

Abstract
Cancer stem cells (CSCs) have been found in many different types of malignant tumors. In our previous study, we found that sphere-forming cells (SFCs) from the renal cell carcinoma (RCC) cell line SK-RC-42 are rich in CSCs. However, our previous reports are based on only one human RCC cell line, which makes it difficult to determine whether the findings from this cell line represent the general mechanisms in human RCC. Therefore, in this study, we attempted to evaluate whether the sphere culture method could enrich for CSCs from another human RCC cell line, 786-O, and to characterize their immunological phenotype. We discovered that a small population of 786-O cells was capable of growing as tumor spheres. The SFCs had many properties similar to CSCs, including greater ability to self-renew in vitro and in vivo, higher mRNA expression levels of several ‘stemness’ genes, stronger tumorigenicity and resistance to chemotherapeutic agents than monolayer adherent cells (MACs). The SFCs expressed low levels of MHC-I, HLA-DR, CD80, CD86, CD152 and CD154. Additionally, the SFCs had lower expression levels of Her2 and hTERT, FasL, Fas, the transcription factor forkhead box protein 3 (FoxP3) and activating natural killer cell receptors than did the MACs. In addition, both 786-O SFCs and MACs were weakly positive for B7-H4 expression, while the expression level of B7-H1 in 786-O SFCs was lower than that in MACs. Furthermore, 786-O SFCs and MACs both expressed substantial and comparable levels of membrane complement regulatory proteins (mCRPs). Finally, we found that 786-O SFCs triggered T cell apoptosis. These findings suggested that tumor spheres from 786-O cells are rich in CSCs. The immunological phenotype of the SFCs described in our study suggests that CSCs might play an important role in tumor immune evasion.
Keywords: Renal cell carcinoma, tumor spheres, cancer stem cells, immunological phenotype, tumor immune evasion
Introduction
Renal cell carcinoma (RCC) is one of the most common types of urologic tumors, representing more than 80% of all malignancies of the kidney. RCC is also known to be highly vascular and relatively radioresistant [1]. Emerging evidence shows that cancer stem cells (CSCs) play an important role in cancer initiation, progression and recurrence [2,3]. A useful approach for the identification and purification of CSCs, specifically in the absence of suitable surface marker expression, is based on the phenomenon that stem cells have the ability to form spheres in serum-free medium (SFM) [4].
Sphere-forming cells (SFCs) were first isolated from the central nervous system and were able to generate neurons and astrocytes in culture [5]. The sphere culture method has since been employed to isolate and characterize adult stem cells. Under similar environmental conditions, a subpopulation of tumor-derived cells have been found to behave similarly to endogenous stem cells and are thus referred to as CSCs [4].
In our previous study, we successfully used the sphere culture method to identify and enrich for CSCs from an established human RCC cell line SK-RC-42 [6]. However, our previous reports are based on only one human RCC cell line, which makes it difficult to determine whether the observations based on the cell line represent general mechanisms in human RCC. Thus, in this study, we attempted to evaluate whether the sphere culture method could enrich for CSCs from another human RCC cell line, 786-O, and to further characterize their immunological phenotype.
Materials and methods
Ethics statement
Mice were housed in facilities accredited by the Experimental Animal Center of Central South University (Changsha, Hunan, China). Our study was conducted in accordance with the recommendations of the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health. The study protocol was approved by the Animal Experimental Ethics Committee of Hunan Province and the Research Ethics Committee of our hospital. Informed consent was obtained from healthy donors.
Culture of RCC cells, tumor spheres and tumor subspheres
The human RCC cell line 786-O was obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). Monolayer adherent cells (MACs) were maintained in Dulbecco’s modified Eagle’s medium (DMEM)/F-12 medium with 10% FBS. Sphere cell culture was performed according to published protocols with modifications [5,6]. Briefly, single cells were plated in ultra-low attachment plates (Corning, NY) at a density of 10,000 viable cells/ml in the initial passage and 500 cells/ml in subsequent passages. The cells were grown in SFM consisting of DMEM/F-12 medium supplemented with 20 ng/ml EGF (Sigma), 20 ng/ml bFGF (Sigma) and B27 (Invitrogen). To passage sphere cells, we dissociated cells with 0.1% trypsin and 1 mM EDTA (Invitrogen), strained them through a 40-μm nylon mesh (Falcon) and microscopically analyzed single cells and used them for subsequent experiments.
Side population (SP) analysis, proliferation assay, cell cycle analysis, semiquantitative reverse transcription polymerase chain reaction (RT-PCR), and drug sensitivity assay
The detailed procedures have been described in our recent paper [6].
Flow cytometry
The following anti-human monoclonal antibodies (mAbs) were used for flow cytometry: anti-CD133-PE (Miltenyi-Biotec); anti-OX40-PE, anti-CD44-FITC and anti-CD24-PE (BD Biosciences); anti-CD105-PE, anti-HLA-I-FITC, anti-CD95-FITC, anti-CD95L-PE, anti-CD155-PE, anti-CD112-PE, anti-B7-H1-PE, anti-B7-H4-PE, anti-FoxP3-PE, anti-HLA-E-PE, anti-HLA-G-PE, anti-CD46-PE, anti-CD55-PE and anti-CD59-PE (eBioscience); anti-CD40-PE, anti-HLA-DR-PE, anti-CD34-PE, anti-CD80-FITC and anti-CD86-PE (Immunotech); anti-CD200-FITC (AbD Serotec); and anti-Her2-FITC (Bender Medsystems). Unconjugated mouse anti-hTERT antibodies and FITC-conjugated goat anti-mouse IgM were also used. Appropriate isotype controls were used for each antibody. Intracellular forkhead box protein 3 (FoxP3) and hTERT staining was performed after fixation and permeabilization of cells with Fix/Perm buffers (BD Pharmingen) according to the manufacturer’s instructions. Flow cytometry was performed using a COULTER EPICS XL flow cytometer (Beckman Coulter) equipped with the Expo32 software. Side-scatter and forward-scatter profiles were used to eliminate cell doublets. The results are expressed as the mean fluorescence intensity (MFI) and % of positive cells.
Tumorigenicity and histological analysis
Five-week-old BALB/c female nude mice were purchased from the Animal Institute of Peking Union Medical College (PUMC). Approximately 5 × 104 786-O SFCs and MACs were subcutaneously injected into the left and right axilla, respectively, of the same nude mouse (n = 5) to comparatively evaluate their tumorigenic potential. Hematoxylin and eosin (H&E) staining was performed on 5-μm paraffin-embedded sections following standard protocols.
Apoptosis assay
Peripheral blood mononuclear cells (PBMCs) were prepared from healthy donor blood by centrifugation on a Ficoll-Hypaque density gradient (Sigma-Aldrich). Supernatants from 786-O SFCs and MACs were collected after 3 days in culture. T cell apoptosis assays were performed with the Annexin V/7-AAD staining kit (BD Pharmingen). Healthy donor PBMCs were cultured in medium, 786-O SFC supernatant or 786-O MAC supernatant and harvested by centrifugation after 24 h. The cells were stained with PE-conjugated anti-CD3 antibodies. Next, FITC-conjugated Annexin V and 7-AAD were added, and CD3+ T cell apoptosis was analyzed by flow cytometry.
Statistical methods
The results are expressed as the mean ± SD. Student’s t-test was used to compare the differences among groups. P < 0.05 was considered significant. Statistical analyses were performed using SPSS 19.0 software (SPSS Inc., Chicago, IL, USA).
Results
786-O RCC cells can form self-renewing tumor spheres
To determine if 786-O cells can form tumor spheres, we cultured the cells as monolayers in the presence of 10% FBS and subsequently in SFM containing EGF and bFGF. Large tumor spheres were formed 12 days later (Figure 1A). The self-renewal capacity of 786-O SFCs was assessed by dissociating the primary tumor spheres into single cells and growing them at a clonal density of 500 cells/ml in SFM containing EGF and bFGF. Tumor subspheres were formed after 14 days (Figure 1A), displaying the self-renewal capacity of the 786-O tumor spheres.
Figure 1.

Tumor spheres derived from 786-O cells share relevant features with stem cells. (A) Examples of primary spheres in 786-O and subspheres re-formed after single 786-O sphere cells were re-seeded at a clonal density and grown in DMEM/F-12 medium supplemented with 20 ng/ml EGF, 20 ng/ml bFGF and B27. (B) 786-O SFCs and MACs were incubated with antibodies against CD44, CD24, CD105, CD133 and CD34 and analyzed by flow cytometry. 786-O SFCs (C, D) and MACs (E, F) were incubated with Hoechst 33342. Control cells (C, E) were also incubated with verapamil (50 µM, Sigma) to block pumps that eliminate Hoechst 33342. The population selected by the P3 gate represents SP cells. SFCs, sphere-forming cells; MACs, monolayer adherent cells. One representative staining image of three independent experiments is shown.
Expression of putative stem cell markers and the presence of SPs in 786-O SFCs
To explore whether the tumor spheres could be used to enrich for cells expressing stem cell markers [7,8], we stained 786-O SFCs and MACs for CD44, CD24, CD34, CD105 and CD133. However, no significant differences were found regarding the expression of these cell surface markers between the two cell types (Figure 1B).
In many normal tissues or tumors, side population (SP) cells are thought to contain tissue stem cells or CSCs, respectively [9]. Therefore, we attempted to explore whether such SP cells were present in 786-O SFCs. As shown in Figure 1C-F, 786-O SFCs contained more SP cells than MACs (P < 0.01, n = 3).
Cell growth rates and cell cycle distribution
As shown in Figure 2A, 786-O SFCs proliferated at a significantly slower rate than MACs (P < 0.05), which is in accordance with our previous data [6].
Figure 2.
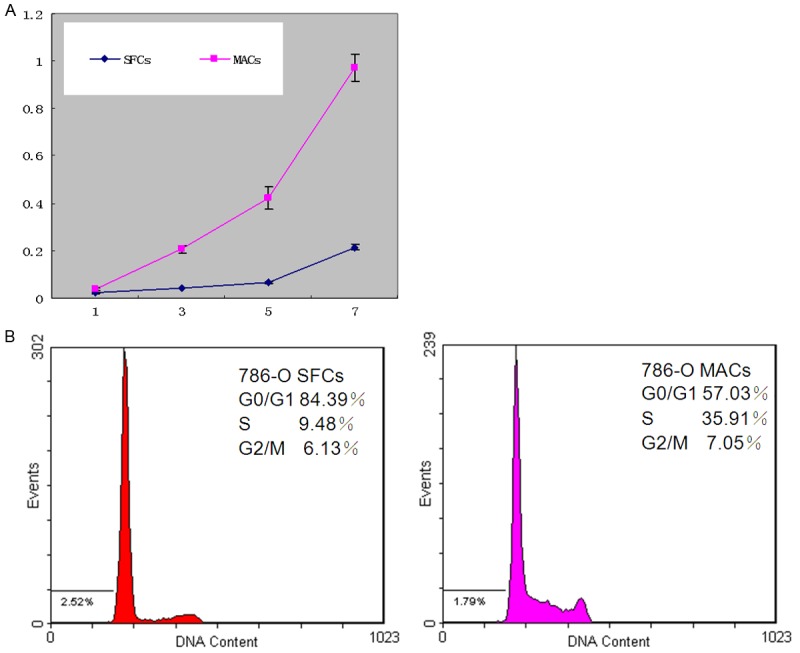
Cell growth curve and cell cycle distribution of 786-O SFCs and MACs. A. Approximately 500 healthy 786-O SFCs and MACs were exposed to DMEM/F-12 medium containing 10% FBS. The absorbance of the cells was measured for 7 days using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) method, and the cell growth curve was constructed accordingly. SFCs proliferate at a significantly slower rate than MACs (P < 0.05). B. For cell cycle analysis, 786-O SFCs and MACs were stained with 40 µg/ml propidium iodide and 1 mg/ml RNase after being fixed with 70% ethanol for 24 h. The cells were then collected using a COULTER EPICS XL flow cytometer (Beckman Coulter, USA). Compared to 786-O MACs, 786-O SFCs show a predominance of the population in G0/G1 phase (n = 3) (P < 0.01). The experiment has been repeated twice.
The cell cycle distributions of 786-O SFCs and MACs are shown in Figure 2B. Compared with 786-O MACs, SFCs showed a predominance of cells in the G0/G1 phase (P < 0.01, n = 3) (Figure 2B).
Drug sensitivity of 786-O SFCs
As shown in Figure 3A-C, 786-O SFCs and MACs were cultured in 96-well plates and treated with various concentrations of doxorubicin, mitomycin C and cisplatin. The SFCs showed significantly greater resistance to these three agents than did the MACs (P < 0.01, n = 3).
Figure 3.
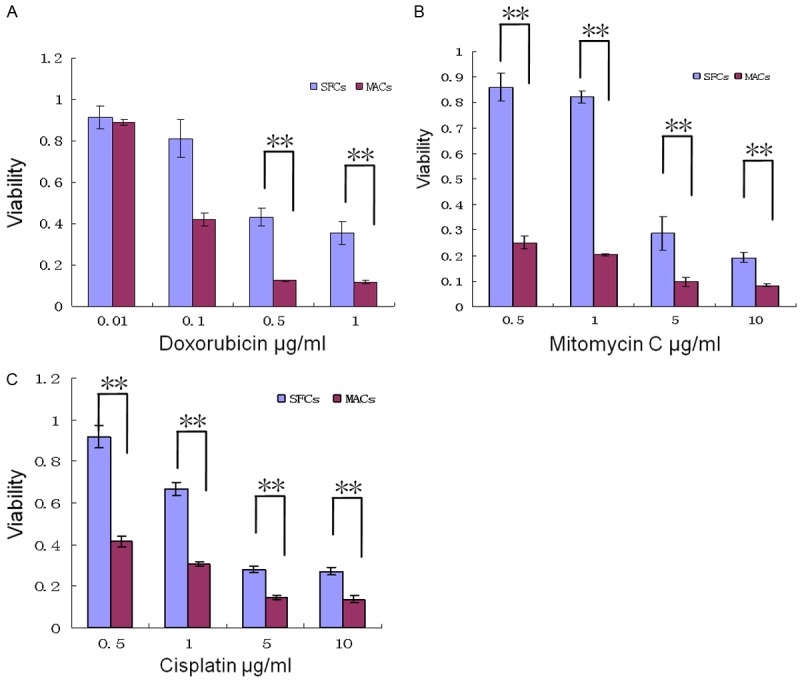
Results of chemotherapeutic drug sensitivity assays in 786-O SFCs and MACs. Approximately 2,000 SFCs and MACs were plated in 96-well flat-bottom plates. Different chemotherapeutic agents along a concentration gradient were added to the plates on the following day and in three replicate wells. Cell viability was evaluated after 2 days of drug treatment by the MTT (Sigma) assay. 786-O SFCs were more resistant to anticancer drugs doxorubicin (A), mitomycin C (B) and cisplatin (C) than were MACs. **P < 0.01. The experiment has been repeated twice.
Tumorigenicity of 786-O SFCs
To determine if 786-O SFCs differed in tumorigenicity from MACs, we implanted 786-O cells of each type into nude mice (n = 5). While the subcutaneous injection of 5 × 104 SFCs invariably gave rise to tumors, the same number of MACs was unable to generate tumors in mice after one month (Figure 4A). H&E staining results confirmed that the tumors formed by 786-O SFCs had typical features of human clear cell RCC. Importantly, more than 106 786-O MACs were required to form tumors in nude mice (data not shown). Thus, the tumor-formation ability of 786-O SFCs was 20 times greater than that of MACs.
Figure 4.
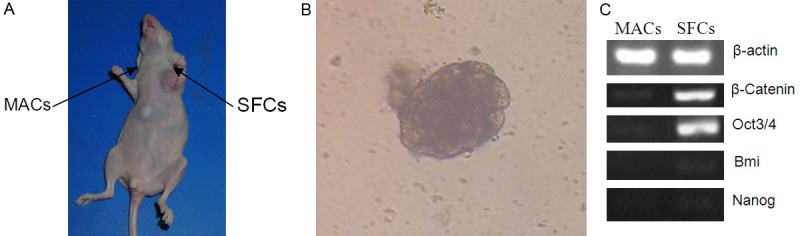
Tumorigenicity and preferential expression of ‘stemness’ genes in 786-O SFCs. A. Approximately 5 × 104 786-O SFCs and MACs were subcutaneously injected into the left and right axilla, respectively, of the same nude mouse, and the mouse was photographed one month after injection. The experiment has been repeated once with similar outcomes. B. When the mice were sacrificed 45 days after implantation, RCC cells were isolated from tumors grown from SFCs and cultured in SFM plus EGF and bFGF. Typical tumor spheres were formed within 2 to 3 weeks. C. Semiquantitative RT-PCR analysis of ‘stemness’ genes using 786-O SFCs and MACs. The experiment has been repeated twice with similar outcomes.
When mice were sacrificed 45 days after cell implantation, we isolated and cultured RCC cells from tumors grown from SFCs. Typical tumor spheres were formed within 2 to 3 weeks in SFM plus EGF and bFGF (Figure 4B). Furthermore, 5 × 104 tumor sphere cells were subcutaneously injected into the axilla of nude mice (n = 4). After 4 weeks, all injected mice developed tumors. These results indicate that a stem cell population exists in 786-O SFCs after in vivo transplantation.
Expression of ‘stemness’ genes by 786-O SFCs
As shown in Figure 4C and Supplemental Data, we found that 786-O SFCs expressed significantly higher mRNA levels of Oct3/4 and β-catenin than MACs, although neither 786-O SFCs not MACs displayed Bmi or Nanog expression, indicating that 786-O SFCs could be enriched for cells possessing stem cell-like properties.
Expression of MHC molecules, costimulatory molecules and tumor-associated antigens (TAAs) by 786-O SFCs
To characterize the immunologic phenotype of 786-O SFCs, we assessed the expression of MHC-I, MHC-II (HLA-DR), CD80, CD86, CD40, CD154, CD152 and OX40 by flow cytometry. As shown in Figure 5A and 5B, very few SFCs and MACs expressed MHC-I, HLA-DR, CD80, CD86, CD152 and CD154. In contrast, CD40 was consistently and significantly expressed by 786-O SFCs and MACs (Figure 5B). In addition, compared to MACs, SFCs showed preferential expression of OX40 (Figure 5B). These results suggested that 786-O SFCs and MACs do not possess the capacity to deliver signals for T cell activation and cannot be recognized by activated CD8+ cytotoxic T lymphocytes (CTLs).
Figure 5.

Immunophenotypes of 786-O SFCs and MACs. 786-O SFCs and MACs were incubated with Abs against MHC class I, MHC class II and costimulatory molecules (A and B), TAAs (C) and immune evasion-associated molecules (D), respectively. Then, the expression of MHC class I, MHC class II and costimulatory molecules (A and B), TAAs (C) and immune evasion-associated molecules (D) by 786-O SFCs and MACs was determined by flow cytometry. Proportions of the positive populations are shown. One representative staining image of three independent experiments is shown.
Next, flow cytometric analysis was used to explore the expression of tumor-associated antigens (TAAs) reported to be expressed by RCC cells [10]. Our results revealed Her2 expression in 74.97 ± 6.49% (mean ± SEM, n = 3) of MACs. In contrast, only 2.23 ± 0.84% (mean ± SEM, n = 3) of SFCs expressed Her2 (Figure 5C). In addition, hTERT was consistently expressed by 786-O SFCs and MACs (Figure 5C). Nonetheless, the expression level of hTERT in SFCs was lower than that in MACs (28.07 ± 3.43 versus 55.33 ± 4.61 MFI, n = 3). These results are in line with our previous data [6]. Taken together, these data suggest that 786-O SFCs likely downregulate the expression of TAAs to escape T cell surveillance.
Expression of immune evasion-associated molecules by 786-O SFCs
Recent reports suggest that CD200 expression by tumor cells suppresses antitumor responses [11]. However, CD200 was only expressed by very few 786-O SFCs and MACs (Figure 5D). Interestingly, we discovered that the expression levels of CD95L and CD95 on 786-O SFCs were lower than those on MACs (Figure 5D).
The expression of coinhibitory B7 family members, such as B7-H1 and B7-H4, by tumor cells has been shown to inhibit tumor-specific T cell-mediated immunity [12]. Both 786-O SFCs and MACs were weakly positive for B7-H4 expression (Figure 6A), but the expression level of B7-H1 in 786-O SFCs was lower than that in MACs (Figure 6A).
Figure 6.
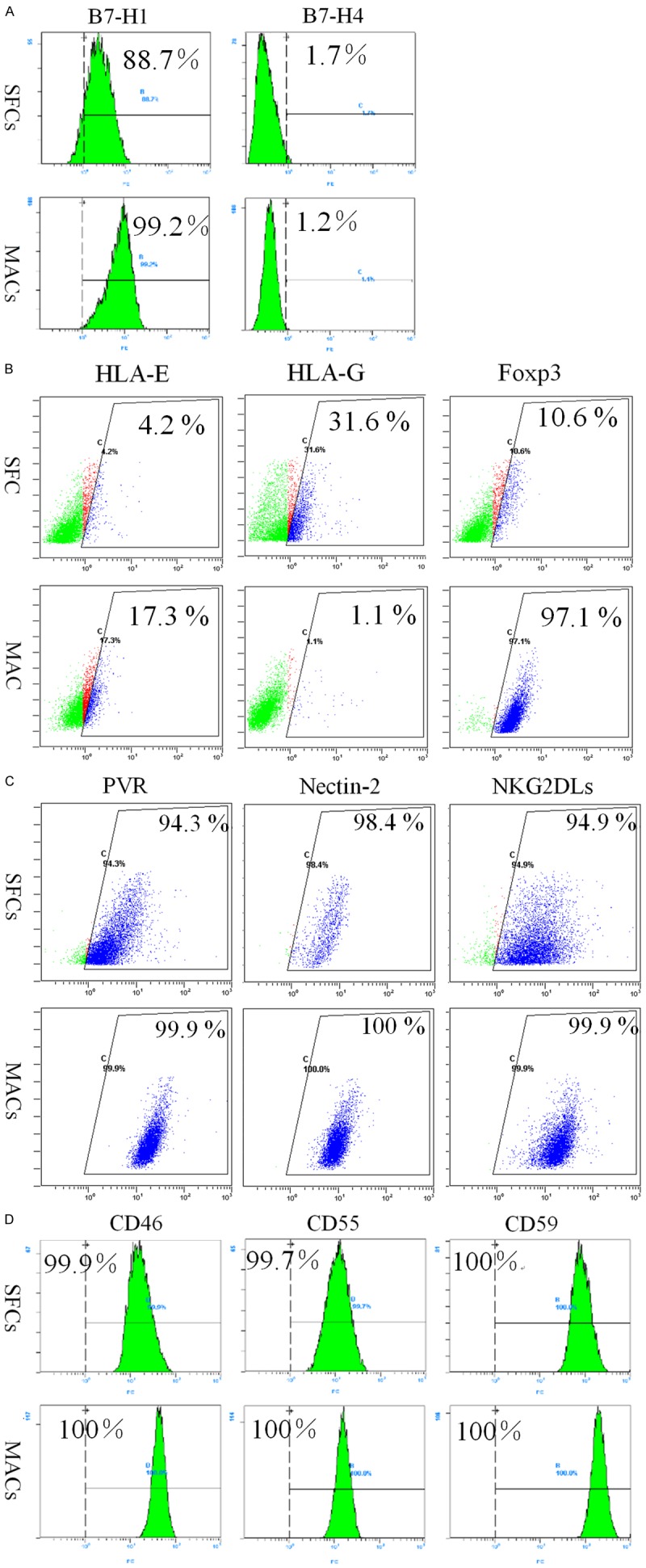
Expression of immune evasion-associated molecules on 786-O SFCs and MACs. Representative flow cytometric analyses of 786-O SFCs and MACs stained for coinhibitory B7 family members (A), non-classical MHC class I molecules (B) and ligands for activating NK receptors (C). (D) Single-color flow cytometric analyses of 786-O SFCs and MACs for the expression of mCRPs. One representative staining image of three independent experiments is shown.
HLA-E and HLA-G are non-classical MHC class I molecules, and accumulating evidence indicates their biological roles in inactivating immune responses [13]. Indeed, the expression level of HLA-E was lower and the expression level of HLA-G was higher in 786-O SFCs than in 786-O MACs (Figure 6B).
More interestingly, flow cytometric analysis revealed that 786-O MACs expressed substantial levels of the transcription factor FoxP3 (Figure 6B). However, the expression level of FoxP3 in 786-O SFCs was lower than that in MACs (Figure 6B).
Expression of ligands specific for natural killer (NK) cell receptors and membrane complement regulatory proteins (mCRPs) on 786-O SFCs
Natural killer (NK) cells have been shown to play important roles in tumor immunity. Thus, we detected the expression of ligands for activating NK cell receptors on 786-O SFCs and MACs. As shown in Figure 6C, the ligands for DNAM-1, such as poliovirus receptor (PVR) and Nectin-2, were expressed by nearly all 786-O SFCs and MACs. However, the expression of these two molecules on SFCs was much lower than that on MACs. Moreover, lower expression levels of cell surface NKG2DL were observed on 786-O SFCs than on MACs (Figure 6C), which is in line with our previous data [6] and findings of other investigators [14].
Furthermore, membrane complement regulatory proteins (mCRPs), such as CD46, CD55 and CD59, can be overexpressed by tumor cells to limit immune surveillance by the complement system [15]. Nearly all 786-O SFCs and MACs expressed CD46, CD55 and CD59 (Figure 6D), which is in line with our previous data [6].
786-O SFCs induce T cell apoptosis
As shown in Figure 7, compared with MAC supernatant or medium alone, 786-O SFC supernatant could significantly induce T cell apoptosis in PBMCs from healthy donors (P < 0.01, n = 3). These results are in line with previous data obtained by other investigators [16].
Figure 7.
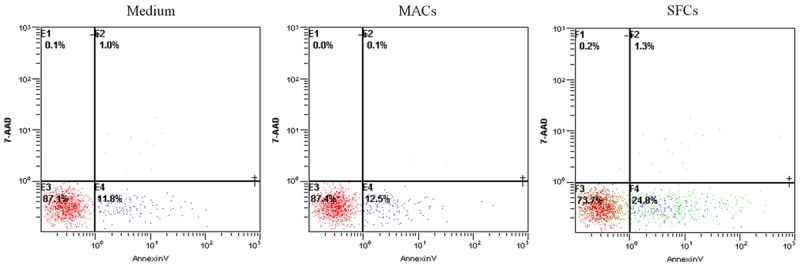
786-O SFC supernatant induces T cell apoptosis after 24 h of exposure. After culturing with 786-O SFCs supernatant, MAC supernatant or medium alone, T cells were stained with 7-AAD and Annexin V. Compared with 786-O MAC supernatant or medium alone, 786-O SFC supernatant enhanced T cell apoptosis.
Discussion
In this study, we found that another human RCC cell line, 786-O, was also capable of forming tumor spheres when grown in defined media, consistent with our previous reports [6]. The 786-O SFCs maintained their ability to proliferate and to self-renew. The SFCs displayed higher tumorigenicity than the MACs in vivo.
Thus far, many reports have shown that SP cells are rich in CSCs [17,18]. More recently, one research group reported that the Rh123high population in cultured RCC 786-O cells displayed stem-like characteristics in vitro [19]. Here, we found that 786-O SFCs contained almost 9-fold more SP cells than 786-O MACs, which may provide an additional explanation for the higher tumorigenicity of 786-O SFCs than MACs. However, we could not find significant differences between SFCs and MACs regarding the expression of putative stem cell markers, including CD44, CD24, CD34, CD105 and CD133 [4,20], indicating that these cell surface markers cannot substitute the sphere formation technique for isolating 786-O CSCs.
In our study, we discovered that in vitro, the 786-O SFCs were more resistant to commonly used chemotherapeutic agents, including doxorubicin, mitomycin C and cisplatin, than were the 786-O MACs. Additionally, we demonstrated that 786-O SFCs expressed higher mRNA levels of several ‘stemness’ genes [21], including Oct-3/4 and β-catenin, indicating that 786-O SFCs are rich in cells possessing stem cell-like properties.
Furthermore, we investigated the immunological phenotype of 786-O SFCs and MACs. On one hand, our results revealed almost complete absence of MHC class I molecule and HLA-DR expression in both 786-O SFCs and MACs, which has been considered the most common strategy exploited by tumors cells to escape T cell control [22]. In addition, the expression of very low levels of CD80 and CD86 was observed in both 786-O SFCs and MACs, which can be anticipated to induce T cell anergy [16]. On the other hand, by downregulating the expression of TAAs (such as Her2 and hTERT), 786-O SFCs could escape the surveillance of tumor-specific CTLs.
The FasL (CD95L) counterattack hypothesis is one of the most controversial mechanisms of tumor immune evasion [22]. Interestingly, we discovered that the expression level of FasL was lower in 786-O SFCs than in MACs. In addition, with lower expression of Fas (CD95), 786-O SFCs may be less sensitive to cytotoxicity mediated by tumor-specific T cells. Recently, one research group reported that CD200 was expressed on approximately threefold more CSCs than on non-CSCs in several cancer cell lines [23]. Nonetheless, CD200 was only expressed by very few 786-O SFCs and MACs, suggesting that the expression of CD200 was not a major mechanism used by 786-O SFCs to evade antitumor responses.
The selective expression of inhibitory B7 molecules, including B7-H1 and B7-H4, on tumor cells has been recognized as an important immunosuppressive mechanism [12]. Despite expressing low levels of B7-H4, most of the 786-O SFCs expressed B7-H1, indicating that 786-O SFCs probably induce T cell apoptosis and resist immune-mediated destruction [12].
Strong evidence supports the role of non-classical MHC class I molecules, most notably HLA-E and HLA-G, in tumor immune escape [13]. With higher expression of HLA-G than MACs, 786-O SFCs might have the ability to prevent antitumor responses.
FoxP3 is a key transcription factor regulating the development and function of regulatory T (Treg) cells [24]. However, recent reports have demonstrated that FOXP3 expressed in pancreatic ductal adenocarcinoma serves as a prognostic biomarker and a crucial determinant of an immunosuppressive microenvironment by recruiting Treg cells by directly transactivating CCL5 [25]. Inconsistent with our previous data [6], the expression level of FoxP3 was markedly lower in 786-O SFCs than in MACs, indicating that FoxP3 probably does not play a major role in immune evasion mediated by 786-O SFCs.
NK cells have potent antitumor activity. The function of NK cells is regulated by the activation or inhibition of receptors present on their surfaces. The activation of NK cells results in cytotoxic activity on target cells through the release of toxic granules and inflammatory cytokines [26]. With reduced expression levels of ligands of DNAM-1 and NKG2D, 786-O SFCs may display decreased sensitivity to NK cell-mediated killing.
mCRPs impose an obvious obstacle to anticancer antibody-based therapy [15]. In line with our previous data [6], we discovered high expression levels of mCRPs, such as CD46, CD55 and CD59, on 786-O SFCs, indicating that the therapeutic potential of anticancer antibodies may be significantly limited in these cells due to the inhibition of complement-dependent cellular cytotoxicity (CDCC).
Tumor-induced T cell apoptosis is an important mechanism of immune evasion by RCC [27]. To avoid allogeneic responses that could confound the interpretation of the data, we used supernatants from 786-O SFCs and MACs in immunologic assays with T cells from healthy donors to determine the effects of CSCs in the absence of preexisting T cell immunosuppression. We found that the supernatant from 786-O SFCs could increase immune cell apoptosis in PBMCs from healthy donors, indicating that 786-O SFCs can mediate immunosuppression by the apoptotic elimination of immune cells likely by secretion of factor(s). However, the exact factor(s) need to be validated in future experiments.
In summary, our present study suggested that SFCs derived from 786-O cells possess CSC characteristics. More importantly, our study highlights a detailed analysis of the immunologic features of CSCs enriched from an RCC cell line, suggesting that CSCs in RCC may contribute to immune suppression by multiple mechanisms. Further research will be carried out using primary RCC tumor samples to confirm and expand on the findings of this study.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81402453 to Y.Z.); the Natural Science Foundation of Hunan Province (2015JJ4058 to Y.Z.); the Project of Hunan Development and Reform Commission ([2013]1199 to Y.Z.), China; the Elite Program of Xiangya hospital (to Y.Z.).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Teyssonneau D, Gross-Goupil M, Domblides C, Haaser T, Pointillart V, Daste A, Hauger O, Ravaud A. Treatment of spinal metastases in renal cell carcinoma: a critical review. Crit Rev Oncol Hematol. 2018;125:19–29. doi: 10.1016/j.critrevonc.2018.02.017. [DOI] [PubMed] [Google Scholar]
- 2.Kaur G, Sharma P, Dogra N, Singh S. Eradicating cancer stem cells: concepts, issues, and challenges. Curr Treat Options Oncol. 2018;19:20. doi: 10.1007/s11864-018-0533-1. [DOI] [PubMed] [Google Scholar]
- 3.Bussolati B, Dekel B, Azzarone B, Camussi G. Human renal cancer stem cells. Cancer Lett. 2013;338:141–146. doi: 10.1016/j.canlet.2012.05.007. [DOI] [PubMed] [Google Scholar]
- 4.Abbaszadegan MR, Bagheri V, Razavi MS, Momtazi AA, Sahebkar A, Gholamin M. Isolation, identification, and characterization of cancer stem cells: a review. J Cell Physiol. 2017;232:2008–2018. doi: 10.1002/jcp.25759. [DOI] [PubMed] [Google Scholar]
- 5.Reynolds BA, Weiss S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science. 1992;255:1707–1710. doi: 10.1126/science.1553558. [DOI] [PubMed] [Google Scholar]
- 6.Zhong Y, Guan K, Guo S, Zhou C, Wang D, Ma W, Zhang Y, Li C, Zhang S. Spheres derived from the human SK-RC-42 renal cell carcinoma cell line are enriched in cancer stem cells. Cancer Lett. 2010;299:150–160. doi: 10.1016/j.canlet.2010.08.013. [DOI] [PubMed] [Google Scholar]
- 7.Sagrinati C, Netti GS, Mazzinghi B, Lazzeri E, Liotta F, Frosali F, Ronconi E, Meini C, Gacci M, Squecco R, Carini M, Gesualdo L, Francini F, Maggi E, Annunziato F, Lasagni L, Serio M, Romagnani S, Romagnani P. Isolation and characterization of multipotent progenitor cells from the Bowman’s capsule of adult human kidneys. J Am Soc Nephrol. 2006;17:2443–2456. doi: 10.1681/ASN.2006010089. [DOI] [PubMed] [Google Scholar]
- 8.Bussolati B, Bruno S, Grange C, Ferrando U, Camussi G. Identification of a tumor-initiating stem cell population in human renal carcinomas. FASEB J. 2008;22:3696–3705. doi: 10.1096/fj.08-102590. [DOI] [PubMed] [Google Scholar]
- 9.Shimoda M, Ota M, Okada Y. Isolation of cancer stem cells by side population method. Methods Mol Biol. 2018;1692:49–59. doi: 10.1007/978-1-4939-7401-6_5. [DOI] [PubMed] [Google Scholar]
- 10.Rittig SM, Haentschel M, Weimer KJ, Heine A, Muller MR, Brugger W, Horger MS, Maksimovic O, Stenzl A, Hoerr I, Rammensee HG, Holderried TA, Kanz L, Pascolo S, Brossart P. Long-term survival correlates with immunological responses in renal cell carcinoma patients treated with mRNA-based immunotherapy. Oncoimmunology. 2016;5:e1108511. doi: 10.1080/2162402X.2015.1108511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ring EK, Markert JM, Gillespie GY, Friedman GK. Checkpoint proteins in pediatric brain and extracranial solid tumors: opportunities for immunotherapy. Clin Cancer Res. 2017;23:342–350. doi: 10.1158/1078-0432.CCR-16-1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. 2013;13:227–242. doi: 10.1038/nri3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zeestraten EC, Reimers MS, Saadatmand S, Goossens-Beumer IJ, Dekker JW, Liefers GJ, van den Elsen PJ, van de Velde CJ, Kuppen PJ. Combined analysis of HLA class I, HLA-E and HLA-G predicts prognosis in colon cancer patients. Br J Cancer. 2014;110:459–468. doi: 10.1038/bjc.2013.696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kohga K, Tatsumi T, Takehara T, Tsunematsu H, Shimizu S, Yamamoto M, Sasakawa A, Miyagi T, Hayashi N. Expression of CD133 confers malignant potential by regulating metalloproteinases in human hepatocellular carcinoma. J Hepatol. 2010;52:872–879. doi: 10.1016/j.jhep.2009.12.030. [DOI] [PubMed] [Google Scholar]
- 15.Afshar-Kharghan V. The role of the complement system in cancer. J Clin Invest. 2017;127:780–789. doi: 10.1172/JCI90962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wei J, Barr J, Kong LY, Wang Y, Wu A, Sharma AK, Gumin J, Henry V, Colman H, Priebe W, Sawaya R, Lang FF, Heimberger AB. Glioblastoma cancer-initiating cells inhibit T-cell proliferation and effector responses by the signal transducers and activators of transcription 3 pathway. Mol Cancer Ther. 2010;9:67–78. doi: 10.1158/1535-7163.MCT-09-0734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ho MM, Ng AV, Lam S, Hung JY. Side population in human lung cancer cell lines and tumors is enriched with stem-like cancer cells. Cancer Res. 2007;67:4827–4833. doi: 10.1158/0008-5472.CAN-06-3557. [DOI] [PubMed] [Google Scholar]
- 18.Wu C, Wei Q, Utomo V, Nadesan P, Whetstone H, Kandel R, Wunder JS, Alman BA. Side population cells isolated from mesenchymal neoplasms have tumor initiating potential. Cancer Res. 2007;67:8216–8222. doi: 10.1158/0008-5472.CAN-07-0999. [DOI] [PubMed] [Google Scholar]
- 19.Lu J, Cui Y, Zhu J, He J, Zhou G, Yue Z. Biological characteristics of Rh123(high) stem-like cells in a side population of 786-O renal carcinoma cells. Oncol Lett. 2013;5:1903–1908. doi: 10.3892/ol.2013.1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matak D, Brodaczewska KK, Szczylik C, Koch I, Myszczyszyn A, Lipiec M, Lewicki S, Szymanski L, Zdanowski R, Czarnecka AM. Functional significance of CD105-positive cells in papillary renal cell carcinoma. BMC Cancer. 2017;17:21. doi: 10.1186/s12885-016-2985-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alberti L, Losi L, Leyvraz S, Benhattar J. Different effects of BORIS/CTCFL on stemness gene expression, sphere formation and cell survival in epithelial cancer stem cells. PLoS One. 2015;10:e0132977. doi: 10.1371/journal.pone.0132977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol. 2007;25:267–296. doi: 10.1146/annurev.immunol.25.022106.141609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kawasaki BT, Mistree T, Hurt EM, Kalathur M, Farrar WL. Co-expression of the toleragenic glycoprotein, CD200, with markers for cancer stem cells. Biochem Biophys Res Commun. 2007;364:778–782. doi: 10.1016/j.bbrc.2007.10.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kitagawa Y, Sakaguchi S. Molecular control of regulatory T cell development and function. Curr Opin Immunol. 2017;49:64–70. doi: 10.1016/j.coi.2017.10.002. [DOI] [PubMed] [Google Scholar]
- 25.Wang X, Lang M, Zhao T, Feng X, Zheng C, Huang C, Hao J, Dong J, Luo L, Li X, Lan C, Yu W, Yu M, Yang S, Ren H. Cancer-FOXP3 directly activated CCL5 to recruit FOXP3(+)Treg cells in pancreatic ductal adenocarcinoma. Oncogene. 2017;36:3048–3058. doi: 10.1038/onc.2016.458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee HH, Kang H, Cho H. Natural killer cells and tumor metastasis. Arch Pharm Res. 2017;40:1037–1049. doi: 10.1007/s12272-017-0951-9. [DOI] [PubMed] [Google Scholar]
- 27.Ng CS, Novick AC, Tannenbaum CS, Bukowski RM, Finke JH. Mechanisms of immune evasion by renal cell carcinoma: tumor-induced T-lymphocyte apoptosis and NFkappaB suppression. Urology. 2002;59:9–14. doi: 10.1016/s0090-4295(01)01503-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.