

Abstract
Patient: Female, 49
Final Diagnosis: Essential thrombocythemia with CML
Symptoms: Decreased appetite • fatigue • weight loss
Medication: —
Clinical Procedure: —
Specialty: Hematology
Objective:
Unusual clinical course
Background:
Essential thrombocythemia (ET) is one of the BCR-ABL gene fusion negative chronic myeloproliferative disorders (MPDs), which also include polycythemia vera (PV), and myelofibrosis. Few clinical cases have reported the progression of ET to chronic myelogenous leukemia (CML) with the expression of the BCR-ABL gene. This report describes such a case and includes a review of other reported cases of CML co-occurring with BCR-ABL-negative chronic MPDs.
Case Report:
A 49-year-old woman was diagnosed with ET in 2007. Cytogenetic testing was negative for expression of the JAK2 or BCR-ABL1 genes. Eight years later, in January 2015, she presented with excessive fatigue, poor appetite, unintentional weight loss, a white blood cell (WBC) count of 24,700 per mL, hemoglobin of 9.9 g/dl, and a platelet count of 557,000 per mL, with blasts and basophils in the blood film. Cytogenetic analysis with fluorescent in situ hybridization (FISH) confirmed a 9: 22 chromosomal translocation (Philadelphia chromosome), and quantitative reverse transcription polymerase chain reaction (qRT-PCR) detected the expression of the BCRABL gene, confirming a diagnosis of CML. In February 2015, first-line therapy commenced with nilotinib, which was changed to imatinib after three months. During the following nine months, qRT-PCR confirmed a trend to deep molecular remission (MR5). However, she developed early myelofibrosis, and myelosuppressive therapy was resumed.
Conclusions:
This rare case highlights the importance of cytogenetic testing in cases of CMPD that transform to CML, not only to confirm the diagnosis but to plan treatment, as Philadelphia chromosome-positive and -negative cases differ in their management.
MeSH Keywords: Hydroxyurea; Leukemia, Myelogenous, Chronic, BCR-ABL Positive; Myeloproliferative Disorders; Thrombocythemia, Essential
Background
Chronic myeloproliferative disorders (MPDs) include polycythemia vera (PV), primary myelofibrosis (PMF), and essential thrombocythemia (ET) [1]. According to the current guidelines from the World Health Organization (WHO), BCR-ABL1 gene positivity is diagnostic for chronic myelogenous leukemia (CML). BCR-ABL1-negative MPNs include PV, ET, and PMF. MPDs and myeloproliferative neoplasms (MPNs) can all progress to leukemia with time [2,3].
This report is of a case of the development of CML after eight years in a patient who was initially diagnosed with ET and who was initially negative for the BCR-ABL1 fusion gene. The patient was also negative for other cytogenetic abnormalities during the ET phase. ET commonly transforms into PMF and AML [4,5], but a review of the literature showed that this case was one among only five reported cases where ET transformed into CML with the presence of the BCR-ABL1 fusion oncogene.
Case Report
A 49-year-old woman was diagnosed with essential thrombocythemia (ET) in 2007, with a platelet count of 106 per µL of blood. Following the current guidelines from the World Health Organization (WHO) and other guidelines, mutation testing was negative for Janus kinase 2 (JAK2) [6] or BCR-ABL1 genes [7]. Aspirin at 81 mg once a day was prescribed. In the absence of other high-risk features that would indicate the need for myelosuppressive therapy, she was managed with aspirin alone and with routine monitoring of blood counts every three months. A year later, in January 2015, she presented with excessive fatigue, poor appetite, and unintentional weight loss. Her hematology report showed that her white blood cell (WBC) count was 24,700 per µL, her hemoglobin was 9.9 g/dl, with a platelet count 557,000 per µL (Figure 1). A peripheral blood film confirmed the presence of blasts and basophils. Cytogenetic analysis with fluorescence in situ hybridization (FISH) confirmed a 9: 22 chromosomal translocation, indicating the presence of the Philadelphia chromosome.
Figure 1.
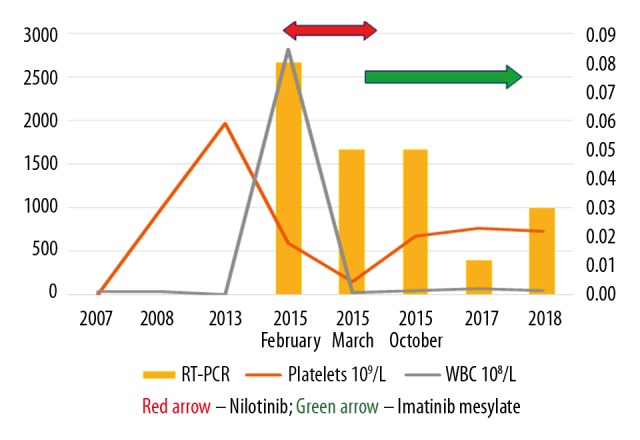
Combined line graph and bar graph representing the white blood cell (WBC) counts and platelet counts and the expression levels of the BCR-ABL gene by quantitative reverse transcription polymerase chain reaction (qRT-PCR). The line graph represents the white blood cell (WBC) counts and platelet counts plotted against the Y-axis. The bar graph, plotted against the secondary Y-axis, represents the expression levels of the BCR-ABL gene determined by quantitative reverse transcription polymerase chain reaction (qRT-PCR). A deep molecular response (DMR) with an MR of 5.0 indicated treatment-free remission (TFR).
In February 2015, nilotinib was commenced as first-line therapy for CML. Over the next three months, although she had a good response, her treatment was switched to imatinib due to myalgia and cytopenia (grade 3 adverse event), both of which improved rapidly following change of therapy, and she tolerated imatinib well, with the only adverse event being mild peripheral edema (a grade 1 adverse event). On follow-up over the next nine months and with continued imatinib treatment, the patient responded very well. Quantitative reverse transcription polymerase chain reaction (qRT-PCR) showed a downward trend to deep molecular remission with an MR of 5.0, indicated treatment-free remission (TFR). Her weight gain due to peripheral edema was managed successfully with diuretics.
Although she had a good response to therapy, recently, her platelet levels began to increase resulting in a concern for disease progression. A repeat bone marrow aspiration and biopsy were performed that showed the persistence of the ET clone and early myelofibrosis. She was recommenced on myelosuppressive therapy and continues to respond well to imatinib and cytoreductive therapy for ET.
Discussion
Chronic myeloproliferative disorders (MPDs) including polycythemia vera (PV), primary myelofibrosis (PMF), and essential thrombocythemia (ET) can progress to myeloproliferative neoplasms (MPNs). The classification and diagnosis of MPNs have been revised with the addition of cytogenetic features as determining factors. The 2008 World Health Organization (WHO) classification of MPNs indicated the importance of histologic categories and genetic characterization of myeloid neoplasms for treatment planning. MPNs usually have a normal bone marrow with terminal myeloid expansion, leading to an abnormal increase in one or more cell lines [8].
Essential thrombocytosis (ET) is one of the chronic MPDS that results in increased production of platelets, which can be dangerous, with complications including stroke and myocardial infarction. Mutations leading to platelet proliferation in ET have been identified in several prospective studies [9]. ET has by far the best prognosis among the three MPDs, with an almost normal life expectancy. The most common mutations seen in ET are in the JAK2 (V617F) and calreticulin genes (CALR) [10]. The risk of thrombosis and bleeding are the basis for risk stratification of ET, and current therapy in ET aims at lowering these risks. The presence of high-risk features, including age >60 years and a previous history of thrombosis, determine the necessity for cytoreductive therapy. In most cases, ET has been associated with a normal life expectancy.
Some new prognostic scoring systems have been proposed for ET, based on additional risk factors, such as a high leukocyte count (>11×109/L), cardiac risk factors, and JAK2 gene mutation status, but they have yet to be validated. The most important cause of morbidity and mortality in PV and ET are thrombosis and bleeding, and progression to myelofibrosis and acute leukemic transformation. In a few instances, ET can transform to myelofibrosis and such a transformation is termed ‘post-ET myelofibrosis’ [11,12]. Although coexisting BCR-ABL-positive MPNs are rare, few cases have been reported on the co-occurrence of BCR-ABL fusion at the initial stages of the disease [13–16].
This case report describes a case of ET with a negative JAK2 mutation, and BCR-ABL rearrangement, which transformation into CML over a course of eight years. Patients with ET progress to myelofibrosis [12] and rarely to AML after treatment, but the emergence of CML is rare. To our knowledge, only five previous cases of this association have been reported in the literature. [17–21] (Table 1). The patient described in the present case report continues to have ET, while CML is in remission, indicating a persistence of an ET clone which is unique and was not reported in any of the previously published cases.
Table 1.
Previously reported cases of essential thrombocythemia (ET) with transformation to chronic myelogenous leukemia (CML) with the emergence of the Philadelphia chromosome.
| No. | Age/Sex | CMPD | JAK2 V617F | Chromosomal translocation | Initial treatment for the CMPD | Transformation to CML (duration in years) | Chromosomal translocation | CML treatment | Response for CML treatment | Outcome | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 58/F | ET | ND | Negative | HU | 18 | Positive | Interferon alpha, UR-PBSCT | CCyR | Improve | [20] |
| 2 | 65/F | ET | ND | Negative | HU | 6 | Positive | Imatinib mesylate | PCyR | Improve | [17] |
| 3 | 73/M | ET | ND | Negative | HU | 12 | Positive | Imatinib mesylate | PCyR | Progress | [18] |
| 4 | 38/F | ET | ND | Negative Monosomy 6,22 | Aspirin | 13 | Positive + monosom7 | Imatinib mesylate + RIST | CCyR | Improve | [19] |
| 5 | 82/M | ET | ND | Negative | HU | 9 | Positive double Ph1 | Imatinib mesylate Nilotinib | CCyR | Progress | [21] |
| 6 | 49/F | ET | ND | Negative | Aspirin | 7 | Positive | HU + Nilotinib, after three months, Nilotinib replaced with IM | CCyR | Improve | Current case |
F – Female; M – Male; ET – essential thrombocythemia; HU – hydroxyurea; ND – not detected; IM – imatinib mesylate; CCyR – complete cytogenetic remission; RIST – reduced intensity allogeneic stem cell transplant; UR-PBSCT – unrelated donor peripheral blood stem cell transplantation; CML – chronic myelogenous leukemia. The first reported case [17] was treated initially with HU. Anagrelide was then used to treat the increasing platelet counts, while the CML was managed with imatinib. Almost all reported cases [18–21] have been treated with HU and imatinib. The time between initial diagnosis of ET and transformation to CML was six, 12, 13, nine, and 18 years, respectively. Our case had a history that covered a period of seven years.
On the emergence of CML, previously reported cases used treatment with imatinib mesylate (in four cases) and interferon-alpha (in one case). Although our patient started therapy with a second-generation tyrosine kinase inhibitor (TKI), the adverse events resulted in her being switched to imatinib. A previous study has reported that ruxolitinib, presently approved for the management of patients with intermediate or high-risk myelofibrosis, when synergistically used with imatinib, eradicated BCR-ABL cells and may help in overcoming drug resistance in CML patients, and this is currently being explored in clinical trials for the eradication of CML stem cell [22].
In addition to the reported cases of ET transforming to CML, a number of cases of transformation of PV to CML have also been reported in the literature. In the past, prior to the availability of TKIs, many of these patients received 32phosphorus or chemotherapy for the treatment of PV, which raises the possibility that irradiation and alkylating agents may have played a vital role in the transformation to CML [23,24].
There may be two possible models for the transformation of PV to CML, as the transformation could have occurred in a stem cell that was dependent or independent of a PV clone, and also, as coexisting Philadelphia chromosome (Ph)-negative clonal disorders are revealed in a subset of CML patients who have been treated with imatinib, emergence of CML is a secondary event leading to expansion of a clone with proliferative potential [24]. Previous epidemiologic data has indicated that the pathogenesis of the chronic phase of CML is due to two or more genetic events and not due to a single hit in many situations [24]. Because of these findings, Mauro et al. proposed field carcinogenesis (inherent genomic instability) in patients with coexisting Ph-positive and Ph-negative disorders [24].
A myeloproliferative disorder which is a separate entity from both Ph-positive CML and Ph-negative ET is Ph-positive ET, which is important to mention in view of the present case report. Ph-positive ET is characterized by a BCR-ABL-induced maturation defect of hematopoietic stem cells, and not clustered and enlarged mature megakaryocytes as seen in activated JAK2V617F or MPL515 mutations in Ph-negative ET. Ph-positive ET differs from CML in that it is more common in women and there is no splenomegaly, and there is an absence of features of CML in peripheral blood and bone marrow. The prognosis of BCR-ABL-positive ET is poor although hemorrhagic complications are comparatively low. Eventually, features of classic Ph-positive CML with a high risk for blast transformation and myelofibrosis may develop after several years. The BCR-ABL-positive ET may be considered a third variant of CML, known as CML-megakaryocyte predominant (CML-MP) [6], as described in the Hannover Bone Marrow Classification of MPD [6]. The use of tyrosine kinase inhibitors (TKIs) is justified in the treatment of BCR-ABL-positive ET as TKIs target cells that are BCRABL-positive [25].
Conclusions
A case of the rare association between essential thrombocythemia (ET) and the progression to chronic myelogenous leukemia (CML) has been reported with the expression of the BCR-ABL gene. The patient was successfully treated with imatinib. Only five previous cases have been identified in the literature. This rare case highlights the importance of cytogenetic testing in cases of chronic myeloproliferative disorders (MPDs) that transform to CML, not only to confirm the diagnosis but to plan treatment, as Philadelphia chromosome-positive and -negative cases differ in their management.
Footnotes
Conflict of interest
None.
References:
- 1.Tefferi A, Thiele J, Vardiman JW. The 2008 World Health Organization classification system for myeloproliferative neoplasms: Order out of chaos. Cancer. 2009;115(17):3842–47. doi: 10.1002/cncr.24440. [DOI] [PubMed] [Google Scholar]
- 2.Tefferi A, Pardanani A. Myeloproliferative neoplasms – a contemporary review. JAMA Oncol. 2015;1(1):97–105. doi: 10.1001/jamaoncol.2015.89. [DOI] [PubMed] [Google Scholar]
- 3.Lantblom AR, Bower H, Therese M, et al. Second malignancies in patients with myeloproliferative neoplasms: A population-based cohort study of 9379 patients. Leukemia. 2018;32(10):2203–10. doi: 10.1038/s41375-018-0027-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bhatt VR. Leukemic transformation in essential thrombocythemia. Future Oncol. 2014;10(16):2593–602. doi: 10.2217/fon.14.239. [DOI] [PubMed] [Google Scholar]
- 5.Tabata M, Imagawa S, Tarumoto T, et al. Essential thrombocythemia transformed to acute myelogenous leukemia with t(3;17)(p24; q12), del(5) (q13q34) after treatment with carboquone and hydroxyurea. Jpn J Clin Oncol. 2000;30(7):310–12. doi: 10.1093/jjco/hyd084. [DOI] [PubMed] [Google Scholar]
- 6.Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352(17):1779–90. doi: 10.1056/NEJMoa051113. [DOI] [PubMed] [Google Scholar]
- 7.Stoll DB, Peterson P, Exten R, et al. Clinical presentation and natural history of patients with essential thrombocythemia and the Philadelphia chromosome. Am J Hematol. 1988;27(2):77–83. doi: 10.1002/ajh.2830270202. [DOI] [PubMed] [Google Scholar]
- 8.Levine RL, Gilliland DG. Myeloproliferative disorders. Blood. 2008;112(6):2190–98. doi: 10.1182/blood-2008-03-077966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao L, Wu S, Huang E, et al. Integrated micro/messenger RNA regulatory networks in essential thrombocytosis. PLoS One. 2018;13(2):e0191932. doi: 10.1371/journal.pone.0191932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nangalia J, Grienfeld J, Green AR. Pathogenesis of myeloproliferative disorders. Annu Rev Pathol. 2016;11:101–26. doi: 10.1146/annurev-pathol-012615-044454. [DOI] [PubMed] [Google Scholar]
- 11.Finazzi G, Barbui T. Evidence and expertise in the management of polycythemia vera and essential thrombocythemia. Leukemia. 2008;22:1494–502. doi: 10.1038/leu.2008.177. [DOI] [PubMed] [Google Scholar]
- 12.Verstovsek S. Therapeutic potential of Janus-activated kinase-2 inhibitors for the management of myelofibrosis. Clin Cancer Res. 2010;16(7):1988–96. doi: 10.1158/1078-0432.CCR-09-2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cappetta M, Pérez V, Zubillaga MN, et al. Concomitant detection of BCR–ABL translocation and JAK2 V617F mutation in five patients with myeloproliferative neoplasm at diagnosis. Int J Lab Hematol. 2013;35(1):e4–e5. doi: 10.1111/ijlh.12010. [DOI] [PubMed] [Google Scholar]
- 14.Gomez-Gelvez JC, Ivan E, Betz BL, Lim MS. Concomitant BCR-ABL1 positive chronic myelogenous leukemia emerging in a patient with MPL W515L associated primary myelofibrosis. Human Pathology: Case Reports. 2016;3:6–11. [Google Scholar]
- 15.Hummel JM, Kletecka MC, Sanks JK, et al. Concomitant BCR-ABL1 translocation and JAK2(V617F) mutation in three patients with myeloproliferative neoplasms. Diagn Mol Pathol. 2012;21(3):176–83. doi: 10.1097/PDM.0b013e318246975e. [DOI] [PubMed] [Google Scholar]
- 16.Pagnano KB, Delamain MT, Magnus MM, et al. Concomitant essential thrombocythemia with JAK2 V617F mutation in a patient with chronic myeloid leukemia with major molecular response with imatinib and long-term follow-up. Oncol Lett. 2016;12(1):485–87. doi: 10.3892/ol.2016.4631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cesar JM, Cabello P, Ferro T, Navarro JL. Emergence of chronic myelogenous leukemia in a patient with primary thrombocythemia and absence of BCR/ABL rearrangement. Cancer Genet Cytogenet. 2006;167(1):74–77. doi: 10.1016/j.cancergencyto.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 18.Curtin NJ, Campbell PJ, Green AR. The Philadelphia translocation and preexisting myeloproliferative disorders. Br J Haematol. 2005;128(5):734–36. doi: 10.1111/j.1365-2141.2005.05396.x. [DOI] [PubMed] [Google Scholar]
- 19.Martin SE, DellaValla J. Untreated essential thrombocythemia evolving to biphenotypic leukemia, Philadelphia chromosome positive with monosomy 7: Response to imatinib and reduced-intensity allogeneic stem cell transplant. Leukemia. 2005;19(6):1095–96. doi: 10.1038/sj.leu.2403725. [DOI] [PubMed] [Google Scholar]
- 20.Wahlin A, Golovleva I. Emergence of Philadelphia positive chronic myeloid leukaemia during treatment with hydroxyurea for Philadelphia negative essential thrombocythaemia. Eur J Haematol. 2003;70(4):240–41. doi: 10.1034/j.1600-0609.2003.00043.x. [DOI] [PubMed] [Google Scholar]
- 21.Mizutani S, Kuroda J, Shimizu D, et al. Emergence of chronic myelogenous leukemia during treatment for essential thrombocythemia. Int J Hematol. 2010;91(3):516–21. doi: 10.1007/s12185-010-0502-3. [DOI] [PubMed] [Google Scholar]
- 22.Lurlo A, Gianelli U, Rapezzi D, et al. Imatinib and ruxolitinib association: First experience in two patients. Haematologica. 2014;99(6):76–77. doi: 10.3324/haematol.2013.102525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jantunen E, Nousiainen T. Ph-positive chronic myelogenous leukemia evolving after polycythemia vera. Am J Hematol. 1991;37(3):212. doi: 10.1002/ajh.2830370318. [DOI] [PubMed] [Google Scholar]
- 24.Mirza I, Frantz C, Clarke G, et al. Transformation of polycythemia vera to chronic myelogenous leukemia. Arch Pathol Lab Med. 2007;131(11):1719–24. doi: 10.5858/2007-131-1719-TOPVTC. [DOI] [PubMed] [Google Scholar]
- 25.Zafar L, Alam F, Parvez A, Behera S. BCR/ABL positive thrombocythemia: A diagnostic dilemma. Egypt J Intern Med. 2017;29:83–85. [Google Scholar]