

Abstract
Incense burning is common in Asian countries due to the religious beliefs. Environmental exposure to incense burning smoke is a potential risk factor for tumor development and progression of non-small cell lung cancer (NSCLC). Eastern Asia ethnic origin is strongly associated the clinical benefits of epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) in NSCLC patients. However, the impact of the oriental custom of incense burning on the cancer progression and the EGFR TKI-sensitivity of NSCLC remains unclear. Our results showed that long-term exposure to incense burning extract (IBE) increases the cellular proliferation with S phase accumulation and the motility activity of NSCLCs. Interestingly, IBE enhances EGFR signaling activity without affecting its genetic status, and increases the cellular sensitivity of NSCLC cell lines to EGFR TKIs. Auramine, a yellow dye for making incense sticks, was identified as a residual composition in the burning incense smoke, and showed similar EGFR TKI-sensitizing effects. Furthermore, IBE or auramine transcriptionally induce EGFR ligand amphiregulin (AREG) expression for the enhancement of EGFR activity. Neutralization of AREG reduced the viability of IBE-treated cells. These results indicated that exposure to incent smoke may enhance NSCLC progression and their sensitivity to EGFR TKIs through increasing their oncogenic addiction to AREG-induced EGFR signaling.
Keywords: Non-small cell lung cancer, EGFR, AREG, incent burning smoke
Introduction
Lung cancer is the most common cancer type worldwide both in terms of numbers of cases and deaths, and has high case fatality (ratio of mortality to incidence, 0.87) [1,2]. Most of lung cancers are carcinomas originated from epithelial cells. Lung cancer is broadly divided into small-cell lung cancer (SCLC, comprising 15-20% of lung cancers), and non-small-cell lung cancer (NSCLC, comprising 80-85% of lung cancers) [3]. Non-small-cell lung cancer is the major type of epithelial lung cancer. The most common sub-types of NSCLC are adenocarcinoma, squamous cell carcinoma, and large cell carcinoma. NSCLC can be treated with surgery, radiotherapy, chemotherapy, or a combination, depending on their stage. Some patients with advanced lung cancer may be responsive to tyrosine kinase inhibitors (TKIs) of epidermal growth factor receptor (EGFR).
EGFR is a member of cell-surface ErbB receptor family, which includes EGFR (ErbB-1), HER2/c-neu (ErbB-2), HER3 (ErbB-3), and HER4 (ErbB-4). EGFR is activated by binding with its specific ligands, such as epidermal growth factor (EGF), transforming growth factor α (TGF-α), Heparin-binding EGF-like growth factor (HB-EGF), and Amphiregulin (AREG) [4]. Upon activation, EGFR goes through a transition from an inactive form to an active homo-dimer. In addition to forming homo-dimers after ligand binding, EGFR may also create an activated heterodimer by pairing with another member of ErbB receptor family and this dimerization stimulates its intracellular protein-tyrosine kinase activity. As a result, autophosphorylations in the C-terminal domain of EGFR occur at several tyrosine (Y) residues, including Y992, Y1045, Y1068, Y1148 and Y1173. These autophosphorylations lead to the activation of downstream signaling by recruiting several proteins which associate with these phosphorylated tyrosines through their SH2 or PTB domains. These downstream signaling proteins initiate several signal transduction cascades, mainly the MAPK (RAS-RAF-MEK-ERK), PI3K (PI3K-AKT-mTOR), and JNK pathways, leading to DNA synthesis and cell proliferation. Such signaling pathways also mediate other malignant phenotypes such as cell migration and adhesion [4]. The identification of EGFR as an oncogene leads to the development of anticancer therapeutics directly against EGFR, including gefitinib, erlotinib, afatinib, and icotinib for lung cancer targeted therapies [5]. Clinical trials have indicated the significant variability in response to EGFR TKIs, and found higher response rate in Japanese patients than in European-derived population (27.5% vs. 10.4%) [6]. The better clinical responses to EGFR TKIs have been observed most frequently in women, in non-smokers, and in NSCLC patients with adenocarcinomas [7-9]. Consequently, lung cancer patients with these specific characteristics are associated with EGFR mutations, and showed better clinical outcome through therapies with these drugs [10,11].
EGFR mutations mostly are identified in these NSCLC patients, and lead to EGFR over activation and association with the poor prognosis of lung cancer patients [12]. Most of the somatic activating EGFR mutations were found in the adenosine triphosphate (ATP)-binding pocket of the receptor tyrosine kinase domain. Sequencing of the EGFR gene revealed that a majority of tumors responding to EGFR TKIs harbored mutations in the TK domain [13,14]. Overall, the frequency of EGFR mutations is 5-40% [15]. For patients whose tumors bearing EGFR mutations, the response rate to erlotinib and gefitinib is approximately 75%, suggesting that these mutations play a critical role in driving malignant transformation [15,16]. Approximately 45% of NSCLC patients in the East Asia and 10% of NSCLC patients in US bear EGFR mutations [13,17,18]. These somatic mutations, particularly exon 18 G719X point mutation, exon 19 deletion, and exon 21 L858R point mutation, occur within the kinase domain of EGFR [13], and increase the kinase activity and subsequent hyper-activation of its downstream pro-survival signaling pathways [19]. These activating EGFR mutations are commonly observed in patients with the characters of adenocarcinomas, female, non-smoker and Asian offspring, and are positively associated with the responsiveness to EGFR TKIs [20,21]. By contrast, the exon 20 T790M mutation of EGFR is associated with acquired resistance to TKI targeted therapy [22]. Therefore, EGFR mutations are the criteria to decide the use of EGFR TKIs for lung cancer patients, and have been used as the predictive biomarker for the therapeutic efficacy. However, it remains unknown why Asian patients express activating EGFR mutants at high incidence.
There are many religions practiced in Asia, and the most common types are Hinduism (centered around India), Buddhism (centered around China), Islam (mainly in the Middle East), and Christianity (in Russia). Especially, Buddhism, Taoism, and Shinto are widely distributed religions in Asian region, and incense burning is the common way by which people delivered their faith. A typical composition of stick incense consists of herbal and wood powder, fragrance material, staining matter, adhesive powder, and bamboo stick [23]. Upon combustion of incense stick, smoke from incense burning contains particulate matter (PM), gas products and volatile organic compounds (VOCs). Particulate products from incense burning (45 mg/g) on averagely are greater than that from cigarette burning (10 mg/g) [23]. The gas products from incense burning include CO, CO2, NOx, SO2 etc.; smoke from incense stick combustion in religious and ritual places also produces a large number of health-damaging and carcinogenic air pollutant such as benzene, toluene, xylene, formaldehyde, 1.3 butadiene, styrene, as well as aldehyde and polycyclic aromatic hydrocarbons (PAHs) [23-27].
Because of the region factor, religion in Asian and incense burning are inseparable. In addition, some studies showed that incense smoke induces blood lead level and cardiovascular disease mortality [28,29]. Long-term use of incense is also associated with the increasing risk of PAHs toxicity, including DNA adducts formation and development of squamous cell carcinoma [30-32]. Some studies even demonstrated that cigarette smoking and frequency of exposure to incense facilitates the development of lung cancer [33]. However, it remains unclear whether exposure to incense burning smoke impacts the lung cancer incidence and tumor progression. Also, Japanese research also reported that the response rate of Asian lung cancer patients bearing EGFR mutant to EGFR inhibitor-gefitinib was approximately 75-90% [34-37]. Although EGFR mutation has been implicated in the responsiveness of Asian NSCLC patients to anti-cancer EGFR targeted therapies, it is still not clear whether incense-burning smoke is related to sensitivity of these patients to EGFR targeted therapies.
Materials and methods
Preparation of incense burning extract (IBE) medium
One incense stick was used to prepare 10 mL of IBE medium. Incense-burning smoke was dissolved into RPMI-HEPES medium by suction with pump. The pH value of IBE medium was adjusted to 7.4. The IBE medium was filtrated by 0.22 μm filter to remove large particles, and was defined as 100% IBE stock medium storage at -30°C before use.
Cell culture
Human H292 lung cancer cell line, derived from a lymph node metastasis of a pulmonary mucoepidermoid carcinoma, was cultured in RPMI (Roswell Park Memorial Institute) 1640 (HyClone) supplemented with 10 mM HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid), 10 mM sodium pyruvate, 10% heat-inactivated FBS (fetal bovine serum) (GeneDriex), 100 units/ml penicillin, and 100 μg/ml streptomycin (Thermo). HCC827 lung adenocarcinoma cell line expressing EGFR mutant (E746-A750 deletion) was cultured in RPMI 1640 (HyClone) supplemented with 10% heat-inactivated FBS, 100 units/ml penicillin, and 100 μg/ml streptomycin (Thermo). Both of them were gifts from Prof. Mien-Chie Hung (M.D. Anderson Cancer Center, Tx). Lung cancer cell lines were treated with IBE medium (H292 IBE 1%, H292 IBE 5%) or with 1 μM auramine for over one month to establish the stable clones. All cells were incubated at 37°C in a humidified incubator containing 5% CO2.
Cell counting
After wash with PBS, cells were detached by incubation with trypsin-EDTA, and then collected and centrifuged at 1,000 rpm for 5 minutes. The cells were re-suspended in fresh medium and loaded onto hemocytometer. The cell numbers were counted under an Inversion Microscope.
MTT and WST-1 assay
Cells were trypsinized to seed at density of 4 × 103-8 × 103 cells/well in 96-well plate followed by treatment with EGFR TKIs for indicated time. After TKI treatment, the culture medium was removed and cells were washed and incubated with 100 μL of serum-free medium with 5 mg/mL MTT solution (Sigma) or WST-1 for 3 hours. Then, 100 μL of DMSO was added to lyse the cells and the absorption was detected by using ELISA reader at the wavelength of O.D. 550.
Western blot
The concentration of total protein lysate was measured by Bradford protein assay (Bio-Rad protein assay). Protein lysates (10-40 μg) were heated in 1X sample buffer at 95°C for 5 minutes, and loaded into SDS-PAGE under 300 V, 30 mA/each gel until protein sample separated to the bottom. The used 1X running buffer was composed of 3.03 g Tris-base, 14.4 g Glycine, and 0.1% SDS. Then, the separated protein was transferred to a PVDF membrane (0.45 μM, millipore) or a NC membrane (0.22 μM, GE Healthcare) in transfer buffer under 150 V, 300 mA for 3 hour. The transfer buffer composed of 700 mL of ddH2O, 200 mL of Methanol, 100 mL of 10X running buffer, and 3.75 mL of SDS. After these steps, the transferred membrane was blocked in 5% milk in Tris-bufferd saline-Tween (10 mL of 2 μM Tris-HCl pH 7.4, 100 mL of 5 M NaCl, 0.5 mL of 100% Tween-20, and 890 mL of ddH2O) for one hour at room temperature. The membranes were hybridized with primary antibodies at 4°C overnight, and then incubated with HRP-labeled secondary antibodies at room temperature for 1 hour. The chemoluminescence signal was catalyzed by ECL (GE Healthcare or millipore) to detect the level of protein expression.
Clonogenic formation assay
Cells were seeded less than 15000 cells/well and then treated with gefitinib or erlotinib for 7 days. 1% Crystal violet (buffered with 30% ethanol) staining was then performed.
Migration and invasion assay
The migration and invasion assays were preformed using trans-wells plate (0.8 µm costar) without or with matrix gel. Approximately 2 × 105 cells in 200 ul of serum free medium were placed in the upper-chamber, 300 ul of different condition medium were placed in the lower-chamber. The plate was incubated for 24 hours at 37°C in 5% CO2, and then cells were fixed in 4% formaldehyde for 15 minutes and stained 0.05% crystal violet in PBS for 30 minutes. Cells on the upper slide of the filters were removed with cotton-tripped swabs, and the filters were washed with PBS. Cells on the down slide of the filters were examined and counted under a microscope.
Flow cytometry
H292 cells were treated with mitomycin C to arrest cell cycle for 3 hours. After treatment, cells were washed with PBS for two times and trypsinized. Next, cells were collected and centrifuged at 1,000 rpm for 5 minutes, and then were fixed with 75% Ethanol at room temperature followed by staining with PI buffer (1% Triton X-100, 0.1 mg/mL PureLinkTM RNase A (Cat no. 12091-021), and 20 μg/mL Propidium iodide in 1X PBS) for 30 minutes. The cell cycle population was analyzed by using Flow Cytometry.
RNA extraction and reverse transcription (RT)
After treatments, cells were washed with PBS for two times and total RNA was isolated with 0.5 mL of TrizolTM reagent (Roche) followed by addition of 0.1 mL of chloroform and centrifugation at 13,000 rpm for 15 minutes to separate aqueous phase, interphase, and organic phase. Next, the precipitated RNA from the aqueous phase was mixed with 0.25-0.5 mL of isopropanol and centrifuged at 13,000 rpm for 15 minutes. After removal of supernatant, gel-like pellet was washed with 1 mL of 75% ethanol twice after centrifugation at 7500 rpm for 15 minutes. Finally, after removal of supernatant, RNA pellet was air-dried for about 30 minutes and then dissolved in DEPC-treated water for storage at 4°C for 24 hours. Sample concentration and purity were determined at OD 260 nm and 280 nm. The OD260/280 ratio should be in the range between 1.8-2.0. Then, the first-strand cDNA synthesis was performed with 500 ng RNA, dNTP, Oligo-dT and distilled DEPC H2O. The mixture was heated to 65°C for 5 minutes and quick chilled to 4°C for 5 minutes. Then 5X First-Strand Buffer, DTT, M-MLV RT Enzyme were added to the mixture, incubated at 37°C, 25°C then back to 37°C again for 2, 10, 50 minutes, respectively, and then at 70°C for 15 minutes. The cDNA products were used for amplification in PCR.
Statistic analysis
Data were displayed as mean ± S.E.M. The significance of difference between the experimental and control groups was assessed by Student’s t test. The difference is significant if p value is < 0.05.
Results
IBE induced proliferation, migration, and invasion of NSCLCs
To investigate the effect of incense burning on NSCLC cancer cell progression and drug sensitivity, wild-type EGFR-expressing H292 lung cancer cell line was treated with different concentrations of IBE medium for 24 hours followed by the examination of cell viability. The results of MTT assay showed that H292 cells were died significantly in a dose-dependent manner after exposure to 20% IBE medium (Supplementary Figure 1). To mimic the scenario of religious belief, H292 cells were chronically treated with the sub-lethal concentration (1% and 5%) of IBE medium for at least one month to establish IBE stable clone. The data from both crystal violet staining and cell counting assays showed that IBE stable clones showed much higher cell proliferation ability as compared with their parental cells (Figure 1A and 1B). In parallel, the S phase distribution of IBE stable clones after 12 hours of release from synchronization by mitomycin C was also higher than that of parental cells (Figure 1C). Next, we examined the effect of IBE on the motility of lung cancer cells. To exclude the enhancement of cell growth by IBE, IBE stable clones were treated with mitomycin C for 3 hours to arrest cell cycle at G1 phase prior to migration and invasion assays. The migration (Figure 2A) and invasion (Figure 2B) abilities of 1% and 5% IBE clones are dramatically higher than that of parental cells within 24 hours. Taken together, these findings suggest that exposure to IBE renders cancer cells much more malignant.
Figure 1.
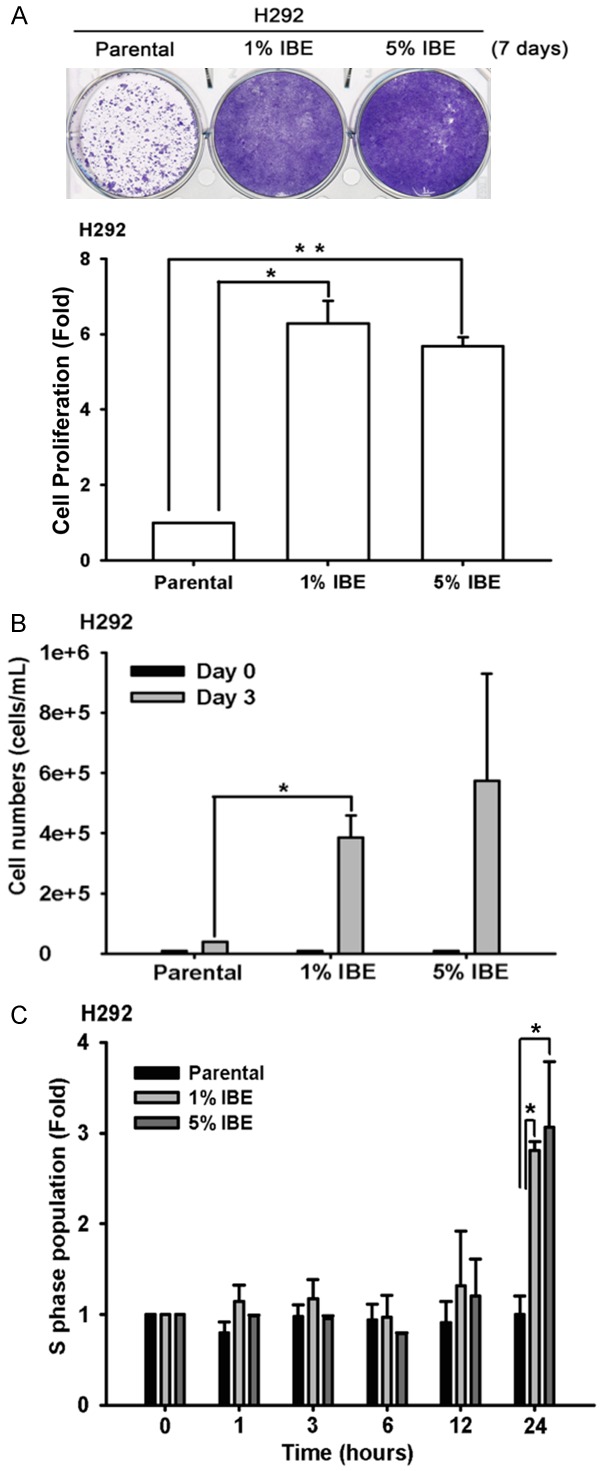
IBE treatment induced cell proliferation in NCI-H292 cells. A. H292 parental and IBE stable clone were seeded in 6-well for 7 days followed by 1% crystal violet staining. The result was quantitated according to the absorbance at 570 nm followed by dissolving the crystal violet staining with acetic acid. B. H292 parental and IBE stable clone were seeded in 12-well for 3 days, and then cell viabilities were determined by automated cell counter. C. The cell cycle status of H292 parental and IBE stable cells were examined by FACS. Cells were pre-treated with mitomycin C for 3 hours for synchronization followed by PI staining for 1, 3, 6, 12, 24 hours, respectively. Results were expressed as mean ± S.E.M. of two independent experiments. *: P < 0.05; **: P < 0.01 as compared with control group.
Figure 2.
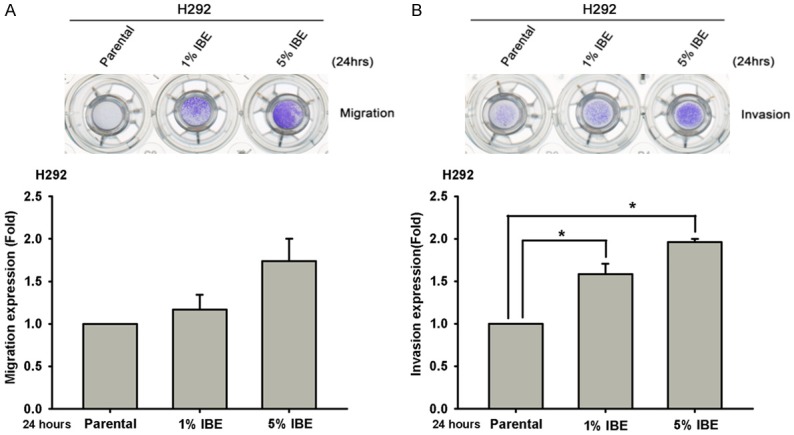
IBE induced cell migration and invasion abilities of H292 cells. H292 parental cells and their IBE stable clone were pretreated with mitomycin C for 3 hours to synchronize their cell cycle at G1 phase, and then their cell migration and invasion abilities after 24 hours of culture were determined in Boyden chamber migration (A) and invasion assays (B). Results were expressed as mean ± S.E.M. of two independent experiments. * or #: P < 0.05 as compared with control group.
IBE and its ingredient auramine induced EGFR kinase activation through upregulating AREG expression
Tumor progression involves diverse regulatory events especially those signaling pathways driven by receptor tyrosine kinases. To elucidate the underlying mechanisms of IBE-induced tumor progression, we tested whether any receptor tyrosine kinase was activated in response to the IBE exposure. To this end, general tyrosine phosphorylation was examined by using 4G10 anti-phosphotyrosine antibody, and the results showed that some increased signals were observed around 100-190 kDa in H292 IBE clones (Figure 3A). EGFR is a 185 kDa receptor tyrosine kinase, and its signaling pathway is frequently amplified and crucial for malignancy in many cancer types, including NSCLC. Therefore, the roles of EGFR and its downstream signaling in IBE-induced tumor progression were then examined, and we found that IBE induced the kinase activations of EGFR and its downstream effectors Akt and Erk by detecting their activating phosphorylations at Y1068, Y845 for EGFR, at T202 and Y204 for Erk1/2, and T308 for Akt in the absence or presence of EGF stimulation (Figure 3B). We also examined the tyrosine kinase activity of c-Met and ErbB2 (HER2), whose molecular weight are 140/170 and 185 kDa, respectively, but their kinase activities were suppressed and not increased respectively by IBE treatment (Figure 3C). These data suggested that IBE induced kinase activation of EGFR signaling for tumor progression, but meanwhile suppressed c-Met in H292 cells. Short-term treatment (3 days) with IBE also slightly induced kinase activation of EGFR signaling in H292 and H322 cells (Figure 3D). In addition to lung cancer cell expressing wild-type EGFR, we also examine the effect of IBE on EGFR activation in lung cancer cells expressing EGFR mutants. We treated EGFR exon19 deletion HCC827 cells and EGFR exon21/T790M double mutation H1975 cells with IBE for 7 days. However, the data showed that IBE did not induce EGFR kinase activation significantly in these two cells (Figure 3E), suggesting that IBE treatment may specifically induce EGFR signaling pathway in wild-type NSCLC cells but not mutant EGFR-expressing NSCLC cells.
Figure 3.
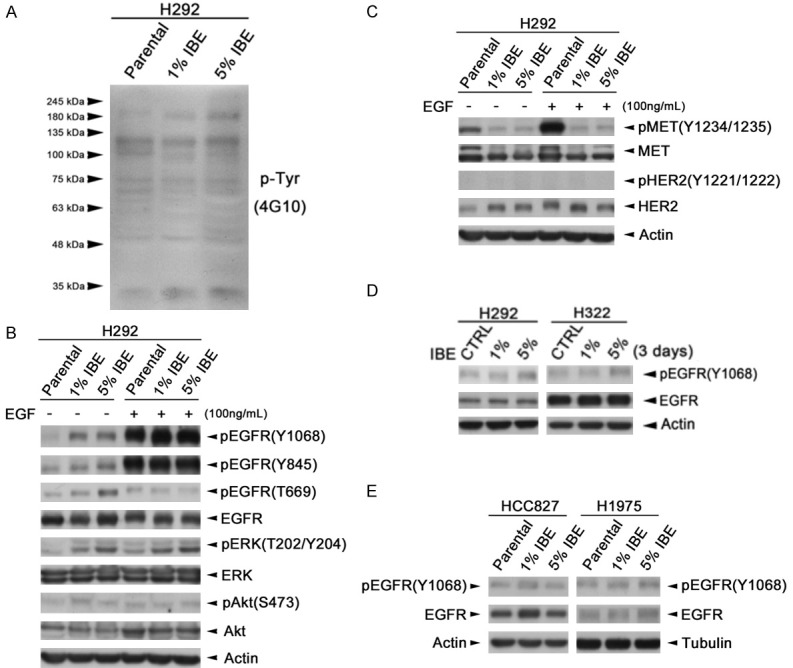
IBE induced EGFR tyrosine kinase activation in wt-EGFR-expressing NSCLC cells. (A) Whole cell lysates extracted from H292 parental and IBE stable clones were subjected to western blot analysis with 4G10 anti-phosphotyrosine antibody. (B, C) H292 parental and IBE stable clone cells were stimulated with or without EGF for 30 minutes. (D, E) EGFR wild-type H292, H322 cells (D) and EGFR mutant HCC827, H1975 cells (E) were treated with concentration 1% and 5% IBE for 3 days and 7 days, respectively. Total protein was extracted and subjected to western blot analysis with indicated antibody.
Next, we addressed whether IBE cells release cytokines to activate EGFR. The IBE conditioned medium was collected from the 1% IBE clones, which were cultured in IBE-free medium for 24 hours before the harvest. The parental H292 lung cancer cells were then cultured with the parental or IBE conditioned medium for 30 minutes as illustrated in Figure 4A. Our data from Western blot analysis showed that, like direct IBE treatment, the conditioned medium from IBE clones induced EGFR, Akt and Erk phosphorylations, suggesting an autocrine regulation to activate EGFR by IBE exposure (Figure 4B). We examined the effect of IBE treatment on the expressions of some common EGFR ligands including EGF, TGF, HB-EGF and AREG, and found that the mRNA level of AREG was increased in IBE-treated H292 cells (Figure 4C). The secretion of AREG from IBE cells was also detected (Figure 4D). These findings suggest that IBE may activate EGFR signaling pathway through induction of AREG.
Figure 4.
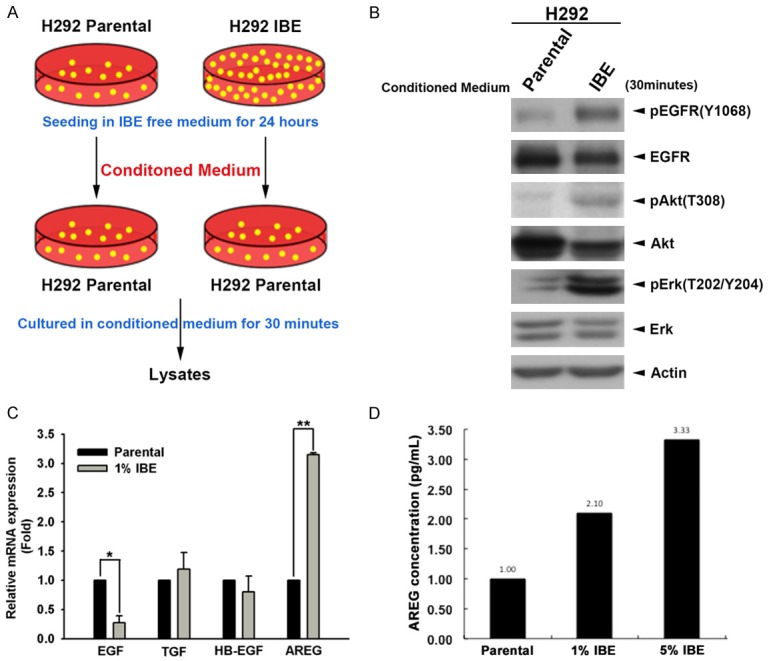
The secreted AREG mediates IBE-induced EGFR kinase activation. A. H292 parental and 1% IBE stable clone cells were seeded in IBE free medium for 24 hours, then conditioned medium was collected and used for culture of H292 parental cells for 30 minutes. B. Total lysates extracted from cells cultured with conditioned medium were performed with western blot analysis by with indicated antibodies. C. The expression of EGF, TGF, HB-EGF and AREG mRNA in H292 cells and their 1% IBE clone were analyzed by real-rime PCR. D. The levels of AREG in the culture medium of H292 parental cells and their 1% and 5% IBE stable clones were measured in the ELISA assays. Results were expressed as mean ± S.E.M. of two independent experiments. *: P < 0.05; **: P < 0.01 as compared with control group.
In the process of making incense stick, auramine O is a dye diarylmethane used for surface staining to make incense stick better looking. We found that auramine, a residual composition from the smoke after burning incense sticks in mass spectrometry analysis (Supplementary Figure 2 and [38]), increases the metastatic abilities and stemness characters of NSCLCs [38]. Also, treatment with auramine increased the proliferation of H292 cells (Figure 5A). Similar to IBE, auramine also induced AREG secretion of H292 cells into the culture medium, and the induction was suppressed by the anti-AREG neutralizing antibody (Figure 5B). Activations of EGFR signaling and its downstream Akt and ERK activities by auramine treatment were also found in H292 cells (Figure 5C). Treatment with anti-AREG antibody further suppressed the auramine-induced EGFR and ERK activations (Figure 5D), indicating that AREG mediated IBE- and auramine-induced EGFR activation. Silence of AREG also reduced the viability of auramine-treated H292 clone (Figure 5E).
Figure 5.
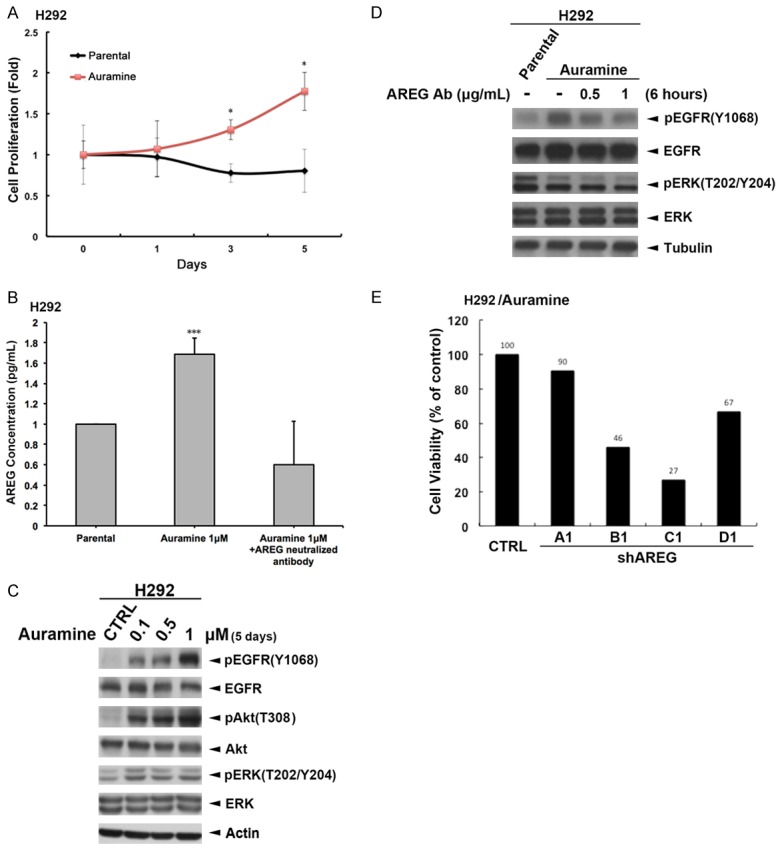
Auramine induced the cell proliferation of H292 cells through AREG-dependent EGFR signaling. (A) The cell viabilities of H292 cells and its auramine-stable clone were measured by MTT assays. (B) Total lysates prepared from H292 cells treated with 0.1, 0.5, 1 mM auramine for 5 days were subjected to Western blot with indicated antibodies. (C, D) Auramine stable clone of H292 cells were treated with indicated concentration of anti-AREG antibody for 6 hours. The level of secreted AREG in the culture medium was determined by ELISA assay (C). The total lysates of these cells were prepared and subjected into Western blot analysis with indicated antibody (D). (E) Auramine stable clone of H292 cells were infected with lentivirus expressing AREG shRNA, and their viabilities were then determined by MTT assays.
Exposure to IBE enhances EGFR-TKI sensitivity in H292 cells
Previous studies suggested that Asian NSCLC patients bearing EGFR mutants showed better responses to EGFR TKIs therapies [1,34,36]. But it is not clear whether incense-use custom in Asia is associated with the higher sensitivity of lung cancer cells to EGFR TKIs. In comparison to parental cells, IBE- or aruamine-treated H292 cells were much more sensitive to EGFR TKI gefitinib (Figure 6A) or erlotinib (Figure 6B and 6C) in MTT assays. To examine the possibility of EGFR mutation in IBE-treated cells, gene sequencing of EGFR was be performed. However, after alignment of the EGFR sequence expressed in 1% IBE clones and the parental H292 cells, activating mutations, including exon 19 deletion or L858R substitutions, were not found in H292/IBE clones (Supplementary Figure 3). Furthermore, TKI treatment induced PARP and Caspase 3 cleavages in IBE clones more than in their parental cells (Figure 6D). Also, erlotinib-induced sub-G1 population was higher in IBE clones than in parental cells (Figure 6E), indicating that IBE treatment sensitizes H292 cells to TKI-induced apoptosis. The data from clonogenic formation (Figure 6F) and cell counting (Figure 6G) assays also showed that IBE-treated H292 cells were much more sensitive to various EGFR TKIs including gefitinib, erlotinib or afatinib. In addition, we also found treatment with gefitinib can suppress the spheroid formation of IBE-treated H292 clones but not their parental cells (Figure 6H). These results demonstrated that exposure to IBE may render lung cancer cells more sensitive to EGFR TKIs. However, the effect of IBE on TKI sensitization was not found in HCC827 cells (data not shown), suggesting that exposure to IBE enhances EGFR-TKI sensitivity in wild-type NSCLC cells but not in mutant EGFR NSCLC cells.
Figure 6.
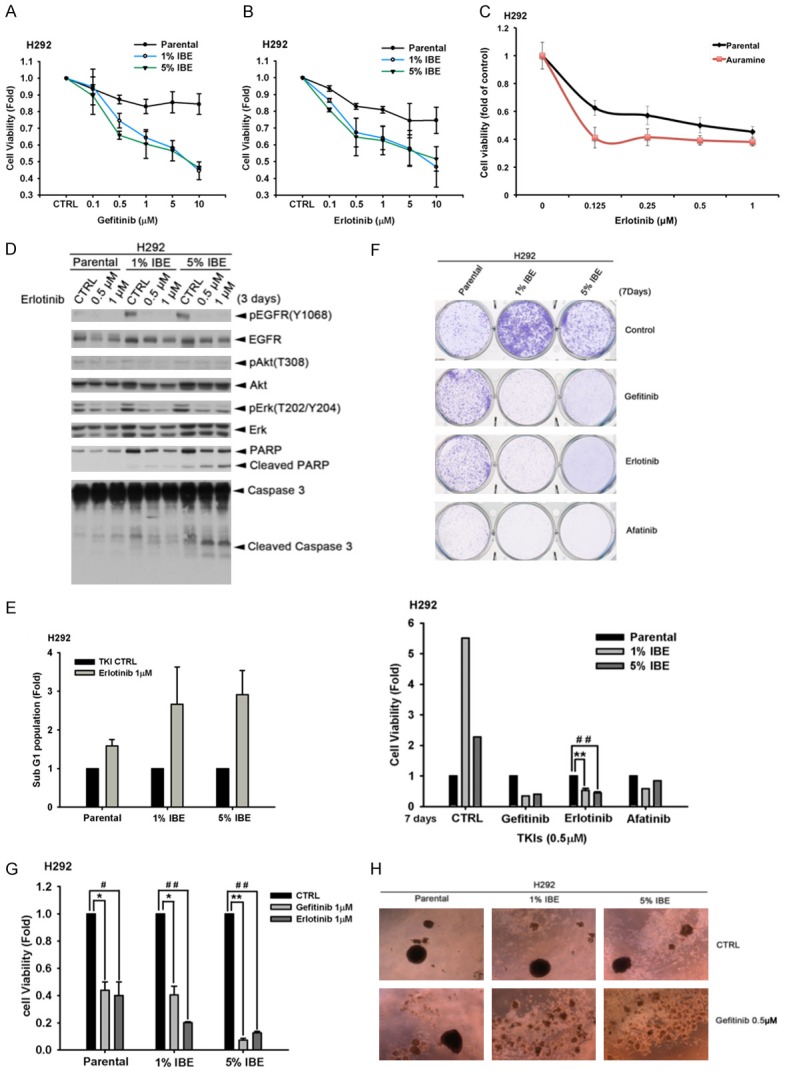
IBE/Auramine treatments render EGFR wt-expressing lung cancer cells more sensitive to EGFR TKI. (A and B) H292 parental and IBE stable clone cells were seeded at the density of 2500 cells/well and treated with 0.1, 0.5, 1, 5, 10 μM of gefitinib (A) or erlotinib (B) for 3 days. Cell viabilities were determined by WST-1 assay. (C) H292 parental cells and auramine stable clone were seeded at the density of 2500 cells/well and treated with indicated concentration of erlotinib for 3 days. Cell viabilities were determined by WST-1 assay. (D) H292 parental cells and IBE clones were treated with 1 μM of EGFR TKI erlotinib for 3 days. Whole-lysates extracted from these cells were subjected to Western blot analysis with indicated antibodies. (E) H292 parental cells and their IBE stable clone were treated with 1 μM of EGFR TKI erlotinib for 3 days. The sub-G1 population was determined by using flow cytometry analysis with PI staining. (F) H292 parental cells and IBE stable clone were treated with 0.5 μM EGFR-TKI gefitinib, erlotinib or afatinib for 7 days followed by 1% crystal violet staining. The result was quantitated determined according to the absorbance at 570 nm followed by dissolving crystal violet with acetic acid. (G) H292 parental cells and IBE stable clone were seeded in 12-well and then treated with 1 μM of gefitinib or erlotinib for 3 days. Cell numbers were counted. (H) H292 parental cells and IBE clones were seeded and suspended in sphere formation medium in non-coated dishes with 0.5 μM EGFR-TKI gefitinib for 7 days. Results were expressed as mean ± S.E.M. of two independent experiments. * or #: P < 0.05; ** or ##: P < 0.01 as compared with control group.
Discussion
Lung cancer is one of the most common cancer types worldwide, and its incidence and mortality are very high, especially in the developed countries. It might attribute to multiple risk factors, such as lung cancer family gene history, cigarette smoke, air pollution or cooking smoke, and incense-burning smoke, which can induce lung cancer formation. Among these risk factors, incense-burning smoke, a part of daily life of Asian people, might be a very important factor in lung cancer tumorigenesis [30-32] and other diseases [28,29]. However, it is not clear whether incense-burning smoke can induce tumor progression of lung cancer. In this study, we demonstrated that IBE induced cell progression in proliferation, migration and invasion abilities. These findings are paralleled to some case reports that people long-term exposure to incense smoke environment, such as temple worker or Buddhism, Taoism and Shinto believer, may increase the risk of squamous cell carcinoma of the respiratory tract or lung cancer, and cigarette smoke may further increase their risk [39]. Similar to the inducing effect of cigarette smoke on DNA synthesis [40], our data that population of NSCLC in S phase was increased by IBE also supports the role of incense smoking in promoting lung cancer proliferation.
Cell proliferation as a result of S phase accumulation involving various reasons, and overexpression of growth factor receptors on cell membranes was the main event contributing to cell survival [41]. EGFR is overexpressed approximately in 40-80% of lung cancers and drives important signaling pathways for cancer progression [42]. EGFR mutations lead to its kinase activation and are associated with development of non-small cell lung cancer [43]. Our data indicated that IBE not only induced the cell progression and spheroid formation but also the sensitivity to EGFR TKI gefitinib or erlotinib. Previous studies showed that EGFR activating mutation is associated with EGFR TKIs sensitivity [15,16]. Our data showed that EGFR and its downstream signaling Akt and Erk activations in IBE- and auramine-treated cells. However, EGFR mutations were not found in the IBE-treated cells in this study. On the other hand, EGFR mutants were found in many NSCLC cell types, and may represent loss-of-function of some negatively regulatory domains or enhance tyrosine kinase activity intrinsically [44,45]. Somatic mutations of EGFR, such as exon19 deletions or L858R substitution can be detected in NSCLC cells, which are highly responsive to EGFR TKIs [46]. However, EGFR secondary T790M mutation was determined to cause TKIs resistance [46]. In this study, EGFR-activating and TKI-sensitizing effects of IBE was not observed in EGFR mutant-expressing NSCLC cells, suggesting that EGFR activity in these cells is too high to be further enhanced by IBE.
Autocrine regulation in cancer is recognized as a major mechanism providing self-sufficient growth signals, including EGFR activation [47]. Several ligands, including EGF, transforming growth factor α (TGF-α), amphiregulin (AREG), heparin-binding EGF-like growth factor (HB-EGF), betacellulin, and epiregulin, were identified for EGFR activation [47]. The involvement of EGFR activation in diseases or normal tissue homeostasis depends on the expression levels of both ligand and receptor [47]. Among these well-known EGFR ligands, our data from this study showed that autocrine cytokines AREG mediates the IBE-induced activation of EGFR signaling, leading to NSCLC progression and sensitization to EGFR TKIs. AREG is associated with lower survival rate of patients with NSCLC and poor prognosis [48,49]. In contrast to our findings, however, high serum level of AREG has also been proposed as a diagnostic marker for predicting a poor response of advance NSCLC patients to gefitinib [50]. The anti-apoptotic activity of AREG through inactivation of BAX has further been proposed to mediate the gefitinib resistance in NSCLCs [51]. In addition to the classic ligands for EGFR, angiogenin/Ribonuclease 5 has been recently identified as a novel EGFR ligand and a serum biomarker for erlotinib sensitivity in pancreatic cancer [52]. Interestingly, our data from mass spectrum analysis (see Supplementary Materials and Methods) showed the significant changes in the gene expressions, which are involved in RNA process, in response to IBE and Auramine treatment (Supplementary Figure 4). It is worthy to further address whether Ribonuclease 5 or other potential molecules are also regulated by IBE to affect the sensitivity to EGFR TKIs.
In addition to EGFR, co-regulation of other pathways by IBE and auramine may account for sensitizing effect of AREG on the response to EGFR TKIs. Although AREG has also been reported to activate HER2 activity through dimerization with EGFR for tumor proliferation and migration of breast cancer cells [53], HER2 tyrosine kinase activity was not affected by IBE treatment in NSCLCs in our study. c-Met amplification has been demonstrated as one of the reasons causing the acquired EGFR TKIs resistance, and therefore combination with c-Met inhibitors was employed to improve survival rate of lung cancer patients who failed to EGFR TKIs [54,55]. Interestingly, our data showed that IBE not only increases EGFR activation but also suppresses the kinase activity of c-MET. It seemingly exhibits the switch of survival dependence from multiple oncogenic drivers, including c-MET, to EGFR mainly, revealing a possible “Achilles’ heel” that can be targeted therapeutically in incent smoke-associated NSCLC cells. Thus, exposure to IBE or auramine may render H292 cells more oncogenic addictive to EGFR signaling, resulting in EGFR TKIs sensitization. However, it remains further studies to further investigate how IBE and auramine induce AREG expression and suppress c-MET activation.
In summary, IBE and its ingredient auramine increased EGFR activation through AREG induction but suppressed c-Met activity to not only promote tumor progression but also enhance EGFR TKIs sensitivity in EGFR wild-type cells (Figure 7). These findings provide another plausible explanation of high response rate to EGFR TKIs in eastern Asian NSCLC patients, but further clinical studies are required to prove this notion.
Figure 7.
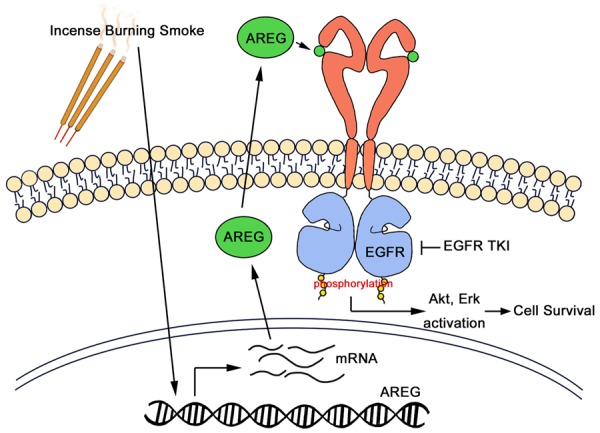
Hypothetical working model of incense burning smoke render NSCLC more aggressive and sensitive to EGFR TKIs. Exposure to IBE or its residue composition auramine can induce tumor progression and enhance EGFR TKI sensitivity through modulation of EGFR signaling. AREG is upregulated by IBE and auramine transcriptionally and is secreted as an autocrine ligand for EGFR activation, rendering NSCLC cells more addicted to the increased EGFR signaling for survival.
Acknowledgements
This work was supported by grants from the Ministry of Science Technology, Taiwan (MOST105-2320-B-039-056-MY3, MOST-105-2314-B-039-035-MY3, MOST105-2314-B-040-011-MY3), China Medical University Hospital (DMR-107-174), and the Ministry of Health and Welfare (MOHW105-TDU-B-212-13400). This work was also financially supported by the “Drug Development Center, China Medical University” from The Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE) in Taiwan. The grantee had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Gazdar AF. Activating and resistance mutations of EGFR in non-small-cell lung cancer: role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene. 2009;28(Suppl 1):S24–31. doi: 10.1038/onc.2009.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 3.Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer. 2007;7:169–181. doi: 10.1038/nrc2088. [DOI] [PubMed] [Google Scholar]
- 4.Lo HW, Hsu SC, Hung MC. EGFR signaling pathway in breast cancers: from traditional signal transduction to direct nuclear translocalization. Breast Cancer Res Treat. 2006;95:211–218. doi: 10.1007/s10549-005-9011-0. [DOI] [PubMed] [Google Scholar]
- 5.Liang W, Wu X, Fang W, Zhao Y, Yang Y, Hu Z, Xue C, Zhang J, Zhang J, Ma Y, Zhou T, Yan Y, Hou X, Qin T, Dinglin X, Tian Y, Huang P, Huang Y, Zhao H, Zhang L. Network meta-analysis of erlotinib, gefitinib, afatinib and icotinib in patients with advanced non-small-cell lung cancer harboring EGFR mutations. PLoS One. 2014;9:e85245. doi: 10.1371/journal.pone.0085245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fukuoka M, Yano S, Giaccone G, Tamura T, Nakagawa K, Douillard JY, Nishiwaki Y, Vansteenkiste J, Kudoh S, Rischin D, Eek R, Horai T, Noda K, Takata I, Smit E, Averbuch S, Macleod A, Feyereislova A, Dong RP, Baselga J. Multi-institutional randomized phase II trial of gefitinib for previously treated patients with advanced non-small-cell lung cancer (the IDEAL 1 trial) [corrected] . J. Clin. Oncol. 2003;21:2237–2246. doi: 10.1200/JCO.2003.10.038. [DOI] [PubMed] [Google Scholar]
- 7.Janne PA, Gurubhagavatula S, Yeap BY, Lucca J, Ostler P, Skarin AT, Fidias P, Lynch TJ, Johnson BE. Outcomes of patients with advanced non-small cell lung cancer treated with gefitinib (ZD1839, “Iressa”) on an expanded access study. Lung Cancer. 2004;44:221–230. doi: 10.1016/j.lungcan.2003.12.014. [DOI] [PubMed] [Google Scholar]
- 8.Kris MG, Natale RB, Herbst RS, Lynch TJ Jr, Prager D, Belani CP, Schiller JH, Kelly K, Spiridonidis H, Sandler A, Albain KS, Cella D, Wolf MK, Averbuch SD, Ochs JJ, Kay AC. Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non-small cell lung cancer: a randomized trial. JAMA. 2003;290:2149–2158. doi: 10.1001/jama.290.16.2149. [DOI] [PubMed] [Google Scholar]
- 9.Miller VA, Kris MG, Shah N, Patel J, Azzoli C, Gomez J, Krug LM, Pao W, Rizvi N, Pizzo B, Tyson L, Venkatraman E, Ben-Porat L, Memoli N, Zakowski M, Rusch V, Heelan RT. Bronchioloalveolar pathologic subtype and smoking history predict sensitivity to gefitinib in advanced non-small-cell lung cancer. J. Clin. Oncol. 2004;22:1103–1109. doi: 10.1200/JCO.2004.08.158. [DOI] [PubMed] [Google Scholar]
- 10.Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, Halmos B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 11.Peled N, Yoshida K, Wynes MW, Hirsch FR. Predictive and prognostic markers for epidermal growth factor receptor inhibitor therapy in non-small cell lung cancer. Ther Adv Med Oncol. 2009;1:137–144. doi: 10.1177/1758834009347923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.da Cunha Santos G, Shepherd FA, Tsao MS. EGFR mutations and lung cancer. Annu Rev Pathol. 2011;6:49–69. doi: 10.1146/annurev-pathol-011110-130206. [DOI] [PubMed] [Google Scholar]
- 13.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, Haber DA. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 14.Russo A, Franchina T, Ricciardi GR, Picone A, Ferraro G, Zanghi M, Toscano G, Giordano A, Adamo V. A decade of EGFR inhibition in EGFR-mutated non small cell lung cancer (NSCLC): old successes and future perspectives. Oncotarget. 2015;6:26814–26825. doi: 10.18632/oncotarget.4254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Riely GJ, Pao W, Pham D, Li AR, Rizvi N, Venkatraman ES, Zakowski MF, Kris MG, Ladanyi M, Miller VA. Clinical course of patients with non-small cell lung cancer and epidermal growth factor receptor exon 19 and exon 21 mutations treated with gefitinib or erlotinib. Clin Cancer Res. 2006;12:839–844. doi: 10.1158/1078-0432.CCR-05-1846. [DOI] [PubMed] [Google Scholar]
- 16.Jackman DM, Yeap BY, Sequist LV, Lindeman N, Holmes AJ, Joshi VA, Bell DW, Huberman MS, Halmos B, Rabin MS, Haber DA, Lynch TJ, Meyerson M, Johnson BE, Janne PA. Exon 19 deletion mutations of epidermal growth factor receptor are associated with prolonged survival in non-small cell lung cancer patients treated with gefitinib or erlotinib. Clin Cancer Res. 2006;12:3908–3914. doi: 10.1158/1078-0432.CCR-06-0462. [DOI] [PubMed] [Google Scholar]
- 17.Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, Singh B, Heelan R, Rusch V, Fulton L, Mardis E, Kupfer D, Wilson R, Kris M, Varmus H. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101:13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, Eck MJ, Sellers WR, Johnson BE, Meyerson M. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 19.Sordella R, Bell DW, Haber DA, Settleman J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science. 2004;305:1163–1167. doi: 10.1126/science.1101637. [DOI] [PubMed] [Google Scholar]
- 20.Kerr KM. Clinical relevance of the new IASLC/ERS/ATS adenocarcinoma classification. J Clin Pathol. 2013;66:832–838. doi: 10.1136/jclinpath-2013-201519. [DOI] [PubMed] [Google Scholar]
- 21.Shim HS, Lee DH, Park EJ, Kim SH. Histopathologic characteristics of lung adenocarcinomas with epidermal growth factor receptor mutations in the International Association for the Study of Lung Cancer/American Thoracic Society/European Respiratory Society lung adenocarcinoma classification. Arch Pathol Lab Med. 2011;135:1329–1334. doi: 10.5858/arpa.2010-0493-OA. [DOI] [PubMed] [Google Scholar]
- 22.Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin TC, Krishnaswamy G, Chi DS. Incense smoke: clinical, structural and molecular effects on airway disease. Clin Mol Allergy. 2008;6:3. doi: 10.1186/1476-7961-6-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin TC, Chang FH, Hsieh JH, Chao HR, Chao MR. Characteristics of polycyclic aromatic hydrocarbons and total suspended particulate in indoor and outdoor atmosphere of a Taiwanese temple. J Hazard Mater. 2002;95:1–12. doi: 10.1016/s0304-3894(02)00146-2. [DOI] [PubMed] [Google Scholar]
- 25.See SW, Wang YH, Balasubramanian R. Contrasting reactive oxygen species and transition metal concentrations in combustion aerosols. Environ Res. 2007;103:317–324. doi: 10.1016/j.envres.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 26.Chuang HC, BeruBe K, Lung SC, Bai KJ, Jones T. Investigation into the oxidative potential generated by the formation of particulate matter from incense combustion. J Hazard Mater. 2013;244-245:142–150. doi: 10.1016/j.jhazmat.2012.11.034. [DOI] [PubMed] [Google Scholar]
- 27.Dewangan S, Chakrabarty R, Zielinska B, Pervez S. Emission of volatile organic compounds from religious and ritual activities in India. Environ Monit Assess. 2013;185:9279–9286. doi: 10.1007/s10661-013-3250-z. [DOI] [PubMed] [Google Scholar]
- 28.Pan A, Clark ML, Ang LW, Yu MC, Yuan JM, Koh WP. Incense use and cardiovascular mortality among Chinese in Singapore: the Singapore Chinese health study. Environ Health Perspect. 2014;122:1279–1284. doi: 10.1289/ehp.1307662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hwang YH, Lin YS, Lin CY, Wang IJ. Incense burning at home and the blood lead level of preschoolers in Taiwan. Environ Sci Pollut Res Int. 2014;21:13480–13487. doi: 10.1007/s11356-014-3273-1. [DOI] [PubMed] [Google Scholar]
- 30.Chiang KC, Liao CM. Heavy incense burning in temples promotes exposure risk from airborne PMs and carcinogenic PAHs. Sci Total Environ. 2006;372:64–75. doi: 10.1016/j.scitotenv.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 31.Friborg JT, Yuan JM, Wang R, Koh WP, Lee HP, Yu MC. Incense use and respiratory tract carcinomas: a prospective cohort study. Cancer. 2008;113:1676–1684. doi: 10.1002/cncr.23788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dalibalta S, Elsayed Y, Alqtaishat F, Gomes I, Fernandes N. A health risk assessment of Arabian incense (Bakhour) smoke in the United Arab Emirates. Sci Total Environ. 2015;511:684–691. doi: 10.1016/j.scitotenv.2014.12.024. [DOI] [PubMed] [Google Scholar]
- 33.Tang L, Lim WY, Eng P, Leong SS, Lim TK, Ng AW, Tee A, Seow A. Lung cancer in Chinese women: evidence for an interaction between tobacco smoking and exposure to inhalants in the indoor environment. Environ Health Perspect. 2010;118:1257–1260. doi: 10.1289/ehp.0901587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Asahina H, Yamazaki K, Kinoshita I, Sukoh N, Harada M, Yokouchi H, Ishida T, Ogura S, Kojima T, Okamoto Y, Fujita Y, Dosaka-Akita H, Isobe H, Nishimura M. A phase II trial of gefitinib as first-line therapy for advanced non-small cell lung cancer with epidermal growth factor receptor mutations. Br J Cancer. 2006;95:998–1004. doi: 10.1038/sj.bjc.6603393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yoshida K, Yatabe Y, Park JY, Shimizu J, Horio Y, Matsuo K, Kosaka T, Mitsudomi T, Hida T. Prospective validation for prediction of gefitinib sensitivity by epidermal growth factor receptor gene mutation in patients with non-small cell lung cancer. J Thorac Oncol. 2007;2:22–28. [PubMed] [Google Scholar]
- 36.Inoue A, Suzuki T, Fukuhara T, Maemondo M, Kimura Y, Morikawa N, Watanabe H, Saijo Y, Nukiwa T. Prospective phase II study of gefitinib for chemotherapy-naive patients with advanced non-small-cell lung cancer with epidermal growth factor receptor gene mutations. J. Clin. Oncol. 2006;24:3340–3346. doi: 10.1200/JCO.2005.05.4692. [DOI] [PubMed] [Google Scholar]
- 37.Tamura K, Okamoto I, Kashii T, Negoro S, Hirashima T, Kudoh S, Ichinose Y, Ebi N, Shibata K, Nishimura T, Katakami N, Sawa T, Shimizu E, Fukuoka J, Satoh T, Fukuoka M West Japan Thoracic Oncology Group. Multicentre prospective phase II trial of gefitinib for advanced non-small cell lung cancer with epidermal growth factor receptor mutations: results of the West Japan Thoracic Oncology Group trial (WJTOG0403) Br J Cancer. 2008;98:907–914. doi: 10.1038/sj.bjc.6604249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tung JC, Huang WC, Yang JC, Chen GY, Fan CC, Chien YC, Lin PS, Candice Lung SC, Chang WC. Auramine O, an incense smoke ingredient, promotes lung cancer malignancy. Environ Toxicol. 2017;32:2379–2391. doi: 10.1002/tox.22451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tse LA, Yu IT, Qiu H, Au JS, Wang XR. A case-referent study of lung cancer and incense smoke, smoking, and residential radon in Chinese men. Environ Health Perspect. 2011;119:1641–1646. doi: 10.1289/ehp.1002790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krayzler E, Nagler RM. Cigarette smoke-induced effects on the cell cycle in oral cancer cells: reduction of G2/M fraction. Cancer Genomics Proteomics. 2015;12:73–76. [PubMed] [Google Scholar]
- 41.Collin de L’hortet A, Gilgenkrantz H, Guidotti JE. EGFR: a master piece in G1/S phase transition of liver regeneration. Int J Hepatol. 2012;2012:476910. doi: 10.1155/2012/476910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ettinger DS. Clinical implications of EGFR expression in the development and progression of solid tumors: focus on non-small cell lung cancer. Oncologist. 2006;11:358–373. doi: 10.1634/theoncologist.11-4-358. [DOI] [PubMed] [Google Scholar]
- 43.Sequist LV, Bell DW, Lynch TJ, Haber DA. Molecular predictors of response to epidermal growth factor receptor antagonists in non-small-cell lung cancer. J. Clin. Oncol. 2007;25:587–595. doi: 10.1200/JCO.2006.07.3585. [DOI] [PubMed] [Google Scholar]
- 44.Pines G, Kostler WJ, Yarden Y. Oncogenic mutant forms of EGFR: lessons in signal transduction and targets for cancer therapy. FEBS Lett. 2010;584:2699–2706. doi: 10.1016/j.febslet.2010.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Red Brewer M, Yun CH, Lai D, Lemmon MA, Eck MJ, Pao W. Mechanism for activation of mutated epidermal growth factor receptors in lung cancer. Proc Natl Acad Sci U S A. 2013;110:E3595–3604. doi: 10.1073/pnas.1220050110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Engelman JA, Mukohara T, Zejnullahu K, Lifshits E, Borras AM, Gale CM, Naumov GN, Yeap BY, Jarrell E, Sun J, Tracy S, Zhao X, Heymach JV, Johnson BE, Cantley LC, Janne PA. Allelic dilution obscures detection of a biologically significant resistance mutation in EGFR-amplified lung cancer. J Clin Invest. 2006;116:2695–2706. doi: 10.1172/JCI28656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.DeWitt AE, Dong JY, Wiley HS, Lauffenburger DA. Quantitative analysis of the EGF receptor autocrine system reveals cryptic regulation of cell response by ligand capture. J Cell Sci. 2001;114:2301–2313. doi: 10.1242/jcs.114.12.2301. [DOI] [PubMed] [Google Scholar]
- 48.Busser B, Coll JL, Hurbin A. The increasing role of amphiregulin in non-small cell lung cancer. Pathol Biol (Paris) 2009;57:511–512. doi: 10.1016/j.patbio.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 49.Taverna S, Pucci M, Giallombardo M, Di Bella MA, Santarpia M, Reclusa P, Gil-Bazo I, Rolfo C, Alessandro R. Amphiregulin contained in NSCLC-exosomes induces osteoclast differentiation through the activation of EGFR pathway. Sci Rep. 2017;7:3170. doi: 10.1038/s41598-017-03460-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ishikawa N, Daigo Y, Takano A, Taniwaki M, Kato T, Hayama S, Murakami H, Takeshima Y, Inai K, Nishimura H, Tsuchiya E, Kohno N, Nakamura Y. Increases of amphiregulin and transforming growth factor-alpha in serum as predictors of poor response to gefitinib among patients with advanced non-small cell lung cancers. Cancer Res. 2005;65:9176–9184. doi: 10.1158/0008-5472.CAN-05-1556. [DOI] [PubMed] [Google Scholar]
- 51.Busser B, Sancey L, Josserand V, Niang C, Favrot MC, Coll JL, Hurbin A. Amphiregulin promotes BAX inhibition and resistance to gefitinib in non-small-cell lung cancers. Mol Ther. 2010;18:528–535. doi: 10.1038/mt.2009.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang YN, Lee HH, Chou CK, Yang WH, Wei Y, Chen CT, Yao J, Hsu JL, Zhu C, Ying H, Ye Y, Wang WJ, Lim SO, Xia W, Ko HW, Liu X, Liu CG, Wu X, Wang H, Li D, Prakash LR, Katz MH, Kang Y, Kim M, Fleming JB, Fogelman D, Javle M, Maitra A, Hung MC. Angiogenin/Ribonuclease 5 is an EGFR ligand and a serum biomarker for erlotinib sensitivity in pancreatic cancer. Cancer Cell. 2018;33:752–769. e758. doi: 10.1016/j.ccell.2018.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schmucker H, Blanding WM, Mook JM, Wade JF, Park JP, Kwist K, Shah H, Booth BW. Amphiregulin regulates proliferation and migration of HER2-positive breast cancer cells. Cell Oncol (Dordr) 2018;41:159–168. doi: 10.1007/s13402-017-0363-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brugger W, Thomas M. EGFR-TKI resistant non-small cell lung cancer (NSCLC): new developments and implications for future treatment. Lung Cancer. 2012;77:2–8. doi: 10.1016/j.lungcan.2011.12.014. [DOI] [PubMed] [Google Scholar]
- 55.McDermott U, Pusapati RV, Christensen JG, Gray NS, Settleman J. Acquired resistance of non-small cell lung cancer cells to MET kinase inhibition is mediated by a switch to epidermal growth factor receptor dependency. Cancer Res. 2010;70:1625–1634. doi: 10.1158/0008-5472.CAN-09-3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.