

Supplemental Digital Content is available in the text.
Keywords: atherosclerosis; cholesterol; diet; fatty acids, volatile; feces; inflammation
Abstract
Rationale:
Several studies have suggested a role for the gut microbiota in inflammation and atherogenesis. A causal relation relationship between gut microbiota, inflammation, and atherosclerosis has not been explored previously.
Objective:
Here, we investigated whether a proinflammatory microbiota from Caspase1−/− (Casp1−/−) mice accelerates atherogenesis in Ldlr−/− mice.
Method and Results:
We treated female Ldlr−/− mice with antibiotics and subsequently transplanted them with fecal microbiota from Casp1−/− mice based on a cohousing approach. Autologous transplantation of fecal microbiota of Ldlr−/− mice served as control. Mice were cohoused for 8 or 13 weeks and fed chow or high-fat cholesterol–rich diet. Fecal samples were collected, and factors related to inflammation, metabolism, intestinal health, and atherosclerotic phenotypes were measured. Unweighted Unifrac distances of 16S rDNA (ribosomal DNA) sequences confirmed the introduction of the Casp1−/− and Ldlr−/− microbiota into Ldlr−/− mice (referred to as Ldlr−/−(Casp1−/−) or Ldlr−/−(Ldlr−/−) mice). Analysis of atherosclerotic lesion size in the aortic root demonstrated a significant 29% increase in plaque size in 13-week high-fat cholesterol–fed Ldlr−/−(Casp1−/−) mice compared with Ldlr−/−(Ldlr−/−) mice. We found increased numbers of circulating monocytes and neutrophils and elevated proinflammatory cytokine levels in plasma in high-fat cholesterol–fed Ldlr−/−(Casp1−/−) compared with Ldlr−/−(Ldlr−/−) mice. Neutrophil accumulation in the aortic root of Ldlr−/−(Casp1−/−) mice was enhanced compared with Ldlr−/−(Ldlr−/−) mice. 16S-rDNA-encoding sequence analysis in feces identified a significant reduction in the short-chain fatty acid–producing taxonomies Akkermansia, Christensenellaceae, Clostridium, and Odoribacter in Ldlr−/−(Casp1−/−) mice. Consistent with these findings, cumulative concentrations of the anti-inflammatory short-chain fatty acids propionate, acetate and butyrate in the cecum were significantly reduced in 13-week high-fat cholesterol–fed Ldlr−/−(Casp1−/−) compared with Ldlr−/−(Ldlr−/−) mice.
Conclusions:
Introduction of the proinflammatory Casp1−/− microbiota into Ldlr−/− mice enhances systemic inflammation and accelerates atherogenesis.
Atherosclerosis, the main underlying cause of cardiovascular disease, is traditionally considered a lipid-driven disease. However, numerous studies have shown that atherosclerosis is influenced by the innate and adaptive immune system with cytokines involved in all stages of atherogenesis.1,2 Moreover, the CANTOS-trial (Canakinumab Anti-inflammatory Thrombosis Outcomes Study) demonstrated that an antibody against IL (interleukin)-1β reduced recurrent cardiovascular events in patients with a previous myocardial infarction, indicating that inflammation enhances cardiovascular risk in humans.3
Editorial, see p 12
In This Issue, see p 2
Gut microbiota is known to be involved in the shaping of the immune system during early life. Recent studies have suggested a role for the gut microbiota in the regulation of inflammation by influencing differentiation of inflammatory cell types, cytokine production and hematopoiesis.4–6 A leaky gut and alterations in gut microbiota composition can both lead to leakage of endotoxins into the circulation that promotes systemic inflammation and to the development of obesity and related metabolic diseases.7,8 Symptomatic atherosclerosis is associated with an altered gut metagenome in the human population,9,10 and bacterial DNA has been detected in atherosclerotic plaques.11 Furthermore, a high blood concentration of the microbiota-dependent metabolite trimethyl-amine-N-oxide (TMAO) has been linked to an increased risk of atherosclerosis,12–14 indicating a pivotal role for the gut microbiota in atherogenesis. In addition, germ-free ApoE-deficient (ApoE−/−) mice showed lower circulating lipopolysaccharide levels, reduced systemic inflammation, and decreased atherogenesis compared with conventionally raised ApoE−/− mice.15 Taken together, these findings suggest a triangular relationship between the gut microbiota, host immunity, and atherogenesis; however, proof to support a proinflammatory role for the gut microbiota in atherogenesis is lacking.
To examine whether introduction of a proinflammatory gut microbiota accelerates atherogenesis, we exposed female Ldlr−/− mice to the proinflammatory gut microbiota of Casp1−/− mice,7 as previous reports have demonstrated that alterations in their microbiota sensitize mice to the development of several inflammatory diseases.7,16 The gut microbiota of Casp1−/− mice promoted atherosclerosis and increased blood leukocyte numbers, proinflammatory plasma cytokines, and neutrophil accumulation in atherosclerotic plaques, whereas plasma lipid and TMAO levels, and gut integrity were unaffected. The Casp1−/− microbiota reduced microbiota-derived anti-inflammatory short-chain fatty acids (SCFAs).
Methods
The authors declare that all data supporting the findings of this study are available in its Online Data Supplement.
Results
Casp1−/− Microbiota Successfully Introduced into Ldlr−/− Mice
To study whether a proinflammatory microbiota accelerates atherogenesis, we exposed antibiotic-treated Ldlr−/− mice to the gut microbiota of Casp1−/− mice through fecal microbiota transplantation via a cohousing approach7 (Figure 1A). Autologous transplantation of fecal microbiota from Ldlr−/− mice into antibiotic-treated Ldlr−/− mice via a cohousing approach7 served as control. Analysis of fecal microbiota composition at time of sacrifice revealed both cohousing and diet-associated changes in gut microbial ecology (Figure 1B). Unweighted UniFrac distances of 16S-rDNA (ribosomal DNA) sequences, a measure for β-diversity, demonstrated clustering between the Ldlr−/− mice receiving Casp1−/− microbiota (referred to as Ldlr−/−[Casp1−/−] mice) and the Casp1−/− donor mice (Figure 1B; Online Table II). Analogously, we observed clustering between Ldlr−/− mice receiving the autologous microbiota transplantation (referred to as Ldlr−/−(Ldlr−/−) mice) and their respective donor mice. We also observed a clear separation between mice fed chow or high-fat cholesterol (HFC) diet (Figure 1B; Online Table II), and this was consistent for all donor and recipient mice. As expected, α-diversity was not different between Ldlr−/−(Ldlr−/−) and Ldlr−/−(Casp1−/−) mice (Online Figure IB). Altogether, these data demonstrate that Casp1−/− and Ldlr−/− microbiota were successfully transferred into Ldlr−/− mice.
Figure 1.
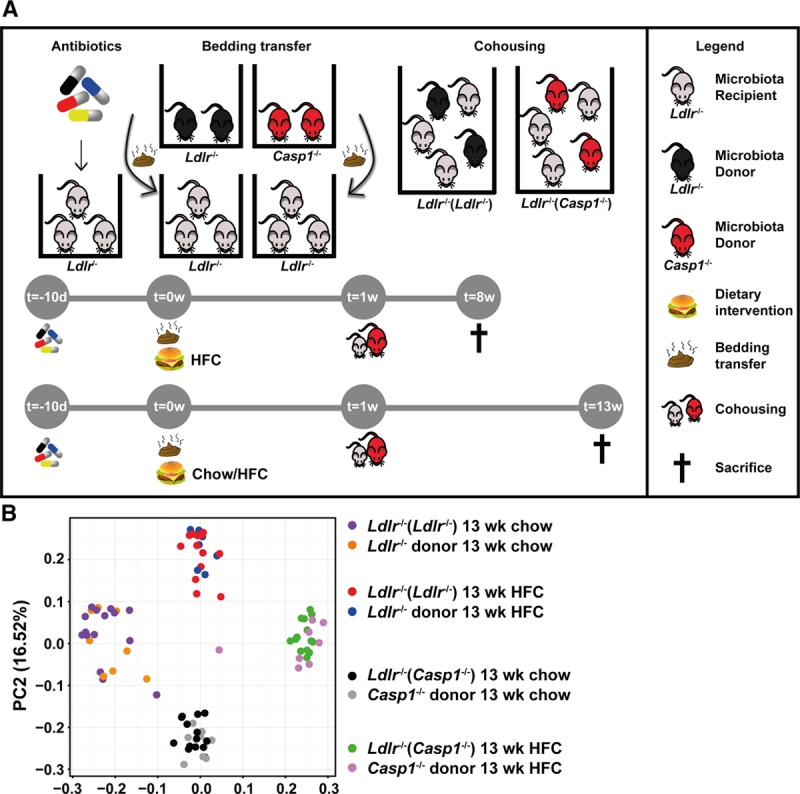
Transplantation of Casp1−/− microbiota into Ldlr−/− mice via a cohousing approach. Female Ldlr−/− mice aged 12 wk were exposed to fecal microbiota derived from Casp1−/− or Ldlr−/− mice for 8 or 13 wk while fed a chow diet or high-fat cholesterol (HFC) diet. A, Experimental setup of the cohousing approach. Female Ldlr−/− mice were orally gavaged with a cocktail of broad-spectrum antibiotics for a period of 10 d to suppress intestinal microbes. This was followed by daily transfer of used bedding material from cages housing nonantibiotic-treated Ldlr−/− (donor) or Casp1−/− (donor) mice to cages housing the antibiotic-treated Ldlr−/− mice for 1 wk. During this period the mice were kept on chow diet or switched to an HFC diet for the remainder of the study. The antibiotic-treated Ldlr−/− mice were then cohoused with nonantibiotic-treated Casp1−/− mice (referred to as Ldlr−/−(Casp1−/−) mice) or Ldlr−/− mice (autologous transplantation, referred to as Ldlr−/−(Ldlr−/−) mice) in a 3:2 ratio for a period of 8 or 13 wk. B, Principal coordinate analysis plot of Unweighted UniFrac distance on the basis of 16S-rDNA (ribosomal DNA)-encoding sequences in feces collected from chow- and HFC-fed Ldlr−/− mice exposed to Casp1−/− or Ldlr−/− microbiota for 13 wk. Chow: Ldlr−/− mice (donor), n=8; Ldlr−/−(Ldlr−/−) mice, n=15; Casp1−/− mice (donor), n=9; Ldlr−/−(Casp1−/−) mice, n=14. HFC: Ldlr−/− mice (donor), n=7; Ldlr−/−(Ldlr−/−) mice, n=13; Casp1−/− mice (donor), n=8; Ldlr−/−(Casp1−/−) mice, n=14. PC indicates principal coordinate.
Casp1−/− Dysbiosis Promotes Atherosclerosis in Ldlr−/− Mice Fed an HFC Diet
We analyzed atherosclerotic lesion size in the aortic root, and we found that Casp1−/− microbiota did not affect atherosclerotic lesion size in Ldlr−/− mice fed chow or an HFC diet for 8 weeks (Figure 2A and 2B). However, atherosclerotic lesion size was increased by 29% in Ldlr−/−(Casp1−/−) mice compared with Ldlr−/−(Ldlr−/−) mice after 13 weeks of HFC feeding (Figure 2A and 2B; P<0.05). The collagen and macrophage content in aortic root sections was not different between the mice (Online Figure IIA), indicating that lesion size but not severity was increased. In the aortic arches, gene expression of several macrophage-related and inflammatory markers was similar between HFC-fed mice with the exception of a significant increase in Il-10 expression in Ldlr−/−(Casp1−/−) mice (Online Figure IIB). Body weight, plasma triglyceride, and cholesterol levels (Online Figure IIIA–IIID) also did not differ, and no alteration was observed in plasma levels of TMAO, its TMA precursors (choline, l-carnitine, betaine, and γ-butyrobetaine; Online Figure IIIE), and TMAO-producing taxonomies (Online Figure IIIF). Altogether, these results exclude plasma lipid levels and TMAO as factors that contribute to the increased atherosclerotic lesions in Ldlr−/−(Casp1−/−) mice.
Figure 2.
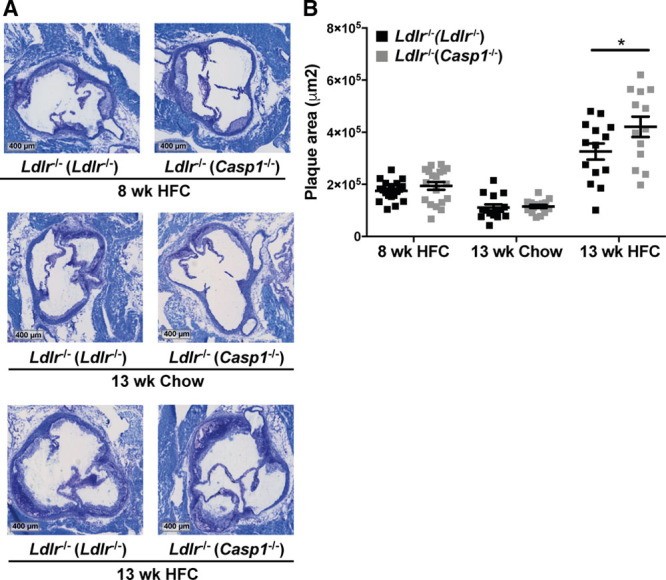
Casp1−/− microbiota promotes atherosclerosis development in Ldlr−/− mice fed high-fat cholesterol (HFC) diet. A, Representative toluidine blue stained slides of the aortic root. Scale bars, 400 μm. B, Quantification of atherosclerotic root lesion area. Chow (13 wk): Ldlr−/−(Ldlr−/−) mice, n=15; Ldlr−/−(Casp1−/−) mice, n=16. HFC (8 wk): Ldlr−/−(Ldlr−/−) mice, n=19; Ldlr−/−(Casp1−/−) mice, n=19. HFC (13 wk): Ldlr−/−(Ldlr−/−) mice, n=14; Ldlr−/−(Casp1−/−) mice, n=13. In bar graphs, data represent number of observations. For the scatter plot, the midline represents the mean±SEM. *P<0.05 by unpaired 1-tailed Student t test.
Casp1−/− Dysbiosis Promotes Inflammation
Next, we assessed whether Casp1−/− dysbiosis accelerates atherosclerosis by increasing plasma inflammatory cytokines. We found a significant elevation in the plasma levels of IL-1β, IL-2, IL-10, and IFN (interferon)-γ in Ldlr−/−(Casp1−/−) mice after 13 weeks of HFC diet (Figure 3A), whereas CXCL1 (chemokine [C-X-C motif] ligand 1), TNF (tumor necrosis factor)-α, IL-5 and IL-6 were not affected (Online Figure IVB). Flow cytometry analysis showed an increase in the number of blood Ly6Clo and Ly6Chi monocytes and neutrophils in Ldlr−/−(Casp1−/−) mice compared with Ldlr−/−(Ldlr−/−) mice accompanied by an increase in white blood cell count (Figure 3B; Online Figure IVA), with leukocyte percentages being unchanged (Online Figure IVC). We also observed increased neutrophil accumulation in atherosclerotic plaques of Ldlr−/−(Casp1−/−) mice compared with Ldlr−/−(Ldlr−/−) fed an HFC diet for 13 weeks (Figure 3C and 3D). These data suggest that Casp1−/− dysbiosis enhances IL-1β plasma levels resulting in monocytosis and neutrophilia and increased neutrophil accumulation in atherosclerotic plaques.
Figure 3.
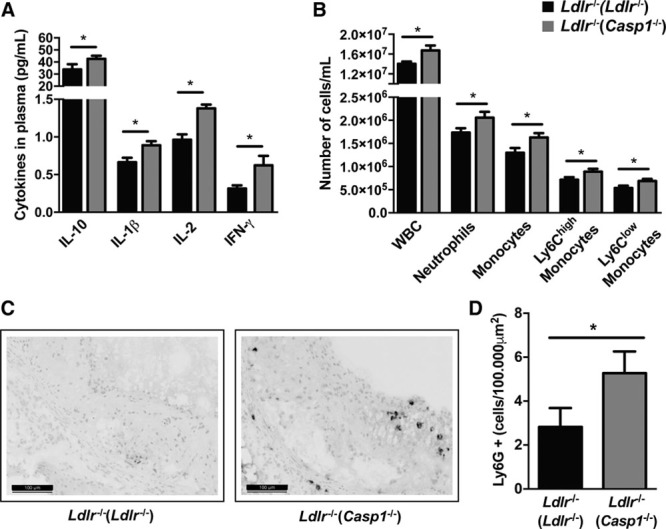
Casp1−/− dysbiosis leads to systemic inflammation. A, Plasma cytokines at time of sacrifice. n=10 per group. B, White blood cell (WBC) count and immune subsets during week 5 of cohousing. Ldlr−/−(Ldlr−/−) mice, n=18; Ldlr−/−(Casp1−/−) mice, n=17. C–D, Female Ldlr−/− mice aged 12 wk were exposed to fecal microbiota derived from Casp1−/− or Ldlr−/− mice for 13 wk while fed high-fat cholesterol (HFC) diet. C, Representative Ly6G-stained slides of the aortic root. Scale bars, 100 μm. D, Number of infiltrated neutrophils per 100.000 μm2 characterized by Ly6G-stained slides of the aortic root. Ldlr−/−(Ldlr−/−) mice, n=13; Ldlr−/−(Casp1−/−) mice, n=12. Data represent mean±SEM. *P<0.05 as determined by unpaired 1-tailed Student t test. IFN indicates interferon; and IL, interleukin.
Exposure to Casp1−/− Microbiota Does Not Impair Intestinal Barrier Function in Ldlr−/− Mice Fed an HFC Diet
A disturbance in microbiota composition may affect intestinal integrity and subsequently promote systemic inflammation7,8 To investigate the effect of Casp1−/− microbiota on intestinal barrier function, we analyzed the gut microbiota composition using the linear discriminant analysis (LDA) effect size (LEfSe) method. We identified 34 microbial taxonomies that differed in abundance between Ldlr−/−(Ldlr−/−) and Ldlr−/−(Casp1−/−) mice (Online Figure VA). Casp1−/− dysbiosis resulted in a significant expansion of the genera Bilophila, Streptococcus, and Mucispirillum (Online Figure VB–VD) under both chow- and HFC-diet conditions. Although these genera are associated with intestinal inflammation, and are known to expand under inflammatory conditions,17 we did not observe any differences in intestinal barrier function, for example, inflammation and epithelial injury (Online Figure VIA–VIC). In addition, mucus layer thickness of the colon (Online Figure VID and VIE) and Muc-2 expression (Online Figure VIF) in the colon were not altered between groups, suggesting that the integrity of the mucus layer of the colon was not different between Ldlr−/−(Ldlr−/−) and Ldlr−/−(Casp1−/−) mice. Although intestinal permeability was significantly impaired by HFC feeding, only Ldlr−/−(Casp1−/−) mice fed chow diet displayed increased permeability compared with Ldlr−/−(Ldlr−/−) mice (Online Figure VIG). These results indicate that Casp1−/− microbiota does not change the intestinal barrier function under HFC-diet conditions and, therefore, cannot explain the increase in plasma inflammatory cytokines.
Exposure to Casp1−/− Microbiota Lowers SCFA-Producing Microbial Taxonomies and Cecum Concentration of SCFAs
We observed a significant reduction in the abundance of the SCFA-producing taxonomies Akkermansia (Figure 4A), Christensenellaceae (Figure 4B), Clostridium (Figure 4C), and Odoribacter (Figure 4D) in Ldlr−/−(Casp1−/−) mice. As previous studies have shown that SCFAs reduce inflammation,18,19 we measured the concentrations of acetate, propionate, and butyrate in the cecum of the mice. Consistent with the lower abundance of SCFA-producing taxonomies, a significant reduction was observed in the cumulative levels of these SCFAs in Ldlr−/−(Casp1−/−) mice compared with Ldlr−/−(Ldlr−/−) mice (Figure 4E) and this was mainly because of lower acetate levels in the Ldlr−/−(Casp1−/−) mice. Thus, it is conceivable that a reduction in the anti-inflammatory SCFAs may have contributed to the increased levels of inflammatory plasma cytokines of mice exposed to Casp1−/− microbiota.
Figure 4.
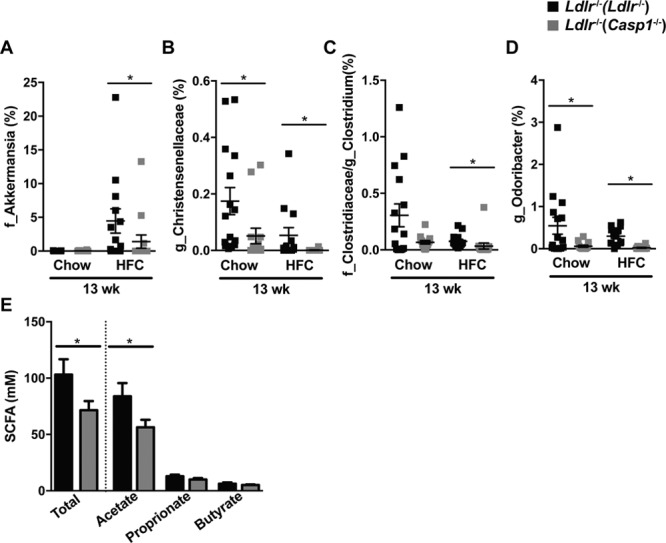
Casp1−/−-induced alterations in the gut microbiota. Female Ldlr−/− mice were exposed to fecal microbiota derived from Casp1−/− or Ldlr−/− mice by means of cohousing for 13 wk while fed chow and high-fat cholesterol (HFC) diet. A–D, Abundance of microbiota taxonomies based on LEfSe analysis of 16S-rDNA (ribosomal DNA)-encoding sequences in feces collected at time of sacrifice. A, Family Akkermansia. B, Genus Christensenellaceae. C, Genus Clostridium. D, Genus Odoribacter. E, Cecum concentration of propionate, acetate, and butyrate in HFC-fed mice. A–D, Chow: Ldlr−/−(Ldlr−/−) mice, n=15; Ldlr−/−(Casp1−/−) mice, n=14; HFC: Ldlr−/−(Ldlr−/−) mice, n=13; Ldlr−/−(Casp1−/−) mice, n=14. E, Ldlr−/−(Ldlr−/−) mice, n=8; Ldlr−/−(Casp1−/−) mice, n=9. Data represent mean±SEM. *P<0.05 as determined by Kruskal-Wallis test (A–D) and unpaired 1-tailed Student t test (E). SCFA indicates short-chain-fatty acid.
Discussion
We examined whether a proinflammatory microbiota accelerates atherogenesis in female Ldlr−/− mice, a mouse model exhibiting dyslipidemia, inflammation, and atherosclerosis, when fed a western style diet.20 We found that Casp1−/− microbiota increased atherosclerosis in the aortic root in HFC-fed Ldlr−/− mice (Figure 1A and 1B). This was accompanied by increased proinflammatory plasma cytokines (Figure 3A), increased blood leukocyte numbers, particularly monocytes and neutrophils (Figure 3B), increased neutrophil accumulation in atherosclerotic plaques (Figure 3C and 3D), and reduced levels of SCFAs in the cecum (Figure 4E). These results imply a causal relationship between microbiota composition, inflammation, and atherosclerosis.
We found that in particular the plasma levels of IFN-γ, IL-2, and IL-1β were increased in Ldlr−/− mice with Casp1−/− dysbiosis, suggesting that accelerated atherosclerosis in these mice is partially driven by these cytokines, which is supported by previous studies.21–23 Furthermore, we showed an increase in peripheral blood leukocytes, which have previously been linked to cardiovascular disease.24 Within the leukocyte population, neutrophils and monocytes are important contributors to atherogenesis.25 Increased monocytes and neutrophils in the circulation may lead to infiltration of monocytes and neutrophils into atherosclerotic plaques and further promoting plaque growth.25
We observed that exposure to Casp1−/− microbiota lowers SCFA-producing taxonomies and cumulative cecum concentrations of SCFAs. SCFAs have anti-inflammatory properties and can suppress NF-κB (nuclear factor κB) activity in immune cells,18 resulting in reduced production of proinflammatory cytokines including IFN-γ, IL-1β, and IL-2.26 Furthermore, SCFAs may act as modulators of immune homeostasis by acting as HDAC (histone deacetylase) inhibitors.19 Oral butyrate supplementation has recently been shown to attenuate the adhesion and migration of macrophages and to decrease proinflammatory cytokines in atherosclerotic plaques.27 Thus, it is tempting to speculate that the reduction in SCFAs in Ldlr−/− mice after exposure to Casp1−/− microbiota may have contributed to increased levels of proinflammatory cytokines and leukocytes in the circulation and neutrophil accumulation in the atherosclerotic plaque.
It is well recognized that microbial transplantation can be transient.13 Thus, we cannot exclude that certain effects on TMAO, although not present at time of sacrifice, may have been lost throughout the length of the study. In line with this, the possibility exists that the inflammatory effects may have been dampened over time. Future studies, therefore, should include more frequent and earlier time points to rule out these possibilities.
Whereas previous studies have shown a decreased abundance of Akkermansia muciniphila on high-fat diet feeding, our data show on opposing effect on A muciniphila after HFC feeding. A similar effect on high-fat and high-carbohydrate diet feeding in mice was recently shown28 and warrants further investigation. Nevertheless, promising results have been obtained with the administration of A muciniphila resulting in protection against atherogenesis in ApoE−/− mice by strengthening the gut barrier and preventing metabolic endotoxemia-induced inflammation.29 Likewise, metformin’s reported beneficial effects on atherosclerosis in humans with type I and II diabetes mellitus and nondiabetic dysglycaemia may be related to its ability to enhance the growth of A muciniphila and promote SCFA production.30 Together with our findings, this indicates that manipulation of the gut microbiota composition is an interesting treatment strategy to protect against inflammation and atherosclerosis and reduce cardiovascular disease risk.
Acknowledgments
We dedicate this article to M.H. Hofker. His ideas laid the groundwork for this article. We thank Mihai Netea (Radboud University Nijmegen Medical Center, Department of General Internal Medicine, Nijmegen, the Netherlands) for the generous gift of the Casp1−/− mice and Theo Boer (University of Groningen, University Medical Center Groningen, Department of Laboratory Medicine, Groningen, the Netherlands) for technical assistance. Editorial services were provided by Kate Mc Intyre (University of Groningen, University Medical Center Groningen, Department of Genetics, Groningen, the Netherlands). Additional Information: Coauthor M.H. Hofker died September 12, 2016.
Sources of Funding
E. Brandsma, J. Fu, F. Kuipers, M. Hofker, and D.P.Y. Koonen are supported by a grant from CardioVasculair Onderzoek Nederland (CVON2012-03). Additional financial support was provided by the Jan Kornelis de Cock Foundation to E. Brandsma and the Graduate School for Drug Exploration, University of Groningen (E. Brandsma, M. Ríos Morales). J. Fu is supported by the Netherlands Organization for Scientific Research VIDI-grant (NWO-VIDI 864.13.013). M. Westerterp is supported by Netherlands Organization for Scientific Research VIDI-grant 917.15.350 and a Rosalind Franklin Fellowship from the University Medical Center Groningen.
Disclosures
None.
Supplementary Material
In September 2018, the average time from submission to first decision for all original research papers submitted to Circulation Research was 14.06 days.
These authors contributed equally to this article.
The online-only Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/CIRCRESAHA.118.313234.
Nonstandard Abbreviations and Acronyms
- HFC
- high-fat cholesterol-rich
- IL
- interleukin
- NF-κB
- nuclear factor κB
- TMAO
- trimethylamine-N-oxide
- TNF
- tumor necrosis factor
- rDNA
- ribosomal DNA
- SCFAs
- short-chain fatty acids
Novelty and Significance
What Is Known?
Atherosclerosis, the main underlying cause of cardiovascular disease, is influenced by both—the innate and adaptive immune systems.
Gut microbiota shape the immune system during early life and play a role in regulating inflammation by influencing the differentiation of inflammatory cell types, the production of cytokines, and hematopoiesis.
Inflammation and atherosclerosis are linked to changes in gut microbiota composition; however, there is little evidence to support a proinflammatory role of gut microbiota in atherosclerosis.
What New Information Does This Article Contribute?
The presence of a proinflammatory microbiota derived from Caspase1−/− (Casp1−/−) mice is sufficient to promote inflammation and atherosclerosis in antibiotic-treated Ldlr−/− mice, a mouse model with a human-like lipoprotein profile.
The gut microbiota of Casp1−/− mice increases inflammation in antibiotic-treated Ldlr−/− mice, reflected by increased blood leukocyte numbers, particularly monocytes and neutrophils, proinflammatory plasma cytokines, and neutrophil accumulation in atherosclerotic plaques.
The gut microbiota of Casp1−/− mice reduces the microbiota-derived anti-inflammatory short-chain fatty acids in antibiotic-treated Ldlr−/− mice, whereas plasma lipid, trimethylamine-N-oxide levels, and gut integrity are unaffected.
Several human studies have provided evidence that links the gut microbiota to cardiovascular disease. Nevertheless, the evidence supporting a causal role of the gut microbiota in cardiovascular disease is limited to the understanding of the importance of trimethylamine-N-oxide in atherogenesis. Recent findings suggest a pivotal role of the gut microbiota in regulating inflammation. Here, we provide a novel, alternative mechanism by which the gut microbiota may contribute to atherogenesis, independent of plasma lipids and trimethylamine-N-oxide levels. We show that introduction of a proinflammatory gut microbiota into a mouse model with a human-like lipoprotein profile increases systemic inflammation and accelerates atherogenesis. This was associated with a reduction in microbiota-derived anti-inflammatory short-chain fatty acids, implying a causal relationship between microbiota composition, inflammation, and atherosclerosis. Collectively, these findings indicate that manipulation of the gut microbiota composition may be potentially effective treatment strategy to protect against inflammation and atherosclerosis and thereby reduce the risk of cardiovascular disease.
References
- 1.Moss JW, Ramji DP. Cytokines: roles in atherosclerosis disease progression and potential therapeutic targets. Future Med Chem. 2016;8:1317–1330. doi: 10.4155/fmc-2016-0072. doi: 10.4155/fmc-2016-0072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473:317–325. doi: 10.1038/nature10146. doi: 10.1038/nature10146. [DOI] [PubMed] [Google Scholar]
- 3.Ridker PM, Everett BM, Thuren T, et al. CANTOS Trial Group. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377:1119–1131. doi: 10.1056/NEJMoa1707914. doi: 10.1056/NEJMoa1707914. [DOI] [PubMed] [Google Scholar]
- 4.Schirmer M, Smeekens SP, Vlamakis H, et al. Linking the human gut microbiome to inflammatory cytokine production capacity. Cell. 2016;167:1897. doi: 10.1016/j.cell.2016.11.046. doi: 10.1016/j.cell.2016.11.046. [DOI] [PubMed] [Google Scholar]
- 5.Geva-Zatorsky N, Sefik E, Kua L, Pasman L, Tan TG, Ortiz-Lopez A, Yanortsang TB, Yang L, Jupp R, Mathis D, Benoist C, Kasper DL. Mining the human gut microbiota for immunomodulatory organisms. Cell. 2017;168:928.e–943.e11. doi: 10.1016/j.cell.2017.01.022. doi: 10.1016/j.cell.2017.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Khosravi A, Yáñez A, Price JG, Chow A, Merad M, Goodridge HS, Mazmanian SK. Gut microbiota promote hematopoiesis to control bacterial infection. Cell Host Microbe. 2014;15:374–381. doi: 10.1016/j.chom.2014.02.006. doi: 10.1016/j.chom.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, Thaiss CA, Kau AL, Eisenbarth SC, Jurczak MJ, Camporez JP, Shulman GI, Gordon JI, Hoffman HM, Flavell RA. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 2012;482:179–185. doi: 10.1038/nature10809. doi: 10.1038/nature10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cani PD, Osto M, Geurts L, Everard A. Involvement of gut microbiota in the development of low-grade inflammation and type 2 diabetes associated with obesity. Gut Microbes. 2012;3:279–288. doi: 10.4161/gmic.19625. doi: 10.4161/gmic.19625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karlsson FH, Fåk F, Nookaew I, Tremaroli V, Fagerberg B, Petranovic D, Bäckhed F, Nielsen J. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat Commun. 2012;3:1245. doi: 10.1038/ncomms2266. doi: 10.1038/ncomms2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jie Z, Xia H, Zhong SL, et al. The gut microbiome in atherosclerotic cardiovascular disease. Nat Commun. 2017;8:845. doi: 10.1038/s41467-017-00900-1. doi: 10.1038/s41467-017-00900-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koren O, Spor A, Felin J, Fåk F, Stombaugh J, Tremaroli V, Behre CJ, Knight R, Fagerberg B, Ley RE, Bäckhed F. Human oral, gut, and plaque microbiota in patients with atherosclerosis. Proc Natl Acad Sci USA. 2011;108(suppl 1):4592–4598. doi: 10.1073/pnas.1011383107. doi: 10.1073/pnas.1011383107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koeth RA, Wang Z, Levison BS, et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med. 2013;19:576–585. doi: 10.1038/nm.3145. doi: 10.1038/nm.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gregory JC, Buffa JA, Org E, Wang Z, Levison BS, Zhu W, Wagner MA, Bennett BJ, Li L, DiDonato JA, Lusis AJ, Hazen SL. Transmission of atherosclerosis susceptibility with gut microbial transplantation. J Biol Chem. 2015;290:5647–5660. doi: 10.1074/jbc.M114.618249. doi: 10.1074/jbc.M114.618249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schiattarella GG, Sannino A, Toscano E, Giugliano G, Gargiulo G, Franzone A, Trimarco B, Esposito G, Perrino C. Gut microbe-generated metabolite trimethylamine-N-oxide as cardiovascular risk biomarker: a systematic review and dose-response meta-analysis. Eur Heart J. 2017;38:2948–2956. doi: 10.1093/eurheartj/ehx342. doi: 10.1093/eurheartj/ehx342. [DOI] [PubMed] [Google Scholar]
- 15.Kasahara K, Tanoue T, Yamashita T, Yodoi K, Matsumoto T, Emoto T, Mizoguchi T, Hayashi T, Kitano N, Sasaki N, Atarashi K, Honda K, Hirata KI. Commensal bacteria at the crossroad between cholesterol homeostasis and chronic inflammation in atherosclerosis. J Lipid Res. 2017;58:519–528. doi: 10.1194/jlr.M072165. doi: 10.1194/jlr.M072165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu B, Elinav E, Huber S, Booth CJ, Strowig T, Jin C, Eisenbarth SC, Flavell RA. Inflammation-induced tumorigenesis in the colon is regulated by caspase-1 and NLRC4. Proc Natl Acad Sci USA. 2010;107:21635–21640. doi: 10.1073/pnas.1016814108. doi: 10.1073/pnas.1016814108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Loy A, Pfann C, Steinberger M, Hanson B, Herp S, Brugiroux S, Gomes Neto JC, Boekschoten M V, Schwab C, Urich T, Ramer-Tait AE, Rattei T, Stecher B, Berry D. Lifestyle and horizontal gene transfer-mediated evolution of Mucispirillum schaedleri, a core member of the murine gut microbiota. mSystems. 2017;2:e00171–e00216. doi: 10.1128/mSystems.00171-16. doi: 10.1128/mSystems.00171-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tedelind S, Westberg F, Kjerrulf M, Vidal A. Anti-inflammatory properties of the short-chain fatty acids acetate and propionate : a study with relevance to inflammatory bowel disease. 2007;13:2826–2832. doi: 10.3748/wjg.v13.i20.2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koh A, De Vadder F, Kovatcheva-Datchary P, Bäckhed F. From dietary fiber to host physiology: short-chain fatty acids as key bacterial metabolites. Cell. 2016;165:1332–1345. doi: 10.1016/j.cell.2016.05.041. doi: 10.1016/j.cell.2016.05.041. [DOI] [PubMed] [Google Scholar]
- 20.Subramanian S, Han CY, Chiba T, McMillen TS, Wang SA, Haw A, III, Kirk EA, O’Brien KD, Chait A. Dietary cholesterol worsens adipose tissue macrophage accumulation and atherosclerosis in obese LDL receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2008;28:685–691. doi: 10.1161/ATVBAHA.107.157685. doi: 10.1161/ATVBAHA.107.157685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gupta S, Pablo AM, Jiang Xc, Wang N, Tall AR, Schindler C. IFN-gamma potentiates atherosclerosis in ApoE knock-out mice. J Clin Invest. 1997;99:2752–2761. doi: 10.1172/JCI119465. doi: 10.1172/JCI119465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Upadhya S, Mooteri S, Peckham N, Pai RG. Atherogenic effect of interleukin-2 and antiatherogenic effect of interleukin-2 antibody in apo-E-deficient mice. Angiology. 2004;55:289–294. doi: 10.1177/000331970405500308. doi: 10.1177/000331970405500308. [DOI] [PubMed] [Google Scholar]
- 23.Swirski FK, Nahrendorf M. Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science. 2013;339:161–166. doi: 10.1126/science.1230719. doi: 10.1126/science.1230719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Avanzas P, Arroyo-Espliguero R, Cosín-Sales J, Quiles J, Zouridakis E, Kaski JC. Multiple complex stenoses, high neutrophil count and C-reactive protein levels in patients with chronic stable angina. Atherosclerosis. 2004;175:151–157. doi: 10.1016/j.atherosclerosis.2004.03.013. doi: 10.1016/j.atherosclerosis.2004.03.013. [DOI] [PubMed] [Google Scholar]
- 25.Weber C, Zernecke A, Libby P. The multifaceted contributions of leukocyte subsets to atherosclerosis: lessons from mouse models. Nat Rev Immunol. 2008;8:802–815. doi: 10.1038/nri2415. doi: 10.1038/nri2415. [DOI] [PubMed] [Google Scholar]
- 26.Klampfer L, Huang J, Sasazuki T, Shirasawa S, Augenlicht L. Inhibition of interferon gamma signaling by the short chain fatty acid butyrate. Mol Cancer Res. 2003;1:855–862. [PubMed] [Google Scholar]
- 27.Aguilar EC, Leonel AJ, Teixeira LG, Silva AR, Silva JF, Pelaez JM, Capettini LS, Lemos VS, Santos RA, Alvarez-Leite JI. Butyrate impairs atherogenesis by reducing plaque inflammation and vulnerability and decreasing NFκB activation. Nutr Metab Cardiovasc Dis. 2014;24:606–613. doi: 10.1016/j.numecd.2014.01.002. doi: 10.1016/j.numecd.2014.01.002. [DOI] [PubMed] [Google Scholar]
- 28.Carmody RN, Gerber GK, Luevano JM, Jr, Gatti DM, Somes L, Svenson KL, Turnbaugh PJ. Diet dominates host genotype in shaping the murine gut microbiota. Cell Host Microbe. 2015;17:72–84. doi: 10.1016/j.chom.2014.11.010. doi: 10.1016/j.chom.2014.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li J, Lin S, Vanhoutte PM, Woo CW, Xu A. Akkermansia muciniphila protects against atherosclerosis by preventing metabolic endotoxemia-induced inflammation in apoe−/− mice. Circulation. 2016;133:2434–2446. doi: 10.1161/CIRCULATIONAHA.115.019645. doi: 10.1161/CIRCULATIONAHA.115.019645. [DOI] [PubMed] [Google Scholar]
- 30.Rodriguez J, Hiel S, Delzenne NM. Metformin: old friend, new ways of action-implication of the gut microbiome? Curr Opin Clin Nutr Metab Care. 2018;21:294–301. doi: 10.1097/MCO.0000000000000468. doi: 10.1097/MCO.0000000000000468. [DOI] [PubMed] [Google Scholar]