2F transcription factor to drive expression of genes that promote proliferation and suppress apoptosis. CDK6 can phosphorylate FOXOM1 to enhance FOXOM1 stability, which suppresses senescence. Blue: FDA approved drugs and RED: Active in a clinical trial. B. Intramolecular interactions between PIK3CA and PIK3R1: Domain-specific regulation of PIK3CA (a p110α catalytic subunit of PI3K) by PIK3R1 (p85α regulatory subunit of PI3K) as scaffolds for regulated complex formation. Distribution of some important non-synonymous mutations of PIK3CA and PIK3R1 were depicted.
Recently, alpelisib (BYL719, a PI3K alpha-specific Inhibitor) in combination with letrozole was evaluated in phase Ib study in patients with HR-positive, HER2-negative metastatic BC whose tumor has progressed on endocrine therapies. The combination of alpelisib and letrozole showed clinical activity with a much better safety profile compared with pan P13K inhibitors. The clinical benefit rate (lack of progression [greater than or equal to 6 months] was 35% (44% in patients with PIK3CA mutated and 20% in PIK3CA wild-type tumors; 95% CI: 17%-56%) including five objective responses. Of eight patients remaining on treatment (greater than or equal to) for 12 months, six had tumors with PIK3CA mutation. Those with FGFR1/2 amplification, KRAS, and TP53 mutations did not derive clinical benefit [8]. Results from two phase III trials of more α-selective PI3K inhibitors are awaited: SOLAR-1 (alpelisib and fulvestrant; NCT 02437318) and SANDPIPER (taselisib and fulvestrant; NCT 02340221). During the recent ASCO 2018 meeting, Prof. Baselga revealed the SANDPIPER trial (dated 06-02-18) results. The study enrolled 516 postmenopausal women with locally advanced or metastatic HR-positive, HER2-negative breast cancer that worsened or recurred despite initial hormone treatment with AIs. Women were randomly assigned to receive fulvestrant and placebo (176 women) or fulvestrant and taselisib (GDC-0032) (340 women). Women who received taselisib and fulvestrant had a 30% lower chance of cancer worsening than those who received fulvestrant and a placebo, and taselisib extended the time until cancer worsened by a median of 2 months (7.4 months with taselisib/fulvestrant vs. 5.4 months with fulvestrant/placebo). The response rate to treatment was more than doubled when taselisib was added (28% vs. 11.9%). Overall survival data is not yet available. The most common severe side effects for patients who received taselisib were diarrhea, hyperglycemia, and colitis. Due to side effects, 17% of women who received taselisib stopped treatment early, compared to only 2% of those who did not receive the targeted therapy. Most interestingly, when investigators looked at outcomes by geographic area, they noted that taselisib provided more benefit to study participants who received treatment in North America and Europe, where cancer worsening was delayed by a median of 3.5 months (7.9 with taselisib/fulvestrant vs. 4.5 months with fulvestrant/placebo). In other countries including Eastern Europe and Latin America, taselisib appeared to provide very little or no added benefit. More research is needed to understand the reasons for this discrepancy [62].
Role of cell cycle pathway in endocrine resistance and targeting CYCLIN D-CDK4/6-RB pathway
The mammalian cell cycle is a series of highly regulated processes that cause replication and cell division. The cell cycle is divided into four phases G1, S, G2 and M phase [63,64]. The transition from G1 to S phase is regulated by cyclin-dependent kinases (CDKs). In the presence of mitogenic signals, cyclin-dependent kinases (CDK4 and CDK6) interact with CYCLIN D and form CYCLIN D-CDK4/6 complexes which phosphorylate retinoblastoma protein (RB) and lift repression of E2F transcription genes and causes progression from G1 to S phase (Figure 1A) [65,66]. Activities of CDKs are affected by naturally occurring cyclin-dependent kinase inhibitors (CKI). Depending on target CDK, CKI belongs to two families. INK4 protein family includes p16 INK4a, p18INK4c, p19 INK4d, p15 INK4b. CYCLIN D, E, and A-dependent kinases are inhibited by Cip/kip family of CDKI [67].
Several oncogenic pathways including the RAS-MAPK and PI3K pathways can stimulate CYCLIN D-CDK4/6 complexes which phosphorylate RB (inactivation of RB) and cause cell proliferation. Several mechanisms including amplification of genes encoding CYCLIN D1 (CCND1), CDK4 or CDK6 mutation, loss of tumor suppressor genes (CDKN2A) which encodes p16 INK4a activate CYCLIN D-CDK4/6-INK4-RB pathway. CCND1 amplification is seen in 35% of HR-positive BCs [56,68]. CDK4 amplification is seen in 16% of BCs [56,69]. Loss of p16 INK4a is seen in 49% of BCs [56,69].
Pre-clinical data showed that inhibition of CYCLIN D dependent kinase activity causes tumor shrinkage, reversal of oncogenicity, and in some cases, transformed cell death [10,70]. Selective CDK4/6 inhibitors “turn off” these kinases and dephosphorylated RB (active), resulting in a block of cell cycle progression in mid-G1 phase (Figure 1A). This causes cell cycle arrest and prevents the proliferation of tumor cells [10]. Tumor cells must express wild-type RB for the selective CDK4/6 inhibitor to be effective. Currently, three CDK4/6 inhibitors (Palbociclib, Abemaciclib, Ribociclib) have been approved for the treatment of HR-positive, HER2-negative BC in an advanced and metastatic setting in combination with endocrine therapy. ER+ breast cancer seems to be especially dependent upon CDK4 for proliferation. The tight linkage of the ER signaling pathway to CYCLIN D1-CDK4 may be one reason why BC cells are highly sensitive to the anti-proliferative effects of drugs when they are combined with endocrine therapy. With the combination of a CDK4/6 inhibitor and anti-estrogen therapy, there is an enhancement of G1 and cell-cycle arrest and probably enhanced entry into a senescent state. To date, ER+BC is the malignancy for which this class of drugs has proven most effective and for which we have the most mature data from randomized trials comparing endocrine therapy alone or with endocrine therapy plus CDK4/6 inhibitors.
PALOMA-1 was a double-blind study which included 165 postmenopausal women with HR-positive, HER2-negative BC who had not received systemic treatment for advanced disease. The patients were randomized to receive palbociclib and letrozole or placebo plus letrozole. The median PFS in palbociclib and letrozole group was 20.2 months versus 10.2 months in letrozole group (HR: 0.488, 95% confidence interval (CI) 0.319-0.748, P = 0.0004) [11]. Based on results from the PALOMA-1 trial, FDA granted accelerated approval to palbociclib in combination with letrozole for the treatment of postmenopausal women with ER+/HER2-negative advanced BC [71]. PALOMA-3 (NCT 01942135) compared palbociclib and fulvestrant versus placebo and fulvestrant in (pre and post-menopausal) women with ER+/HER2-negative BC who had relapsed or progressed during or after endocrine therapy. Pre and peri-menopausal women also received goserelin (Zoladex). Palbociclib and fulvestrant showed improved efficacy versus fulvestrant alone (median PFS was 9.5 months versus 4.6 months, HR: 0.46, 95% confidence interval 0.36-0.59, P ≤ 0.001) [72]. FDA approved palbociclib in combination with fulvestrant for the treatment of women with HR+, HER2-negative advanced or metastatic BC with disease progression following endocrine therapy in February 2016 [73].
MONARCH-3 (NCT 02246621) compared abemaciclib and nonsteroidal aromatase inhibitor versus placebo and a nonsteroidal aromatase inhibitor in HR-positive, HER2-negative advanced BC who had no prior systemic therapy in the advanced setting. The combination of abemaciclib and AI was statistically better compared to placebo and AI alone (HR: 0.54; 95% CI, 0.41 to 0.72; P = .000021; median PFS: not reached in the abemaciclib arm, 14.7 months in the placebo arm) [13].
MONALEESA-2 (NCT 01958021), a phase III study compared combination of ribociclib with letrozole versus placebo and letrozole as first-line treatment in postmenopausal women with ER+, HER2-negative advanced BC. The duration of PFS was significantly longer in Ribociclib plus letrozole group than the placebo group (HR: 0.56; 95% CI, 0.43-0.72; P = 3.29 × 10-6 for superiority) [12]. Results of the second interim analysis of MONALEESA-2 were recently published. The median duration of follow-up was 26.4 months. Ribociclib and letrozole was associated with improved median PFS vs. placebo and letrozole alone [median PFS was 25.3 months in ribociclib and letrozole group (95% CI, 23.0-30.3) and 16.0 months (95% CI, 13.4-18.2) in placebo and letrozole group (HR: 0.568; 95% CI, 0.457-0.704; log-rank P = 9.63 × 10-8)]. The objective response rate (ORR) was 42.5% versus 28.7% for all patients treated with ribociclib plus letrozole versus placebo plus letrozole, respectively, and 54.5% versus 38.8%, respectively, for patients with measurable disease. Overall survival data remain immature, with 116 deaths observed; 50 in the ribociclib arm and 66 in the placebo arm (HR: 0.746; 95% CI, 0.517-1.078). The benefit with ribociclib was maintained irrespective of PIK3CA or TP53 mutation status, total RB, Ki67, or p16 protein expression, and CDKN2A, CCND1, or ESR1 mRNA levels. Ribociclib benefit was more pronounced in patients with wild-type receptor tyrosine kinase (RTK) genes as compared to altered RTK genes [74]. Interestingly, biomarker analysis from PALOMA-2 did not show any correlation between expression of RB, Ki67, CYCLIN D1, and p16 in response to palbociclib plus letrozole [75].
Several mechanisms of resistance to CDK4/CDK6 inhibitors have been proposed including CYCLIN E amplification or overexpression, E2F3 amplification, loss of p27 kip1 or p21 cip1, activation of CDK2, acquired CDK6 amplification [76-78]. Herrera-Abreu TM et al. showed that chronic inhibition by CDK4/6 inhibitor causes an increase in AKT phosphorylation which correlates with sustained expression of E2F induced G1-S phase regulators such as CYCLIN E2 or CDK2 which cause failure to complete inhibition of RB phosphorylation [79]. They also showed that combining PI3K and CDK4/6 inhibition resulted in loss of RB phosphorylation with concomitant reduction of CYCLIN E2 and CDK2 expression, resulting in cancer cell apoptosis and tumor regression in vivo and in patient-derived tumor xenograft models, providing rationale for combination of PI3K and CDK4/6 combination treatment for synergistic response and to prevent resistance to CDK4/6 inhibitors when used alone. Yang C et al. found that in cell line models several clones emerged after prolonged exposure to CDK4/6 inhibitor (abemaciclib) that were found to harbor amplification of the CDK6 kinase. Amplification of CDK6 resulted in a marked increase in CDK6 expression and reduced response to CDK4/6 inhibitors. CDK6 overexpression also led to reduced expression of the ER and progesterone receptor (PR), and diminished responsiveness to ER antagonism. Alternative mechanisms of resistance to CDK4/6 inhibitors such as loss of pRB and CYCLIN E1 overexpression also exhibited decreased hormone responsiveness [80]. Recently, Condorelli R et al. analyzed genotyping samples of three patients with metastatic BC via circulating tumor DNA before initiation of treatment with CDK4/6 inhibitor and after disease progression while on CDK4/6 inhibitor. All three patients showed acquired polyclonal RB1 mutation after exposure to the CDK4/6 inhibitor. None of these patients had RB1 mutation before exposure to CDK4/6 inhibitor [81].
Combined inhibition of CDK4/6 and PI3K/AKT/mTOR pathway signaling
Combined CDK4/6 inhibitors and PI3K inhibitors have been investigated pre-clinically primarily in models of BC, where co-activation of the PI3K and cell cycle pathways frequently occur, with the most promising results emerging with PI3K α-isoform-selective drug, since co-alteration of PIK3CA and CYCLIN D1 are often found in ER+BC cohort [82] (See Figure 2). Vora SR et al. showed that combination of CDK4/6 inhibitors and PI3K inhibitor have a synergistic effect of decreasing cell proliferation in PI3KCA mutant BC cell lines which were resistant to PI3K inhibitor [83]. The addition of a PI3K inhibitor to combined CDK4/6 inhibition and hormonal therapy is particularly attractive for PIK3CA mutant ER+BC patients. A recent preclinical data showed that the triplet combination (letrozole/fulvestrant plus ribociclib plus PI3K pathway inhibitor) increased the tumor growth inhibition (TGI) even further, inducing tumor regressions in all xenograft models. The response was independent of PIK3CA or PTEN mutation status. Complete tumor regressions were observed for a subset of mice within each of the triple combination arms. Tumor regressions were maintained for up to four weeks post-interruption of treatment [84]. Interestingly, it has been reported by others that the combined inhibition of CDK4/6 and PI3K alpha isoform is synergistic and immunogenic (synergistically increased tumor infiltrating lymphocytes activation, cytotoxicity and decreased the frequency of myeloid-derived suppressor cells) in triple negative BC models. It simply suggests that this combination is not restricted to ER+/HER2-negative breast cancer [85].
Alterations of PI3K and cell-cycle pathway genes in patients with ER+ breast cancers at Avera
We have included 129 breast cancer patients evaluated at Avera with ER+ disease for the study. A close to 130 genes of different pathways have been identified to be altered in this set of patients. We identified alterations in 14 PI3K-AKT-mTOR signaling genes and 9 cell-cycle pathway genes in 129 patients with ER+ breast cancer at Avera. Alterations of genes were determined from the reports of Foundation Medicine (for DNA sequencing). The genes of the PI3K pathway is altered in 109 patients out of 129 ER+/PR+BC patients (84%; n = 129). The identified genes of the PI3K pathway are PIK3CA, PTEN, MDM2, AKT1, MDM4, TSC2, PIK3C2B, PIK3R1, STK11, AKT3, MTOR, PIK3R2, RICTOR and RPTOR in their descending order of magnitude. The highest number of alterations were observed in the PIK3CA gene (45%; n = 129) which was followed by alterations in the PTEN gene (14%; n = 129) (Figure 2A). Prominent alterations in the PI3K pathway included mutations [e.g. PIK3CA (E542K), AKT1 (E17K)], amplifications (MDM2), frame shifts [TSC2 (R1753fs*58+)] and loss [PTEN] (Table 1). The genes of the cell-cycle pathway are altered in 46 patients out of 129 ER+/PR+BC patients (35.6%; n = 129). The identified genes of the cell-cycle pathway are CCND1, RB1, CDKN1B, CHEK2, CDKN2A/B, CCND2, CDK4, CCNE1 and CHEK1 in their descending order of magnitude. The highest number of alterations were observed in the CCND1 gene (18.6%; n = 129) which was followed by alterations in the RB1 gene (3.87%; n = 129) (Figure 2B). Prominent alterations in the cell-cycle pathway included mutations [e.g. CHEK1 (R160H)], amplifications (CCND1), frame shifts [CHEK2 (T533fs*12+)] and loss [CDKN2A] (Table 1). We also evaluated the co-alteration of the PI3K and the cell-cycle pathway genes in these 129 patients with ER+ breast cancers. The co-alteration was observed in 29 patients (23%; n = 129) (Figure 2C). Interestingly, the majority of the co-alterations were observed in patients with metastatic disease (20) followed by adjuvant (6) disease. The least number of co-alterations were observed in patients with neo-adjuvant disease (3) (Figure 2D). Remarkably, a recent disclosure of the results of the SOLAR 1 study from Novartis at ESMO, October 2018 showed that BYL719 (PI3K alpha-selective inhibitor) is highly effective in patients’ tumors with ER+/PIK3CA mutations who were already exposed to CDK4/6 inhibitor along with fulvestrant. We tested the anti-tumor efficacy of the combination of isoform-specific PI3K inhibitors (e.g., BYL719 and AZD6284) and ribociclib using cell line (PIK3CA mutated T47D, MCF7, and PTEN-null MDA-MB415 cells) based model. Data showed a synergistic effect of PI3K inhibitor and CDK4/6 inhibitor as determined from the changes in proliferative and apoptotic signals as well as cell cycle arrests. Results of our study showed that such a combinatorial strategy with PI3K inhibitor and CDK4/6 inhibitor might be beneficial in RB wt ER+ breast cancer with concurrent genetic alterations of these pathways [82]. Several clinical trials are underway to evaluate the synergistic effect of the PI3K inhibitor with CDK4/6 inhibitor. Phase Ib/II study of combination of PI3K inhibitor, taselisib (GDC-0032), or pictilisib (GDC-0941) with palbociclib with subsequent addition of fulvestrant (NCT 02389842) or a combination buparlisib (BKM120)/alpelisib (BYL719) plus ribociclib with fulvestrant in patients with PIK3CA mutant breast cancer is ongoing (NCT 02088684). Michaloglou C et al. showed that mTORC1/2 inhibitor (AZD2014) causes modulation of E2F mediated transcription and cooperate with CDK4/6 inhibitor (palbociclib) in inhibition of E2 function in HR-positive BC [86]. mTOR inhibitor in combination with CDK4/6 inhibitor and exemestane is being studied in a clinical trial (NCT 01857193). In recently disclosed results of PALOMA-3 trial data, PIK3CA alterations levels (from ctDNA) above the median cut off were associated with approximately a fivefold increased risk for development of resistance to this CDK4/6 inhibitor within several months [87]. This recent data clearly indicate that patients with advanced ER+; high PIK3CA mutant burden BC could benefit from the introduction of alpha-specific PI3K inhibitor along with CDK4/6 inhibitor and fulvestrant.
Table 1.
Specific co-alterations in the PI3K and the cell-cycle pathway genes in ER+ BC patients in Avera
| Metastatic | Adjuvant | NeoAdjuvant |
|---|---|---|
| AKT1 (E17K) & CCND1 (Amp) | PIK3CA (E545K) & CCND1 (Amp) | PIK3CA (E545K) & CCND1 (Amp) |
| PIK3CA (E542K) & CCND1 (Amp) | PIK3CA (H1047R) & CDKN2A (Loss) | PIK3CA (E970K, H1047R)/PTEN (R130Q) & RB1 (E31*) |
| CDK4 (Amp) & MDM2 (Amp) | PIK3CA (K111 del)/TSC2 (R1753fs*58+) & CCNE1 (Amp) | PIK3CA (E542K) & CCND1 (Amp) |
| PIK3CA (Amp, H1047R)/MDM4 (Amp) & CCND1 (Amp) | PIK3CA (C420R) & CCND1 (Amp) | |
| PIK3CA (Amp, H1047R)/MDM4 (Amp) & CCND1 (Amp) | TSC2 (A607T) & CHK1 (R160H) | |
| PIK3CA (E724K, H1047L) & CCND1 (Amp)/CHK2 (T533fs*12+) | PIK3CA (E545K) & CHK2 (Splice site 444+1G>A) | |
| PIK3CA (E545K)/MDM2 (Amp)/RICTOR (Amp) & CCND1 (Amp)/CDK4 (Amp) | ||
| PIK3CA (E545K)/PTEN (Loss) & RB1 (E580*) | ||
| MTOR (I1973F-subclonal)/MDM2 (Amp) & CCND1 (Amp) | ||
| AKT1 (E17K) & CDKN1 B (Q65*) | ||
| AKT3 (Amp)/PIK3CA (Amp) & CCND1 (Amp) | ||
| PIK3CA (E726K, Q546R) & RB1 (R556*) | ||
| PIK3CA (E545K) & CCND1 (Amp) | ||
| PIK3CA (H1047L)/PTEN (R233*) & CHK2 (Loss exons 9-10) | ||
| STK11 (Loss)/RPTOR (Amp) & CDKN2A (Loss) | ||
| PIK3CA (C420R) & CDKN1B (F33fs*9) | ||
| PIK3CA (E545K)/PIK3R1 (D464H) & CCND1 (Amp) | ||
| PIK3R2 (Rearrangement intron 13) & CDKN2A/B (Loss) | ||
| PIK3CA (E545K) & CCND1 (Amp) | ||
| PIK3CA (E545K)/PTEN (C134F) & CCND1 (Amp) |
Specific types of alterations in genes of the PI3K and the cell-cycle pathways co-altering in ER+ BC patients with metastatic, adjuvant and neo-adjuvant diseases are presented in separate columns with different colors.
Conclusion
Hormone receptor-positive breast cancer is a very dynamic field and clinical advances in the last 20 years have significantly improved the outcome of this disease. Progression-free survival and overall survival has significantly improved in patients with HR-positive BC. There has been significant research to understand the mechanism of hormone resistance and develop strategies to overcome hormone resistance in BC. Most recently, CDK4/CDK6 inhibitors were added in the armamentarium for the treatment of HR-positive, HER2-negative advanced BC and are being studied in a clinical setting. PI3K alpha specific inhibitors are also being evaluated in clinical trials. Clinical trials are exploring the efficacy of the triplet combination of hormonal therapy, CDK4/CDK6 inhibitor, and inhibitor of the PI3K pathway in advanced HR-positive, HER2-negative breast cancers. The role of immunotherapy is being evaluated in several clinical trials in advanced BC and have shown a more objective response in triple negative and HER2-positive BCs and less in HR-positive breast cancer [88]. The role of immunotherapy, fibroblast growth factor inhibitors in the treatment of HR-positive BC are also being explored, and several clinical trials are underway to evaluate the efficacy of FGFR inhibitors and immunotherapy. Recent studies in the mouse model of breast cancer demonstrated that CDK4/6 inhibitors not only induces tumor cell arrest but also promote anti-tumor immunity in two different ways: 1) CDK4/6 inhibitors activate tumor cell expression of endogenous retroviral elements, increasing intracellular double-stranded RNA, which stimulates the production of interferon III, which then enhance tumor antigen presentation and 2) CDK4/6 inhibitors suppress proliferation of regulatory T cells (Tregs) [89]. Eventually, these events activate cytotoxic T cell-mediated tumor cell clearance.
Next step would be to identify the optimal sequence of combination treatments and to identify predictive biomarkers which would help clinicians to tailor the treatment for the individual patient. The question that which patients with HR-positive BC could be treated by hormonal therapy alone at the time of initial diagnosis and which patients would benefit from combination treatment at the beginning of treatment has yet to be answered. The results from the ongoing clinical trials would help us to better answer these questions and to personalize treatment for each patient with HR-positive BC.
Acknowledgements
The authors acknowledge Genomic Oncology Institute, Department of Molecular and Experimental Medicine, Avera Cancer Institute, Sioux Falls, SD for their support. The authors also acknowledge Foundation Medicine for DNA sequencing.
Disclosure of conflict of interest
None.
References
- 1.Nadji M, Gomez-Fernandez C, Ganjei-Azar P, Morales AR. Immunohistochemistry of estrogen and progesterone receptors reconsidered: experience with 5,993 breast cancers. Am J Clin Pathol. 2005;123:21–27. doi: 10.1309/4wv79n2ghj3x1841. [DOI] [PubMed] [Google Scholar]
- 2.Lipton A, Leitzel K, Ali SM, Demers L, Harvey HA, Chaudri-Ross HA, Evans D, Lang R, Hackl W, Hamer P, Carney W. Serum HER-2/neu conversion to positive at the time of disease progression in patients with breast carcinoma on hormone therapy. Cancer. 2005;104:257–263. doi: 10.1002/cncr.21202. [DOI] [PubMed] [Google Scholar]
- 3.Meng S, Tripathy D, Shete S, Ashfaq R, Haley B, Perkins S, Beitsch P, Khan A, Euhus D, Osborne C, Frenkel E, Hoover S, Leitch M, Clifford E, Vitetta E, Morrison L, Herlyn D, Terstappen LW, Fleming T, Fehm T, Tucker T, Lane N, Wang J, Uhr J. HER-2 gene amplification can be acquired as breast cancer progresses. Proc Natl Acad Sci U S A. 2004;101:9393–9398. doi: 10.1073/pnas.0402993101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crowder RJ, Phommaly C, Tao Y, Hoog J, Luo J, Perou CM, Parker JS, Miller MA, Huntsman DG, Lin L, Snider J, Davies SR, Olson JA Jr, Watson MA, Saporita A, Weber JD, Ellis MJ. PIK3CA and PIK3CB inhibition produce synthetic lethality when combined with estrogen deprivation in estrogen receptor-positive breast cancer. Cancer Res. 2009;69:3955–3962. doi: 10.1158/0008-5472.CAN-08-4450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miller TW, Hennessy BT, Gonzalez-Angulo AM, Fox EM, Mills GB, Chen H, Higham C, Garcia-Echeverria C, Shyr Y, Arteaga CL. Hyperactivation of phosphatidylinositol-3 kinase promotes escape from hormone dependence in estrogen receptor-positive human breast cancer. J Clin Invest. 2010;120:2406–2413. doi: 10.1172/JCI41680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baselga J, Campone M, Piccart M, Burris HA 3rd, Rugo HS, Sahmoud T, Noguchi S, Gnant M, Pritchard KI, Lebrun F, Beck JT, Ito Y, Yardley D, Deleu I, Perez A, Bachelot T, Vittori L, Xu Z, Mukhopadhyay P, Lebwohl D, Hortobagyi GN. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 2012;366:520–529. doi: 10.1056/NEJMoa1109653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baselga J, Im SA, Iwata H, Cortes J, De Laurentiis M, Jiang Z, Arteaga CL, Jonat W, Clemons M, Ito Y, Awada A, Chia S, Jagiello-Gruszfeld A, Pistilli B, Tseng LM, Hurvitz S, Masuda N, Takahashi M, Vuylsteke P, Hachemi S, Dharan B, Di Tomaso E, Urban P, Massacesi C, Campone M. Buparlisib plus fulvestrant versus placebo plus fulvestrant in postmenopausal, hormone receptor-positive, HER2-negative, advanced breast cancer (BELLE-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2017;18:904–916. doi: 10.1016/S1470-2045(17)30376-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mayer IA, Abramson VG, Formisano L, Balko JM, Estrada MV, Sanders ME, Juric D, Solit D, Berger MF, Won HH, Li Y, Cantley LC, Winer E, Arteaga CL. A phase Ib study of alpelisib (BYL719), a PI3Kalpha-specific inhibitor, with letrozole in ER+/HER2-metastatic breast cancer. Clin Cancer Res. 2017;23:26–34. doi: 10.1158/1078-0432.CCR-16-0134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miller TW, Balko JM, Fox EM, Ghazoui Z, Dunbier A, Anderson H, Dowsett M, Jiang A, Smith RA, Maira SM, Manning HC, Gonzalez-Angulo AM, Mills GB, Higham C, Chanthaphaychith S, Kuba MG, Miller WR, Shyr Y, Arteaga CL. ERalpha-dependent E2F transcription can mediate resistance to estrogen deprivation in human breast cancer. Cancer Discov. 2011;1:338–351. doi: 10.1158/2159-8290.CD-11-0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Finn RS, Dering J, Conklin D, Kalous O, Cohen DJ, Desai AJ, Ginther C, Atefi M, Chen I, Fowst C, Los G, Slamon DJ. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009;11:R77. doi: 10.1186/bcr2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Finn RS, Crown JP, Lang I, Boer K, Bondarenko IM, Kulyk SO, Ettl J, Patel R, Pinter T, Schmidt M, Shparyk Y, Thummala AR, Voytko NL, Fowst C, Huang X, Kim ST, Randolph S, Slamon DJ. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol. 2015;16:25–35. doi: 10.1016/S1470-2045(14)71159-3. [DOI] [PubMed] [Google Scholar]
- 12.Hortobagyi GN, Stemmer SM, Burris HA, Yap YS, Sonke GS, Paluch-Shimon S, Campone M, Blackwell KL, Andre F, Winer EP, Janni W, Verma S, Conte P, Arteaga CL, Cameron DA, Petrakova K, Hart LL, Villanueva C, Chan A, Jakobsen E, Nusch A, Burdaeva O, Grischke EM, Alba E, Wist E, Marschner N, Favret AM, Yardley D, Bachelot T, Tseng LM, Blau S, Xuan F, Souami F, Miller M, Germa C, Hirawat S, O’Shaughnessy J. Ribociclib as first-line therapy for hr-positive, advanced breast cancer. N Engl J Med. 2016;375:1738–1748. doi: 10.1056/NEJMoa1609709. [DOI] [PubMed] [Google Scholar]
- 13.Goetz MP, Toi M, Campone M, Sohn J, Paluch-Shimon S, Huober J, Park IH, Tredan O, Chen SC, Manso L, Freedman OC, Garnica Jaliffe G, Forrester T, Frenzel M, Barriga S, Smith IC, Bourayou N, Di Leo A. MONARCH 3: abemaciclib as initial therapy for advanced breast cancer. J. Clin. Oncol. 2017;35:3638–3646. doi: 10.1200/JCO.2017.75.6155. [DOI] [PubMed] [Google Scholar]
- 14.Anbalagan M, Rowan BG. Estrogen receptor alpha phosphorylation and its functional impact in human breast cancer. Mol Cell Endocrinol. 2015;418:264–72. doi: 10.1016/j.mce.2015.01.016. [DOI] [PubMed] [Google Scholar]
- 15.Nilsson S, Makela S, Treuter E, Tujague M, Thomsen J, Andersson G, Enmark E, Pettersson K, Warner M, Gustafsson JA. Mechanisms of estrogen action. Physiol Rev. 2001;81:1535–1565. doi: 10.1152/physrev.2001.81.4.1535. [DOI] [PubMed] [Google Scholar]
- 16.Giuliano M, Schifp R, Osborne CK, Trivedi MV. Biological mechanisms and clinical implications of endocrine resistance in breast cancer. Breast. 2011;20(Suppl 3):S42–49. doi: 10.1016/S0960-9776(11)70293-4. [DOI] [PubMed] [Google Scholar]
- 17.Vrtacnik P, Ostanek B, Mencej-Bedrac S, Marc J. The many faces of estrogen signaling. Biochem Med (Zagreb) 2014;24:329–342. doi: 10.11613/BM.2014.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Osborne CK, Schiff R. Mechanisms of endocrine resistance in breast cancer. Annu Rev Med. 2011;62:233–247. doi: 10.1146/annurev-med-070909-182917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Musgrove EA, Sutherland RL. Biological determinants of endocrine resistance in breast cancer. Nat Rev Cancer. 2009;9:631–643. doi: 10.1038/nrc2713. [DOI] [PubMed] [Google Scholar]
- 20.Thomas RS, Sarwar N, Phoenix F, Coombes RC, Ali S. Phosphorylation at serines 104 and 106 by Erk1/2 MAPK is important for estrogen receptor-alpha activity. J Mol Endocrinol. 2008;40:173–184. doi: 10.1677/JME-07-0165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lannigan DA. Estrogen receptor phosphorylation. Steroids. 2003;68:1–9. doi: 10.1016/s0039-128x(02)00110-1. [DOI] [PubMed] [Google Scholar]
- 22.Sakamoto T, Eguchi H, Omoto Y, Ayabe T, Mori H, Hayashi S. Estrogen receptor-mediated effects of tamoxifen on human endometrial cancer cells. Mol Cell Endocrinol. 2002;192:93–104. doi: 10.1016/s0303-7207(02)00086-2. [DOI] [PubMed] [Google Scholar]
- 23.Early Breast Cancer Trialists’ Collaborative Group (EBCTCG) Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet. 2005;365:1687–1717. doi: 10.1016/S0140-6736(05)66544-0. [DOI] [PubMed] [Google Scholar]
- 24.International Breast Cancer Study Group. Colleoni M, Gelber S, Goldhirsch A, Aebi S, Castiglione-Gertsch M, Price KN, Coates AS, Gelber RD. Tamoxifen after adjuvant chemotherapy for premenopausal women with lymph node-positive breast cancer: International Breast Cancer Study Group Trial 13-93. J. Clin. Oncol. 2006;24:1332–1341. doi: 10.1200/JCO.2005.03.0783. [DOI] [PubMed] [Google Scholar]
- 25.Davies C, Pan H, Godwin J, Gray R, Arriagada R, Raina V, Abraham M, Medeiros Alencar VH, Badran A, Bonfill X, Bradbury J, Clarke M, Collins R, Davis SR, Delmestri A, Forbes JF, Haddad P, Hou MF, Inbar M, Khaled H, Kielanowska J, Kwan WH, Mathew BS, Mittra I, Müller B, Nicolucci A, Peralta O, Pernas F, Petruzelka L, Pienkowski T, Radhika R, Rajan B, Rubach MT, Tort S, Urrútia G, Valentini M, Wang Y, Peto R Adjuvant Tamoxifen: Longer Against Shorter (ATLAS) Collaborative Group. Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years after diagnosis of oestrogen receptor-positive breast cancer: ATLAS, a randomised trial. Lancet. 2013;381:805–816. doi: 10.1016/S0140-6736(12)61963-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fisher B, Costantino J, Redmond C, Poisson R, Bowman D, Couture J, Dimitrov NV, Wolmark N, Wickerham DL, Fisher ER, et al. A randomized clinical trial evaluating tamoxifen in the treatment of patients with node-negative breast cancer who have estrogen-receptor-positive tumors. N Engl J Med. 1989;320:479–484. doi: 10.1056/NEJM198902233200802. [DOI] [PubMed] [Google Scholar]
- 27.Fisher B, Dignam J, Bryant J, DeCillis A, Wickerham DL, Wolmark N, Costantino J, Redmond C, Fisher ER, Bowman DM, Deschenes L, Dimitrov NV, Margolese RG, Robidoux A, Shibata H, Terz J, Paterson AH, Feldman MI, Farrar W, Evans J, Lickley HL. Five versus more than five years of tamoxifen therapy for breast cancer patients with negative lymph nodes and estrogen receptor-positive tumors. J Natl Cancer Inst. 1996;88:1529–1542. doi: 10.1093/jnci/88.21.1529. [DOI] [PubMed] [Google Scholar]
- 28.Bonneterre J, Thurlimann B, Robertson JF, Krzakowski M, Mauriac L, Koralewski P, Vergote I, Webster A, Steinberg M, von Euler M. Anastrozole versus tamoxifen as first-line therapy for advanced breast cancer in 668 postmenopausal women: results of the tamoxifen or Arimidex randomized group efficacy and tolerability study. J. Clin. Oncol. 2000;18:3748–3757. doi: 10.1200/JCO.2000.18.22.3748. [DOI] [PubMed] [Google Scholar]
- 29.Baum M, Budzar AU, Cuzick J, Forbes J, Houghton JH, Klijn JG, Sahmoud T ATAC Trialists’ Group. Anastrozole alone or in combination with tamoxifen versus tamoxifen alone for adjuvant treatment of postmenopausal women with early breast cancer: first results of the ATAC randomised trial. Lancet. 2002;359:2131–2139. doi: 10.1016/s0140-6736(02)09088-8. [DOI] [PubMed] [Google Scholar]
- 30.Cuzick J, Sestak I, Baum M, Buzdar A, Howell A, Dowsett M, Forbes JF ATAC/LATTE investigators. Effect of anastrozole and tamoxifen as adjuvant treatment for early-stage breast cancer: 10-year analysis of the ATAC trial. Lancet Oncol. 2010;11:1135–1141. doi: 10.1016/S1470-2045(10)70257-6. [DOI] [PubMed] [Google Scholar]
- 31.Nabholtz JM, Buzdar A, Pollak M, Harwin W, Burton G, Mangalik A, Steinberg M, Webster A, von Euler M. Anastrozole is superior to tamoxifen as first-line therapy for advanced breast cancer in postmenopausal women: results of a North American multicenter randomized trial. Arimidex study group. J. Clin. Oncol. 2000;18:3758–3767. doi: 10.1200/JCO.2000.18.22.3758. [DOI] [PubMed] [Google Scholar]
- 32.Paridaens RJ, Dirix LY, Beex LV, Nooij M, Cameron DA, Cufer T, Piccart MJ, Bogaerts J, Therasse P. Phase III study comparing exemestane with tamoxifen as first-line hormonal treatment of metastatic breast cancer in postmenopausal women: the European organisation for research and treatment of cancer breast cancer cooperative group. J. Clin. Oncol. 2008;26:4883–4890. doi: 10.1200/JCO.2007.14.4659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gibson L, Lawrence D, Dawson C, Bliss J. Aromatase inhibitors for treatment of advanced breast cancer in postmenopausal women. Cochrane Database Syst Rev. 2009:CD003370. doi: 10.1002/14651858.CD003370.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Toy W, Shen Y, Won H, Green B, Sakr RA, Will M, Li Z, Gala K, Fanning S, King TA, Hudis C, Chen D, Taran T, Hortobagyi G, Greene G, Berger M, Baselga J, Chandarlapaty S. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat Genet. 2013;45:1439–1445. doi: 10.1038/ng.2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jeselsohn R, Yelensky R, Buchwalter G, Frampton G, Meric-Bernstam F, Gonzalez-Angulo AM, Ferrer-Lozano J, Perez-Fidalgo JA, Cristofanilli M, Gomez H, Arteaga CL, Giltnane J, Balko JM, Cronin MT, Jarosz M, Sun J, Hawryluk M, Lipson D, Otto G, Ross JS, Dvir A, Soussan-Gutman L, Wolf I, Rubinek T, Gilmore L, Schnitt S, Come SE, Pusztai L, Stephens P, Brown M, Miller VA. Emergence of constitutively active estrogen receptor-alpha mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin Cancer Res. 2014;20:1757–1767. doi: 10.1158/1078-0432.CCR-13-2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fribbens C, O’Leary B, Kilburn L, Hrebien S, Garcia-Murillas I, Beaney M, Cristofanilli M, Andre F, Loi S, Loibl S, Jiang J, Bartlett CH, Koehler M, Dowsett M, Bliss JM, Johnston SR, Turner NC. Plasma ESR1 mutations and the treatment of estrogen receptor-positive advanced breast cancer. J. Clin. Oncol. 2016;34:2961–2968. doi: 10.1200/JCO.2016.67.3061. [DOI] [PubMed] [Google Scholar]
- 37.Ellis MJ, Llombart-Cussac A, Feltl D, Dewar JA, Jasiowka M, Hewson N, Rukazenkov Y, Robertson JF. Fulvestrant 500 mg Versus Anastrozole 1 mg for the first-line treatment of advanced breast cancer: overall survival analysis from the phase II FIRST study. J. Clin. Oncol. 2015;33:3781–3787. doi: 10.1200/JCO.2015.61.5831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Robertson JFR, Bondarenko IM, Trishkina E, Dvorkin M, Panasci L, Manikhas A, Shparyk Y, Cardona-Huerta S, Cheung KL, Philco-Salas MJ, Ruiz-Borrego M, Shao Z, Noguchi S, Rowbottom J, Stuart M, Grinsted LM, Fazal M, Ellis MJ. Fulvestrant 500 mg versus anastrozole 1 mg for hormone receptor-positive advanced breast cancer (FALCON): an international, randomised, double-blind, phase 3 trial. Lancet. 2016;388:2997–3005. doi: 10.1016/S0140-6736(16)32389-3. [DOI] [PubMed] [Google Scholar]
- 39.Turner NC, Ro J, André F, Loi S, Verma S, Iwata H, Harbeck N, Loibl S, Huang Bartlett C, Zhang K, Giorgetti C, Randolph S, Koehler M, Cristofanilli M PALOMA3 Study Group. Palbociclib in hormone-receptor-positive advanced breast cancer. N Engl J Med. 2015;373:209–219. doi: 10.1056/NEJMoa1505270. [DOI] [PubMed] [Google Scholar]
- 40.Hu XF, Veroni M, De Luise M, Wakeling A, Sutherland R, Watts CK, Zalcberg JR. Circumvention of tamoxifen resistance by the pure anti-estrogen ICI 182,780. Int J Cancer. 1993;55:873–876. doi: 10.1002/ijc.2910550529. [DOI] [PubMed] [Google Scholar]
- 41.Takai O, Sasaki T, Muryoi T, Harata N, Hashimoto H, Yoshinaga K. Incidence of anti-DNA idiotype-positive cells in human peripheral blood. J Clin Immunol. 1989;9:313–321. doi: 10.1007/BF00918663. [DOI] [PubMed] [Google Scholar]
- 42.Hamilton EP, Patel MR, Armstrong AC, Baird RD, Jhaveri K, Hoch M, Klinowska T, Lindemann JPO, Morgan SR, Schiavon G, Weir HM, Im SA. A first-in-human study of the new oral selective estrogen receptor degrader azd9496 for ER(+)/HER2(-) advanced breast cancer. Clin Cancer Res. 2018;24:3510–3518. doi: 10.1158/1078-0432.CCR-17-3102. [DOI] [PubMed] [Google Scholar]
- 43.Dickler MN, Villanueva R, Perez Fidalgo JA, Mayer IA, Boni V, Winer E, Hamilton EP, Bellet M, Urruticoechea A, Gonzalez-Martin A, Cortes J, Martin M, Giltnane J, Gates M, Cheeti S, Fredrickson J, Wang X, Friedman LS, Spoerke JM, Metcalfe C, Liu L, Li R, Morley R, McCurry U, Chan IT, Mueller L, Milan S, Lauchle J, Humke EW, Bardia A. A first-in-human phase I study to evaluate the oral selective estrogen receptor degrader (SERD), GDC-0927, in postmenopausal women with estrogen receptor positive (ER+), HER2-negative metastatic breast cancer (BC) Cancer Research. 2018:78. [Google Scholar]
- 44.Gluck S. Consequences of the convergence of multiple alternate pathways on the estrogen receptor in the treatment of metastatic breast cancer. Clin Breast Cancer. 2017;17:79–90. doi: 10.1016/j.clbc.2016.08.004. [DOI] [PubMed] [Google Scholar]
- 45.Miller TW, Rexer BN, Garrett JT, Arteaga CL. Mutations in the phosphatidylinositol 3-kinase pathway: role in tumor progression and therapeutic implications in breast cancer. Breast Cancer Res. 2011;13:224. doi: 10.1186/bcr3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 47.Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- 48.Dey N, De P, Leyland-Jones B. PI3K-AKT-mTOR inhibitors in breast cancers: from tumor cell signaling to clinical trials. Pharmacol Ther. 2017;175:91–106. doi: 10.1016/j.pharmthera.2017.02.037. [DOI] [PubMed] [Google Scholar]
- 49.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yamnik RL, Digilova A, Davis DC, Brodt ZN, Murphy CJ, Holz MK. S6 kinase 1 regulates estrogen receptor alpha in control of breast cancer cell proliferation. J Biol Chem. 2009;284:6361–6369. doi: 10.1074/jbc.M807532200. [DOI] [PubMed] [Google Scholar]
- 51.Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–619. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 52.Polivka J Jr, Janku F. Molecular targets for cancer therapy in the PI3K/AKT/mTOR pathway. Pharmacol Ther. 2014;142:164–175. doi: 10.1016/j.pharmthera.2013.12.004. [DOI] [PubMed] [Google Scholar]
- 53.Ciruelos Gil EM. Targeting the PI3K/AKT/mTOR pathway in estrogen receptor-positive breast cancer. Cancer Treat Rev. 2014;40:862–871. doi: 10.1016/j.ctrv.2014.03.004. [DOI] [PubMed] [Google Scholar]
- 54.Yardley DA, Noguchi S, Pritchard KI, Burris HA 3rd, Baselga J, Gnant M, Hortobagyi GN, Campone M, Pistilli B, Piccart M, Melichar B, Petrakova K, Arena FP, Erdkamp F, Harb WA, Feng W, Cahana A, Taran T, Lebwohl D, Rugo HS. Everolimus plus exemestane in postmenopausal patients with HR(+) breast cancer: BOLERO-2 final progression-free survival analysis. Adv Ther. 2013;30:870–884. doi: 10.1007/s12325-013-0060-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hortobagyi GN, Chen D, Piccart M, Rugo HS, Burris HA 3rd, Pritchard KI, Campone M, Noguchi S, Perez AT, Deleu I, Shtivelband M, Masuda N, Dakhil S, Anderson I, Robinson DM, He W, Garg A, McDonald ER 3rd, Bitter H, Huang A, Taran T, Bachelot T, Lebrun F, Lebwohl D, Baselga J. Correlative analysis of genetic alterations and everolimus benefit in hormone receptor-positive, human epidermal growth factor receptor 2-negative advanced breast cancer: results from BOLERO-2. J. Clin. Oncol. 2016;34:419–426. doi: 10.1200/JCO.2014.60.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chagpar RB, Links PH, Pastor MC, Furber LA, Hawrysh AD, Chamberlain MD, Anderson DH. Direct positive regulation of PTEN by the p85 subunit of phosphatidylinositol 3-kinase. Proc Natl Acad Sci U S A. 2010;107:5471–5476. doi: 10.1073/pnas.0908899107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cheung LW, Hennessy BT, Li J, Yu S, Myers AP, Djordjevic B, Lu Y, Stemke-Hale K, Dyer MD, Zhang F, Ju Z, Cantley LC, Scherer SE, Liang H, Lu KH, Broaddus RR, Mills GB. High frequency of PIK3R1 and PIK3R2 mutations in endometrial cancer elucidates a novel mechanism for regulation of PTEN protein stability. Cancer Discov. 2011;1:170–185. doi: 10.1158/2159-8290.CD-11-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen L, Yang L, Yao L, Kuang XY, Zuo WJ, Li S, Qiao F, Liu YR, Cao ZG, Zhou SL, Zhou XY, Yang WT, Shi JX, Huang W, Hu X, Shao ZM. Characterization of PIK3CA and PIK3R1 somatic mutations in Chinese breast cancer patients. Nat Commun. 2018;9:1357. doi: 10.1038/s41467-018-03867-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bosch A, Li Z, Bergamaschi A, Ellis H, Toska E, Prat A, Tao JJ, Spratt DE, Viola-Villegas NT, Castel P, Minuesa G, Morse N, Rodon J, Ibrahim Y, Cortes J, Perez-Garcia J, Galvan P, Grueso J, Guzman M, Katzenellenbogen JA, Kharas M, Lewis JS, Dickler M, Serra V, Rosen N, Chandarlapaty S, Scaltriti M, Baselga J. PI3K inhibition results in enhanced estrogen receptor function and dependence in hormone receptor-positive breast cancer. Sci Transl Med. 2015;7:283ra251. doi: 10.1126/scitranslmed.aaa4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chopra N, Turner NC. Targeting PIK3CA-mutant advanced breast cancer in the clinical setting. Lancet Oncol. 2017;18:842–843. doi: 10.1016/S1470-2045(17)30430-8. [DOI] [PubMed] [Google Scholar]
- 62.Baselga J, Dent SF, Cortés J, Im YH, Diéras V, Harbeck N, Krop IE, Verma S, Wilson TR, Jin H, Wang L, Schimmoller F, Hsu JY, He Y, DeLaurentiis M, Drullinsky P, Jacot W. Phase III study of taselisib (GDC-0032) + fulvestrant (FULV) v FULV in patients (pts) with estrogen receptor (ER)-positive, PIK3CA-mutant (MUT), locally advanced or metastatic breast cancer (MBC): primary analysis from SANDPIPER. J. Clin. Oncol. 2018:36. [Google Scholar]
- 63.Lundberg AS, Weinberg RA. Control of the cell cycle and apoptosis. Eur J Cancer. 1999;35:531–539. [PubMed] [Google Scholar]
- 64.Caldon CE, Daly RJ, Sutherland RL, Musgrove EA. Cell cycle control in breast cancer cells. J Cell Biochem. 2006;97:261–274. doi: 10.1002/jcb.20690. [DOI] [PubMed] [Google Scholar]
- 65.Sherr CJ, Beach D, Shapiro GI. Targeting CDK4 and CDK6: from discovery to therapy. Cancer Discov. 2016;6:353–367. doi: 10.1158/2159-8290.CD-15-0894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shapiro GI. Cyclin-dependent kinase pathways as targets for cancer treatment. J. Clin. Oncol. 2006;24:1770–1783. doi: 10.1200/JCO.2005.03.7689. [DOI] [PubMed] [Google Scholar]
- 67.Ortega S, Malumbres M, Barbacid M. Cyclin D-dependent kinases, INK4 inhibitors and cancer. Biochim Biophys Acta. 2002;1602:73–87. doi: 10.1016/s0304-419x(02)00037-9. [DOI] [PubMed] [Google Scholar]
- 68.Musgrove EA, Caldon CE, Barraclough J, Stone A, Sutherland RL. Cyclin D as a therapeutic target in cancer. Nat Rev Cancer. 2011;11:558–572. doi: 10.1038/nrc3090. [DOI] [PubMed] [Google Scholar]
- 69.Geradts J, Wilson PA. High frequency of aberrant p16 (INK4A) expression in human breast cancer. Am J Pathol. 1996;149:15–20. [PMC free article] [PubMed] [Google Scholar]
- 70.Sauter ER, Nesbit M, Litwin S, Klein-Szanto AJ, Cheffetz S, Herlyn M. Antisense cyclin D1 induces apoptosis and tumor shrinkage in human squamous carcinomas. Cancer Res. 1999;59:4876–4881. [PubMed] [Google Scholar]
- 71.Beaver JA, Amiri-Kordestani L, Charlab R, Chen W, Palmby T, Tilley A, Zirkelbach JF, Yu J, Liu Q, Zhao L, Crich J, Chen XH, Hughes M, Bloomquist E, Tang S, Sridhara R, Kluetz PG, Kim G, Ibrahim A, Pazdur R, Cortazar P. FDA approval: palbociclib for the treatment of postmenopausal patients with estrogen receptor-positive, HER2-negative metastatic breast cancer. Clin Cancer Res. 2015;21:4760–4766. doi: 10.1158/1078-0432.CCR-15-1185. [DOI] [PubMed] [Google Scholar]
- 72.Cristofanilli M, Turner NC, Bondarenko I, Ro J, Im SA, Masuda N, Colleoni M, DeMichele A, Loi S, Verma S, Iwata H, Harbeck N, Zhang K, Theall KP, Jiang Y, Bartlett CH, Koehler M, Slamon D. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2016;17:425–439. doi: 10.1016/S1470-2045(15)00613-0. [DOI] [PubMed] [Google Scholar]
- 73.Walker AJ, Wedam S, Amiri-Kordestani L, Bloomquist E, Tang S, Sridhara R, Chen W, Palmby TR, Fourie Zirkelbach J, Fu W, Liu Q, Tilley A, Kim G, Kluetz PG, McKee AE, Pazdur R. FDA approval of palbociclib in combination with fulvestrant for the treatment of hormone receptor-positive, HER2-negative metastatic breast cancer. Clin Cancer Res. 2016;22:4968–4972. doi: 10.1158/1078-0432.CCR-16-0493. [DOI] [PubMed] [Google Scholar]
- 74.Hortobagyi GN, Stemmer SM, Burris HA, Yap YS, Sonke GS, Paluch-Shimon S, Campone M, Petrakova K, Blackwell KL, Winer EP, Janni W, Verma S, Conte P, Arteaga CL, Cameron DA, Mondal S, Su F, Miller M, Elmeliegy M, Germa C, O’Shaughnessy J. Updated results from MONALEESA-2, a phase III trial of first-line ribociclib plus letrozole versus placebo plus letrozole in hormone receptor-positive, HER2-negative advanced breast cancer. Ann Oncol. 2018;29:1541–1547. doi: 10.1093/annonc/mdy155. [DOI] [PubMed] [Google Scholar]
- 75.Finn R, Jiang Y, Rugo H, Moulder SL, Im SA, Gelmon KA, et al. Biomarker analyses from the phase 3 PALOMA-2 trial of palbociclib (P) with letrozole (L) compared with placebo (PLB) plus L in postmenopausal women with ER+/HER2-advanced breast cancer (ABC) Ann Oncol. 2016;27 [Google Scholar]
- 76.Malumbres M, Sotillo R, Santamaria D, Galan J, Cerezo A, Ortega S, Dubus P, Barbacid M. Mammalian cells cycle without the D-type cyclin-dependent kinases Cdk4 and Cdk6. Cell. 2004;118:493–504. doi: 10.1016/j.cell.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 77.Sherr CJ, Roberts JM. Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 2004;18:2699–2711. doi: 10.1101/gad.1256504. [DOI] [PubMed] [Google Scholar]
- 78.Kozar K, Ciemerych MA, Rebel VI, Shigematsu H, Zagozdzon A, Sicinska E, Geng Y, Yu Q, Bhattacharya S, Bronson RT, Akashi K, Sicinski P. Mouse development and cell proliferation in the absence of D-cyclins. Cell. 2004;118:477–491. doi: 10.1016/j.cell.2004.07.025. [DOI] [PubMed] [Google Scholar]
- 79.Herrera-Abreu MT, Palafox M, Asghar U, Rivas MA, Cutts RJ, Garcia-Murillas I, Pearson A, Guzman M, Rodriguez O, Grueso J, Bellet M, Cortes J, Elliott R, Pancholi S, Baselga J, Dowsett M, Martin LA, Turner NC, Serra V. Early adaptation and acquired resistance to CDK4/6 inhibition in estrogen receptor-positive breast cancer. Cancer Res. 2016;76:2301–2313. doi: 10.1158/0008-5472.CAN-15-0728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yang C, Li Z, Bhatt T, Dickler M, Giri D, Scaltriti M, Baselga J, Rosen N, Chandarlapaty S. Acquired CDK6 amplification promotes breast cancer resistance to CDK4/6 inhibitors and loss of ER signaling and dependence. Oncogene. 2017;36:2255–2264. doi: 10.1038/onc.2016.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Condorelli R, Spring L, O’Shaughnessy J, Lacroix L, Bailleux C, Scott V, Dubois J, Nagy RJ, Lanman RB, Iafrate AJ, Andre F, Bardia A. Polyclonal RB1 mutations and acquired resistance to CDK4/6 inhibitors in patients with metastatic breast cancer. Ann Oncol. 2018;29:640–645. doi: 10.1093/annonc/mdx784. [DOI] [PubMed] [Google Scholar]
- 82.De P, Williams C, Krie A, Solomon B, AL E. Genomic landscape of the PI3K pathway and cell-cycle pathway in ER+BC: a treatment strategy. St. Gallen International Breast Cancer Conference. 2015 [Google Scholar]
- 83.Vora SR, Juric D, Kim N, Mino-Kenudson M, Huynh T, Costa C, Lockerman EL, Pollack SF, Liu M, Li X, Lehar J, Wiesmann M, Wartmann M, Chen Y, Cao ZA, Pinzon-Ortiz M, Kim S, Schlegel R, Huang A, Engelman JA. CDK4/6 inhibitors sensitize PIK3CA mutant breast cancer to PI3K inhibitors. Cancer Cell. 2014;26:136–149. doi: 10.1016/j.ccr.2014.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.O’Brien NA, Tomaso ED, Ayala R, Tong L, AL E. In vivo efficacy of combined targeting of CDK4/6, ER and PI3K signaling in ER+ breast cancer. AACR Annual Metting. 2014 [Google Scholar]
- 85.Teo ZL, Versaci S, Dushyanthen S, Caramia F, Savas P, Mintoff CP, Zethoven M, Virassamy B, Luen SJ, McArthur GA, Phillips WA, Darcy PK, Loi S. Combined CDK4/6 and PI3Kalpha inhibition is synergistic and immunogenic in triple-negative breast cancer. Cancer Res. 2017;77:6340–6352. doi: 10.1158/0008-5472.CAN-17-2210. [DOI] [PubMed] [Google Scholar]
- 86.Michaloglou C, Crafter C, Siersbaek R, Delpuech O, Curwen JO, Carnevalli LS, Staniszewska AD, Polanska UM, Cheraghchi-Bashi A, Lawson M, Chernukhin I, McEwen R, Carroll JS, Cosulich SC. Combined inhibition of mTOR and CDK4/6 is required for optimal blockade of E2F function and long-term growth inhibition in estrogen receptor-positive breast cancer. Mol Cancer Ther. 2018;17:908–920. doi: 10.1158/1535-7163.MCT-17-0537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.O’Leary B, Hrebien S, Morden JP, Beaney M, Fribbens C, Huang X, Liu Y, Bartlett CH, Koehler M, Cristofanilli M, Garcia-Murillas I, Bliss JM, Turner NC. Early circulating tumor DNA dynamics and clonal selection with palbociclib and fulvestrant for breast cancer. Nat Commun. 2018;9:896. doi: 10.1038/s41467-018-03215-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pusztai L, Karn T, Safonov A, Abu-Khalaf MM, Bianchini G. New strategies in breast cancer: immunotherapy. Clin Cancer Res. 2016;22:2105–2110. doi: 10.1158/1078-0432.CCR-15-1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Goel S, DeCristo MJ, Watt AC, BrinJones H, Sceneay J, Li BB, Khan N, Ubellacker JM, Xie S, Metzger-Filho O, Hoog J, Ellis MJ, Ma CX, Ramm S, Krop IE, Winer EP, Roberts TM, Kim HJ, McAllister SS, Zhao JJ. CDK4/6 inhibition triggers anti-tumour immunity. Nature. 2017;548:471–475. doi: 10.1038/nature23465. [DOI] [PMC free article] [PubMed] [Google Scholar]