

In this article, we review the past applications of in vitro models in identifying human hepatotoxins and then focus on the use of multiscale experimental models in drug development, including the use of zebrafish and human cell-based, 3-dimensional (3D), microfluidic systems of liver functions as key components in applying Quantitative Systems Pharmacology (QSP). We have implemented QSP as a platform to improve the rate of success in the process of drug discovery and development of therapeutics.1,2 Our working definition of QSP is “Determining the mechanism(s) of disease progression and mechanism(s) of action of drugs on multiscale systems through iterative and integrated computational and experimental methods to optimize the development of therapeutic strategies” (Fig. 1).
Fig. 1.

QSP is an approach to drug discovery and development that applies iterative and integrated computational and experimental methods to determine the mechanism(s) of disease progression and mechanism(s) of action of drugs on multiscale systems. QSP starts with patients and patient samples, applies computational and experimental models, and ends with fundamental knowledge that optimizes therapeutic treatments for patients. This article focuses on the use of multiscale experimental models for liver toxicity and efficacy testing, especially phenotypic models.
The stakeholders involved in drug development from academia, industry, and government agencies have long understood the need to improve drug candidate selection by optimizing efficacy while screening out potential toxins so as to concentrate efforts on candidates with favorable chances for market approval. A survey of the number of new drugs released between 2000 and 2009 demonstrated a 25-year low in drug approvals despite increases in research and development (R&D) investment.3 Laverty and colleagues4 reported that 66% of failed clinical trials were due to a lack of efficacy and 21% for unacceptable drug toxicity. However, Sacks and colleagues5 evaluated clinical drug trials between 2000 and 2012 using additional criteria to further refine the analysis and reported that lack of efficacy alone accounted for only 41% of failures, while the combination of poor efficacy and safety accounted for 35%, and safety alone accounted for 19% of drug failures. Together, cardiovascular and liver toxicity accounted for nearly 75% of all postmarket drug withdrawals in the United States between 1975 and 2007.6 Although cardiotoxicity has recently surpassed hepatotoxicity as the main organ toxicity ending clinical trials or causing postmarket drug withdrawal, hepatotoxicity has been the most frequent cause of drug product recalls between 1953 and 2014.7
CURRENT STATUS OF DRUG HEPATOTOXICITY PREDICTION USING MAMMALIAN IN VIVO MODELS
The liver is responsible for a wide range of functions, including xenobiotic detoxification, protein synthesis, synthesis and storage of glucose, production of the bile necessary for digestion, and regulation of blood cholesterol and triglycerides. The organ is positioned downstream of the gastrointestinal tract to enable “first-pass” clearance of orally ingested drugs and toxins. The structural organization of the liver sinusoidal space facilitates close contact between circulating compounds and the transporter-rich hepatocyte membrane proteins that allow for rapid and efficient transport of drugs from the portal blood. The high capacity for biotransformation in the hepatocyte also facilitates the generation of reactive metabolites that can cause liver damage.8
The key data for assessing hepatotoxicity derives from drug safety study protocols approved by regulatory agencies. Although evidence suggests that preclinical animal studies can predict up to approximately 70% of human toxicity, several problems are apparent from this approach.6 First, the traditional animal studies clearly fail to identify all possible adverse liver effects, because many compounds pass safely through animal testing only to be found hepatotoxic in the clinical trials or in the postmarketing. Second, the traditional drug safety protocols were designed only to ask broad questions with a simple yes or no answer; for example, is the compound hepatotoxic? Is the compound a reproductive toxin? Traditional preclinical drug safety assessments essentially ignored the why and the how of toxicity.9 Historically, few efforts were made to link the observational results from drug safety studies to molecular and cell level events, mechanisms of toxicity (MOTs), or to interactions between tissues and organs.
Adherence to the regulatory agencies’ drug safety protocols also failed to account for the poor concordance between animal and human organ toxicity (Table 1). The concordance can be as low as 40% for the liver to better than 90% for drugs with hematological liability.10 A good demonstration of how animal testing failed to identify clinical hepatotoxic drugs is exemplified by a series of structurally similar and marketed drug pairs for the same therapeutic indication (Table 2). In each case, one of the drugs in the pair exhibited no hepatotoxicity during preclinical and clinical trials or in postmarket surveillance, but the other was “silent” during the animal studies, yet induced hepatotoxicity in clinical trials or in the postmarket release. This discordance in the liver findings has been attributed to differences in the metabolism and metabolic clearance pathways between man and animal test species.11
Table 1.
Concordance of animal and human organ toxicity
| Target Organ | Concordance, % |
|---|---|
| Liver | 40–5410,52 |
| Cutaneous/Ophthalmic | 36 |
| Endocrine | 60 |
| Urinary tract | 64 |
| Neurologic | 70 |
| Cardiovascular | 80 |
| Gastrointestinal | 85 |
| Hematologic | 91 |
Data from Olson H, Betton G, Robinson D, et al. Concordance of the toxicity of pharmaceuticals in humans and in animals. Regul Toxicol Pharmacol 2000;32(1):56–67.
Table 2.
Clinical liver effects of structurally similar drugs found safe in animals
| Human Liver Toxic Drug | Structure | Human Liver Safe Drug | Structure |
|---|---|---|---|
| Nefazodone | 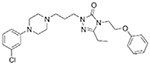 |
Trazodone | 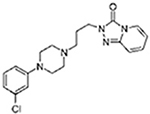 |
| Bromfenac | 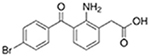 |
Diclofenac | 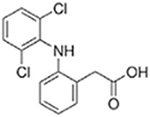 |
| Alpidem | 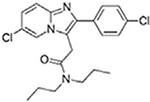 |
Zolpidem | 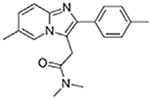 |
| Trovafloxacin | 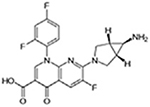 |
Moxifloxacin | 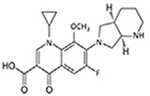 |
| Ibufenac | Ibuprofen | 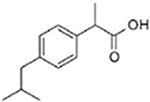 |
|
| Tolcapone | 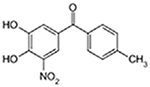 |
Entacapone | 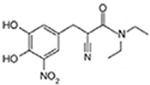 |
After expensive postmarket drug recalls for Troglitazone and Bromfenac and the restrictive labeling for Trovafloxacin and Tolcapone, the pharmaceutical industry initiated strategies to prescreen compounds for liver toxicity. In one example, the industry successfully implemented in vitro and specialized in vivo pharmacokinetic (PK) screening early in the lead optimization process. Although in 1993 nearly 39% of compounds failed in clinical trials due to poor PK, this fell dramatically to 7% by 2003 following implementation of early drug discovery PK profiling.12 However, during the same period of time, the rate of compounds failing in the clinic due to toxicity rose from 10% to 16%.12 Based on the success in PK profiling, it was projected that a strategy similar to early PK screening also would be successful in identifying and eliminating potential hepatotoxins before proceeding into preclinical testing.
IN VITRO MODELS FOR PREDICTING DRUG-INDUCED HEPATOTOXICITY
Many commonly used in vitro hepatotoxicity assays rely on subcellular liver fractions, established hepatoma cell lines, primary animal and human hepatocytes, liver slices, and whole perfused livers. The use of in vitro data from microsomes, primary hepatocytes and S9 subcellular fractions to predict in vivo drug clearance is generally a well-accepted procedure.13,14 Whole perfused livers and liver slices on the other hand are low throughput, require continual usage of animals, are costly, and still suffer the lack of concordance with human toxic liabilities. These models have been reviewed elsewhere.11
Rodent and human primary hepatocytes have become a mainstay of hepatotoxicity testing in the in vitro laboratory.11 Large numbers of healthy hepatocytes can be isolated from a single rat or from a human liver resection or autopsy. This has allowed moderate to high-throughput screening to identify potential hepatotoxins, while at the same time reducing animal use, amount of test agent, cost per compound tested, and the time required to make a toxic liability decision. The primary hepatocyte has been a convenient model for investigating MOT, pharmacokinetics, identification of metabolites, and dose response toxicity. However, an important limitation in the use of isolated primary hepatocytes is a reduction in function and differentiation after 24 to 48 hours in culture.15 To avoid this issue, many in vitro hepatotoxicity tests have been conducted using established human hepatoma cell lines such as HepG2 and HepaRG or with immortalized primary hepatocytes such the Corning HepatoCell (Corning, NY).16–18 The limitation to these cell types, however, is low or absent biotransformation for many of the important cytochrome P450 enzymes and phase II conjugation reactions involved in drug clearance.11,19 The advantages and disadvantages of the more commonly used single-cell and cell fraction in vitro models are compared in Table 3.
Table 3.
Commonly used cell and cellular fraction in vitro models for absorption, distribution, metabolism, excretion, and toxicity (ADMET)
| Model | Application | Advantage | Disadvantage | Reference |
|---|---|---|---|---|
| Hepatocyte cell fractions microsomes | Liver clearance, metabolite ID | Fast, inexpensive, individual variation can be studied | Overestimates in vivo metabolism; only CYP and UGT enzymes | 14,53 |
| S9 | Liver clearance, metabolite ID | Phase I and phase II activity | Lower enzyme activity in the S9 fraction, may miss low level metabolites | 14 |
| 1° hepatocyte suspensions | Liver clearance, metabolite ID | In vivo levels of drug metabolism and transport proteins, cryopreservation | 4-h time limit | 19 |
| Transgenic cell lines for PK | Metabolite ID | Single enzyme reactions generate high levels of metabolites for structural ID | Overestimate involvement of one enzyme species | 14 |
| Established liver cells lines HepG2 | Toxicity testing, MOT, induction | Established cell line, inexpensive | Absence or low expression of most phase I and phase II enzymes | 14,19 |
| HepaRG | Toxicity testing, MOT, induction, metabolite ID | CYP1A2 and 3A4 inducible,established cell line | High CYP3A4, Cyp 7A1 expression, but low in all other CYP levels compared with primary hepatocytes | 19,54 |
| Primary Heps | PK, toxicity testing, metabolite ID | Well characterized, intact metabolism, intact transporters, cryopreservation | Decline in differentiated functions; no immune or fibrosis cells; single donor variation | 14 |
| Spheroids: established cell lines HepG2, HepaRG | PK, toxicity testing, metabolite ID | Extend differentiated cell functions from days to weeks, 3D, improved metabolism | Cell functions lower than primary hepatocytes, low urea, albumin production | 55–57 |
Abbreviations: CYP, cytochrome p450; Heps, hepatocytes; MOT, mechanisms of toxicity; PK, pharmacokinetic; UGT, udp-glucuronosyltransferase
The primary hepatocyte and immortalized hepatocytes have been used in 2D monolayers, 2D co-cultures, 3D single-cell type, and 3D co-cultures depending on the questions posed. Two-dimensional monolayer assays have been applied to collect simple cell death end points to triage large numbers of compounds.11 In recent years, high-throughput screening (HTS) assays have been used to measure MOTs known to be relevant to mechanisms of clinical hepatotoxicity. The latter assay type, which is often referred to as “fit-for-purpose,” tests compounds in a model designed solely to identify a specific mechanism of toxicity. Examples of “fit-for-purpose” hepatocyte assays include mitochondrial dysfunction, oxidative stress, bile salt exporter protein inhibition, covalent binding, and pregnane X receptor nuclear receptor modulation. These 5 mechanisms have a demonstrable association to increased risk for clinical hepatotoxicity.20–24
Although preclinical animal testing remains critical for Investigational New Drug and New Drug Application approval, a significant shift to alternative approaches using quantitative structure-activity relationships (QSAR) computational models, simple in vitro cytotoxicity and “fit-for-purpose” HTS assays have been promoted by governmental agencies such as the Environmental Protection Agency, the National Center for the Advancement of Translational Sciences (NCATS), and the National Toxicology Program. These initiatives have resulted in the development of a number of databases (eg, Tox21, ToxCast) and models for prioritizing compounds based on HTS assay hepatotoxicity, as well as other organ toxicities.25 In addition to the government initiatives, most R&D organizations in academia and the pharmaceutical industry have a slate of in vitro toxicity assays and computational approaches designed to eliminate unfavorable compounds early in the drug discovery process.26
PAST EXPERIENCES WITH IN VITRO LIVER ABSORPTION, DISTRIBUTION, METABOLISM, EXCRETION, AND TOXICITY MODELS PREDICTING HUMAN CLINICAL TOXICITY
The concordance of in vitro toxicity testing with clinical hepatotoxicity varies from only 25% for the simple in vitro assays to nearly 80% for the more complex assays and analyses.17,27 Table 4 presents a subset of computational approaches: in vitro 2D human cell-based HTS assays; a human cell co-culture model; and one in vitro covalent binding assay selected from a joint Food and Drug Administration (FDA) National Center for Toxicology Research, California Institute of Technology, and Hannover Medical School report which cross-validated the concordance of these methods to human drug-induced liver injury (DILI).27 Three interesting findings are noteworthy from the results: (1) multiparametric cell-based models performed better than the computational or cell-free covalent binding assays; (2) the predictive result for DILI-negative drugs (assay specificity) was always higher than the predictive result for DILI-positive drugs (assay sensitivity); and (3) an apparent upper limit to the predicted DILI nearing 75% to 80% was reached with the existing in vitro cell-based systems.27 A likely explanation for the plateau of predictive concordance of 80% with existing in vitro cell models is the reductionist strategy used to simplify the organized, multicellular complexity of the liver microenvironment to single-cell or even 2-cell type testing assays.
Table 4.
Concordance of large-scale in vitro screening to human hepatotoxicitya
| Model | Name/Type | # Compounds | Overallb Concordance, % | True Positives, % | True Negatives, % | Reference |
|---|---|---|---|---|---|---|
| In silico models | ||||||
| Structural alert | DEREKc | 623 | 56 | 46 | 73 | 58 |
| QSAR/Molecular descriptors | 4 commercial programsd | ∼1600 | 63 | 39 | 87 | 59 |
| 2D molecular descriptor | 382 | 76 | 76 | 75 | 60 | |
| PaDel molecular descriptore | 1087 | 69 | 67 | 70 | 27,61 | |
| ECF 6 molecular descriptorsf | 295 | 59 | 53 | 65 | 62 | |
| 2D in vitro cell models | ||||||
| HepG2 | 6 endpointg | 102 | 70 | 40 | 100 | 63 |
| HepG2 | 11 endpointh | 136 | 76 | N/Ai | N/A | 17 |
| 1° Human hepatocytes | 4 endpointj | 344 | 75–80 | 50–60 | 95–100 | 64 |
| Co-cultured 1° human hepatocytes, stromal cells | Micropattern surface 4 endpointk | 45 | 78 | 66 | 90 | 65 |
| In vitro cell-free assays | Covalent binding assay-glutathione adduct formation | 223 | 68 | 45 | 90 | 66 |
Abbreviations: QSAR, quantitative structure-activity relationship; 2D, 2-dimensional.
Cross-validated results.
Concordance = True positives (sensitivity) + true negatives (specificity)/2.
DEREK - toxicity prediction software (Lhasa, Leeds, UK)
MC4PC, MDL-QSAR, BioEpisteme, Predictive Data Miner.
PaDel open source software for calculating molecular descriptors.
ECF (extended connectivity functional fingerprints).
Cell counts, nuclear area, plasma membrane integrity, lysosomal activity, mitochondrial membrane potential and mitochondrial area.
Cell count, DNA degradation, nuclear size, cytoskeletal disruption, DNA damage response, oxidative stress, mitosis, stress kinase, mitochondrial membrane potential and area, cell cycle arrest. Results not cross validated.
Not available.
Cell count, mitochondrial damage, oxidative stress, and intracellular glutathione.
Glutathione levels, ATP levels, albumin, and urea secretion.
Data from Chen M, Bisgin H, Tong L, et al. Toward predictive models for drug-induced liver injury in humans: arewe there yet? Biomark Med 2014;8(2):201–13.
ZEBRAFISH LARVAE AS A LOW-COST, MEDIUM-THROUGHPUT, WHOLE-ORGANISM PLATFORM TO PREDICT DRUG HEPATOTOXICITY
The use of animals for experimentation, especially warm-blooded species, presents ethical concerns, and governing bodies now strive to implement the reduction, refinement, and replacement (“3R”) of animals strategy in research.28 The European Union Directive 2010/63/EU on the protection of animals used for scientific purposes requires the use of species with the lowest capacity to experience pain, suffering, and distress, and mandates that the smallest number of animals be used to obtain scientifically valid results. Studies in rodents are further limited by the high costs for acquisition and maintenance.
In recent years, there has been an increased recognition that in vitro phenotypic experimental cell models, as well as small multicellular organisms, can be used in the multiscale approach described as part of QSP.1 Zebrafish in particular have attracted attention not only as a model for drug discovery, but also as a preclinical model for toxicity assessments.29–32 Their prospective position in drug discovery and toxicity assessment is envisioned to be a bridge between simple cell-based and the still mandated mammalian testing.33
Zebrafish are uniquely positioned for large-scale experimentation. Zebrafish are vertebrate animals with high similarity to mammals, both organotypically and physiologically. They have a tractable, diploid genome that is 70% to 80% similar to humans and that is amenable to both forward and reverse genetics. Because of their small size, zebrafish, at the larval stage, are compatible with multiwell plate formats used in HTS, requiring only small amounts of compounds/drugs. Their high fecundity makes it possible to obtain large numbers of specimens for experimentation, dramatically reducing cost compared with rodent models. The zebrafish embryo therefore provides a cost-effective opportunity to discover potential drug liabilities using functional assays in a living animal as a complement to the emerging human tissue models.
The zebrafish embryonic liver is completely developed and functional by 72 hours post fertilization (hpf), as judged by organ appearance and functional markers, such as phase 1 and phase 2 biotransformation capabilities, serum protein secretion, glycogen storage, and lipogenesis.34–36 Importantly, transgenic zebrafish larvae expressing human Cyp3A4 have been developed and will find use in PK and toxicity testing.37
Assays for zebrafish hepatotoxicity have thus far mainly been observational. In zebrafish larvae, necrotic cells can be visually identified by a change in appearance from translucency to opaque black.31 A major shortcoming of macroscopic cell death assays is that they are not sensitive enough to detect early toxicity.38 Nonetheless, a variant of this methodology using liver degeneration, changes in size, and yolk sac retention as endpoints has recently been published and shown to predict 8 of 8 known hepatotoxicants.39 Changes in liver appearance, for example, cellular organization, interactions, and shape, also can be detected by histopathology from tissue slices, although this method is time-consuming, requires a trained pathologist, and therefore is usually reserved to validate observations by other measurements. Last, hepatotoxicity can be assessed in the adult zebrafish using canonical liver enzyme assays (eg, alanine aminotransferase), although the use of adults eliminates the convenience, ethical impact, and high-throughput compatibility that embryos offer.40 Our own data suggest that even gross organism toxicity, assessed by visual inspection of morphologic changes in 72 hpf larvae (ie, bent tails, distended peritoneum and edema, pericardial congestion) can distinguish hepatotoxic from nontoxic agents (Table 5).
Table 5.
Gross morphologic observations in zebrafish larvae identify known toxicants including hepatotoxic amiodarone
| Compound/Dose Range | Gross Morphology | Concentration, μM | ||||||
|---|---|---|---|---|---|---|---|---|
| Dose range | 200 | 66 | 20 | 6.6 | 2 | 0.66 | DMSO | |
| Menadione | Live/deada | 0/4 | 0/4 | 0/4 | 4/0 | 4/0 | 4/0 | 4/0 |
| Visual toxicityb | 1/4 | 2/4 | 0/4 | 0/4 | ||||
| Amiodarone | Live/dead | 0/4 | 0/4 | 4/0 | 4/0 | 4/0 | 4/0 | 4/0 |
| Visual toxicity | 2/4 | 3/4 | 0/4 | 1/4 | 0/4 | |||
| Dose range | 1000 | 300 | 100 | 30 | 10 | 3 | DMSO | |
| Caffeine | Live/dead | 4/0 | 4/0 | 4/0 | 4/0 | 4/0 | 4/0 | 4/0 |
| Visual toxicity | 3/4 | 4/4 | 0/4 | 0/4 | 0/4 | 0/4 | 0/4 | |
| Dose range | 20 | 6.6 | 2 | 0.66 | 0.2 | 0.07 | DMSO | |
| CCCP | Live/dead | 1/4 | 4/0 | 4/0 | 4/0 | 4/0 | 4/0 | 4/0 |
| Visual toxicity | 0/1 | 2/4 | 3/4 | 3/4 | 4/4 | 2/4 | 0/4 | |
| Rotenone | Live/dead | 1/4 | 3/4 | 4/0 | 4/0 | 4/0 | 4/0 | 4/0 |
| Visual toxicity | 1/1 | 0/3 | 1/4 | 2/4 | 3/4 | 2/4 | 0/4 | |
Abbreviations: CCCP, Carbonyl cyandide m-chorophenyl hydrazone; DMSO, dimethyl sulfoxide.
Translucent (live), Opaque (dead).
Bent tail, distended peritoneum, edema, pericardial congestion.
Zebrafish toxicity research is now shifting from observational to mechanism-based toxicity assays. Mesens and colleagues41 explored a molecular endpoint that captures effects on lipid metabolism because liver injury is frequently associated with perturbations in lipid metabolism. Hence, the group looked at expression of liver-specific fatty acid binding protein 10a (L-FABP 10a) as a molecular biomarker for hepatotoxicity and found that changes in expression were predictive of specific mechanisms. A corollary example was recently published by Verstraelen and colleagues,42 in which they evaluated the expression of 5 liver-specific genes (including 2 apoptosis, and 2 metabolism-related) following exposure with 5 known toxicants. Their results confirmed those of Mesens and colleagues41 with L-FABP 10a and further documented that biomarker responses are compound-dependent, mechanism-dependent, and concentration-dependent. At the present time, the utility of biomarkers for prediction of toxicity appears to have potential in “fit-for-purpose” studies. If highly predictive biomarkers can be found, the zebrafish offers the opportunity to generate transgenic reporter lines that would greatly increase throughput.
Our own work has embraced adaptation of in vitro, human mechanism-based toxicity models and screening in zebrafish. In addition to morphologic observations (see Table 5), this suite of assays includes measurements of reactive oxygen species (ROS) and mitochondrial membrane perturbations because they are very good predictors of clinical toxicity.24 Fig. 2 illustrates the utility of ROS measurements in zebrafish larvae using menadione. Menadione is a naphthoquinone that generates ROS in cells through redox cycling. Menadione caused time-dependent and dose-dependent generation of ROS in zebrafish that correlated with embryonal toxicity and ROS induction and death in cultured hepatocytes, although there were quantitative differences between these types of models, likely due to differences in drug uptake, glutathione levels, and possibly metabolism. Additional development of these methods is required.
Fig. 2.

ROS generation correlates with death in zebrafish larvae and rat hepatocytes. (A) Zebrafish larvae at 72 hpf were arrayed in 96-well microplates, loaded with dihydroethidium (DHE) for 30 minutes, and treated with menadione (50 μM). At the indicated time points, plates were scanned and analyzed for red oxyethidium fluorescence on an ArrayScan VTi high-content reader (ThermoFisher, Waltham, MA, USA). (B) Fluorescence micrographs of dose-dependent menadione-induced oxyethidium generation at 48 hpf. (C) Zebrafish larvae at 72 hpf (closed circles) or cultured rat hepatocytes (open circles) were treated in 96-well plates with various concentrations of menadione and oxyethidium fluorescence quantified. Toxicity correlated with production of ROS in both zebrafish embryos and hepatocytes.
CASE STUDY: USING “FIT-FOR-PURPOSE” ASSAY EVALUATIONS TO RANK-ORDER COMPOUNDS
Given the limits to predicting human hepatotoxicity from current in vitro and in vivo methods, additional improvements to the test systems and analytical methods are needed to select better compounds for preclinical testing. A case study is presented to illustrate one strategy used at the University of Pittsburgh Drug Discovery Institute to rank-order compounds for hepatotoxicity risk. Compounds are screened through zebrafish embryos and a set of in vitro “fit-for-purpose” cytotoxicity and mechanism of toxicity assays are then applied (Fig. 3). Rank ordering does not rely on any one single assay, but as a profile of risk factors calculated from the safety margin (the ratio of toxic level to therapeutic level) categorically binned into high, moderate, or low risk. The development of this approach was an extension of the time-dependent and concentration-dependent multiplexed MOT endpoint assays for mitochondrial function, oxidative stress, and cytotoxicity analyses developed and validated in the CellCiphr HepG2 and primary hepatocyte toxicity panels. Our use of the safety margin to classify risk, taken together with a Pfizer study that reported an increase in concordance between in vitro assays and clinical hepatotoxicity with drugs that induced 2 or more MOTs, suggested that some improvements are possible.17,23,43 In the case study described here, the chance of clinical hepatotoxicity increases in compounds with more high and moderate risk factors.
Fig. 3.

Case use of multiple “fit-for-purpose” assays to determine hepatotoxicity risk. The concentration of inhibitor in which the response is reduced by half (IC50) results from 6 different assays in zebrafish, HepG2 and primary hepatocytes are used to calculate the safety margin, defined as the toxic IC50 response/Cmax blood concentration. The lower the safety window the higher the risk for hepatotoxicity. To rank-order compounds, the safety margin is categorized into high (red), moderate (yellow), or low (green) risk to generate the heat map. The overall rank order is determined by the number of high and moderate risks. a Acute lethal dose at which one-half of zebrafish embryos are dead by 24-hour exposure. b Drug quantity not sufficient (QNS) to repeat study to calculate safety margin.
EMERGENCE OF HUMAN TISSUE AND ORGAN MODELS
For the reasons presented previously, new strategies are required to better identify hepatotoxic compounds, especially chronic toxicity, as well as to develop better human efficacy and disease models. The development of biomimetic, multicellular, 3D, microfluidic microphysiology models of the human liver and other organs are in development.44,45
Cellular responses to drugs in the intact human organ are more accurately represented by 3D human cell cultures than the traditional static 2D cell cultures, with additional advantages provided by the inclusion of media perfusion to provide nutrients, oxygen, chemicals, and remove waste products.46–48 Researchers are now capitalizing on the increased availability of human primary, immortalized, or induced pluripotent stem cell (iPSC)-derived hepatocytes, new bioengineering materials, microfabrication techniques, and microfluidic devices to construct reasonable representations of the adult human liver acinus in 3D multicellular microphysiological systems (MPS).48,49 These MPS can be maintained for a month or longer, allowing chronic, as well as acute, responses to drug challenges. In our recent study, we demonstrated acute and chronic drug effects, including the induction of fibrosis by methotrexate and the induction of immune-mediated hepatotoxicity.48 A comparison of some current static and perfused 3D, multicell models are presented in Table 6.
Table 6.
Examples of multicellular static and microfluidic liver models for absorption, distribution, metabolism, excretion, and toxicity (ADMET) testing
| Model | Application | Advantage | Disadvantage | References |
|---|---|---|---|---|
| Static models | ||||
| Co-culture spheroids | Stellate cell activation | Long term 3D culturing with improved drug metabolism and output of albumin, urea | Specialized plates to from spheroids, specialized culturing techniques | 67 |
| Co-culture micropatterned primary hepatocytes with fibroblasts | Hepatotoxicity Metabolite ID Disease models | Hepatocytes maintain differentiated function 2–3 wk | 2D cultures, specialized plates | 65,68 |
| Four-cell spheroids primary hepatocytes, primary liver NPC | Hepatotoxicity Metabolite ID | Hepatocytes maintain differentiated function >3 wk, 3D, immune-mediated toxicity | Specialized culturing techniques | 69 |
| 3D microfluidic, multicellular liver models | ||||
| Primary hepatocytes, endothelial cells | Hepatotoxicity | Hepatocytes maintain differentiated function >3 wk, microfluidic improves function | Specialized culturing techniques, perfusion system | 70 |
| Hepatocytes, endothelial cells, stellate cells, Kupffer-like immune cells | PK, toxicity, therapeutic intervention, liver disease model | Hepatocytes maintain differentiated function >3 wk, immune-mediated toxicity, fibrosis activation microfluidic improves function | Specialized culturing techniques, perfusion system | 48,51,71 |
Abbreviations: 2D, 2 dimensional; 3D, 3 dimensional; NPC, non parenchymal cells; PK, pharmacokinetic.
Of particular interest to those who study hepatotoxicity is the creation of a “liver on a chip” with iPSC-derived adult hepatocytes from patients who have susceptibility to DILI events or other defined genetic and disease backgrounds. This would place liver MPS platforms at the center of personalized medicine and in the continuum of the QSP approach to drug discovery and evaluation of disease progression.1,2 The potential ramifications and promises of this new paradigm for drug discovery, disease progression, and toxicity assessments are discussed in more detail in the Prospectus.
PROSPECTUS: MOVING TO THE FUTURE: INTEGRATING THE HUMAN LIVER ON A CHIP, COMPUTATIONAL MODELS, AND QUANTITATIVE SYSTEMS PHARMACOLOGY
Collectively, the limited concordance of laboratory animal drug safety testing with human safety, the apparent 80% limit of success of human-based 2D in vitro models and the 65% to 75% rate of success with computational models to predict drug-induced clinical hepatotoxicity has shifted the focus to the creation of human, 3D, microfluidic systems, referred to as MPS. One such platform has been developed at the University of Pittsburgh Drug Discovery Institute and integrates an MPS liver model using 4 human liver cell types organized into a microfluidic, 3D, sinusoidal complex, with the capacity for live cell monitoring of MOT using fluorescence-based biosensors over a period of several weeks. Secreted proteins, cytokines, and metabolites collected from the efflux are analyzed by biochemical assays and mass spectrometry along with the results from imaging biosensors for parameters such as apoptosis, ROS production, and free calcium levels. All of the data are linked in a database designed to collect, manage, and model the data (Fig. 4). Integrated human MPS platforms that are biomimetics of normal organ structure and function have the potential to improve on the current predictive limit (approximately 80%) and diminish the odds of “silent” human hepatotoxins from being introduced into clinical trials or the market.
Fig. 4.

Overview of the Human Liver Microphysiology Platform for studying human liver physiology, disease models and drug safety testing. The platform is composed of the following: (A) the Sequentially Layered, Self-Assembly Liver model (SQL-SAL) constructed from a microfluidic device and 4 human cell types, a fraction of which are “sentinel” cells expressing fluorescence-based biosensors, and that can include disease-specific cells, such as cancer cells. Data are collected from the model via (B) high-content imaging readouts of transmitted light contrast and fluorescence; and (C) biochemical and mass spectrometry readouts.48,51 (D) The multiplexed data are uploaded into the Microphysiology Systems Database (MPS-Db) to manage data, associate external data sources, and build predictive models of human efficacy and toxicity.50
Continued improvements to the MPS liver models will include the application of renewable cells (eg, human iPSC-derived adult hepatocytes, as well as the nonparenchymal cells) to permit the investigation of the heterogeneous human genetic backgrounds, as well as specific diseases (eg, nonalcoholic fatty liver disease, hepatocarcinoma, and rare childhood liver diseases). Further advances also will include liver metabolic zonation and higher throughput arrays of MPS. The microphysiology database also will continue to evolve as a tool to manage, mine, and model the experimental data, as well as public sources of preclinical and clinical findings, expert-based drug knowledge, physical properties, pharmacology targets, adverse event reporting, and large datasets from “omics,” including toxicogenomics, metabolomics, proteomics, reactive metabolite proteomics, and transcriptomics.50 Furthermore, QSP is expected to increase our understanding of the integrated and interacting cellular, tissue, and organ networks; genes; proteins; and metabolic processes that give rise to liver disease progression, therapeutic efficacy, and drug-induced hepatotoxicity.1,50
KEY POINTS.
Quantitative Systems Pharmacology is a multiscale, iterative, and integrated computational and experimental approach for optimizing the development of therapeutic strategies.
Limited efficacy and drug safety methods continue to limit the efficiency of the discovery and development of therapeutics.
Mammalian in vivo models have low concordance to clinical hepatotoxicity.
The predictive concordance between the existing in vitro experimental models coupled with computational modeling and clinical hepatotoxicity has reached a limit.
Three-dimensional, multicellular, microfluidic, human tissue and organ experimental models are a promising alternative to whole animal and cultured cell methods for efficacy and safety testing.
Acknowledgments
Research reported in Clinics in Liver Disease was supported by the NIH National Center for Advancing translational Sciences (NCATS) of the National Institutes of Health under award number UH2TR000503.
Footnotes
The authors have nothing to disclose.
REFERENCES
- 1.Stern AM, Schurdak ME, Bahar I, et al. A perspective on implementing a quantitative systems pharmacology platform for drug discovery and the advancement of personalized medicine. J Biomol Screen 2016;21(6):521–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sorger PK, Allerheiligen SBR, Abernathy DR, et al. Quantitative and systems pharmacology in the post-genomic era: new approaches to discovering drugs and understanding therapeutic mechanisms In an NIH white paper by the QSP workshop group – October, 2011. Bethesda (MD): NIH; 2011. R Ward, Editor. [Google Scholar]
- 3.Kaitin KI, DiMasi JA. Pharmaceutical innovation in the 21st century: new drug approvals in the first decade, 2000–2009. Clin Pharmacol Ther 2011;89(2):183–8. [DOI] [PubMed] [Google Scholar]
- 4.Laverty H, Benson C, Cartwright E, et al. How can we improve our understanding of cardiovascular safety liabilities to develop safer medicines? Br J Pharmacol 2011;163(4):675–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sacks LV, Shamsuddin HH, Yasinskaya YI, et al. Scientific and regulatory reasons for delay and denial of FDA approval of initial applications for new drugs, 2000–2012. JAMA 2014;311(4):378–84. [DOI] [PubMed] [Google Scholar]
- 6.Stevens JL, Baker TK. The future of drug safety testing: expanding the view and narrowing the focus. Drug Discov Today 2009;14(3–4):162–7. [DOI] [PubMed] [Google Scholar]
- 7.Onakpoya IJ, Heneghan CJ, Aronson JK. Post-marketing withdrawal of 462 medicinal products because of adverse drug reactions: a systematic review of the world literature. BMC Med 2016;14:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yuan L, Kaplowitz N. Mechanisms of drug-induced liver injury. Clin Liver Dis 2013;17(4):507–18, vii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doull J, Bruce M. General principles of toicology In: Klaassen CD, editor. Cassertt and Doull’s toxicology: the basic science of poisons. New York: Macmillan Publishing Company; 1986. p. 11–32. [Google Scholar]
- 10.Olson H, Betton G, Robinson D, et al. Concordance of the toxicity of pharmaceuticals in humans and in animals. Regul Toxicol Pharmacol 2000;32(1):56–67. [DOI] [PubMed] [Google Scholar]
- 11.Godoy P, Hewitt NJ, Albrecht U, et al. Recent advances in 2D and 3D in vitro systems using primary hepatocytes, alternative hepatocyte sources and non-parenchymal liver cells and their use in investigating mechanisms of hepatotoxicity, cell signaling and ADME. Arch Toxicol 2013;87(8):1315–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kubinyi H Drug research: myths, hype and reality. Nat Rev Drug Discov 2003; 2(8):665–8. [DOI] [PubMed] [Google Scholar]
- 13.Ito K, Houston JB. Comparison of the use of liver models for predicting drug clearance using in vitro kinetic data from hepatic microsomes and isolated hepatocytes. Pharm Res 2004;21(5):785–92. [DOI] [PubMed] [Google Scholar]
- 14.Brandon EF, Raap CD, Meijerman I, et al. An update on in vitro test methods in human hepatic drug biotransformation research: pros and cons. Toxicol Appl Pharmacol 2003;189(3):233–46. [DOI] [PubMed] [Google Scholar]
- 15.Soldatow VY, Lecluyse EL, Griffith LG, et al. In vitro models for liver toxicity testing. Toxicol Res (Camb) 2013;2(1):23–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mennecozzi M, et al. Hepatotoxicity screening taking a mode-of-action approach using HepaRG cells and HCA. Altex Proceedings 2012;1:193–204. [Google Scholar]
- 17.Vernetti L, et al. Cellular systems biology applied to preclinical safety testing: a case study of CellCiphr profiling In: Ekins S, Xu JJ, editors. Drug efficacy, safety, and biologics discovery: Emerging technologies and tools. Hoboken (NJ): John Wiley & Sons; 2009. p. 53–73. [Google Scholar]
- 18.Faris RA, Hong YL, Liu J, et al. Corning (R) hepatocell-cells closely model the behavior of parental cells for predicting hepatotoxicity In drug metabolism reviews. Abingdon (United Kingdom): Taylor & Francis Ltd; 2015. [Google Scholar]
- 19.Bi YA, Kazolias D, Duignan DB. Use of cryopreserved human hepatocytes in sandwich culture to measure hepatobiliary transport. Drug Metab Dispos 2006; 34(9):1658–65. [DOI] [PubMed] [Google Scholar]
- 20.Wang YM, Chai SC, Brewer CT, et al. Pregnane X receptor and drug-induced liver injury. Expert Opin Drug Metab Toxicol 2014;10(11):1521–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Usui T, Mise M, Hashizume T, et al. Evaluation of the potential for drug-induced liver injury based on in vitro covalent binding to human liver proteins. Drug Metab Dispos 2009;37(12):2383–92. [DOI] [PubMed] [Google Scholar]
- 22.Will Y, Dykens J. Mitochondrial toxicity assessment in industry–a decade of technology development and insight. Expert Opin Drug Metab Toxicol 2014;10(8): 1061–7. [DOI] [PubMed] [Google Scholar]
- 23.Aleo MD, Luo Y, Swiss R, et al. Human drug-induced liver injury severity is highly associated with dual inhibition of liver mitochondrial function and bile salt export pump. Hepatology 2014;60(3):1015–22. [DOI] [PubMed] [Google Scholar]
- 24.Pereira CV, Nadanaciva S, Oliveira PJ, et al. The contribution of oxidative stress to drug-induced organ toxicity and its detection in vitro and in vivo. Expert Opin Drug Metab Toxicol 2012;8(2):219–37. [DOI] [PubMed] [Google Scholar]
- 25.Collins FS, Gray GM, Bucher JR. Toxicology. Transforming environmental health protection. Science 2008;319(5865):906–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blomme EA, Will Y. Toxicology strategies for drug discovery: present and future. Chem Res Toxicol 2016;29(4):473–504. [DOI] [PubMed] [Google Scholar]
- 27.Chen M, Bisgin H, Tong L, et al. Toward predictive models for drug-induced liver injury in humans: are we there yet? Biomark Med 2014;8(2):201–13. [DOI] [PubMed] [Google Scholar]
- 28.Russell WMS, Burch RL. The principles of humane experimental technique. London: Methuen; 1959. p. 238. [Google Scholar]
- 29.Rubinstein AL. Zebrafish assays for drug toxicity screening. Expert Opin Drug Metab Toxicol 2006;2(2):231–40. [DOI] [PubMed] [Google Scholar]
- 30.McGrath P, Li CQ. Zebrafish: a predictive model for assessing drug-induced toxicity. Drug Discov Today 2008;13(9–10):394–401. [DOI] [PubMed] [Google Scholar]
- 31.Hill A, Mesens N, Steemans M, et al. Comparisons between in vitro whole cell imaging and in vivo zebrafish-based approaches for identifying potential human hepatotoxicants earlier in pharmaceutical development. Drug Metab Rev 2012; 44(1):127–40. [DOI] [PubMed] [Google Scholar]
- 32.Zon LI, Peterson RT. In vivo drug discovery in the zebrafish. Nat Rev Drug Discov 2005;4(1):35–44. [DOI] [PubMed] [Google Scholar]
- 33.Parng C In vivo zebrafish assays for toxicity testing. Curr Opin Drug Discov Devel 2005;8(1):100–6. [PubMed] [Google Scholar]
- 34.Chu J, Sadler KC. New school in liver development: lessons from zebrafish. Hepatology 2009;50(5):1656–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Behra M, Etard C, Cousin X, et al. The use of zebrafish mutants to identify secondary target effects of acetylcholine esterase inhibitors. Toxicol Sci 2004; 77(2):325–33. [DOI] [PubMed] [Google Scholar]
- 36.Jones HS, Panter GH, Hutchinson TH, et al. Oxidative and conjugative xenobiotic metabolism in zebrafish larvae in vivo. Zebrafish 2010;7(1):23–30. [DOI] [PubMed] [Google Scholar]
- 37.Poon KL, Wang X, Ng AS, et al. Humanizing the zebrafish liver shifts drug metabolic profiles and improves pharmacokinetics of CYP3A4 substrates. Arch Toxicol 2016. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 38.Wolf JC, Wolfe MJ. A brief overview of nonneoplastic hepatic toxicity in fish. Toxicol Pathol 2005;33(1):75–85. [DOI] [PubMed] [Google Scholar]
- 39.He JH, Guo SY, Zhu F, et al. A zebrafish phenotypic assay for assessing drug-induced hepatotoxicity. J Pharmacol Toxicol Methods 2013;67(1):25–32. [DOI] [PubMed] [Google Scholar]
- 40.Murtha JM, Qi W, Keller ET. Hematologic and serum biochemical values for zebrafish (Danio rerio). Comp Med 2003;53(1):37–41. [PubMed] [Google Scholar]
- 41.Mesens N, Crawford AD, Menke A, et al. Are zebrafish larvae suitable for assessing the hepatotoxicity potential of drug candidates? J Appl Toxicol 2015;35(9): 1017–29. [DOI] [PubMed] [Google Scholar]
- 42.Verstraelen S, Peers B, Maho W, et al. Phenotypic and biomarker evaluation of zebrafish larvae as an alternative model to predict mammalian hepatotoxicity. J Appl Toxicol 2016;36:1194–206. [DOI] [PubMed] [Google Scholar]
- 43.Abraham VC, Towne DL, Waring JF, et al. Application of a high-content multiparameter cytotoxicity assay to prioritize compounds based on toxicity potential in humans. J Biomol Screen 2008;13(6):527–37. [DOI] [PubMed] [Google Scholar]
- 44.Bhatia SN, Ingber DE. Microfluidic organs-on-chips. Nature 2014;201:4. [DOI] [PubMed] [Google Scholar]
- 45.Wikswo J Annual thematic issue: the biology and medicine of microphysiological systems [special issue]. Exp Biol Med 2014;239(9):1061–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nam KH, Smith AS, Lone S, et al. Biomimetic 3D tissue models for advanced high-throughput drug screening. J Lab Autom 2015;20(3):201–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ouattara DA, Choi SH, Sakai Y, et al. Kinetic modelling of in vitro cell-based assays to characterize non-specific bindings and ADME processes in a static and a perfused fluidic system. Toxicol Lett 2011;205(3):310–9. [DOI] [PubMed] [Google Scholar]
- 48.Vernetti LA, Senutovitch N, Boltz R, et al. A human liver microphysiology platform for investigating physiology, drug safety, and disease models. Exp Biol Med (Maywood) 2016;241(1):101–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sarkar U, Rivera-Burgos D, Large EM, et al. Metabolite profiling and pharmacokinetic evaluation of hydrocortisone in a perfused three-dimensional human liver bioreactor. Drug Metab Dispos 2015;43(7):1091–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gough A, Vernetti L, Bergenthal L, et al. The microphysiology systems database for analyzing and modeling compound interactions with human and animal organ models. Applied In Vitro Toxicology 2016;2(2):103–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Senutovitch N, Vernetti L, Boltz R, et al. Fluorescent protein biosensors applied to microphysiological systems. Exp Biol Med (Maywood) 2015;240(6):795–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cross H, Tower B. Pre-market evaluation of hepatotoxicity in health products,Health Canada Guidance document # 12–104742–88, 2012.
- 53.Jia L, Liu X. The conduct of drug metabolism studies considered good practice (II): in vitro experiments. Curr Drug Metab 2007;8(8):822–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kanebratt KP, Andersson TB. Evaluation of HepaRG cells as an in vitro model for human drug metabolism studies. Drug Metab Dispos 2008;36(7):1444–52. [DOI] [PubMed] [Google Scholar]
- 55.Ramaiahgari SC, den Braver MW, Herpers B, et al. A 3D in vitro model of differentiated HepG2 cell spheroids with improved liver-like properties for repeated dose high-throughput toxicity studies. Arch Toxicol 2014;88(5):1083–95. [DOI] [PubMed] [Google Scholar]
- 56.Wang Z, Luo X, Anene-Nzelu C, et al. HepaRG culture in tethered spheroids as an in vitro three-dimensional model for drug safety screening. J Appl Toxicol 2015;35(8):909–17. [DOI] [PubMed] [Google Scholar]
- 57.Lubberstedt M, Müller-Vieira U, Mayer M, et al. HepaRG human hepatic cell line utility as a surrogate for primary human hepatocytes in drug metabolism assessment in vitro. J Pharmacol Toxicol Methods 2011;63(1):59–68. [DOI] [PubMed] [Google Scholar]
- 58.Greene N, Fisk L, Naven RT, et al. Developing structure-activity relationships for the prediction of hepatotoxicity. Chem Res Toxicol 2010;23(7):1215–22. [DOI] [PubMed] [Google Scholar]
- 59.Matthews EJ, Ursem CJ, Kruhlak NL, et al. Identification of structure-activity relationships for adverse effects of pharmaceuticals in humans: part B. Use of (Q) SAR systems for early detection of drug-induced hepatobiliary and urinary tract toxicities. Regul Toxicol Pharmacol 2009;54(1):23–42. [DOI] [PubMed] [Google Scholar]
- 60.Cheng A, Dixon SL. In silico models for the prediction of dose-dependent human hepatotoxicity. J Comput Aided Mol Des 2003;17(12):811–23. [DOI] [PubMed] [Google Scholar]
- 61.Liew CY, Lim YC, Yap CW. Mixed learning algorithms and features ensemble in hepatotoxicity prediction. J Comput Aided Mol Des 2011;25(9):855–71. [DOI] [PubMed] [Google Scholar]
- 62.Ekins S, Williams AJ, Xu JJ. A predictive ligand-based Bayesian model for human drug-induced liver injury. Drug Metab Dispos 2010;38(12):2302–8. [DOI] [PubMed] [Google Scholar]
- 63.Persson M, Løye AF, Mow T, et al. A high content screening assay to predict human drug-induced liver injury during drug discovery. J Pharmacol Toxicol Methods 2013;68(3):302–13. [DOI] [PubMed] [Google Scholar]
- 64.Xu JJ, Henstock PV, Dunn MC, et al. Cellular imaging predictions of clinical drug-induced liver injury. Toxicol Sci 2008;105(1):97–105. [DOI] [PubMed] [Google Scholar]
- 65.Khetani SR, Kanchagar C, Ukairo O, et al. Use of micropatterned cocultures to detect compounds that cause drug-induced liver injury in humans. Toxicol Sci 2013;132(1):107–17. [DOI] [PubMed] [Google Scholar]
- 66.Sakatis MZ, Reese MJ, Harrell AW, et al. Preclinical strategy to reduce clinical hepatotoxicity using in vitro bioactivation data for >200 compounds. Chem Res Toxicol 2012;25(10):2067–82. [DOI] [PubMed] [Google Scholar]
- 67.Basu A, Saito K, Meyer K, et al. Stellate cell apoptosis by a soluble mediator from immortalized human hepatocytes. Apoptosis 2006;11(8):1391–400. [DOI] [PubMed] [Google Scholar]
- 68.Khetani SR, Bhatia SN. Microscale culture of human liver cells for drug development. Nat Biotechnol 2008;26(1):120–6. [DOI] [PubMed] [Google Scholar]
- 69.Messner S, Agarkova I, Moritz W, et al. Multi-cell type human liver microtissues for hepatotoxicity testing. Arch Toxicol 2013;87(1):209–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schutte J, Hagmeyer B, Holzner F, et al. Artificial micro organs–a microfluidic device for dielectrophoretic assembly of liver sinusoids. Biomed Microdevices 2011;13(3):493–501. [DOI] [PubMed] [Google Scholar]
- 71.Prodanov L, Jindal R, Bale SS, et al. Long-term maintenance of a microfluidic 3D human liver sinusoid. Biotechnol Bioeng 2016;113(1):241–6. [DOI] [PMC free article] [PubMed] [Google Scholar]