

Abstract
Background
Optimum flowering time is a key agronomic trait in Brassica napus. To investigate the genetic architecture and genetic regulation of flowering time in this important crop, we conducted quantitative trait loci (QTL) analysis of flowering time in a recombinant inbred line (RIL) population, including lines with extreme differences in flowering time, in six environments, along with RNA-Seq analysis.
Results
We detected 27 QTLs distributed on eight chromosomes among six environments, including one major QTL on chromosome C02 that explained 11–25% of the phenotypic variation and was stably detected in all six environments. RNA-Seq analysis revealed 105 flowering time-related differentially expressed genes (DEGs) that play roles in the circadian clock/photoperiod, autonomous pathway, and hormone and vernalization pathways. We focused on DEGs related to the regulation of flowering time, especially DEGs in QTL regions.
Conclusions
We identified 45 flowering time-related genes in these QTL regions, eight of which are DEGs, including key flowering time genes PSEUDO RESPONSE REGULATOR 7 (PRR7) and FY (located in a major QTL region on C02). These findings provide insights into the genetic architecture of flowering time in B. napus.
Electronic supplementary material
The online version of this article (10.1186/s12864-018-5356-8) contains supplementary material, which is available to authorized users.
Keywords: Flowering time, QTL, Gene expression, RNA-Seq, Brassica napus
Background
In flowering plants, the transition from the vegetative stage to the reproductive stage helps to ensure reproductive success, including successful seed production [1]. This trait is especially important in crop plants, as it can determine crop cultivation ranges and ensure high productivity. Thus, flowering time is a vital trait that is a target of selection during crop breeding. Flowering time is sensitive to various environmental signals (such as day length and temperature) and endogenous signals (e.g., developmental stage and age) [2, 3]. To date, much is known about candidate genes controlling flowering time in Arabidopsis thaliana. More than 300 flowering time genes have been identified, and several key regulators that function in pathways that control flowering time have been detected [4, 5]. Six major pathways control flowering time in Arabidopsis: vernalization, the photoperiod/circadian clock, and the ambient temperature, gibberellin, autonomous, and endogenous pathways [1, 4, 6–8]. In Arabidopsis, FLOWERING LOCUS C (FLC) and FRIGIDA (FRI) are key genes in the vernalization response, whereas CONSTANS (CO) functions in the response to photoperiod [9, 10]. FLOWERING LOCUS T (FT) encodes a mobile signal long described as “florigen”, which functions as a central floral integrator in the control of flowering [11].
Oilseed rape (Brassica napus L., also known as rapeseed or canola) is one of the most important oil crops worldwide. Many important and complex agronomic traits such as yield [12], plant height [13], oil content [14], seed weight [15], and flowering time [16] have been mapped in this crop. Flowering time in rapeseed not only has a crucial impact on yield, but it also influences the sowing time of other rotation crops [16]. Quantitative trait locus (QTL) analysis and genome-wide associated mapping (GWAS) have been used to identify candidate flowering time genes in oilseed rape. Many QTLs related to flowering time have been identified in this crop. For example, one major QTL was identified that explains 50% of the total phenotypic variation for flowering time in B. napus. This QTL is related to VFN2, a major vernalization-responsive flowering time gene in Arabidopsis [17]. Raman et al. (2013) performed QTL analysis for flowering time using a doubled haploid (DH) population [18]. Liu et al. (2016) identified 22 QTLs (including four major QTLs) for flowering time in B. napus using a DH population [19]. GWAS was also recently used to screen for candidate flowering time genes in B. napus. Xu et al. (2016) identified 41 SNPs associated with flowering time using GWAS of 523 B. napus cultivars [20]. Raman et al. (2016) obtained 69 SNP markers associated with flowering time using GWAS approaches and detected several candidate flowering time genes, such as FT, FRUITFUL, FLC, CO, FRI, and PHYTOCHROME B, within 20 Kb regions [21]. QTLs or genes have also been identified in other Brassica crops, such as B. rapa [22, 23] and B. oleracea [24].
Although much effort has focused on investigating flowering time, stable QTLs for this trait have not yet been identified, and global transcriptome analysis of different rapeseed genotypes has not yet been performed. Therefore, in this study, we performed joint QTL mapping and RNA-Seq analysis to uncover the genetic architecture of flowering time in B. napus.
Materials and methods
Plant materials and growth conditions
A recombinant inbred line (RIL) population consisting of 172 lines was constructed from a cross between GH06 (female parent, late flowering, semi-winter) and P174 (male parent, early flowering, semi-winter). The GH06 × P174 RIL population was previously used to map seed fiber content in oilseed rape [25]. The population was obtained from Chongqing Engineering Research Center for Rapeseed,Southwest University.
The flowering time trait was evaluated in six environments (the temperature data in each environment was shown in Additional file 1: Table S1), including Giessen (E8.76/N50.56), Germany in 2009 (09Gi) and Beibei (E106.26/N29.82), Chongqing, China in 2012–2016 (12Cq, 13Cq, 14Cq, 15Cq, and 16Cq, respectively). In Giessen, the seeds were sown directly in the spring of 2009. In Chongqing, seeds from the RILs and the parental lines were sown in nursery beds on September 18, 2012, 2013, 2014, 2015, and 2016 and transplanted to the field one month later. Each line of the RIL population was grown in a 4.5 m2 (1.5 × 3) plot with 80–90 plants (in Giessen environment) or 50–60 plants (in Chongqing environment). Flowering time data were recorded for each line from the sowing day to the day when 50% of the plants showed the first blooming floret.
Genetic and QTL mapping
A high-density SNP genetic map was constructed using the Brassica 60 K BeadChip Array [25]. A genetic map containing 2795 SNP markers with a mean distance of 0.66 cM between adjacent SNP markers was used for QTL mapping.
Windows QTL Cartographer version 2.5 with default settings was used to detect QTLs for flowering time via the composite interval mapping method [26]. The logarithm of the odds (LOD) threshold for detecting a significant QTL was established by permutation analysis with 1000 permutations. The linkage map and QTL position was generated using MapChart software [27].
To screen candidate genes in QTL regions, following procedures were conducted: (1) 1-LOD likelihood intervals surrounding the peak of the QTL likelihood plot were regarded as the QTL interval; (2) Ten SNPs located within and at each end of each interval were considered, selecting the SNP with either the largest or smallest physical distance at each end to maximize the physical size of the region, based on previously published physical locations of each SNP [28]; (3), Genes located in the intervals were selected as candidate genes based on published annotation of the B. napus genome [29].
RNA isolation and transcriptome sequencing
Five early-flowering lines (marked “E”) and five late-flowering lines (marked “L”) were selected from the RIL population based on the flowering time in six environments. To detect candidate genes involved in regulating the days to flowering, shoot tissues (S) and leaves (L) were collected from both E and L lines at 10 o’clock am in the vegetative stage at 20 weeks after germination in 16Cq environment. For both the E and L lines, shoot tissues (ES and LS) or leaves (EL and LL) from five lines were pooled, immediately frozen in liquid nitrogen, and stored at − 80 °C until use.
Total RNA was isolated from each sample using a Plant RNA Mini Kit (Tiangen, Inc., China) according to the manufacturer’s protocol. Four cDNA libraries were constructed and RNA-Seq was performed on an Illumina HiSeq 2500 platform by Novogene Bioinformatics Technology Co. Ltd. (Beijing, China) according to the manufacturer’s instructions. Moreover, these paired end sequencing reads were immediately uploaded to NCBI with accession number SRP108958.
RNA sequencing data analysis
High-quality reads were obtained after the adapter sequences and low quality sequences were filtered out from the raw data using the NGS QC toolkit [30]. The clean reads were mapped to the B. napus genome (http://www.genoscope.cns.fr/brassicanapus/data/) using TopHat v2.0.11. Unique reads were further analyzed and gene expression levels were calculated using Cufflinks v2.2.0 [31]. Gene expression levels were estimated by the FPKM (fragments per kilobase of exon per million mapped fragments) method, and DEGs were identified using the criteria FDR ≤ 0.01 and |log2 (FPKM early/FPKM late)| ≥ 1.
To further investigate the potential functions of the DEGs, KEGG enrichment analysis was performed using the KOBAS2.0 website (http://kobas.cbi.pku.edu.cn/home.do).
Identification of B. napus homologs of flowering time-related genes
To discover flowering time genes in B. napus, 306 flowering-time related (FTR) genes in A. thaliana were downloaded from the Flowering Interactive Database (http://www.phytosystems.ulg.ac.be/florid/). Homologs of these genes in B. napus were identified by BLASTN analysis against the B. napus reference genome. Top hits with E-values ≤1E− 20 and identity ≥80% were used to screen for the corresponding homologous genes.
qRT-PCR confirmation of RNA-Seq data
To confirm the RNA-Seq data and the DEGs identified in the early- and late-flowering lines, 47 genes were subjected to qRT-PCR analysis. One microgram of total RNA per sample (the same samples used for RNA-seq) was used to synthesize cDNA using the M-MuLV RT kit (Takara Biotechnology, Japan) according to the manufacturer’s instructions (TransGen, China). The qPCR was performed as described previously [32]. BnACTIN7 and BnUBC21 were used as internal controls, and the 2-ΔΔCt method was used to evaluate relative gene expression levels. The gene-specific primers are shown in Additional file 2: Table S2. Each PCR was performed with three technical replicates.
Results
Analysis of flowering time in six environments
We analyzed flowering time traits in a population of 172 RILs. The flowering time values of the two parental lines, as well as the mean, maximum, and minimum values of the RIL population for flowering time in six environments, were summarized in Table 1. The transgressive segregation of flowering time traits in all six environments was shown in Fig. 1. We detected a great difference between the two parental lines and within the RIL populations. The correlation coefficients of flowering time among the six environments are shown in Additional file 3: Table S3. Our results indicate that flowering time is positively and significantly correlated among the six environments (r2 = 0.255–0.766, P < 0.01). The correlation between the German and the Chinese locations (r2 < 0.4) are lower than among the Chinese environments (r2 > 0.6) because of the great difference between German environment and Chinese environment.
Table 1.
Phenotypic variation in flowering time in the RILs and their parents
| Environment | Parents | RIL population | ||||
|---|---|---|---|---|---|---|
| GH06 | P174 | Minimum | Maximum | Mean | Std. Deviation | |
| Gi09FT | 88 | 82 | 78 | 92 | 83.76 | 2.555 |
| Cq12FT | 175 | 168 | 165 | 177 | 170.13 | 2.775 |
| Cq13FT | 155 | 145 | 142 | 158 | 149.81 | 4.173 |
| Cq14FT | 165 | 150 | 142 | 167 | 156.01 | 6.014 |
| Cq15FT | 161 | 142 | 134 | 178 | 153.80 | 7.968 |
| Cq16FT | 146 | 134 | 130 | 158 | 142.74 | 6.299 |
09Gi Germany in 2009, 12Cq Chongqing in 2012, 13Cq Chongqing in 2013, 14Cq Chongqing in 2014, 15Cq Chongqing in 2015, 16Cq Chongqing in 2016
Fig. 1.
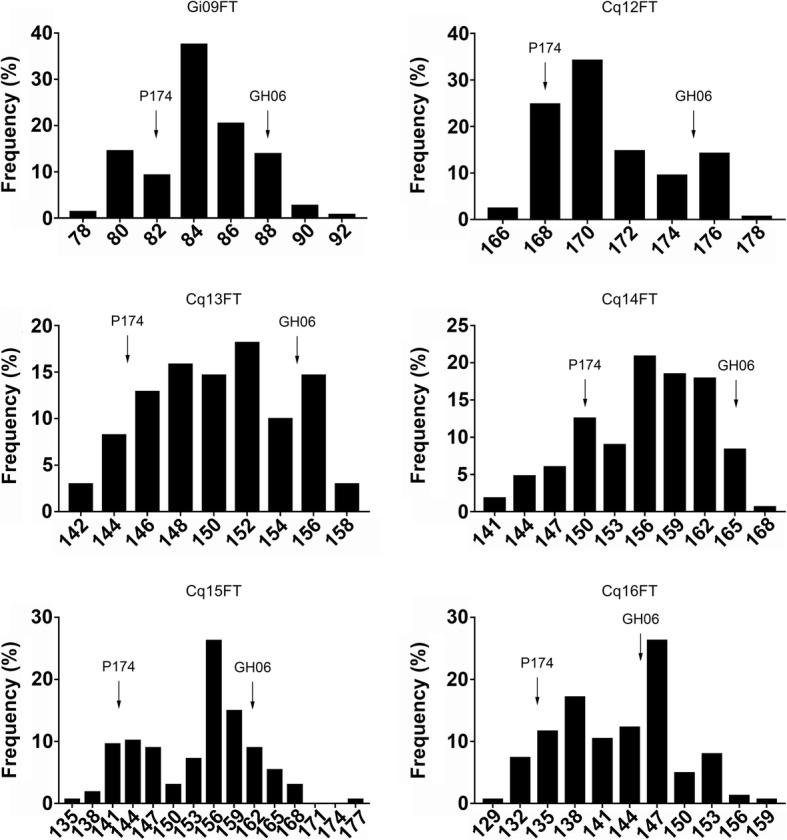
Frequency distribution of the flowering time trait in RILs grown in six different environments. 09Gi: Germany in 2009; 12Cq: Chongqing in 2012; 13Cq: Chongqing in 2013; 14Cq: Chongqing in 2014; 15Cq: Chongqing in 2015; 16Cq: Chongqing in 2016; P174: male parent; GH06: female parent
Mapping of QTLs for flowering time in six environments
We detected 27 QTLs distributed on eight chromosomes in the six environments, with 5.2–25.1% phenotypic variation (PV) and additive effects ranging from − 2.83 to 3.64 (Table 3). Among these QTLs, 1–8 QTLs were detected on eight chromosomes and 3–6 QTLs were identified in each environment (Figs. 2 and 3, Table 2). The values of the additive effects of QTLs on A05, A06, A07, and C04 were negative, whereas those of QTLs on A02, A08, A10, and C02 were positive, indicating that the genetic background of the female parent causes later flowering and that of the male parent causes earlier flowering. By aligning SNP markers in these regions, we identified the physical locations of these QTL regions in the B. napus genome, leading to the detection of 3436 genes (Table 3, Additional file 4: Table S4).
Table 3.
Number of FTR genes in QTL regions
| Name | QTL regions | Gd. (cM) | Pl. (bp) | No. of genes | No. of FTR genes |
|---|---|---|---|---|---|
| QTL-A02 | SNP3549A02-SNP3440A02 | 0–3.2 | A02: 19650440–20,741,204 | 148 | 0 |
| QTL-A06 | SNP13822A06-SNP13882A06 | 92.398–96.47 | A06: 7212328–21,686,640 | 1909 | 33 |
| QTL-A07 | SNP16177A07-SNP16216A07 | 85.8–88.7 | A07: 19645576–19,981,239 | 55 | 0 |
| QTL-A08 | SNP18612A08-SNP18586A08 | 0.408–4.458 | A08: 18395488–18,767,730 | 89 | 2 |
| QTL-A10 | SNP22584A10-SNP22402A10 | 23.7–31.736 | A10: 13337887–14,487,871 | 255 | 3 |
| QTL-C02 | SNP35881C02-SNP25258C02 | 44.191–50.702 | CO2: 15599–5,545,697 | 980 | 7 |
In total, 3436 genes were detected in QTL regions, including 45 FTR genes. No FTR genes were detected in QTL regions on chromosome A02 or A07
Fig. 2.

Graphs of QTLs for flowering time in an RIL population throughout the genome in plants grown in six different environments. 09Gi: Germany in 2009; 12Cq: Chongqing in 2012; 13Cq: Chongqing in 2013; 14Cq: Chongqing in 2014; 15Cq: Chongqing in 2015; 16Cq: Chongqing in 2016
Fig. 3.

Locations of significant QTLs for flowering time on a high-density SNP map. For simplicity, only markers in QTL confidence intervals, along with the two terminal markers at each end of each QTL-containing chromosome, are shown. Full map data are provided in Liu et al. (2013). 09Gi: Germany in 2009; 12Cq: Chongqing in 2012; 13Cq: Chongqing in 2013; 14Cq: Chongqing in 2014; 15Cq: Chongqing in 2015; 16Cq: Chongqing in 2016
Table 2.
Significant QTLs associated with flowering time in the RIL population
| QTL names | Trait | Chromosome | Position (cM) | Additive | LOD | QTL region (cM) | R2 |
|---|---|---|---|---|---|---|---|
| qFTA02 | Cq14FT | A02 | 2.01 | 1.55 | 3.28 | 0–2.8 | 0.05 |
| Cq15FT | A02 | 2.61 | 1.84 | 3.53 | 2.4–2.9 | 0.06 | |
| qFTA05 | Gi09FT | A05 | 93.01 | −0.59 | 4.02 | 92.7–93.8 | 0.07 |
| qFTA06–1 | Cq12FT | A06 | 96.31 | −0.95 | 6.10 | 94.5–96.5 | 0.12 |
| Cq15FT | A06 | 97.51 | −2.83 | 7.63 | 96.5–101.7 | 0.10 | |
| Cq16FT | A06 | 96.31 | −1.85 | 4.11 | 95.5–96.5 | 0.07 | |
| qFTA06–2 | Cq12FT | A06 | 105.81 | −0.76 | 4.16 | 104.4–106.4 | 0.06 |
| qFTA06–3 | Cq15FT | A06 | 109.21 | −2.34 | 6.87 | 108.7–111.5 | 0.07 |
| qFTA07 | Gi09FT | A07 | 87.31 | −0.78 | 4.67 | 85.8–88.7 | 0.09 |
| qFTA08–1 | Cq13FT | A08 | 1.31 | 1.16 | 4.33 | 0.4–6.1 | 0.07 |
| Cq15FT | A08 | 1.31 | 2.99 | 8.48 | 0.6–4.4 | 0.14 | |
| Cq14FT | A08 | 2.71 | 1.86 | 4.30 | 2–7.2 | 0.09 | |
| Cq16FT | A08 | 2.71 | 1.68 | 3.50 | 1.9–7.2 | 0.06 | |
| Cq13FT | A08 | 7.61 | 1.00 | 3.09 | 7–8.7 | 0.05 | |
| qFTA08–2 | Cq15FT | A08 | 9.71 | 2.25 | 3.98 | 8.7–11.3 | 0.07 |
| Cq14FT | A08 | 12.51 | 1.90 | 5.52 | 10.4–13.5 | 0.11 | |
| Cq16FT | A08 | 12.71 | 1.83 | 4.72 | 10.3–13.5 | 0.09 | |
| Cq12FT | A10 | 27.51 | 0.83 | 4.73 | 23.7–31.8 | 0.07 | |
| qFTC02–1 | Cq12FT | C02 | 42.41 | 1.12 | 7.12 | 42.3–42.5 | 0.17 |
| Cq15FT | C02 | 43.71 | 3.26 | 8.46 | 42.7–44.2 | 0.16 | |
| Gi09FT | C02 | 43.71 | 0.87 | 4.82 | 42.5–44.2 | 0.11 | |
| Cq16FT | C02 | 43.71 | 2.16 | 5.65 | 43.5–44.2 | 0.12 | |
| qFTC02–2 | Cq13FT | C02 | 46.21 | 2.06 | 11.70 | 44.8–47.1 | 0.21 |
| Cq14FT | C02 | 49.11 | 3.64 | 8.91 | 45.9–50.7 | 0.19 | |
| Cq12FT | C02 | 48.11 | 1.43 | 13.30 | 45.7–50.7 | 0.25 | |
| Gi09FT | C02 | 50.11 | 1.14 | 8.57 | 45.8–50.7 | 0.19 | |
| qFTC04 | Gi09FT | C04 | 127.91 | −0.68 | 6.23 | 127.2–129 | 0.09 |
09Gi Germany in 2009, 12Cq Chongqing in 2012, 13Cq Chongqing in 2013, 14Cq Chongqing in 2014, 15Cq Chongqing in 2015, 16Cq Chongqing in 2016. Stable QTL were represented bold
Illumina sequencing and global analysis of gene expression
To gain insights into the transcriptomic changes in the early- and late-flowering lines, we performed RNA-Seq analysis of four samples, representing leaf and shoot tissues from early- and late-flowering lines. After removing 0.82–1.41% of the sequences, including low-quality reads and adapter sequences, 29.87 Gb of clean data were obtained and used for quantitative analysis of gene expression. We mapped these clean reads to the reference B. napus genome using TopHat software; 69.75–71.62% of the clean reads were mapped to the genome, including 62.16–65.46% and 5.54–8.43% uniquely mapped and multi-mapped reads, respectively, while the remaining reads (28.38–30.25%) were unmapped (Table 4).
Table 4.
Summary of read numbers from the RNA-Seq data for the four samples
| Sample ID | Total Reads | Mapped Reads | Unique Mapped Reads | Multiple Mapped Reads | Unmapped Reads |
|---|---|---|---|---|---|
| EL | 50,590,768 | 35,840,072 (70.84%) | 31,573,071 (62.41%) | 4,267,001 (8.43%) | 14,750,696 (29.16%) |
| LL | 46,020,194 | 32,432,370 (70.47%) | 28,605,892 (62.16%) | 3,826,478 (8.31%) | 13,588,824 (29.53%) |
| ES | 51,678,990 | 36,044,431 (69.75%) | 33,179,361 (64.20%) | 2,865,070 (5.54%) | 15,634,559 (30.25%) |
| LS | 51,644,744 | 36,989,817 (71.62%) | 33,808,757 (65.46%) | 3,181,060 (6.16%) | 14,654,927 (28.38%) |
EL leaves of early-flowering bulks, LL leaves of late-flowering bulks, ES shoots of late-flowering bulks, LS shoots of late-flowering bulks
Using FPKM analysis, 58,266 genes with values of FPKM≥0.1 were identified in the four libraries. Additionally, 19.08–19.97% of the genes in the four libraries had very low expression levels (FPKM< 1.0), 20.08–20.23% had low expression levels (1.0 ≤ FPKM< 3.0), 37.60–38.79% had moderate expression levels (3.0 ≤ FPKM< 15.0), 15.54–16.15% had high expression levels (15.0 ≤ FPKM< 60.0), and 5.81–6.76% had very high expression levels (FPKM≥60.0) (Fig. 4a). The distribution of expressed genes in the four libraries is shown in Fig. 4b 44,225 (76.0%) genes were expressed in all four libraries, and 613–1250 genes were uniquely expressed in one of the four libraries (Fig. 4b).
Fig. 4.
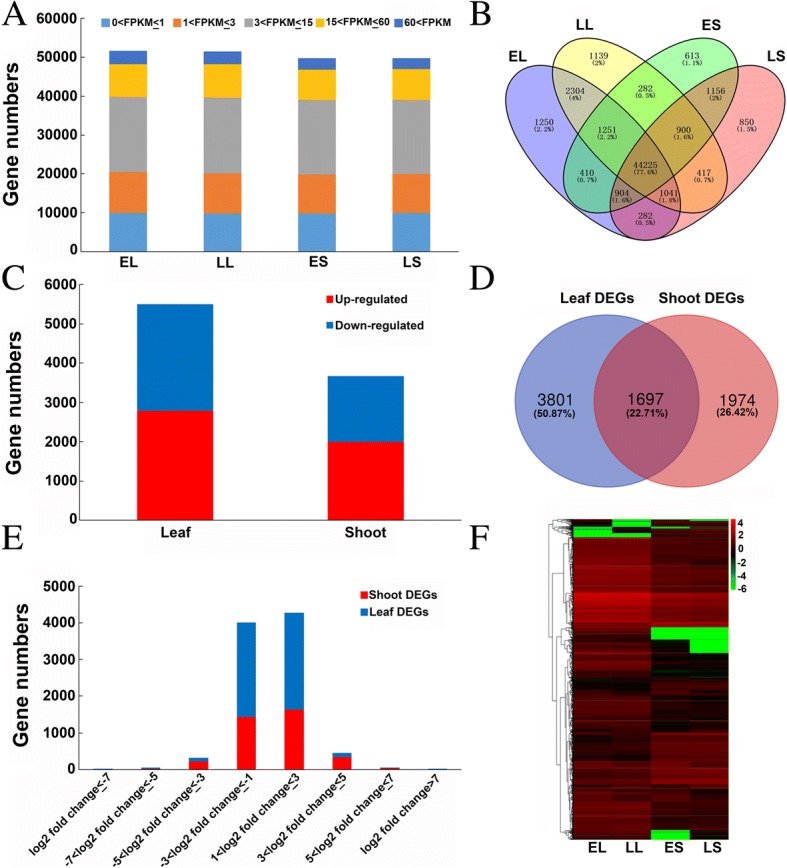
Gene expression profiles and DEGs identified between early- and late-flowering bulks. a Statistical analysis of data from the four samples, i.e. early- and late-flowering bulks in leaf and shoot tissues: b Venn diagram of the number of genes detected in the four samples. c Identification of DEGs in leaf and shoot tissues between early- and late-flowering bulks. d Venn diagram of DEGs in leaf and shoot tissues. e Fold changes in the expression of DEGs detected between early- and late-flowering bulks in leaf and shoot tissues, respectively. f Heat map of gene expression levels in the four samples. Fold change: FPKM early/FPKM late
Transcriptome differences between early- and late-flowering lines
To identify important genes responsible for flowering time variation, we selected 5498 and 3671 significant DEGs based on the criteria |log2 (FPKM early/FPKM late)| ≥ 0.58 and FDR ≤ 0.01 in leaf and shoot tissues, respectively. Of the 5498 DEGs in leaves, 2707 (49.2%) genes were downregulated and 2791 (50.8%) were upregulated. A total of 3671 DEGs, including 1673 (45.6%) downregulated genes and 1998 (54.4%) upregulated genes, were detected in shoot tissues (Fig. 4c). In addition, 1697 DEGs were common to both leaf and shoot tissues, whereas 3801 and 1974 DEGs were specific to leaf and shoot tissue, respectively (Fig. 4d). Moreover, the fold changes in the expression (up- or downregulation) of most DEGs in both leaf and shoot tissues were approximately 2–8 (Fig. 4e). We constructed a heatmap of the expression patterns of these DEGs in the four samples using MeV4.9 software (Fig. 4f).
Transcription factors (TFs) play crucial roles in many biological processes, including flowering time regulation [33]. In the current study, we identified 78 genes encoding TFs among the common DEGs in leaf and shoot tissues. These genes were divided into 29 TF families, including ERF, NAC, bHLH, bZIP, and C3H2 genes, with the same expression patterns detected in both leaves and shoots (Fig. 5a). Plant hormones also help regulate flowering time [1]. In this study, 116 hormone-related genes were identified from among the common DEGs in leaf and shoot tissues (Fig. 5b). The top three such genes were related to abscisic acid (33), auxin (27), and ethylene (24).
Fig. 5.

Heat maps of TF (a) and hormone-related (b) genes identified among the DEGs. ABA: abscisic acid; BR: brassinolide; CK: cytokinin; ET: ethylene; GA: gibberellin; JA: jasmonic acid
Functional classification of common DEGs involved in flowering time pathways
To further explore the roles of the common DEGs identified in both leaf and shoot tissues, 99 important biological pathways in B. napus were identified in the KEGG pathway database (Additional file 5: Table S5). Among these significant pathways, ribosome, biosynthesis of amino acids, carbon metabolism, oxidative phosphorylation, and ubiquitin-mediated proteolysis were the most highly represented pathways to which common DEGs were assigned. Important pathways including RNA transport and plant hormone signal transduction were also identified in this study (Additional file 5: Table S5).
Expression analysis of homologous genes influencing flowering time in Arabidopsis
We identified 1172 homologs of FTR genes in the B. napus genome using BLASTN analysis (Additional file 6: Table S6). The B. napus FTR genes were classified into nine flowering-related pathways (number of genes shown in parentheses): aging (43), ambient temperature (25), circadian clock/photoperiod (401), flower development and meristem identity (58), flowering time integrator (38), autonomous (454), hormones (98), vernalization (67), and sugar pathways (46). Many genes are involved in more than one pathways (Additional file 6: Table S6).
To identify DEGs related to the flowering pathway, we screened DEGs between two bulks with extreme differences in flowering time among these putative FTR genes. In total, 105 flowering time genes were identified as DEGs using the criteria: |log2 fold change| > 0.58 (|fold change| > 1.5), FDR < 0.05 (later-flowering lines as a control) (Additional file 7: Table S7). Of these, 60 and 72 DEGs were identified in leaf and shoot tissues, respectively. Furthermore, 19 upregulated and eight downregulated DEGs were commonly identified both in leaf and shoot tissues (Fig. 6). The differentially expressed FTR genes mainly belong to the autonomous (27), circadian clock/photoperiod (38), and flower development and meristem identity pathways (14). BnaC02g04790D, a homolog of FY located in a major QTL on chromosome C02, plays crucial roles in the autonomous pathway. Genes including FVE, UBIQUITIN CARRIER PROTEIN 1 (UBC1), UBIQUITIN-SPECIFIC PROTEASE 13 (UBP13), and LSD1-LIKE 2 (LDL2) were also detected, as well as an upregulated gene in the circadian clock/photoperiod pathway, BnaC02g03470D, encoding PRR7 (which promotes flowering). Several other important circadian clock/photoperiod pathway genes were also detected, such as PRR3, PRR9, TIME FOR COFFEE (TIC), TIMING OF CAB EXPRESSION 1 (TOC1), LATE ELONGATED HYPOCOTYL (LHY), CYCLING DOF FACTOR (CDF), and CONSTANS-LIKE 5 (COL5). Key genes in the flower development and meristem identity pathways were detected, including AGAMOUS-LIKE 14 (AGL14), APETALA 1/2 (AP1/2), and LFY. Many crucial genes in other pathways, such as the aging (TOE1), ambient temperature (AGL31), hormone (GA2ox1), sugar (SUS4), and vernalization pathways (FLC and VIN3), as well as flowering time integrator genes (FLC, SOC1, and FT) were also identified (Additional file 7: Table S7).
Fig. 6.
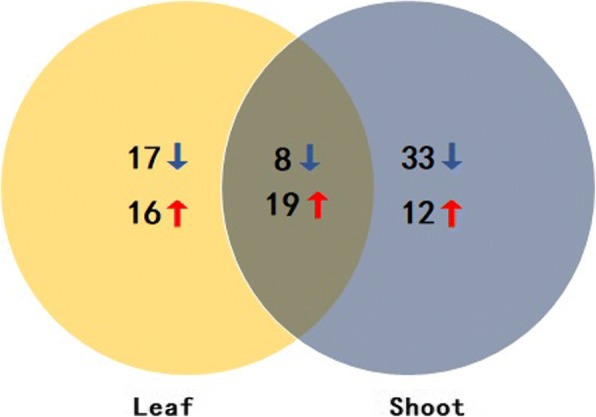
Venn diagram comparing the number of DEGs in leaf and shoot tissues between early- and late-flowering bulks. Blue and red arrows indicate downregulation and upregulation, respectively. Numbers indicate genes differentially expressed in early-flowering bulks compared to late-flowering bulks
Screening for candidate flowering time genes by integrating QTL mapping and RNA sequencing data
As mentioned above, we detected 3436 genes in QTL regions and determined their expression levels via RNA-Seq (Additional file 8: Table S8). Of these genes, 45 are FTR genes in oilseed rape (Fig. 7). Based on the criteria |log2 fold change| ≥ 1.0, FDR < 0.01 (later-flowering lines as a control), 471 genes were differentially expressed between the early- and late-flowering lines (Additional file 9: Table S9). Of these, seven flowering time-related genes were also detected (Table 5). BnaA06g24000D, an ortholog of AGL31 that functions in the ambient temperature pathway, was upregulated in leaves but downregulated in shoot tissues. BnaC02g03470D, located in a major QTL region and encoding PRR7, plays key roles in the circadian clock pathway and was upregulated in both leaf and shoot tissues. Three autonomous pathway genes, BnaC02g04790D, BnaA06g29740D, and BnaC02g01940D, encoding protein transducin/WD40 repeat-like superfamily protein (FY), arginine methyltransferase 4A (PRMT4A), and ubiquitin-specific protease 13 (UBP13), respectively, were differentially expressed between the early- and late-flowering lines. These two genes function in chromatin modification and protein stability control, respectively. Of the photoperiod pathway genes, BnaA06g16420D and BnaA06g30130D, BnaA06g16420D was downregulated in leaves, whereas BnaA06g30130D was upregulated in leaves, with no mRNA detected in shoot tissues (Table 5).
Fig. 7.

Distribution pattern of FTR genes in QTL regions. FTR genes were identified on chromosome A06, A08, A10, and C02. For simplicity, only markers in QTL confidence boundaries, along with the two terminal markers at each end of the chromosome, were shown
Table 5.
Seven differentially expressed FTR genes in QTL regions
| Gene ID | TAIR ID | Gene | L-log2 Fold change | S-log2 Fold change | Regulator | Pathway | Conditions |
|---|---|---|---|---|---|---|---|
| BnaA06g30130D | AT5G48560 | CIB2 | 1.310 | – | Pos. | Circadian Clock/Photoperiod | LD [no data under SD] |
| BnaA06g16420D | AT5G63470 | NF-YC4 | −1.778 | – | Pos. | Circadian Clock/Photoperiod | LD only |
| BnaA06g29740D | AT5G49020 | PRMT4A | −2.060 | – | Pos. | Autonomous pathway | SD and LD |
| BnaA06g24000D | AT5G65050 | MAF2, AGL31 | – | −2.916 | Neg. | Ambient temperature | SD and LD |
| BnaC02g04790D | AT5G13480 | FY | 1.338 | 2.312 | Pos. | Autonomous pathway | SD and LD |
| BnaC02g03470D | AT5G02810 | PRR7 | 2.471 | 3.165 | Pos. | Circadian Clock/Photoperiod | LD only |
| BnaC02g01940D | AT5G06600 | UBP13 | 0.734 | 1.198 | Neg. | Autonomous pathway | SD and LD |
L-log2 Fold change: fold changes in leaves early- and late-flowering bulks; S-log2 Fold change: fold changes in shoots between early- and late-flowering bulks; SD short-day conditions, LD long-day conditions, Pos. positive, Neg. negative
Verification of transcriptome sequencing data
To confirm the transcriptome data and to explore selected FTR genes that were differentially expressed between the early- and late-flowering bulks, we subjected 47 randomly selected genes to qRT-PCR analysis, including TF genes, hormone-related genes, and candidate genes in QTL regions (PRR7 and FY) (Additional file 2: Table S2). We detected high correlations (R2 = 0.853 and 0.861 in leaf and shoot tissues, respectively) between the qRT-PCR and RNA-Seq data (Fig. 8 and Additional file 10: Figure S1), suggesting that the RNA-Seq data are reliable.
Fig. 8.
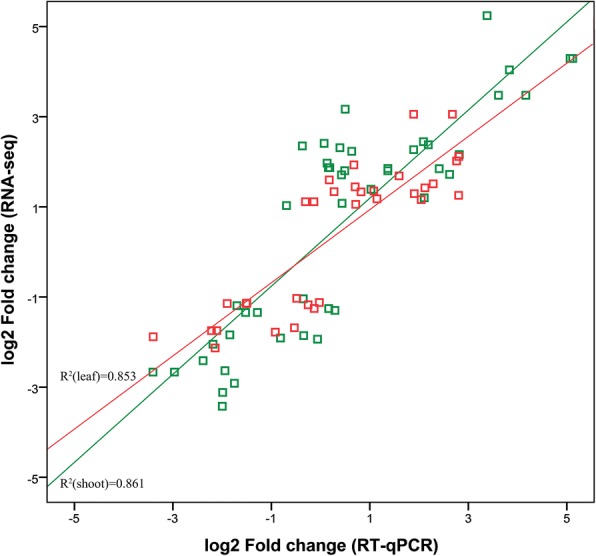
qRT-PCR validation of the expression patterns of 47 randomly selected DEGs identified by transcriptome sequencing. Red and green lines indicate regression lines for leaves and shoots, respectively
Discussion
Like many other important traits, flowering time is conditioned by the interaction of genes, endogenous signals, and environmental factors [2, 5]. In the present study, we investigated the variation in flowering time among an RIL population in six environments and in leaf and shoot tissues from early- and late-flowering lines via RNA-Seq analysis.
In the current study, 23 significant QTLs were identified under at least two conditions (Fig. 3; Table 2), suggesting that these are stable QTLs in our RIL population. Four QTLs located on chromosome A02, A05, A10, and C04 were identified under only one condition, suggesting that these QTLs are environment-specific. Flowering time QTLs located on chromosome A02, A03, A10, C02, and C03 were previously identified in B. napus and B. rapa populations [17, 34–37]. In addition, all of these regions are homologous with the top of chromosome 5 in Arabidopsis [38], a region harboring many flowering time genes such as FLC [39], CO [40], LFY [41], and FY [42]. In B. napus, nine homologs of FLC genes were detected [43]. BnCOs were identified on chromosome A02, A10, and C02, whereas BnFYs were only detected on A02 and A03 [44]. In the current study, flowering time QTLs were detected on all of the abovementioned regions except A03 and C03, and additional QTLs were identified on chromosome A05, A06, A07, A08, and C04. Finally, two QTLs located on A06 were detected in three environments, and 33 flowering time genes (e.g., CO, PRR9, and AGL31, Fig. 7) were identified in 7,212,328–21,686,640 (92.39–96.47 cM).
Differential expression of FTR genes regulates flowering time in two contrasting bulks of RILs
In the present study, we detected important genes involved in flowering time and explored the mechanisms that regulate the flowering pathway in oilseed rape using RNA-Seq technology. We subjected leaf and shoot tissues from early- and late-flowering time lines at the vegetative stage to RNA-Seq analysis. We performed BLASTN analysis against the B. napus genome using 306 sequences of known FTR genes in Arabidopsis. We identified 1172 rapeseed FTR genes, 105 of which were differentially expressed between two contrasting bulks of RILs. Most of genes encoding negative regulators of flowering, such as BnFLC, BnLHY, and BnTIC, were downregulated in the early- versus late-flowering lines, with 35 of 51 negative regulatory genes downregulated. In addition, 22 out of 46 genes encoding positive regulators of flowering were upregulated in the early- versus late-flowering lines. The expression patterns of these genes were correlated with the corresponding phenotypes. However, there were some exceptions. For example, BnaC03g05900D and BnaA02g01670D, two orthologous genes of FY, were downregulated in early-flowering plants compared to late-flowering plants, whereas another FY ortholog, BnaC02g04790D, was upregulated in early-flowering plants and is located in a major QTL region. These three genes are positive regulators in the autonomous pathway, suggesting that this pathway may be partially responsible for the differences in flowering time between the two types of plants.
CDF1 (CYCLING DOF FACTOR 1) negatively regulates flowering time in Arabidopsis [45]. CDF1 suppresses the expression of CO, leading to the downregulation of FT. The expression of FT is positively regulated by GI (GIGANTEA) [46]. Overexpression of CDF1 leads to later flowering, whereas the downregulation of CDF1 (using RNAi technology) leads to early flowering under LD conditions [45]. In the current study, we identified BnaA08g19870D and BnaC03g42040D as orthologous genes of CDF1. BnaA08g19870D was downregulated in leaves, while BnaC03g42040D was upregulated both in leaf and shoot tissues. These results suggest that the functions of these genes differ from those in Arabidopsis.
Candidate genes involved in flowering time through four major pathways
FT DEGs involved in four major flowering pathways, including the circadian clock/photoperiod, autonomous, hormone, and vernalization pathways were detected. Photoperiod is an important environmental factor that regulates flowering [47]. Genes in the circadian clock/photoperiod pathway, including LHY, PRR, CIRCADIAN CLOCK ASSOCIATED 1 (CCA1), CASEIN KINASE II BETA SUBUNIT 4 (CKB4), CDF, COL, CALCIUM-DEPENDENT PROTEIN KINASE 33 (CPK33), AS, and GI play critical roles as floral enhancers by regulating the expression of CO [48, 49]. Although BnCOs were not differentially expressed in the present study, most positive regulators in the circadian clock/photoperiod pathway, such as PRR7, CPK33, and COL5 were upregulated in the early-flowering versus late-flowering lines, whereas negative regulators such as LHY, CDF1, and CDF2 were downregulated. We propose that the circadian clock/photoperiod pathway is closely associated with the differences in flowering time between two contrasting bulks of RILs. However, sampling time can significantly influence gene expression involved in clock-dependent processes. In this study, we sampled in the morning, many genes such as CO and FT accumulated in the evening could not identified as DEGs.
Like the circadian clock/photoperiod pathway, the expression of autonomous pathway-associated genes corresponded with the differences in flowering time between two contrasting bulks of RILs. Key genes involved in this pathway, including FPA, FY, FLOWERING LOCUS D (FLD), FLOWERING TIME CONTROL PROTEIN (FCA), FVE, FLOWERING LOCUS KH DOMAIN (FLK), and RELATIVE OF EARLY FLOWERING 6 (REF6), were previously characterized in Arabidopsis [50, 51]. All of the proteins encoded by these genes promote flowering by repressing FLC expression [52]. In the current study, we identified 27 FT DEGs involved in the autonomous pathway in oilseed rape, including FVE, LDL2, FY, UBP13, EMF1, and AGL6. Notably, BnaC02g04790D, a homolog of FY located in the major QTL region on chromosome C02, was upregulated in the early-flowering versus late-flowering lines (Table 5).
The phytohormone gibberellin promotes flowering by increasing the expression of SOC1 [53]. Other critical genes involved in the response to GA signaling include GID1, GA, and DELLA [54]. In the current study, we identified ten FT DEGs in B. napus, including eight downregulated negative regulators of flowering. These downregulated genes include two RGL3 genes encoding DELLA proteins and five genes encoding GA2ox1s, which are involved in the catabolism of bioactive gibberellins. Interestingly, the Arabidopsis ga2ox1 single mutant does not display an altered flowering-time phenotype, but a quintuple ga2ox mutant, ga2ox1;2;3;4;6, flowers early under both short-day (SD) and long-day (LD) conditions [55].
Like the autonomous pathways, many genes involved in the vernalization pathway promote flowering by repressing the expression of FLC [56], as FLC suppresses flowering, with the help of its activator FRI [57]. Several FLC orthologs have been isolated and characterized in B. rapa [22, 58], orange [59], and B. napus [60]. In the present study, we detected four FTR DEGs in B. napus involved in the vernalization pathway, including VRN1 and VIN3, encoding two components of the PRC2 complex, WDR5A, encoding a component of the COMPASS complex, and AGL19. In detail, VRN1 and VIN3, encoding positive regulators of flowering time in the vernalization pathway, were upregulated in shoot tissues and leaves, respectively. AGL19, encoding a positive regulator of the vernalization pathway, was downregulated in shoot tissues, whereas WDR5A, encoding a negative regulator of this pathway, was upregulated in B. napus. These results suggest that the vernalization pathway may not be the main factor influencing the variation in flowering time investigated in our study.
In addition to FLC, other key floral integrators include SOC1, LFY, and FT [61]. In the current study, positive regulatory genes SOC1 and FT were upregulated in both shoot tissues and leaves, while FLC and LFY were upregulated only in leaf and shoot tissues, respectively. Moreover, the key positive floral integrator gene, LHY, was downregulated in shoot tissues. Together, our RNA-Seq analysis identified candidate genes involved in flowering time variance in B. napus.
Integration of QTL mapping and RNA-Seq results
As mentioned above, we identified 3436 genes in QTL regions, including 45 flowering time genes. We combined QTL mapping data with expression analysis of these genes via RNA-Seq. Seven FTR genes were differentially expressed in leaf or shoot tissues between two contrasting bulks of RILs (Table 5). Positive regulatory genes BnaC02g04790D and BnaC02g03470D, which are involved in the autonomous pathway and the circadian clock/photoperiod, respectively, were upregulated in both leaf and shoot tissues and are located in major QTLs on chromosome C02. BnaC02g04790D encodes an mRNA processing factor that regulates FCA expression. In addition, the expression of FLC is higher in fy single mutants than in wild type, leading to a late-flowering phenotype under both SD and LD conditions. Overexpression of FY in fy complements the mutant phenotype, leading to a normal flowering-time phenotype [42, 62, 63]. BnaC02g03470D encodes a component of the circadian clock in the PRR family. Functional analysis showed that the prr7 single mutant is late flowering under LD conditions only [64, 65]. PRR7, a transcriptional repressor of CCA1 and LHY, is involved in both positive and negative feedback loops of the circadian clock, thereby influencing flowering time [66]. Another positive regulator of flowering time, BnaA06g30130D (CIB2), which is involved in the circadian clock/photoperiod pathway, was upregulated in B. napus leaves in the current study. CIB2 is a bHLH TF that positively regulates the expression of FT [67]. Indeed, overexpression of CIB2 leads to early flowering under LD [68]. BnaA06g24000D encodes MADS AFFECTING FLOWERING 2 (MAF2, also known as AGAMOUS-LIKE 31 [AGL31]), a negative regulator in the ambient temperature pathway. Overexpression of MAF2 leads to late flowering under both SD and LD conditions, and the maf2 single mutant has an early-flowering phenotype under SD and LD conditions [69–72]. MAF2 suppresses flowering in response to short cold periods [70]. BnaA06g16420D and BnaA06g29740D, encoding positive regulators of flowering, were downregulated in leaves, whereas BnaC02g01940D, encoding a negative regulator of flowering, was upregulated in both leaf and shoot tissues. Together, these findings highlight the complexity of the regulatory mechanisms controlling flowering time in rapeseed.
Conclusion
In this study, we detected 27 QTLs distributed on eight chromosomes among six environments, including one major QTL on chromosome C02 that explained 11–25% of the phenotypic variation and was stably detected in all six environments. RNA-Seq analysis revealed 105 flowering time-related differentially expressed genes (DEGs) that play roles in the circadian clock/photoperiod, autonomous pathway, and hormone and vernalization pathways. We focused on DEGs related to the regulation of flowering time, especially DEGs in QTL regions. We identified 45 flowering time-related genes in these QTL regions, eight of which are DEGs, including key flowering time genes PSEUDO RESPONSE REGULATOR 7 (PRR7) and FY (located in a major QTL region on C02). These findings provide insights into the genetic architecture of flowering time in B. napus.
Additional files
Table S1. Temperature data of each environment. Maximum and minimal temperature data of each environment in growing period were collected. (XLSX 36 kb)
Table S2. Primers used for qRT-PCR verification. In total, 47 DEGs were selected to confirm the accuracy and reliability of RNA-Seq data used in this study. (XLSX 12 kb)
Table S3. Correlation of flowering times among the six environments. 09Gi: Germany in 2009; 12Cq: Chongqing in 2012; 13Cq: Chongqing in 2013; 14Cq: Chongqing in 2014; 15Cq: Chongqing in 2015; 16Cq: Chongqing in 2016. **Represents significance at the P = 0.01 level. (XLSX 8 kb)
Table S4. Genes detected in QTL regions. In total, 3436 genes were detected in QTL regions. (XLSX 199 kb)
Table S5. Significantly enriched pathways among DEGs in leaves and shoots between early- and late-flowering bulks. In total, 100 pathways were detected among these DEGs. (XLSX 16 kb)
Table S6. Flowering time-related (FTR) genes in B. napus detected using Arabidopsis FTR genes as queries via BLASTP analysis. In total, 1173 genes in B. napus were detected using 306 A. thaliana flowering time genes as queries. (XLSX 67 kb)
Table S7. Identification of differentially expressed FTR genes in B. napus in leaves and shoots between early- and late-flowering bulks. In total, 105 FTR genes were differentially expressed between two bulks. (XLSX 20 kb)
Table S8. Expression analysis of genes of QTL regions in the four samples using RNA-Seq data. EL: leaves of early-flowering bulks; LL: leaves of early-flowering bulks; ES: shoots of late-flowering bulks; LS: shoots of late-flowering bulks. (XLSX 242 kb)
Table S9. Identification of differentially expressed genes in QTL regions. L-log2 Fold change: fold changes in leaves between early- and late-flowering bulks; S-log2 Fold change: fold changes in shoots between early- and late-flowering bulks. (XLSX 50 kb)
Figure S1. Confirmation of RNA-Seq data using qRT-PCR technology. In total, 47 DEGs were selected to confirm the accuracy and reliability of RNA-Seq data used in this study. (TIF 192 kb)
Acknowledgements
We thank Benjamin Wittkop for supporting temperature data of Giessen, Germany in 2009.
Funding
This work was supported by the National Natural Science Foundation of China (31771830, 31701335), the Fundamental Research Funds for Central Universities (XDJK2017A009), the Chongqing Science and Technology Commission (cstc2016shmszx80083) and the “111” Project (B12006). The funding bodies didn’t include in the design of the study nor in collection, analysis, and interpretation of data, and also not in writing of the manuscript.
Availability of data and materials
The sequencing data were uploaded in NCBI with accession number SRP108958.
Abbreviations
- AGL14
AGAMOUS-LIKE 14
- AP1/2
APETALA 1/2
- CCA1
CIRCADIAN CLOCK ASSOCIATED 1
- CDF
CYCLING DOF FACTOR
- CDF1
CYCLING DOF FACTOR 1
- CKB4
CASEIN KINASE II BETA SUBUNIT 4
- CO
CONSTANS
- COL5
CONSTANS-LIKE 5
- CPK33
CALCIUM-DEPENDENT PROTEIN KINASE 33
- DEGs
Differentially expressed genes
- DH
doubled haploid
- FCA
FLOWERING TIME CONTROL PROTEIN
- FLC
FLOWERING LOCUS C
- FLD
FLOWERING LOCUS D
- FLK
FLOWERING LOCUS KH DOMAIN
- FPKM
fragments per kilobase of exon per million mapped fragments
- FRI
FRIGIDA
- FT
FLOWERING LOCUS T
- FTR
Flowering-time related
- GO
Gene Ontology
- GWAS
Genome-wide associated mapping
- LD
long-day
- LDL2
LSD1-LIKE 2
- LHY
LATE ELONGATED HYPOCOTYL
- LOD
Logarithm of the odds
- MAF2
MADS AFFECTING FLOWERING 2
- PRR7
PSEUDO RESPONSE REGULATOR 7
- PV
Phenotypic variation
- QTL
Quantitative trait locus
- REF6
RELATIVE OF EARLY FLOWERING 6
- RIL
Recombinant inbred line
- SD
Short-day
- TIC
TIME FOR COFFEE
- TOC1
TIMING OF CAB EXPRESSION 1
- UBC1
UBIQUITIN CARRIER PROTEIN 1
- UBP13
Ubiquitin-specific protease 13
- UBP13
UBIQUITIN-SPECIFIC PROTEASE 13
Author’s contribution
LL conceived the project; LL and HJ designed the experiments; HJ, AZ, JM, TW and BY performed the field experiments; LS, ML, JL and XX provided technical assistance; HJ, AZ and JM analyzed the transcriptome data; HJ wrote the article; and AP and LL supervised and complemented the writing. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declared no conflict of interest in the authorship and publication of this document.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Hongju Jian, Email: jianhongju1989@126.com.
Aoxiang Zhang, Email: 1029630781@qq.com.
Jinqi Ma, Email: jinqima1996@163.com.
Tengyue Wang, Email: 1084191053@qq.com.
Bo Yang, Email: 1007423827@qq.com.
Lan Shuan Shuang, Email: lanshuan@gmail.com.
Min Liu, Email: 2013204024@njau.edu.cn.
Jiana Li, Email: ljn1950@swu.edu.cn.
Xinfu Xu, Email: xinfuxu@126.com.
Andrew H. Paterson, Email: paterson@uga.edu
Liezhao Liu, Email: liezhao2003@126.com.
References
- 1.Srikanth A, Schmid M. Regulation of flowering time: all roads lead to Rome. Cell Mol Life Sci. 2011;68(12):2013–2037. doi: 10.1007/s00018-011-0673-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhai QZ, Zhang X, Wu FM, Feng HL, Deng L, Xu L, Zhang M, Wang QM, Li CY. Transcriptional mechanism of Jasmonate receptor COI1-mediated delay of flowering time in Arabidopsis. Plant Cell. 2015;27(10):2814–2828. doi: 10.1105/tpc.15.00619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andres F, Coupland G. The genetic basis of flowering responses to seasonal cues. Nat Rev Genet. 2012;13(9):627–639. doi: 10.1038/nrg3291. [DOI] [PubMed] [Google Scholar]
- 4.Fornara F, de Montaigu A, Coupland G. SnapShot: control of flowering in Arabidopsis. Cell. 2010;141(3):550, e1-2. 10.1016/j.cell.2010.04.024. [DOI] [PubMed]
- 5.Bouche F, Lobet G, Tocquin P, Perilleux C. FLOR-ID: an interactive database of flowering-time gene networks in Arabidopsis thaliana. Nucleic Acids Res. 2016;44(D1):D1167–D1171. doi: 10.1093/nar/gkv1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alonso-Blanco C, Aarts MG, Bentsink L, Keurentjes JJ, Reymond M, Vreugdenhil D, Koornneef M. What has natural variation taught us about plant development, physiology, and adaptation? Plant Cell. 2009;21(7):1877–1896. doi: 10.1105/tpc.109.068114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Putterill J, Laurie R, Macknight R. It's time to flower: the genetic control of flowering time. Bioessays. 2004;26(4):363–373. doi: 10.1002/bies.20021. [DOI] [PubMed] [Google Scholar]
- 8.Roux F, Touzet P, Cuguen J, Le Corre V. How to be early flowering: an evolutionary perspective. Trends Plant Sci. 2006;11(8):375–381. doi: 10.1016/j.tplants.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 9.Amasino R. Seasonal and developmental timing of flowering. Plant J. 2010;61(6):1001–1013. doi: 10.1111/j.1365-313X.2010.04148.x. [DOI] [PubMed] [Google Scholar]
- 10.Kumar SV, Lucyshyn D, Jaeger KE, Alos E, Alvey E, Harberd NP, Wigge PA. Transcription factor PIF4 controls the thermosensory activation of flowering. Nature. 2012;484(7393):242–U127. doi: 10.1038/nature10928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Turck F, Fornara F, Coupland G. Regulation and identity of florigen: FLOWERING LOCUS T moves center stage. Annu Rev Plant Biol. 2008;59:573–594. doi: 10.1146/annurev.arplant.59.032607.092755. [DOI] [PubMed] [Google Scholar]
- 12.Quijada PA, Udall JA, Lambert B, Osborn TC. Quantitative trait analysis of seed yield and other complex traits in hybrid spring rapeseed (Brassica napus L.): 1. Identification of genomic regions from winter germplasm. Theor Appl Genet. 2006;113(3):549–561. doi: 10.1007/s00122-006-0323-1. [DOI] [PubMed] [Google Scholar]
- 13.Chen W, Zhang Y, Liu XP, Chen BY, Tu JX, Fu TD. Detection of QTL for six yield-related traits in oilseed rape (Brassica napus) using DH and immortalized F-2 populations. Theor Appl Genet. 2007;115(6):849–858. doi: 10.1007/s00122-007-0613-2. [DOI] [PubMed] [Google Scholar]
- 14.Jiang CC, Shi JQ, Li RY, Long Y, Wang H, Li DR, Zhao JY, Meng JL. Quantitative trait loci that control the oil content variation of rapeseed (Brassica napus L.) Theor Appl Genet. 2014;127(4):957–968. doi: 10.1007/s00122-014-2271-5. [DOI] [PubMed] [Google Scholar]
- 15.Yang P, Shu C, Chen L, Xu JS, Wu JS, Liu KD. Identification of a major QTL for silique length and seed weight in oilseed rape (Brassica napus L.) Theor Appl Genet. 2012;125(2):285–296. doi: 10.1007/s00122-012-1833-7. [DOI] [PubMed] [Google Scholar]
- 16.Li L, Long Y, Zhang LB, Dalton-Morgan J, Batley J, Yu LJ, Meng JL, Li MT. Genome wide analysis of flowering time trait in multiple environments via High-throughput genotyping technique in Brassica napus L. PLoS One. 2015;10(3):e0119425. 10.1371/journal.pone.0119425. [DOI] [PMC free article] [PubMed]
- 17.Osborn TC, Kole C, Parkin IAP, Sharpe AG, Kuiper M, Lydiate DJ, Trick M. Comparison of flowering time genes in Brassica rapa, B-napus and Arabidopsis thaliana. Genetics. 1997;146(3):1123–1129. doi: 10.1093/genetics/146.3.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Raman H, Raman R, Eckermann P, Coombes N, Manoli S, Zou XX, Edwards D, Meng JL, Prangnell R, Stiller J, et al. Genetic and physical mapping of flowering time loci in canola (Brassica napus L.) Theor Appl Genet. 2013;126(1):119–132. doi: 10.1007/s00122-012-1966-8. [DOI] [PubMed] [Google Scholar]
- 19.Liu HD, Du DZ, Guo SM, Xiao L, Zhao ZG, Zhao Z, Xing XR, Tang GY, Xu L, Fu Z, et al. QTL analysis and the development of closely linked markers for days to flowering in spring oilseed rape (Brassica napus L.). Mol Breeding. 2016;36(5)36-52. 10.1007/s11032-016-0477-8.
- 20.Xu LP, Hu KN, Zhang ZQ, Guan CY, Chen S, Hua W, Li JN, Wen J, Yi B, Shen JX et al: Genome-wide association study reveals the genetic architecture of flowering time in rapeseed (Brassica napus L.). DNA Res 2016, 23(1):43–52. [DOI] [PMC free article] [PubMed]
- 21.Raman H, Raman R, Coombes N, Song J, Prangnell R, Bandaranayake C, Tahira R, Sundaramoorthi V, Killian A, Meng J, et al. Genome-wide association analyses reveal complex genetic architecture underlying natural variation for flowering time in canola. Plant Cell Environ. 2016;39(6):1228–1239. doi: 10.1111/pce.12644. [DOI] [PubMed] [Google Scholar]
- 22.Yuan YX, Wu J, Sun RF, Zhang XW, Xu DH, Bonnema G, Wang XW. A naturally occurring splicing site mutation in the Brassica rapa FLC1 gene is associated with variation in flowering time. J Exp Bot. 2009;60(4):1299–1308. doi: 10.1093/jxb/erp010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao JJ, Kulkarni V, Liu NN, Del Carpio DP, Bucher J, Bonnema G: BrFLC2 (FLOWERING LOCUS C) as a candidate gene for a vernalization response QTL in Brassica rapa. J Exp Bot 2010, 61(6):1817–1825. [DOI] [PMC free article] [PubMed]
- 24.Okazaki K, Sakamoto K, Kikuchi R, Saito A, Togashi E, Kuginuki Y, Matsumoto S, Hirai M. Mapping and characterization of FLC homologs and QTL analysis of flowering time in Brassica oleracea. Theor Appl Genet. 2007;114(4):595–608. doi: 10.1007/s00122-006-0460-6. [DOI] [PubMed] [Google Scholar]
- 25.Liu LZ, Qu CM, Wittkop B, Yi B, Xiao Y, He YJ, Snowdon RJ, Li JN. A High-Density SNP map for accurate mapping of seed fibre QTL in Brassica napus L. PLoS One. 2013;8(12). [DOI] [PMC free article] [PubMed]
- 26.Silva Lda C, Wang S, Zeng ZB. Composite interval mapping and multiple interval mapping: procedures and guidelines for using windows QTL cartographer. Methods Mol Biol. 2012;871:75–119. doi: 10.1007/978-1-61779-785-9_6. [DOI] [PubMed] [Google Scholar]
- 27.Voorrips RE. MapChart: software for the graphical presentation of linkage maps and QTLs. J Hered. 2002;93(1):77–78. doi: 10.1093/jhered/93.1.77. [DOI] [PubMed] [Google Scholar]
- 28.Qian LW, Qian W, Snowdon RJ. Sub-genomic selection patterns as a signature of breeding in the allopolyploid Brassica napus genome. BMC Genomics. 2014;15. 10.1186/1471-2164-15-1170. [DOI] [PMC free article] [PubMed]
- 29.Chalhoub B, Denoeud F, Liu SY, Parkin IAP, Tang HB, Wang XY, Chiquet J, Belcram H, Tong CB, Samans B, et al. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science. 2014;345(6199):950–953. doi: 10.1126/science.1253435. [DOI] [PubMed] [Google Scholar]
- 30.Patel RK, Jain M. NGS QC Toolkit: A Toolkit for quality control of next generation sequencing data. PLoS One. 2012;7(2). 10.1371/journal.pone.0030619. [DOI] [PMC free article] [PubMed]
- 31.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28(5):511–U174. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jian H, Wang J, Wang T, Wei L, Li J, Liu L. Identification of rapeseed MicroRNAs involved in early stage seed germination under salt and drought stresses. Front Plant Sci. 2016;7:658. doi: 10.3389/fpls.2016.00658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blumel M, Dally N, Jung C. Flowering time regulation in crops - what did we learn from Arabidopsis? Curr Opin Biotech. 2015;32:121–129. doi: 10.1016/j.copbio.2014.11.023. [DOI] [PubMed] [Google Scholar]
- 34.Schranz ME, Quijada P, Sung SB, Lukens L, Amasino R, Osborn TC. Characterization and effects of the replicated flowering time gene FLC in Brassica rapa. Genetics. 2002;162(3):1457–1468. doi: 10.1093/genetics/162.3.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Udall JA, Quijada PA, Lambert B, Osborn TC. Quantitative trait analysis of seed yield and other complex traits in hybrid spring rapeseed (Brassica napus L.): 2. Identification of alleles from unadapted germplasm. Theor Appl Genet. 2006;113(4):597–609. doi: 10.1007/s00122-006-0324-0. [DOI] [PubMed] [Google Scholar]
- 36.Long Y, Shi J, Qiu D, Li R, Zhang C, Wang J, Hou J, Zhao J, Shi L, Park BS, et al. Flowering time quantitative trait loci analysis of oilseed Brassica in multiple environments and genomewide alignment with Arabidopsis. Genetics. 2007;177(4):2433–2444. doi: 10.1534/genetics.107.080705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shi JQ, Li RY, Qiu D, Jiang CC, Long Y, Morgan C, Bancroft I, Zhao JY, Meng JL. Unraveling the complex trait of crop yield with quantitative trait loci mapping in Brassica napus. Genetics. 2009;182(3):851–861. doi: 10.1534/genetics.109.101642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parkin IAP, Gulden SM, Sharpe AG, Lukens L, Trick M, Osborn TC, Lydiate DJ. Segmental structure of the Brassica napus genome based on comparative analysis with Arabidopsis thaliana. Genetics. 2005;171(2):765–781. doi: 10.1534/genetics.105.042093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Michaels SD, Amasino RM. FLOWERING LOCUS C encodes a novel MADS domain protein that acts as a repressor of flowering. Plant Cell. 1999;11(5):949–956. doi: 10.1105/tpc.11.5.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Putterill J, Robson F, Lee K, Simon R, Coupland G. The Constans gene of Arabidopsis promotes flowering and encodes a protein showing similarities to zinc-finger transcription factors. Cell. 1995;80(6):847–857. doi: 10.1016/0092-8674(95)90288-0. [DOI] [PubMed] [Google Scholar]
- 41.Koornneef M, Blankestijndevries H, Hanhart C, Soppe W, Peeters T. The phenotype of some late-flowering mutants is enhanced by a locus on Chromosome-5 that is not effective in the Landsberg Erecta wild-type. Plant J. 1994;6(6):911–919. doi: 10.1046/j.1365-313X.1994.6060911.x. [DOI] [Google Scholar]
- 42.Simpson GG, Dijkwel PP, Quesada V, Henderson I, Dean C. FY is an RNA 3' end-processing factor that interacts with FCA to control the Arabidopsis floral transition. Cell. 2003;113(6):777–787. doi: 10.1016/S0092-8674(03)00425-2. [DOI] [PubMed] [Google Scholar]
- 43.Schiessl SV, Huettel B, Kuehn D, Reinhardt R, Snowdon RJ. Flowering time gene variation in Brassica species shows evolutionary principles. Front Plant Sci. 2017;8. 10.3389/fpls.2017.01742. [DOI] [PMC free article] [PubMed]
- 44.Fletcher RS, Mullen JL, Heiliger A, McKay JK. QTL analysis of root morphology, flowering time, and yield reveals trade-offs in response to drought in Brassica napus. J Exp Bot. 2015;66(1):245–256. doi: 10.1093/jxb/eru423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Imaizumi T, Schultz TF, Harmon FG, Ho LA, Kay SA. FKF1 F-box protein mediates cyclic degradation of a repressor of CONSTANS in Arabidopsis. Science. 2005;309(5732):293–297. doi: 10.1126/science.1110586. [DOI] [PubMed] [Google Scholar]
- 46.Fornara F, de Montaigu A, Sanchez-Villarreal A, Takahashi Y. Ver Loren van Themaat E, Huettel B, Davis SJ, Coupland G: the GI-CDF module of Arabidopsis affects freezing tolerance and growth as well as flowering. Plant J. 2015;81(5):695–706. doi: 10.1111/tpj.12759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Trevaskis B, Hemming MN, Peacock WJ, Dennis ES. HvVRN2 responds to daylength, whereas HvVRN1 is regulated by vernalization and developmental status. Plant Physiol. 2006;140(4):1397–1405. doi: 10.1104/pp.105.073486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sawa M, Nusinow DA, Kay SA, Imaizumi T. FKF1 and GIGANTEA complex formation is required for day-length measurement in Arabidopsis. Science. 2007;318(5848):261–265. doi: 10.1126/science.1146994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Imaizumi T. Arabidopsis circadian clock and photoperiodism: time to think about location. Curr Opin Plant Biol. 2010;13(1):83–89. doi: 10.1016/j.pbi.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Simpson GG. The autonomous pathway: epigenetic and post-transcriptional gene regulation in the control of Arabidopsis flowering time. Curr Opin Plant Biol. 2004;7(5):570–574. doi: 10.1016/j.pbi.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 51.Noh B, Lee SH, Kim HJ, Yi G, Shin EA, Lee M, Jung KJ, Doyle MR, Amasino RM, Noh YS. Divergent roles of a pair of homologous jumonji/zinc-finger-class transcription factor proteins in the regulation of Arabidopsis flowering time. Plant Cell. 2004;16(10):2601–2613. doi: 10.1105/tpc.104.025353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Koornneef M, Alonso-Blanco C, Blankestijn-de Vries H, Hanhart CJ, Peeters AJM. Genetic interactions among late-flowering mutants of Arabidopsis. Genetics. 1998;148(2):885–892. doi: 10.1093/genetics/148.2.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bernier G, Perilleux C. A physiological overview of the genetics of flowering time control. Plant Biotechnol J. 2005;3(1):3–16. doi: 10.1111/j.1467-7652.2004.00114.x. [DOI] [PubMed] [Google Scholar]
- 54.Harberd NP, Belfield E, Yasumura Y. The angiosperm gibberellin-GID1-DELLA growth regulatory mechanism: how an "inhibitor of an inhibitor" enables flexible response to fluctuating environments. Plant Cell. 2009;21(5):1328–1339. doi: 10.1105/tpc.109.066969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rieu I, Eriksson S, Powers SJ, Gong F, Griffiths J, Woolley L, Benlloch R, Nilsson O, Thomas SG, Hedden P, et al. Genetic analysis reveals that C-19-GA 2-oxidation is a major gibberellin inactivation pathway in Arabidopsis. Plant Cell. 2008;20(9):2420–2436. doi: 10.1105/tpc.108.058818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Amasino RM, Michaels SD. The timing of flowering. Plant Physiol. 2010;154(2):516–520. doi: 10.1104/pp.110.161653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Choi K, Kim J, Hwang HJ, Kim S, Park C, Kim SY, Lee I. The FRIGIDA complex activates transcription of FLC, a strong flowering repressor in Arabidopsis, by Recruiting Chromatin Modification Factors. Plant Cell. 2011;23(1):289–303. doi: 10.1105/tpc.110.075911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xiao D, Zhao JJ, Hou XL, Basnet RK, Carpio DPD, Zhang NW, Bucher J, Lin K, Cheng F, Wang XW, et al. The Brassica rapa FLC homologue FLC2 is a key regulator of flowering time, identified through transcriptional co-expression networks. J Exp Bot. 2013;64(14):4503–4516. doi: 10.1093/jxb/ert264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang JZ, Li ZM, Mei L, Yao JL. Hu CG: PtFLC homolog from trifoliate orange (Poncirus trifoliata) is regulated by alternative splicing and experiences seasonal fluctuation in expression level. Planta. 2009;229(4):847–859. doi: 10.1007/s00425-008-0885-z. [DOI] [PubMed] [Google Scholar]
- 60.Hou JN, Long Y, Raman H, Zou XX, Wang J, Dai ST, Xiao QQ, Li C, Fan LJ, Liu B, et al. A Tourist-like MITE insertion in the upstream region of the BnFLC.A10 gene is associated with vernalization requirement in rapeseed (Brassica napus L.). BMC Plant Biol. 2012;12. 10.1186/1471-2229-12-238. [DOI] [PMC free article] [PubMed]
- 61.Lee J, Lee I. Regulation and function of SOC1, a flowering pathway integrator. J Exp Bot. 2010;61(9):2247–2254. doi: 10.1093/jxb/erq098. [DOI] [PubMed] [Google Scholar]
- 62.Manzano D, Marquardt S, Jones AME, Baurle I, Liu FQ, Dean C. Altered interactions within FY/AtCPSF complexes required for Arabidopsis FCA-mediated chromatin silencing. P Natl Acad Sci USA. 2009;106(21):8772–8777. doi: 10.1073/pnas.0903444106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jiang SL, Kumar S, Eu YJ, Jami SK, Stasolla C, Hill RD. The Arabidopsis mutant, fy-1, has an ABA-insensitive germination phenotype. J Exp Bot. 2012;63(7):2693–2703. doi: 10.1093/jxb/err452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yamamoto Y, Sato E, Shimizu T, Nakamich N, Sato S, Kato T, Tabata S, Nagatani A, Yamashino T, Mizuno T. Comparative genetic studies on the APRR5 and APRR7 genes belonging to the APRR1/TOC1 quintet implicated in circadian rhythm, control of flowering time, and early photomorphogenesis. Plant Cell Physiol. 2003;44(11):1119–1130. doi: 10.1093/pcp/pcg148. [DOI] [PubMed] [Google Scholar]
- 65.Nakamichi N, Kita M, Niinuma K, Ito S, Yamashino T, Mizoguchi T, Mizuno T. Arabidopsis clock-associated pseudo-response regulators PRR9, PRR7 and PRR5 coordinately and positively regulate flowering time through the canonical CONSTANS-dependent photoperiodic pathway. Plant Cell Physiol. 2007;48(6):822–832. doi: 10.1093/pcp/pcm056. [DOI] [PubMed] [Google Scholar]
- 66.Nakamichi N, Kiba T, Henriques R, Mizuno T, Chua NH, Sakakibara H. PSEUDO-RESPONSE REGULATORS 9, 7, and 5 are transcriptional repressors in the Arabidopsis circadian clock. Plant Cell. 2010;22(3):594–605. doi: 10.1105/tpc.109.072892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu HT, Yu XH, Li KW, Klejnot J, Yang HY, Lisiero D, Lin CT. Photoexcited CRY2 Interacts with CIB1 to Regulate Transcription and Floral Initiation in Arabidopsis. Science. 2008;322(5907):1535–1539. doi: 10.1126/science.1163927. [DOI] [PubMed] [Google Scholar]
- 68.Liu YW, Li X, Li KW, Liu HT, Lin CT. Multiple bHLH proteins form heterodimers to mediate CRY2-dependent regulation of flowering-time in Arabidopsis. PLoS Genet. 2013;9(10). [DOI] [PMC free article] [PubMed]
- 69.Gu XF, Le C, Wang YZ, Li ZC, Jiang DH, Wang YQ, He YH. Arabidopsis FLC clade members form flowering-repressor complexes coordinating responses to endogenous and environmental cues. Nat Commun. 2013;4. 10.1038/ncomms2947. [DOI] [PMC free article] [PubMed]
- 70.Ratcliffe OJ, Kumimoto RW, Wong BJ, Riechmann JL. Analysis of the Arabidopsis MADS AFFECTING FLOWERING gene family: MAF2 prevents vernalization by short periods of cold. Plant Cell. 2003;15(5):1159–1169. doi: 10.1105/tpc.009506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rosloski SM, Singh A, Jali SS, Balasubramanian S, Weigel D, Grbic V. Functional analysis of splice variant expression of MADS AFFECTING FLOWERING 2 of Arabidopsis thaliana. Plant Mol Biol. 2013;81(1–2):57–69. doi: 10.1007/s11103-012-9982-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kim DH, Sung S. Coordination of the Vernalization response through a VIN3 and FLC gene family regulatory network in Arabidopsis. Plant Cell. 2013;25(2):454–469. doi: 10.1105/tpc.112.104760. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Temperature data of each environment. Maximum and minimal temperature data of each environment in growing period were collected. (XLSX 36 kb)
Table S2. Primers used for qRT-PCR verification. In total, 47 DEGs were selected to confirm the accuracy and reliability of RNA-Seq data used in this study. (XLSX 12 kb)
Table S3. Correlation of flowering times among the six environments. 09Gi: Germany in 2009; 12Cq: Chongqing in 2012; 13Cq: Chongqing in 2013; 14Cq: Chongqing in 2014; 15Cq: Chongqing in 2015; 16Cq: Chongqing in 2016. **Represents significance at the P = 0.01 level. (XLSX 8 kb)
Table S4. Genes detected in QTL regions. In total, 3436 genes were detected in QTL regions. (XLSX 199 kb)
Table S5. Significantly enriched pathways among DEGs in leaves and shoots between early- and late-flowering bulks. In total, 100 pathways were detected among these DEGs. (XLSX 16 kb)
Table S6. Flowering time-related (FTR) genes in B. napus detected using Arabidopsis FTR genes as queries via BLASTP analysis. In total, 1173 genes in B. napus were detected using 306 A. thaliana flowering time genes as queries. (XLSX 67 kb)
Table S7. Identification of differentially expressed FTR genes in B. napus in leaves and shoots between early- and late-flowering bulks. In total, 105 FTR genes were differentially expressed between two bulks. (XLSX 20 kb)
Table S8. Expression analysis of genes of QTL regions in the four samples using RNA-Seq data. EL: leaves of early-flowering bulks; LL: leaves of early-flowering bulks; ES: shoots of late-flowering bulks; LS: shoots of late-flowering bulks. (XLSX 242 kb)
Table S9. Identification of differentially expressed genes in QTL regions. L-log2 Fold change: fold changes in leaves between early- and late-flowering bulks; S-log2 Fold change: fold changes in shoots between early- and late-flowering bulks. (XLSX 50 kb)
Figure S1. Confirmation of RNA-Seq data using qRT-PCR technology. In total, 47 DEGs were selected to confirm the accuracy and reliability of RNA-Seq data used in this study. (TIF 192 kb)
Data Availability Statement
The sequencing data were uploaded in NCBI with accession number SRP108958.