

Abstract
Cardiac fibrosis is primarily mediated by activated fibroblasts. However, cardiomyocytes have also been implicated in facilitating the fibrotic response. We aimed to explore how cardiomyocyte-derived exosomes affect fibroblasts. We measured cardiac-specific microRNA levels in two rat models of cardiac fibrosis to find out which microRNA was involved in the common mechanisms of cardiac fibrosis. Then, we isolated exosomes from cardiomyocytes and measured their effects on fibroblast proliferation and differentiation into myofibroblasts. We used a microRNA antagomir and an AAV9 microRNA sponge delivery system to inhibit cardiac microRNA in vivo. We then transfused cardiomyocyte-derived exosomes into normal rats to determine the functional effects of the exosomes. miR-208a was upregulated in cardiomyocytes and cardiomyocyte-derived exosomes from both models of cardiac fibrosis and could be transferred into cardiac fibroblasts via the exosomes. The miR-208a-containing exosomes contributed to increased fibroblast proliferation and differentiation into myofibroblasts, an effect that was attenuated by the miR-208a antagomir. When miR-208a was inhibited in vivo, cardiac function improved and cardiac fibrosis was alleviated in post-myocardial-infarction rats. The transfusion of miR-208a-containing exosomes into normal rats resulted in worsened cardiac function. We identified Dyrk2 as the target gene of miR-208a. Cardiomyocytes participate in cardiac fibrosis by secreting exosomes containing miR-208a, which increases fibroblast proliferation and differentiation into myofibroblasts.
Keywords: Cardiomyocyte, fibroblast, exosomes, miR-208a, Dyrk2
Introduction
Several pathological conditions involve abnormal synthesis of cardiac collagen matrix, culminating in excessive cardiac stiffness and abnormal cardiac function [1,2]. Conditions that increase cardiac stress such as excessive haemodynamic pressure and cardiac injuries such as myocardial infarction (MI) drive the development of cardiac fibrosis, a condition marked by an abnormal accumulation of extracellular matrix accumulation that ultimately results in adverse ventricular remodeling and cardiac failure [3,4]. To figure out the mechanism of cardiac fibrosis, scholars have focused on the role of cardiac fibroblasts [5]. However, emerging evidence indicates that the development of cardiac fibrosis may also involve cardiomyocytes [6,7]. Experiments hint that cross-talk between cardiomyocytes and fibroblasts may trigger local angiotensin II (Ang II)-mediated collagen upregulation [7]. Despite growing evidence, studies investigating the role of myocytes in post-MI cardiac fibrosis has not been addressed.
Intracellular gene expression is tightly regulated by microRNAs (miRNAs). Extracellular miRNAs exists in the breast milk, blood, saliva, urine and even cerebrospinal fluid [8]. As such, extracellular miRNAs have been researched as potential disease biomarkers, with certain diseases presenting with unique extracellular miRNA profiles [9-11]. Conditions where extracellular miRNA profiles have been characterized include cardiovascular diseases, diabetes and cancer [10,12,13]. In pathological cardiac states, cardiac-specific miRNA expressions may become deregulated, leading to progressive heart failure associated with myocardial necrosis, fibrosis, left ventricular dilatation, ischemia, hypoxia, arrhythmia and other destructive processes [14]. Indeed, miR-1, miR-133a, miR-208a, and miR-499 have been earmarked as heart-specific miRNAs.
Extracellular miRNAs avoid nuclease degradation either by forming complexes with extracellular proteins or via encapsulation within membrane-derived vesicles (exosomes, microvesicles or apoptotic bodies) [8,12,15,16]. Circulating miRNAs have important functions in gene regulation and intracellular communication [8,17]. Different molecular carriers express unique miRNA signatures [15], and differences can also be detected between parent and carrier cells, indicating that miRNA export to be a highly regulated process [15,18,19]. Exosomes are molecules that measure between 30 nm to 200 nm and are membrane-bound vesicles that ferry a variety of cell signaling substances. Most cells secrete exosomes, including platelets [12], lymphocytes [17], adipocytes [20], and muscle [10,21], tumor [22], glial [23], and stem cells [24-26].
We tested the expression of cardiac-specific miRNAs in ventricle samples from two rat models of cardiac fibrosis to find out which miRNA is involved in the common mechanisms of cardiac fibrosis. We then transferred cardiomyocyte-derived exosomes into cardiac fibroblasts to test their effects on fibroblasts in vitro. Furthermore, we transfused exosomes into miR-208a-inhibited rats with MI evaluate its effects on cardiac fibrosis in vivo. We identified a target gene of miR-208a using bioinformatic analysis.
Materials and methods
Animal studies
The Animal Research Committee of Xin Hua Hospital, Shanghai Jiao Tong University School of Medicine approved all animal experiments performed in this study, which were carried out in accordance to the National Institutes of Health (No. 85-23, revised 1996) guidelines on the care and handling of laboratory animals for biomedical research. A rodent model of cardiomyopathy was developed using adult male (8-12 weeks) Sprague-Dawley rats that were treated chronically with intraperitoneal injections of phosphate-buffered saline (PBS; control) or with 4 mg/kg of doxorubicin (Dox) on days 0, 2, 4 and 6 of the treatment period. MIs were induced in rats that were chosen randomly and in a double-blinded fashion. Briefly, rats were anesthetized by intraperitoneal injections of sodium pentobarbitol (50 mg/kg), intubated and ribs separated to exposed the heart. MIs were induced by ligation of the left anterior descending (LAD) coronary artery. In sham surgery, the chest was opened with no artery ligation performed.
To evaluate the in vivo actions of cardiomyocyte-derived exosomes, we injected the exosomes (100 µg or 200 µg) into myocardium of normal rats by following the guide of echocardiography. 14 days after the procedure, cardiac function was evaluated using a Vevo 2100 imaging system echocardiograph (VisualSonics, Canada) with a 10 MHz central frequency scan head. All functional measurements were taken and analyzed by investigators that were blinded to the treatment groups. After functional evaluation, rat hearts were dissected for histological analysis. Heart fixation was performed with 4% paraformaldehyde, dehydrated with ethanol, paraffin-embedded before sectioned into 7-μm thicknesses.
miR-208a inhibition and the AAV-9 delivery system
miR-208a antagomir or scrambled control (RiboBio, Guangzhou, China) was dissolved in PBS to doses of 80 mg/kg that were injected intravenously (via tail vein) into rats for 7 consecutive days before LAD coronary artery ligation. Two weeks after the ligation, we sacrificed the rats and immediately collected serum and heart samples. Expressions of miR-208a levels in cardiac and serum samples were quantified with qRT-PCR as described below. In order to achieve heart-specific inhibition of miR-208a, we injected adeno-associated virus serotype 9 (AAV9) sponge that carried miR208a sponge (AAV9-miR-208a sponge, 1 × 1012 genome copies per rat; Hanheng Biotechnology, China), or PBS as a control, into the tail vein of wild-type male rats and performed LAD ligation 2 weeks later. AAV9 sponge is a safe, efficient and significantly cardiotropic viral vector that is useful for in vivo gene inhibition [27-30].
Primary cell isolation
For primary cardiomyocyte isolation, adult rats (8-12 weeks) were anesthetized with intraperitoneal Na-pentobarbital, and their heart was isolated and perfused via the aorta. The perfusion buffer consisted of KCl (5.4 mM), NaCl (120 mM), Na2HPO47H2O (0.5 mM), MgSO47H2O (0.5 mM), 2,3 butanedione monoxime (10 mM), taurine (30 mM), HEPES (25 mM), EGTA (0.4 mM) and glucose (22 mM). We perfused the heart with digestion buffer [collagenase II (2.4 mg/ml), protease XIV (0.2 mg/ml), and CaCl2 (50 μM) in perfusion buffer] for approximately 10-15 min. The heart was removed from the cannula with forceps, and the ventricular tissue was quickly triturated in the digestion buffer to ensure most pieces were smaller than 1 mm3. After 5-10 min, we pipetted the dissociated tissue suspension onto a 215 µm mesh and collected the filtrate. We then used 7.5 ml pre-warmed stop buffer [CaCl2 (0.1 mM) and 10% exosome-depleted FBS (SBI, USA) in perfusion buffer] to wash the cardiomyocytes through the mesh, combining them with the first filtrate. The Ca2+ concentration was gradually raised to 1 mM. Cardiomyocytes were then plated on laminin [31]. A 10-10 M solution of Ang II (Sigma, USA) was added to the supernatant to stimulate the cardiomyocytes for 48 h. Hypoxia was induced by decreasing the concentration of oxygen to 1% for 24 h. Ischemia was induced by replacing the culture medium with DMEM without glucose (Gibco, USA) or serum.
A previously described protocol for enzymatic digestion was followed to isolate cardiac fibroblasts [32]. DMEM complete medium was used to culture fibroblasts at 37°C. All cells were cultured with exosome-depleted FBS.
MTS cell proliferation colorimetric assay
An MTS assay (CellTiter 96 AQueous One Solution Cell Proliferation Assay Kit, Promega, USA) was utilized to assess cardiac fibroblast proliferation according to the manufacturer’s protocol. Fibroblasts were seeded at an initial density of 5 × 103 cells/well into 96-well plates. Exosomes were added two hours later, followed by a 24-hour incubation period. Cell proliferation curves were generated based on cell growth measurement at 490 nm using a microplate reader.
Exosome preparation
Exosome isolation was performed as per existing publications [33]. In short, cultured cardiomyocyte supernatant was isolated and centrifuged at 10,000 g for 30 min to remove any cells and cellular debris. Supernatant was then transferred into a fresh tube, filtered with a 0.22 µm membrane, and re-centrifuged at 120,000 g for 2 hours at 4°C. The resulting exosome pellet was subjected to one cycle of washing with sterile PBS before being resuspended in PBS. Alternatively, the cardiomyocte culture supernatant was concentrated with a Amicon Ultra filter (Millipore, Billerica, MA) from 50 mls to 1 ml using a 100,000 molecular-weight cutoff. Exosome isolation was then performed with an ExoQuick kit (SBI, USA) as instructed by the manufacturer. qNano analysis (Izon instrument, UK) was used to determine exosome quantity, based on manufacturer’s instructions [34], while the BCA Protein Assay Kit (Thermo Fisher Scientific, USA) was used to quantify exosome protein concentration.
Malik et al’s protocol for electron microscopy was used for the following experiment [35]. Briefly, a drop of purified exosome pellet was placed on a grid coated with gold, blotted, fixed with 1% glutaraldehyde, and washed with double-distilled water for 2 minutes, and incubated for 5 minutes with uranyl oxylate. A drop of uranyl acetate and methyl cellulose was then used to incubate the purified exosomes twice for 5 minutes each, and for 10 minutes the third time, with a new drop used for each incubation period. Exosomes were then removed for visualization with a standard transmission electron microscopy with a Philips CM120 microscopy.
Exosome labeling
Labeling of exosomes was performed with a phospholipid membrane dye, PKH26 (Sigma, USA) at 37°C for 5 minutes as instructed by the manufacturer. Briefly, the isolated exosomes (1010 particles) were resuspended in 100 μl PBS, 0.5 ml of diluent C added, followed by a mixture of 4 μl PKH26 and another 0.5 ml diluent C after 5 minutes. Exosome-depleted FBS (1 ml) was added to stop the reaction. We then transferred the entire solution to a fresh tube, which was then subjected to filtration with a 0.22 μm membrane, and centrifuged at 120,000 g for 2 hours at 4°C. Basal medium was used to resuspend these PKH26-labeled exosomes before they were incubated to cultured cardiac fibroblasts, which were transfected with a GFP-overexpression lentiviral vector. After incubation for 12 hours, fibroblasts were fixed, washed, and visualized by an Olympus microscope. Alternatively, we transfected CMs with a cel-miR-39-overexpression lentiviral vector. Then the exosomes of these CMs were isolated and added to cardiac fibroblasts. After incubation for 4, 8, 12, or 24 hours, cel-miR-39 expression in fibroblasts was measured.
Exosomal degradation analysis
Exosomes purified from CM culture medium were treated with either Triton X-100, proteinase K or RNase (all Sigma-Aldrich, USA) at 37°C for 45 minutes [36]. Expression of selected miRNAs was determined by qPCR. GW4869 (Sigma-Aldrich) was used as an inhibitor of exosome release.
RNA interference and transfection
A 1% FBS low-serum medium was used as a medium to conduct cardiac fibroblasts assays and transfections. Cardiac fibroblasts were transfected with either 100 nM or 200 nM miR-208a agomir (RiboBio) as per manufacturer’s protocols. 48 hours later, transfected cells were harvested. A Dyrk2 overexpressing plasmid was purchased from Sangon Biotech (Shanghai, China). We purchased siRNAs for Dyrk2 and negative control siRNAs from Invitrogen (Carlsbad, USA). We transfected fibroblasts with Dyrk2 siRNA or control siRNA (50 nM; Invitrogen, USA) following the manufacturer’s guidelines. Lipofectamine 2000 reagent (Invitrogen, USA) was used to perform transfections based on the manufacturer’s protocol. An assay to confirm efficiency and plasmid function was performed 72 h after siRNA transfection.
RNA isolation and quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA extraction was carried out with the TRIzol reagent (Invitrogen, USA) in accordance to manufacturer’s instructions. The Primer-Script one step RT-PCR kit (TaKaRa, Japan) was used for reverse transcription of mRNAs. The SYBR kit (TaKaRa, Japan) was used for detection on the ABI7500 system (Applied Biosystems, USA). The TaqMan MicroRNA Reverse Transcription Kit and the TaqMan MicroRNA Assays (Thermo Fisher, USA) were used to quantitate the expression of cardiac-specific miRNAs. 18S RNA and U6 RNA were used as endogenous controlz for mRNAs and miRNAs, respectively. The relative expression among groups was calculated by the 2-ΔΔCt method. The following primers were used: a-SMA F: 5’-GTCCCAGACATCAGGGAGTAA-3’ and R: 5’-TCGGATACTTCAGCGTCAGGA-3’; Col1a1 F: 5’-GAGCGGAGAGTACTGGATCGA-3’ and R: 5’-CTGACCTGTCTCCATGTTGCA-3’; Col3a1 F: 5’-TGCCATTGCTGGAGTTGGA-3’ and R: 5’-GAAGACATGATCTCCTCAGTGTTGA-3’; CTGF F: 5’-CACAGAGTGGAGCGCCTGTTC-3’ and R: 5’-GATGCACTTTTTGCCCTTCTTAATG-3’; TSP-1 F: 5’-GTGACGGAAAATCAAGTTTGCA-3’ and R: 5’-ACTTGGCACCAGCAAAGCA-3’; 18S F: 5’-TCAAGAACGAAAGTCGGAGG-3’ and R: 5’-GGACATCTAAGGGCATCAC-3’.
Western blotting
SDS-PAGE was used to separate protein into equal amounts. Primary antibody binding was detected by peroxidase-conjugated secondary antibodies and densitometry was used to quantify the resultant bands. The following antibodies were used: anti-Dyrk2, anti-NFATc2, anti-pNFATc2 and anti-GAPDH (Santa Cruz Biotechnology, USA) and anti-CD9, anti-CD68, anti-CD81, and anti-calnexin (Abcam, USA).
Luciferase assay
Lipofectamine RNAiMAX Transfection Reagent (Invitrogen, USA) was used to transfect Dyrk2 overexpression plasmids (50 nM) or siRNAs (50 nM) as previously described [37]. Genomic DNA was served as template for amplification of the entire rat Dyrk2 3’UTR segment by PCR. The PCR product was subcloned into vector pmiR-RB-REPORT (RiboBio, China) following the manufacturer’s protocol. The MutaBest kit (TaKaRa, Japan) was used to generate a dyrk2 mutant-luc plasmid whose DNA was then sequenced to verify its integrity. A 96-well plate was used to culture 293T cells that were transfected with Lipofectamine 2000 reagent (Invitrogen, USA) that contained a 0.2 mg luciferase reporter and miR-208a agomir (100 nM) or control RNAs. Cells were left to incubate for 48 hours before being assayed with a luciferase assay kits (Promega, USA).
Masson’s trichrome staining
Histo-Clear reagent was initially used to remove wax from tissue sample before they were washed with ethanol. The sections were then stained for one hour with Masson’s solution (Sigma, USA) before being subjected to washing with ethanol and acidified water and resin-mounted (Cytoseal, USA). The degree of fibrosis (in percentages) was evaluated with Image J software and was expressed as the ratio of fibrotic tissue to total tissue.
Statistics
The GraphPad Prism software (GraphPad Software, USA) was used for statistical analysis. A Student’s t-test was used to compare two groups when data followed a Gaussian distribution. Otherwise, non-parametric tests were used. Two-tailed P values of less than 0.05 were interpreted to have statistical significance.
Results
miR-208a was consistently increased in two different models of cardiac fibrosis
We first compared the changes in heart tissue between MI rats and sham-operated rats and between Dox-treated rats and PBS-treated rats (Figure 1A). qRT-PCR confirmed that miR-1 and miR-133a were increased in the serum of rats with cardiac fibrosis and decreased in the fibrotic heart tissues from both models. miR-499 was increased in the serum of rats with cardiac fibrosis from both models and in the heart tissue of the rats with MI remodeling; however, it was decreased in the heart tissue of the rats with Dox-induced dilated cardiomyopathy. miR-208a was increased the fibrotic heart tissue and in the serum of rats with cardiac fibrosis from both models (Figure 1B).
Figure 1.

Expression of cardiac-specific miRNAs in different cardiac fibrosis models. A. Changes in heart tissue in two separate models of fibrosis. Hearts subjected to ischemic damage (MI) displayed a decrease in myocardium and an increase in collagen. In the Dox-induced-cardiomyopathy heart, the diameter of the myocardium was increased. B. Expression of the four cardiac-specific miRNAs. miR-1 and miR-133a were upregulated in the heart tissue and downregulated in serum from both models of fibrosis. miR-499 was upregulated in the serum and heart tissue of MI rats and in the heart tissue of Dox-induced rats; miR-499 was downregulated in the serum of Dox-induced rats. miR-208a was upregulated in the serum and heart tissue from both models of fibrosis. Scale bar: 50 µm. *P<0.05, **P<0.01, ***P<0.001 vs. respective controls.
We then tested miR-208a expression in cardiomyocytes and fibroblasts stimulated by hypoxia or treated with Ang II. miR-208a was increased in the cardiomyocytes stimulated by hypoxia (CM-Hypo) or treated with Ang II (CM-Ang II); however, it showed no changes in fibroblasts subjected to either treatment (Figure 2A). Following this, we then cultured supernatant of myocyte cultures together with fibroblasts. This resulted in a marked elevation of miR-208a levels in fibroblasts co-cultured with supernatant from CM-Ang II or CM-Hypo cells (Figure 2B). When we depleted the CM-Ang II or CM-Hypo cells of exosomes by ultracentrifugation or treatment with GW4869 before harvesting the supernatants, the increased expression of miR-208a was abrogated in the target fibroblasts (Figure 2B).
Figure 2.
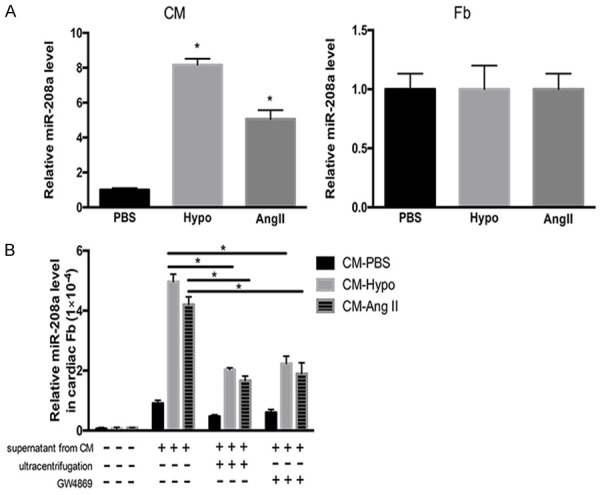
miR-208a expression in hypoxia-stimulated or Ang II-induced cardiomyocytes and fibroblasts. A. miR-208a expression was increased in hypoxia-stimulated (Hypo) or Ang II-induced (AngII) cardiomyocytes (CM) but not in similarly treated fibroblasts (Fb). Exposure to PBS was used as a control. B. Co-culture of fibroblasts with supernatant from hypoxia-stimulated cardiomyocytes (CM-Hypo) or Ang II-induced cardiomyocytes (CM-Ang II) resulted in increased miR-208a levels compared with those in fibroblasts co-cultured with supernatant from PBS-treated cardiomyocytes (CM-PBS). Depletion of cardiomyocyte-derived exosomes from the supernatants by ultracentrifugation or treatment with GW4869 abrogated the increased expression of miR-208a in the target fibroblasts. *P<0.05.
Cardiomyocyte-derived exosomes transferred miR-208a into cardiac fibroblasts
We isolated exosomes from CM-Hypo and CM-Ang II cells to determine the function of exosomes in the cross-talk between cardiomyocytes and fibroblasts. We performed electron microscopy, western blotting, and nanoparticle tracking to identify the function of the exosomes. Electron microscopy showed isolated exosomes as cup-shaped or round-shaped biphospholipid vesicles ranging from approximately 30 to 200 nm in diameter (Figure 3A). Immunoblotting of exosomal preparations and cell lysates revealed that the exosomal preparations were enriched with the exosomal markers CD63, CD9, and CD81 compared with the cell lysates but devoid of calnexin, an endoplasmic reticulum protein (Figure 3B). Hypoxia or Ang II treatment caused cardiomyocytes to produce an increased number of exosomes compared with PBS treatment, as evidenced by qNano analysis (Figure 3C). We next evaluted the capacity of exosomes to be transferred from cardiomyocytes to recipient fibroblasts by labeling isolated cardiomyocyte-derived exosomes with the PKH26 fluorescent dye followed by immersion into a medium of fibroblasts labelled with GFP. After 12 hours of incubation, confocal imaging revealed that the fibroblasts had taken up the fluorescently labeled exosomes (Figure 3E). We sought to confirm this by transfecting cardiomyocytes with the cel-miR-39 miRNA, a miRNA specifically found in Caenorhabditis elegans. After 48 hours, exosomes were isolated from the cel-miR-39-transfected cardiomyocytes and added into fibroblast cultures. Fibroblasts demonstrated increased expression of cel-miR-39, confirming that this miRNA was time-dependently transmitted from the cardiomyocyte-derived exosomes to the fibroblasts (Figure 3D).
Figure 3.

Hypoxia-stimulated or Ang II-induced cardiomyocytes secrete exosomes that are taken up by cardiac fibroblasts. A. Electron micrographs of exosomes. The diameter of an exosome is 106 nm. Scale bar: 50 nm. B. Western blotting showed the exosome markers CD9, CD63, and CD81 and the endoplasmic reticulum marker calnexin. C. qNano analysis of exosomes showed the particle diameter and concentration. The diameter ranged from about 50 nm to 150 nm. Blue, exosomes from PBS-treated cardiomyocytes; green, exosomes from hypoxic cardiomyocytes; red, exosomes from Ang II-induced cardiomyocytes. D. cel-miR-39-transfected cardiomyocytes. Fibroblasts were then exposed to exosomes extracted from cel-miR-39-transfected cardiomyocytes or untransfected (control) for the stated times. *P<0.05 vs. control. E. Confocal images of GFP-overexpressing fibroblasts after incubation with exosomes stained with PKH26 (red). Exosomes were taken up into the cytoplasm of fibroblasts. Scale bar, 10 µm.
Furthermore, miR-208a expression was increased in CM-Ang II cells, CM-Hypo cells as well as in exosomes derived from those cells, in contrast to PBS-treated cells and exosomes (Figure 4A). When we co-cultured cardiac fibroblasts with exosomes derived from CM-Ang II cells (exosomesCM-Ang II) or CM-Hypo cells (exosomesCM-Hypo), the miR-208a level in the fibroblasts increased in a time-dependent manner (Figure 4B). Finally, exosomal degradation analysis using a combination of Triton X-100 (to disrupt phospholipid membranes), proteinase K (to degrade proteins) and ribonuclease (RNase) demonstrated that a large proportion of cardiomyocyte-derived miR-208a was transmitted to fibroblasts as vesicle-protected RNA rather than as soluble ribonucleoprotein complexes (Figure 4C). When interpreted as a whole, these findings indicate that miR-208a was transferred from cardiomyocyte-derived exosomes to fibroblasts.
Figure 4.
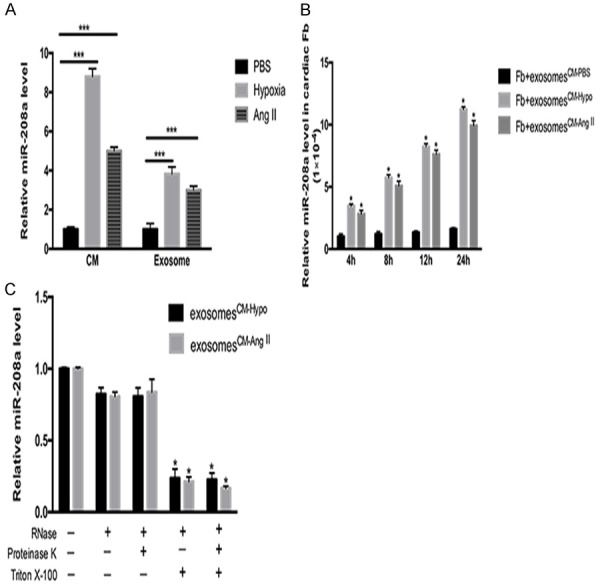
Exosomes transport miR-208a. A. miR-208a expression was increased in hypoxia-stimulated or Ang II-induced cardiomyocytes (CM), as well as in exosomes derived from those cells. ***P<0.001 vs. PBS-treated cardiomyocytes. B. miR-208a was markedly increased in 1 × 105 cardiac fibroblasts in a time-dependent manner when the cells were incubated with 40 µg/ml of cardiomyocyte-derived exosomes and treated with PBS (CM-PBS), Ang II (CM-Ang II) or hypoxic cardiomyocytes (CM-Hypo). *P<0.05 vs. fibroblasts+exosomesCM-PBS. C. CM-Hypo or CM-Ang II exosomes were isolated and co-cultured with the indicated reagents at 37°C for 45 minutes before RNA isolation and measurement of miR-208a expression by qPCR. *P<0.05 vs. respective controls.
MiR-208a promoted cardiac fibroblast proliferation and myofibroblast differentiation in vitro
We determined the effects of exosomesCM-Ang II and exosomesCM-Hypo in fibroblasts. Compared with exosomes from CM-PBS cells (exosomesCM-PBS), the exosomesCM-Ang II and exosomesCM-Hypo promoted the fibroblast differentiation and proliferation in addition to increasing TSP-1, CTGF, Col1a1, Col3a1 and a-SMA expressions. Those exosome-induced effects on the fibroblasts were mitigated when we incubated the fibroblasts with exosomesCM-Ang II or exosomesCM-Hypo in the presence of miR-208a antagomir (Figure 5A and 5B).
Figure 5.

Exosomes containing miR-208a increase fibroblast proliferation in vitro, and miR-208a antagomir alleviates cardiac fibrosis after MI in vivo. A. An miR-208a antagomir effectively decreased the miR-208a level. ***P<0.001 vs. normal control (nc) antagomir. B. miR-208a-containing exosomes derived from hypoxic cardiomyocytes (CM-Hypo) or Ang II-induced cardiomyocytes (CM-Ang II) increased cardiac fibroblast proliferation and differentiation into myofibroblasts compared with exosomes derived from PBS-treated cardiomyocytes (CM-PBS). Those effects were mitigated by miR-208a antagomir. ***P<0.001, *P<0.05 vs. exosomesCM-PBS. C. miR-208a antagomir decreased the miR-208a level in serum and heart tissue from post-myocardial infarction (MI) rats. ***P<0.001. D. Echocardiography showed preserved cardiac function. **P<0.01, ***P<0.001. E. Masson’s staining of heart tissue from post-MI rats showed reduced cardiac fibrosis. ***P<0.001. Scale bar, 50 µm. F. miR-208a antagomir decreased myofibroblast differentiation in vivo. *P<0.05 vs. MI+nc antagomir.
Inhibition of miR-208a improved cardiac function after MI by decreasing cardiac fibrosis in vivo
The miR-208a level was significantly increased in the serum and heart tissue of MI rats compared with that in sham-operated rats, as evidenced by qRT-PCR (Figure 5C). Compared with a scrambled negative-control antagomir, the miR-208a antagomir improved cardiac function after MI, as shown by echocardiography (Figure 5D). Masson’s trichrome staining showed that miR-208a inhibition suppressed cardiac fibrosis in post-MI heart tissues, as evidenced by decreased collagen (Figure 5E) as well as decreased expression of Col1a1, Col3a1 and a-SMA, (Figure 5F).
To confirm the effect of miR-208a attenuation on cardiac fibrosis, we inhibited cardiac miR-208a in vivo with a cardiotropic AAV9 sponge delivery system. One dose of either miR-208a sponge AAV9 or scrambled control was injected into adult rats, which then underwent LAD aortic ligation surgery. The miR-208a sponge significantly inhibited the miR-208a level in serum and heart tissue from post-MI and sham-operated rats (Figure 6A). Furthermore, the AAV9-mediated inhibition of miR-208a maintained cardiac function while reducing collagen deposition in post-MI hearts similarly to the miR-208a antagomir (Figure 6B-D).
Figure 6.

Cardiac inhibition of miR-208a via AAV9 attenuates cardiac fibrosis and improves cardiac function in post-myocardial infarction (MI) rats. A. An miR-208a antagomir (208a sponge AAV9) decreased the miR-208a level in serum and heart tissue from sham-operated and post-MI rats compared with an antagomir for a scrambled microRNA. ***P<0.001. B. Echocardiography showed preserved cardiac function. ***P<0.001. C. Masson’s staining of heart tissue from post-MI rat showed reduced cardiac fibrosis. Scale bar, 50 µm. D. Quantitative analysis of fibrotic heart from post-MI rats showed reduced cardiac fibrosis. ***P<0.001.
To clarify the effect of cardiomyocyte-derived exosomes, we transfused exosomesCM-Hypo (100 µg or 200 µg) into normal rats by ultrasonically guided intramyocardial injection. The exosome treatment dose-dependently increased cardiac miR-208a expression (Figure 7A). Moreover, the injection of exosomesCM-Hypo led to decreased cardiac function, increased cardiac fibrosis, and increased mRNA levels of Col1a1, Col3a1 and α-SMA (Figure 7B-D). Collectively, the results suggest that inhibiting miR-208a alleviated post-MI cardiac fibrosis while improving cardiac function.
Figure 7.

Exosomes derived from hypoxic cardiomyocytes (CM-hypo) increase cardiac fibrosis in normal rats. A. Supplementation with 100 μg or 200 μg exosomesCM-Hypo increased the miR-208a level in heart tissue of normal rats in dose-dependent manner. ***P<0.001 vs. PBS. B. Echocardiography showed exacerbated cardiac function. **P<0.01, ***P<0.001. C. Cardiac tissue from post-myocardial infarction (MI) rats shows increased cardiac fibrosis upon Masson’s staining. Scale bar, 50 µm. **P<0.01, ***P<0.001. D. ExosomesCM-hypo increased myofibroblast differentiation in vivo. *P<0.05 vs. PBS.
Dyrk2 is a target gene of miR-208a
TargetScan bioinformatic analysis returned Dyrk2 as a possible miR-208a target gene. The presence of miR-208a was confirmed to reduce luciferase activity for the wild-type 3’UTR construct of Dyrk2, while no effects were observed in Dyrk2 3’UTR with a mutated miR-208a binding site, strongly suggesting that miR-208a directly targets Dyrk2 (Figure 8A). In order to evaluate if endogenous Dyrk2 expression in cardiac fibroblasts was also regulated by miR-208a, we exposed cardiac fibrobalsts to miR-208a agomir, antagomir, or corresponding negative controls. We found that the miR-208a agomir decreased, whereas the miR-208a antagomir increased Dyrk2 expression, confirming that endogenous Dyrk2 expression falls under the control of miR-208a in cardiac fibroblasts (Figure 8B). Next, we used a plasmid containing an overexpression of Dyrk2 to delineate the functions of Dyrk2 in miR-208a-mediated cardiac fibroblast proliferation and differentiation into myofibroblasts. Overexpression of Dyrk2 alleviated the pro-fibrotic effect of the miR-208a agomir on cardiac fibroblasts (Figure 8C). On the other hand, siRNA-mediated Dyrk2 knockdown fibroblasts were similar to fibroblasts exposed to miR-208a agomir (Figure 8D). Those results identified Dyrk2 as a novel target gene of miR-208a that plays a major role to myofibroblast differentiation.
Figure 8.

miR-208a directly targets the Dyrk2 gene. A. TargetScan and luciferase reporter assays determined Dyrk2 to be gene directly targeted by miR-208a in 293T cells and fibroblasts; *P<0.05 vs. other groups. B. The effectiveness of miR-208a agomir and antagomir and western blotting showed that miR-208a targets Dyrk2. **P<0.01, ***P<0.001 vs. normal control. C. p-EGFP-Dyrk2 mitigated miR-208a effects on fibroblast proliferation and differentiation. *P<0.05, ***P<0.001. D. Dyrk2 siRNA did not further increase the effects of miR-208a on fibroblast proliferation and differentiation. E. Dyrk2 siRNA could decrease NFAT phosphorylation, and overexpression of Dyrk2 could activate NFAT phosphorylation. ***P<0.001 vs. respective controls. NS, not significant.
Dyrk2 counters nuclear factor of activated T-cells (NFAT) dephosphorylation, which triggers fibrosis, by directly phosphorylating the NFAT regulatory domain. To confirm this, determined p-NFAT expression in siRNA-mediated Dyrk2 knockdown with western blotting. The Dyrk2 knockdown significantly decreased p-NFAT expression, whereas plasmids that overexpressed Dyrk2 resulted in increased p-NFAT expression (Figure 8E).
Discussion
Cardiac fibrosis is adverse cardiac remodeling that is a consequence of several different forms of cardiac injury ranging from hemodynamic stress to MI. Cardiac-specific miRNAs are reported to be aberrantly expressed in the fibrotic heart [38]. We measured the expression of four cardiac-specific miRNAs in fibrotic heart tissue and serum and found that miR-208a was consistently upregulated in rats with MI or Dox-induced cardiomyopathy. We then compared miR-208a expression across different types of cells that were exposed to hypoxia or Ang II. There was an elevation in miR-208a expression in induced cardiomyocytes but not in induced fibroblasts; however, when the fibroblasts were co-cultured with supernatant from cultures of induced cardiomyocytes, the miR-208a expression increased in the fibroblasts. That suggests that miR-208a was transferred to the fibroblasts from the stimulated cardiomyocytes.
Several sources of evidence support the phenomenon of extracellular miRNA secretion. In the extracellular space, miRNAs are thought to exist as either bound to proteins or other compounds or encapsulated in microvesicles [39-41]. Kumar and Reddy specified three means of miRNA transport within the peripheral circulation: (1) in a non-vesicle form, bound to high-density lipoprotein (HDL) particles, (2) by forming a complex with the Ago2 protein, or (3) packaged in microparticles such as microvesicles, exosomes and apoptotic bodies [42]. When bound to the small 8-12 nm HDL particle, extracellular plasma miRNAs are able to form a stable ternary structure with its contents apolipoprotein A-I and phosphatidylcholine by means of divalent cation bridging [40]. This form of endogenous miRNA transport only makes up a small proportion of total peripheral miRNA transport. Binding to the Ago2 protein results in a highly stable structure, with miRNAs gaining robust resistance to the nuclease-rich extracellular environment [41]. Encapsulation of miRNAs in exosomes, microvesicles, or apoptotic bodies allow miRNAs to be transported extracellularly without being degraded in the extracellular environment [43]. These microparticles are thought to be a vital means of transport of various bioactive components (miRNA, mRNA, DNA, cytokines, and other proteins) as well as intercellular communication.
Exosomes represent natural transport nanovesicles (30-200 nm) secreted by numerous cell types. These molecules are secreted by the plasma membrane, although they originate from endosomes. Because of their unique properties, exosomes carry out specific functions when utilized for miRNA transport: (1) In pathological conditions, deregulated, disease-specific miRNAs are transported in exosomes, with non-exosomal miRNAs easily cleared from healthy cells; (2) By means of transcytosis, exosomes are able to traverse the several endothelial cell layers involved in the blood barrier, and (3) miRNAs carried by exosomes gain protection from cellular RNases in the circulatory system [44]. Microvesicles are formed by the outward blebbing and budding of the plasma membrane and are slightly larger than exosomes (100-4000 μm in diameter). Platelets are a major source of microvesicle production and secretion [45]. The largest microvesicles are apoptotic bodies, which are discarded from cells during apoptosis and function in miR-126 transport [46,47].
The intercellular transfer of exosomes is a well-established mechanism mediating cell-cell communication [8,48]. We hypothesized that myocyte-derived exosomes transfer miR-208a into fibroblasts during cardiac fibrosis. Our data shows that cardiomyocytes induced by hypoxia or Ang II produced increased numbers of exosomes compared with cardiomyocytes treated with PBS (Figure 3C). We confirmed that those exosomes could be taken up by fibroblasts in a time-dependent manner (Figure 3D and 3E). Furthermore, we discovered that hypoxic or Ang II-treated cardiomyocytes had elevated miR-208a expressions, which is consistent with previous studies [49,50], as well as in exosomes derived from those cells (Figure 4A). When cardiac fibroblasts were co-cultured with the supernatant of cultures of activated myocytes, there was a marked elevation of miR-208a levels in fibroblasts. Those increases were abrogated, however, by the depletion of exosomes from the co-cultured supernatant. Thus, it was the exosomes that transferred miR-208a between the myocytes and the fibroblasts. To confirm our hypothesis, we used a combination of Triton X-100, proteinase K and RNase to demonstrate that the majority of cardiomyocyte-derived miR-208a is transmitted into fibroblasts as vesicle-protected RNA rather than as soluble ribonucleoprotein complexes (Figure 4C).
We determined the function of the miR-208a-containing exosomes in cultured cardiac fibroblasts. Our data suggest that the exosomes promote fibroblast proliferation and myofibroblast differentiation, effects that were mitigated in the presence of an miR-208a antagomir. In order to inhibit miR-208a in vivo in cardiomyocytes, we used an miR-208a antagomir and an AAV9 miR-208a sponge delivery system, which resulted in improved cardiac functions and alleviated cardiac fibrosis in post-MI rats. When we transfused cardiomyocyte-derived exosomes into normal rats, we found that cardiac function worsened.
Transient receptor potential cation channel C6-calcineurin-NFAT signaling is a relatively newly identified module in the regulation of myofibroblast differentiation [51,52]. In resting cell, NFAT proteins usually exist in the cytoplasm in a densely phosphorylated state; conditions that elevate levels of free intracellular Ca2+ levels result in calmodulin-dependent phosphatase calcineurin-mediated NFAT dephosphorylation, allowing it to translocate into the nucleus to trigger fibrosis. NFAT dephosphorylation is opposed by Dyrk2, which directly phosphorylates the conserved serine-proline repeat 3 motif of the NFAT regulatory domain. To confirm this, we knocked down Dyrk2 by siRNA and found that the knockdown significantly decreased p-NFAT expression, whereas the introduction of a Dyrk2 overexpression plasmid resulted in increased p-NFAT expression. We concluded that cardiomyocytes secrete miR-208a-containing exosomes into fibroblasts in the fibrotic heart. The exosomes promote fibroblast proliferation and differentiation into myofibroblasts. Those effects are mediated by miR-208a via the targeting of Dyrk2, which can phosphorylate NFAT and prevent its entry into the nucleus to trigger fibrosis (Figure 9).
Figure 9.
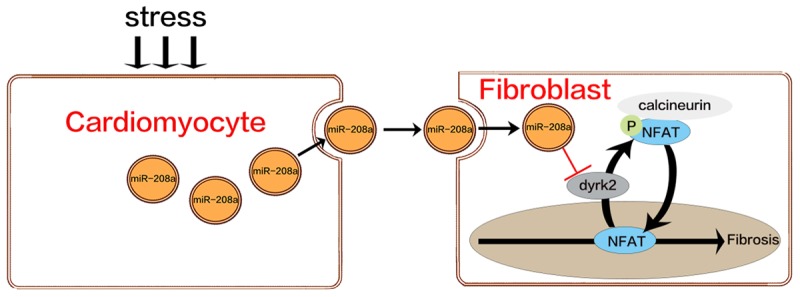
Schematic of the mechanism of Dyrk2 inhibition of NFAT translocation. In resting cells, NFAT proteins reside in the cytoplasm in a densely phosphorylated state. When they are dephosphorylated by calcineurin, they translocate to the nucleus, triggering fibrosis. Dyrk2 counters the calcineurin-facilitated NFAT dephosphorylation by directly phosphorylating the NFAT regulatory domain to stop nuclear translocation.
In summary, our study demonstrates that cross-talk between fibroblasts and myocytes occur in a paracrine fashion to result in cardiac fibrosis. Our results allow a deeper and more insightful understanding of the underlying mechanisms of cardiac fibrosis.
Acknowledgements
All authors have contributed to, read and approved the final manuscript for submission. This work was supported by a grant from the Nature Science Foundations of China Grant (NO.81170124). The funders had no role in study design, data collection and analysis, the decision to publish, or preparation of the manuscript.
Disclosure of conflict of interest
None.
References
- 1.Thum T. Noncoding RNAs and myocardial fibrosis. Nat Rev Cardiol. 2014;11:655–663. doi: 10.1038/nrcardio.2014.125. [DOI] [PubMed] [Google Scholar]
- 2.Leask A. Getting to the heart of the matter: new insights into cardiac fibrosis. Circ Res. 2015;116:1269–1276. doi: 10.1161/CIRCRESAHA.116.305381. [DOI] [PubMed] [Google Scholar]
- 3.Segura AM, Frazier OH, Buja LM. Fibrosis and heart failure. Heart Fail Rev. 2014;19:173–185. doi: 10.1007/s10741-012-9365-4. [DOI] [PubMed] [Google Scholar]
- 4.Thannickal VJ, Zhou Y, Gaggar A, Duncan SR. Fibrosis: ultimate and proximate causes. J Clin Invest. 2014;124:4673–4677. doi: 10.1172/JCI74368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang C, Zhang C, Liu L, A X, Chen B, Li Y, Du J. Macrophage-derived mir-155-containing exosomes suppress fibroblast proliferation and promote fibroblast inflammation during cardiac injury. Mol Ther. 2017;25:192–204. doi: 10.1016/j.ymthe.2016.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pathak M, Sarkar S, Vellaichamy E, Sen S. Role of myocytes in myocardial collagen production. Hypertension. 2001;37:833–840. doi: 10.1161/01.hyp.37.3.833. [DOI] [PubMed] [Google Scholar]
- 7.Sarkar S, Vellaichamy E, Young D, Sen S. Influence of cytokines and growth factors in ANG II-mediated collagen upregulation by fibroblasts in rats: role of myocytes. Am J Physiol Heart Circ Physiol. 2004;287:H107–117. doi: 10.1152/ajpheart.00763.2003. [DOI] [PubMed] [Google Scholar]
- 8.Mittelbrunn M, Sanchez-Madrid F. Intercellular communication: diverse structures for exchange of genetic information. Nat Rev Mol Cell Biol. 2012;13:328–335. doi: 10.1038/nrm3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Skog J, Wurdinger T, van Rijn S, Meijer DH, Gainche L, Sena-Esteves M, Curry WT Jr, Carter BS, Krichevsky AM, Breakefield XO. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol. 2008;10:1470–1476. doi: 10.1038/ncb1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sahoo S, Losordo DW. Exosomes and cardiac repair after myocardial infarction. Circ Res. 2014;114:333–344. doi: 10.1161/CIRCRESAHA.114.300639. [DOI] [PubMed] [Google Scholar]
- 11.Kuwabara Y, Ono K, Horie T, Nishi H, Nagao K, Kinoshita M, Watanabe S, Baba O, Kojima Y, Shizuta S, Imai M, Tamura T, Kita T, Kimura T. Increased microRNA-1 and microRNA-133a levels in serum of patients with cardiovascular disease indicate myocardial damage. Circ Cardiovasc Genet. 2011;4:446–454. doi: 10.1161/CIRCGENETICS.110.958975. [DOI] [PubMed] [Google Scholar]
- 12.Arroyo JD, Chevillet JR, Kroh EM, Ruf IK, Pritchard CC, Gibson DF, Mitchell PS, Bennett CF, Pogosova-Agadjanyan EL, Stirewalt DL, Tait JF, Tewari M. Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc Natl Acad Sci U S A. 2011;108:5003–5008. doi: 10.1073/pnas.1019055108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kroh EM, Parkin RK, Mitchell PS, Tewari M. Analysis of circulating microRNA biomarkers in plasma and serum using quantitative reverse transcription-PCR (qRT-PCR) Methods. 2010;50:298–301. doi: 10.1016/j.ymeth.2010.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chistiakov DA, Orekhov AN, Bobryshev YV. Cardiac-specific miRNA in cardiogenesis, heart function, and cardiac pathology (with focus on myocardial infarction) J Mol Cell Cardiol. 2016;94:107–121. doi: 10.1016/j.yjmcc.2016.03.015. [DOI] [PubMed] [Google Scholar]
- 15.Vickers KC, Palmisano BT, Shoucri BM, Shamburek RD, Remaley AT. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat Cell Biol. 2011;13:423–433. doi: 10.1038/ncb2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhu H, Fan GC. Extracellular/circulating microRNAs and their potential role in cardiovascular disease. Am J Cardiovasc Dis. 2011;1:138–149. [PMC free article] [PubMed] [Google Scholar]
- 17.Mittelbrunn M, Gutierrez-Vazquez C, Villarroya-Beltri C, Gonzalez S, Sanchez-Cabo F, Gonzalez MA, Bernad A, Sanchez-Madrid F. Unidirectional transfer of microRNA-loaded exosomes from T cells to antigen-presenting cells. Nat Commun. 2011;2:282. doi: 10.1038/ncomms1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fichtlscherer S, Zeiher AM, Dimmeler S. Circulating microRNAs: biomarkers or mediators of cardiovascular diseases? Arterioscler Thromb Vasc Biol. 2011;31:2383–2390. doi: 10.1161/ATVBAHA.111.226696. [DOI] [PubMed] [Google Scholar]
- 19.Ohshima K, Inoue K, Fujiwara A, Hatakeyama K, Kanto K, Watanabe Y, Muramatsu K, Fukuda Y, Ogura S, Yamaguchi K, Mochizuki T. Let-7 microRNA family is selectively secreted into the extracellular environment via exosomes in a metastatic gastric cancer cell line. PLoS One. 2010;5:e13247. doi: 10.1371/journal.pone.0013247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Muller G, Jung C, Straub J, Wied S, Kramer W. Induced release of membrane vesicles from rat adipocytes containing glycosylphosphatidylinositol-anchored microdomain and lipid droplet signalling proteins. Cell Signal. 2009;21:324–338. doi: 10.1016/j.cellsig.2008.10.021. [DOI] [PubMed] [Google Scholar]
- 21.Barile L, Gherghiceanu M, Popescu LM, Moccetti T, Vassalli G. Ultrastructural evidence of exosome secretion by progenitor cells in adult mouse myocardium and adult human cardiospheres. J Biomed Biotechnol. 2012;2012:354605. doi: 10.1155/2012/354605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park JE, Tan HS, Datta A, Lai RC, Zhang H, Meng W, Lim SK, Sze SK. Hypoxic tumor cell modulates its microenvironment to enhance angiogenic and metastatic potential by secretion of proteins and exosomes. Mol Cell Proteomics. 2010;9:1085–1099. doi: 10.1074/mcp.M900381-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fruhbeis C, Frohlich D, Kramer-Albers EM. Emerging roles of exosomes in neuron-glia communication. Front Physiol. 2012;3:119. doi: 10.3389/fphys.2012.00119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen L, Wang Y, Pan Y, Zhang L, Shen C, Qin G, Ashraf M, Weintraub N, Ma G, Tang Y. Cardiac progenitor-derived exosomes protect ischemic myocardium from acute ischemia/reperfusion injury. Biochem Biophys Res Commun. 2013;431:566–571. doi: 10.1016/j.bbrc.2013.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sahoo S, Klychko E, Thorne T, Misener S, Schultz KM, Millay M, Ito A, Liu T, Kamide C, Agrawal H, Perlman H, Qin G, Kishore R, Losordo DW. Exosomes from human CD34(+) stem cells mediate their proangiogenic paracrine activity. Circ Res. 2011;109:724–728. doi: 10.1161/CIRCRESAHA.111.253286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vrijsen KR, Sluijter JP, Schuchardt MW, van Balkom BW, Noort WA, Chamuleau SA, Doevendans PA. Cardiomyocyte progenitor cell-derived exosomes stimulate migration of endothelial cells. J Cell Mol Med. 2010;14:1064–1070. doi: 10.1111/j.1582-4934.2010.01081.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wahlquist C, Jeong D, Rojas-Munoz A, Kho C, Lee A, Mitsuyama S, van Mil A, Park WJ, Sluijter JP, Doevendans PA, Hajjar RJ, Mercola M. Inhibition of miR-25 improves cardiac contractility in the failing heart. Nature. 2014;508:531–535. doi: 10.1038/nature13073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aoyama Y, Kobayashi K, Morishita Y, Maeda K, Murohara T. Wnt11 gene therapy with adeno-associated virus 9 improves the survival of mice with myocarditis induced by coxsackievirus B3 through the suppression of the inflammatory reaction. J Mol Cell Cardiol. 2015;84:45–51. doi: 10.1016/j.yjmcc.2015.04.009. [DOI] [PubMed] [Google Scholar]
- 29.Zincarelli C, Soltys S, Rengo G, Rabinowitz JE. Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Mol Ther. 2008;16:1073–1080. doi: 10.1038/mt.2008.76. [DOI] [PubMed] [Google Scholar]
- 30.Karakikes I, Chaanine AH, Kang S, Mukete BN, Jeong D, Zhang S, Hajjar RJ, Lebeche D. Therapeutic cardiac-targeted delivery of miR-1 reverses pressure overload-induced cardiac hypertrophy and attenuates pathological remodeling. J Am Heart Assoc. 2013;2:e000078. doi: 10.1161/JAHA.113.000078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davidson SM, Hausenloy D, Duchen MR, Yellon DM. Signalling via the reperfusion injury signalling kinase (RISK) pathway links closure of the mitochondrial permeability transition pore to cardioprotection. Int J Biochem Cell Biol. 2006;38:414–419. doi: 10.1016/j.biocel.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 32.Stanczyk J, Pedrioli DM, Brentano F, Sanchez-Pernaute O, Kolling C, Gay RE, Detmar M, Gay S, Kyburz D. Altered expression of MicroRNA in synovial fibroblasts and synovial tissue in rheumatoid arthritis. Arthritis Rheum. 2008;58:1001–1009. doi: 10.1002/art.23386. [DOI] [PubMed] [Google Scholar]
- 33.Zhang X, Wang X, Zhu H, Kranias EG, Tang Y, Peng T, Chang J, Fan GC. Hsp20 functions as a novel cardiokine in promoting angiogenesis via activation of VEGFR2. PLoS One. 2012;7:e32765. doi: 10.1371/journal.pone.0032765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang J, Li Y, Xue F, Liu W, Zhang S. Exosomes derived from cardiac telocytes exert positive effects on endothelial cells. Am J Transl Res. 2017;9:5375–5387. [PMC free article] [PubMed] [Google Scholar]
- 35.Malik ZA, Kott KS, Poe AJ, Kuo T, Chen L, Ferrara KW, Knowlton AA. Cardiac myocyte exosomes: stability, HSP60, and proteomics. Am J Physiol Heart Circ Physiol. 2013;304:H954–965. doi: 10.1152/ajpheart.00835.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gray WD, French KM, Ghosh-Choudhary S, Maxwell JT, Brown ME, Platt MO, Searles CD, Davis ME. Identification of therapeutic covariant microRNA clusters in hypoxia-treated cardiac progenitor cell exosomes using systems biology. Circ Res. 2015;116:255–263. doi: 10.1161/CIRCRESAHA.116.304360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cheng J, Du J. Mechanical stretch simulates proliferation of venous smooth muscle cells through activation of the insulin-like growth factor-1 receptor. Arterioscler Thromb Vasc Biol. 2007;27:1744–1751. doi: 10.1161/ATVBAHA.107.147371. [DOI] [PubMed] [Google Scholar]
- 38.Tao L, Bei Y, Chen P, Lei Z, Fu S, Zhang H, Xu J, Che L, Chen X, Sluijter JP, Das S, Cretoiu D, Xu B, Zhong J, Xiao J, Li X. Crucial role of miR-433 in regulating cardiac fibrosis. Theranostics. 2016;6:2068–2083. doi: 10.7150/thno.15007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jung HJ, Suh Y. Circulating miRNAs in ageing and ageing-related diseases. J Genet Genomics. 2014;41:465–472. doi: 10.1016/j.jgg.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Creemers EE, Tijsen AJ, Pinto YM. Circulating microRNAs: novel biomarkers and extracellular communicators in cardiovascular disease? Circ Res. 2012;110:483–495. doi: 10.1161/CIRCRESAHA.111.247452. [DOI] [PubMed] [Google Scholar]
- 41.Turchinovich A, Samatov TR, Tonevitsky AG, Burwinkel B. Circulating miRNAs: cell-cell communication function? Front Genet. 2013;4:119. doi: 10.3389/fgene.2013.00119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kumar S, Reddy PH. Are circulating microRNAs peripheral biomarkers for Alzheimer’s disease? Biochim Biophys Acta. 2016;1862:1617–1627. doi: 10.1016/j.bbadis.2016.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kumar S, Vijayan M, Bhatti JS, Reddy PH. MicroRNAs as peripheral biomarkers in aging and age-related diseases. Prog Mol Biol Transl Sci. 2017;146:47–94. doi: 10.1016/bs.pmbts.2016.12.013. [DOI] [PubMed] [Google Scholar]
- 44.Cheng L, Doecke JD, Sharples RA, Villemagne VL, Fowler CJ, Rembach A, Martins RN, Rowe CC, Macaulay SL, Masters CL, Hill AF Australian Imaging, Biomarkers and Lifestyle (AIBL) Research Group. Prognostic serum miRNA biomarkers associated with Alzheimer’s disease shows concordance with neuropsychological and neuroimaging assessment. Mol Psychiatry. 2015;20:1188–1196. doi: 10.1038/mp.2014.127. [DOI] [PubMed] [Google Scholar]
- 45.Boon RA, Vickers KC. Intercellular transport of microRNAs. Arterioscler Thromb Vasc Biol. 2013;33:186–192. doi: 10.1161/ATVBAHA.112.300139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zernecke A, Bidzhekov K, Noels H, Shagdarsuren E, Gan L, Denecke B, Hristov M, Koppel T, Jahantigh MN, Lutgens E, Wang S, Olson EN, Schober A, Weber C. Delivery of microRNA-126 by apoptotic bodies induces CXCL12-dependent vascular protection. Sci Signal. 2009;2:ra81. doi: 10.1126/scisignal.2000610. [DOI] [PubMed] [Google Scholar]
- 47.Kosaka N, Iguchi H, Yoshioka Y, Takeshita F, Matsuki Y, Ochiya T. Secretory mechanisms and intercellular transfer of microRNAs in living cells. J Biol Chem. 2010;285:17442–17452. doi: 10.1074/jbc.M110.107821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lu A, Pfeffer SR. A CULLINary ride across the secretory pathway: more than just secretion. Trends Cell Biol. 2014;24:389–399. doi: 10.1016/j.tcb.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shyu KG, Wang BW, Cheng WP, Lo HM. MicroRNA-208a increases myocardial endoglin expression and myocardial fibrosis in acute myocardial infarction. Can J Cardiol. 2015;31:679–690. doi: 10.1016/j.cjca.2014.12.026. [DOI] [PubMed] [Google Scholar]
- 50.Shyu KG, Wang BW, Wu GJ, Lin CM, Chang H. Mechanical stretch via transforming growth factor-beta1 activates microRNA208a to regulate endoglin expression in cultured rat cardiac myoblasts. Eur J Heart Fail. 2013;15:36–45. doi: 10.1093/eurjhf/hfs143. [DOI] [PubMed] [Google Scholar]
- 51.Davis J, Molkentin JD. Myofibroblasts: trust your heart and let fate decide. J Mol Cell Cardiol. 2014;70:9–18. doi: 10.1016/j.yjmcc.2013.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Davis J, Burr AR, Davis GF, Birnbaumer L, Molkentin JD. A TRPC6-dependent pathway for myofibroblast transdifferentiation and wound healing in vivo. Dev Cell. 2012;23:705–715. doi: 10.1016/j.devcel.2012.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]