

Abstract
The brain endothelium is a highly specialized vascular structure that maintains the activity and integrity of the central nervous system (CNS). Previous studies have reported that the integrity of the brain endothelium is compromised in a plethora of neuropathologies. Therefore, it is of particular interest to establish a method that enables researchers to investigate and understand the molecular changes in CNS endothelial cells and underlying mechanisms in conjunction with murine models of disease. In the past, approaches to isolate endothelial cells have either involved the use of transgenic reporter mice or suffered from insufficiently pure cell populations and poor yield.
This protocol here is based on well-established protocols that were modified and combined to allow single cell isolation of highly pure brain endothelial cell populations using fluorescence activated cell sorting (FACS). Briefly, after careful removal of the meninges and dissection of the cortex/hippocampus, the brain tissue is mechanically homogenized and enzymatically digested in two steps resulting in a single cell suspension. Cells are stained with a cocktail of fluorochrome-conjugated antibodies identifying not only brain endothelial cells, but also potentially contaminating cell types such as pericytes, astrocytes, and lineage cells. Using flow cytometry, cell populations are separated and sorted directly into either RNA lysis buffer for bulk RNA analyses (e.g., RNA microarray and RNA-Seq) or in pure fetal bovine serum to preserve viability for other downstream applications such as single cell RNA-Seq and Assay for Transposase-Accessible Chromatin using sequencing (ATAC-Seq). The protocol does not require the expression of a transgene to label brain endothelial cells and thus, may be applied to any mouse model. In our hands, the protocol has been highly reproducible with an average yield of 3 x 105 cells from a pool of four adult mice.
Keywords: CNS, Brain endothelium, Pericytes, Blood-brain barrier, Neuro-vascular unit, Single cell isolation, RNA-Seq, RNA microarray
Background
The brain endothelium serves as an interface for systemic factors circulating through the blood. Cerebral capillary endothelial cells constitute the blood-brain barrier (BBB), a physical barrier that limits paracellular flux via unique tight junction protein formations interconnecting cells, to maintain homeostasis of the neuronal environment and thus, neuronal functionality ( Liebner et al., 2011 and 2018). The BBB not only limits the passage of ions and other molecules such as glucose but also prevents uncontrolled exchange of toxins, bacteria, viruses and cells between the blood and the brain parenchyma ( Abbott et al., 2010 ). To accomplish this task, the brain endothelium is dependent on a fine-tuned microenvironment within the neuro-vascular unit (NVU), which consist of endothelial cells with closely associated pericytes and astrocytes as well as extracellular matrix components and microglia (Abbott and Friedman, 2012).
The function and/or integrity of the BBB is compromised in several neuropathologies, such as Alzheimer’s disease (AD), multiple sclerosis, epilepsy, and glioblastoma (Zlokovic, 2008; Marchi et al., 2012 ; Wolburg et al., 2012 ). The fluorescent activated cell sorting (FACS) single cell isolation method described here has been developed in the context of a study of transcriptional changes in brain endothelial cells in AD and inflammation (unpublished). It is a modification of published protocols for brain endothelial cell cultivation ( Liebner et al., 2008 ; Czupalla et al., 2014 ), and is designed to dissociate brain EC into a single cell suspension while preserving many endothelial surface antigens for flow cytometry. We have successfully applied the method in a collaborative investigation of the effects of an aged systemic milieu on hippocampal neurogenesis and microglia activation and the role of vascular cell adhesion molecule 1 (VCAM-1) as a negative regulator of adult neurogenesis and inducer of microglial activity ( Yousef et al., 2018a ). For an alternative brain endothelial cell isolation protocol–slightly faster, but utilizing less gentle enzymatic digestion–see Yousef et al. (2018b) .
In the last decade, several protocols for brain endothelial isolation have been employed predominantly in BBB developmental studies ( Daneman et al., 2010 ; Vanlandewijck et al., 2018 ). However, these techniques are dependent on the presence of transgenic endothelial cell markers and thus, may not easily be applied to transgenic disease mouse models. In addition, depending on the digestion enzyme used, epitopes essential for brain endothelial cell detection, such as CD31 are destroyed. This protocol allows the isolation of highly pure brain endothelial cell population (and pericytes) from any mouse strain or murine animal model using gentle enzymatic digestion steps that preserve endothelial-specific epitopes.
Materials and Reagents
Falcon 15 ml conical centrifuge tubes (Fisher Scientific, Corning, catalog number: 352096)
Falcon 50 ml conical centrifuge tubes (Fisher Scientific, Corning, catalog number: 352070)
Bottle-top vacuum filters, pore size 0.45 μm (MilliporeSigma, Corning, catalog number: CLS430514-12EA)
Bottle-top vacuum filters, pore size 0.22 μm (MilliporeSigma, Corning, catalog number: CLS430513-12EA)
Disposable sterile bottles (Fisher Scientific, Corning, catalog number: 09-761-10)
1.5 ml Snap-Cap Microcentrifuge Safe-LockTM Tubes (Eppendorf, catalog number: 022363204)
1 ml Insulin Syringe with Slip Tip (BD, catalog number: 329654)
1 ml Millex-GV Filter, 0.22 µm (MilliporeSigma, catalog number: SLGV004SL)
Petri Dish 100 x 21 mm (Thermo Fisher Scientific, NuncTM, catalog number: 172931)
Petri Dish 60 x 15 mm (Thermo Fisher Scientific, NuncTM, catalog number: 150326)
Serological pipette 10 ml, sterile (SARSTEDT, catalog number: 86.1254.025)
Optional: 5 ml conical tubes (Eppendorf, catalog number: 0030119487) (see Notes)
Serological pipette 5 ml, sterile (SARSTEDT, catalog number: 86.1253.025)
5 ml Test Tube with Cell Strainer Snap Cap (Falcon, catalog number: 352235)
Disposable Borosilicate Glass Pasteur Pipets, autoclaved (Fisher Scientific, catalog number: 13-678-20C)
Screw-Cap microcentrifuge tubes, 1.5 ml (VWR, catalog number: 89004-290)
Glass or plastic Beaker, 1,000 ml and 200 ml (laboratory specific)
Autoclaved WhatmanTM qualitative filter paper, Grade CF 12 (Sigma-Aldrich, catalog number: WHA10535097) (see Notes)
WhatmanTM Grade 1573-1/2 Qualitative Filter Papers (GE Healthcare, catalog number: 09-927-210)
Disposable scalpel blades, sterile (IntegraTM Miltex®, catalog number: 4-123)
Adult mice, 6-12 weeks, e.g., C57/Bl6 (The Jackson Laboratory, catalog number: 000664)
Deionized, filtered water (dH2O) (Merck Millipore, Milli-Q)
Bovine Serum Albumin, Fraction V, Heat Shock Treated (BSA) (Fisher Scientific, Fisher BioReagentsTM, catalog number: BP1600-1)
Sodium chloride (NaCl) (MilliporeSigma, catalog number: S3014)
Potassium chloride (KCl) (MilliporeSigma, catalog number: PX1405)
Calcium chloride (CaCl2) (MilliporeSigma catalog number: 102391)
Magnesium chloride (MgCl2) (MilliporeSigma catalog number: M8266)
HEPES (Thermo Fisher Scientific, GibcoTM, catalog number: 15630080)
Sodium hydroxide (NaOH), pellets (MilliporeSigma catalog number: S8045)
Water, Molecular Biology Reagent (MilliporeSigma, catalog number: W4502-1L)
Ethanol 70% W/V (MilliporeSigma, catalog number: EX0281-1)
Red Blood Cell Lysis Buffer (MilliporeSigma, catalog number: 11814389001)
Sodium phosphate monobasic (NaH2PO4) (MilliporeSigma, catalog number: S3139)
Potassium phosphate dibasic (KH2PO4) (MilliporeSigma, catalog number: S3264)
Fetal Bovine Serum (FBS), Defined (HyCloneTM, catalog number: SH30070.03)
Hanks' Balanced Salt Solution (HBSS) (Thermo Fisher Scientific, GibcoTM, catalog number: 24020117)
CD31 (PECAM-1) Monoclonal Antibody (390), PE-Cyanine7 (eBioscience, catalog number: 25-0311-82)
MECA-99 (Eugene C. Butcher Laboratory) ( Kruse et al., 1999 ; Lee et al., 2014 )
DyLightTM 488 Antibody Labeling Kit (Thermo Fisher Scientific, catalog number: 53024)
Aminopeptidase N/CD13 Antibody (ER-BMDM1) (NOVUS Biologicals, catalog number: NB100-64843)
ACSA-2 (Miltenyi, catalog number: 130-097-674)
Anti-ACSA-2-PE, mouse (Miltenyi, catalog number: 130-102-365)
Anti-Mouse CD45 PerCP-Cy5.5 (eBioscience, catalog number: 45-0451-82)
Anti-mouse CD11a/CD18 (LFA-1) PerCP/Cy5.5 (BioLegend, catalog number: 141008)
Anti-Mouse CD11b PerCP-Cyanine5.5 (eBioscience, catalog number: 45-0112-80)
Anti-Mouse TER-119 PerCP-Cyanine5.5 (eBioscience, catalog number: 45-5921-82)
UltraComp eBeadsTM (eBioscience, catalog number: 01-2222-42)
Propidium iodide solution (PI) (MilliporeSigma, catalog number: P4864)
Optional: RNeasy Plus Micro Kit (50) (QIAGEN, catalog number: 74034)
Optional: Array, Mouse Gene 1.0 ST ARRAY (Affymetrix, catalog number: 901169)
Collagenase type II (Biochrom Kg, catalog number: C2-22)
Collagenase/Dispase (MilliporeSigma, catalog number: 11097113001)
Deoxyribonuclease I (CellSystems, catalog number: LS006331)
Stock solutions (see Recipes)
PBS for BSA (see Recipes)
Endothelial cell buffer (see Recipes)
Collagenase II solution (see Recipes)
25% BSA (see Recipes)
Collagenase/Dispase solution (see Recipes)
DNase I solution (see Recipes)
FACS Buffer (see Recipes)
Antibody dilutions (see Recipes)
Equipment
Scale and weigh supplies (Laboratory-specific)
DURAN® Erlenmeyer flask, 2,000 ml (DURAN, catalog number: 21 216 63)
-20 °C freezer (laboratory-specific)
Precision GP 10 L General Purpose Water Bath (Precision Scientific, catalog number: TSGP10)
Portable Pipet-Aid® XP Pipette Controller (DRUMMOND, catalog number: 4-000-101)
Thermo ScientificTM NalgeneTM Polypropylene Powder Funnels (Thermo ScientificTM, catalog number: 42520150)
Cylinder, Cylinder, Grad. Cls B, 1,000 ml and 100 ml
Magnetic Stirrer (Thermo Fisher Scientific, catalog number: 90-691-18)
FisherbrandTM Round Stir Bars with Removable Pivot Ring (Thermo Fisher Scientific, fit size to measure column)
FisherbrandTM accumetTM AB15 Basic and BioBasicTM pH/mV/°C Meters or other pH meter
Autoclave (laboratory-specific)
Carbon dioxide chamber (laboratory-specific)
Thermo-isolated container including a lid, filled with ice
Surgical Scissors, Sharp (Fine Science Tools, catalog number: 14002-16)
Fine Scissors, Blunt-Blunt (Fine Science Tools, catalog number: 14108-09)
Semken Forceps, Straight (Fine Science Tools, catalog number: 11008-13)
GSC Go Science Crazy Stainless-Steel Spatula (Fisher Scientific, catalog number: S50788A)
Dumont #3 Forceps (Fine Science Tools, catalog number: 11293-00)
Dumont #5–Ceramic Coated Forceps (Fine Science Tools, catalog number: 11252-50)
Scalpel Handle #4 (Fine Science Tools, catalog number: 10004-13)
Vacuum pump (laboratory-specific)
Centrifuge 5810 R swing bucket for conical tubes (Eppendorf, model: 5810 R)
PIPETMAN Classic P2 (Gilson, catalog number: F144801)
PIPETMAN Classic P10 (Gilson, catalog number: F144802)
PIPETMAN Classic P100 (Gilson, catalog number: F123615)
PIPETMAN Classic P200 (Gilson, catalog number: F123601)
PIPETMAN Classic P1000 (Gilson, catalog number: F123602)
4 °C fridge (laboratory-specific)
Nikon SMZ 745, Stereomicroscope (Nikon, model: SMZ 745)
BD FACSAriaTM II or III cell sorter (BD Biosciences)
Software
BD FACSDIVATM SOFTWARE (BD Biosciences, version: V8.0.1)
FlowJoTM (© FlowJo, LLC, version: 9.9.4 or higher)
Optional: GeneSpring GX (Agilent Technologies, version: 14.8)
Optional: Partek® Genomics Suite® (Partek Incorporated)
Procedure
Notes:
Figure 1 outlines the workflow of the procedure to provide an overview of the individual steps.
This protocol does not require a sterile technique, but sterile equipment and reagents are used whenever possible, to minimize potential contamination and foster cell viability.
Sterilize dissection tools with 70% (vol/vol) ethanol (glass beaker). Let tools dry off before use.
Pre-chill endothelial cell buffer (see Recipes) and 6 cm Petri dish on ice.
Thaw 25% Bovine Serum Albumin (BSA) (see Recipes) in a 37 °C water bath, mix well by gently inverting the tube and immediately put on ice when thawed.
Thaw and pre-warm 2,400 U/ml collagenase II (see Recipes) in a 37 °C water bath for 20 min before use.
Cool centrifuge to 4 °C.
The yield of this protocol is highly dependent on the correct choice of volumes and tube sizes. The protocol described below is designed for isolating cells from a pool of four adult mouse brains (see Notes). For additional brains/experimental groups add extra sample tubes to the process.
Figure 1. Protocol overview for purification of murine brain endothelial cells isolated from cortices/hippocampi of adult mice.
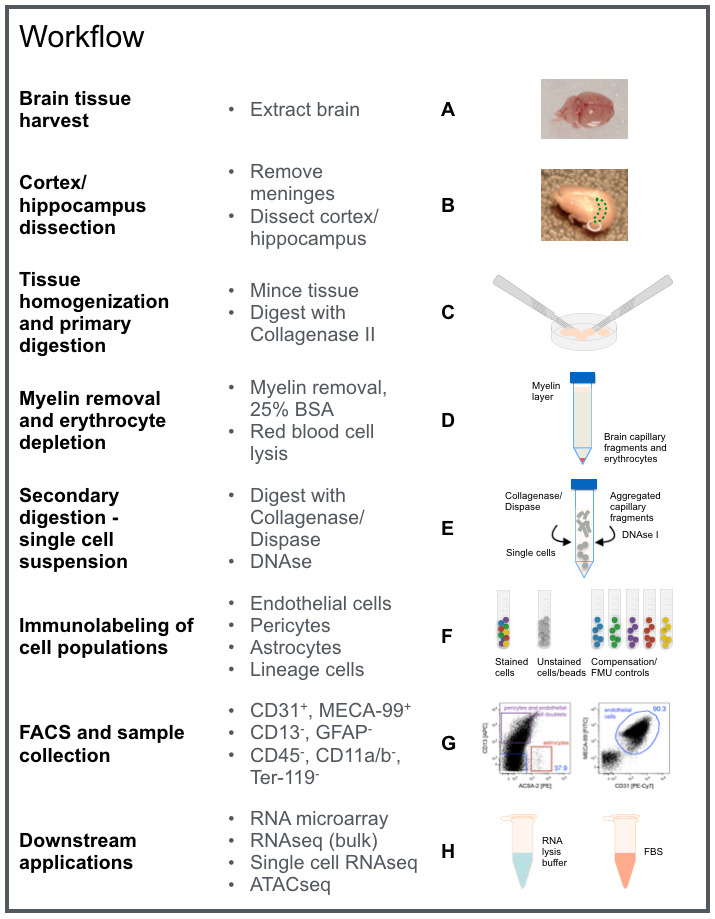
For details refer to Procedure A-H as indicated in this workflow chart. The image contains graphic elements from Motifolio toolkit.
-
Brain tissue harvest
Sacrifice adult mice using carbon dioxide asphyxiation followed by cervical dislocation (adhere to institutional guidelines). Harvest up to four brains at a time.
Spray the head/neck area with 70% (vol/vol) ethanol and remove the head from the torso using sharp surgical scissors (Figure 2).
Carefully dissect the brain from the skull at room temperature. It is critical to preserve the integrity of the brain tissue, as cutting into or puncturing the tissue may lead to difficulties during the meningeal removal and the cortex/hippocampi dissection. Use blunt scissors to cut open the skull along the left and right side (enter through the spinal cord extension), while pushing the tip of the scissors gently outwards (Figure 2).
Carefully lift the upper part of the skull few millimeters and make another cut half way through the middle of the skull and strip away the skull pieces side using forceps (Figure 2).
Use the small spatula to remove the brain from the skull (Figure 2).
Place brains in a 6 cm Petri dish on ice and add ice cold endothelial cell buffer to cover (Figure 2).
Notes:
From here on, process only one brain at a time until noted otherwise.
Ensure that the dissection process described in this section does not take longer than 15 min per brain. While practicing the technique, chill tissue covered in endothelial cell buffer on ice in-between steps to preserve cell viability. Alternatively, tissue dissection may be performed in a Petri dish on ice if the microscope setup allows. However, meninges will be easier to identify on a dark background.
-
Cortex/hippocampi dissection
Transfer the brain to an empty 10 cm Petri dish to remove the cerebellum (including pons and medulla if applicable) and the olfactory bulb using a scalpel, while carefully stabilizing the brain with forceps.
Place the brain onto an autoclaved sheet of Whatman filter paper and gently roll over the surface using a sterile spatula to crudely remove the meningeal layers surrounding the brain (Figure 3). Since brain tissue is soft and sticky, ensure to carefully separate it from the filter paper using a small spatula during rolling to minimize tissue loss. Cortical/hippocampal microvessels have distinct phenotypical properties that differ from those of meningeal vessels and inclusion of the later results in contamination with undesired endothelial cell populations.
Use the small spatula (not forceps) to transfer the tissue onto a 10 cm Petri dish and cover it with ice cold endothelial cell buffer.
Place the dish under a binocular microscope and continue to manually remove the remaining meninges using Ceramic Coated Dumont #5 forceps, while stabilizing the tissue with another set of forceps (Dumont #3 or #5) (Figure 3). Despite the fact that the meninges may be hard to see, as they are delicate and mostly transparent, aim to remove all meningeal structures. Pay particular attention to “red-colored” tissue areas, as they indicate erythrocyte-filled blood vessels, which form a network throughout the meningeal layers and therefore serve as an indicator as to which areas have to be cleaned from meninges.
Separate the hemispheres with a clean cut along the interhemispheric fissure using a sterile scalpel blade (Figure 3).
Transfer one hemisphere into a Petri dish with endothelial cell buffer on ice and continue to dissect the cortex and hippocampus of the remaining half.
Grab a small area of the “whitish” brain tissue–previously enclosed by the cortices including midbrain, thalamus, hypothalamus, basal forebrain, caudate putamen, ventral striatum, anterior olfactory nucleus, and other brain regions not listed here–in the posterior region with Dumont forceps #3 or #5. Then stab onto the tissue below in an almost parallel angle to the bench top, to cause incision using the Dumont forceps #5 like scissors (Figure 3). Keep cutting along the slightly more “pinkish” cortex until the undesired “whitish” tissue is removed. Ideally, you see the rim surface of the cortex and the embedded hippocampus after this dissection (Figure 3). However, if this is not the case, continue to remove undesired tissue parts piece by piece.
Continue to remove meninges in the medial longitudinal fissure and flip over the hemisphere to thoroughly inspect and clean-up the other side of the cortex as well.
Transfer cortex/hippocampus to a clean Petri dish on ice filled with endothelial cell buffer.
Go through Steps B6 to B9 for the second hemisphere and thereafter start over at Step B1 with the next brain until all brains have been processed (~two hours total for 2 x 4 brains; do not exceed 3 h for this part of the procedure, as cell viability will be affected).
Thereafter, process pooled tissue.
-
Tissue homogenization and primary digestion
Wash the pooled brain tissue three times by repeatedly adding endothelial cell buffer, swirling the Petri dish, followed by careful aspiration using a vacuum pump.
After the last aspiration, mince the tissue using two sterile scalpels into small pieces until a homogenous emulsion has formed.
Add 4 ml endothelial cell buffer to the Petri dish and resuspend the tissue by pipetting up and down with a 10 ml pipette.
Transfer the cell suspension into a 15 ml Falcon tube kept on ice.
Repeat Steps C3 and C4 two more times to rinse the Petri dish and top off with endothelial cell buffer to 15 ml.
Centrifuge cell suspension for 7 min at 300 × g at 4 °C and aspirate the supernatant using a vacuum pump.
Estimate the volume of your tissue pellet (typically ~1.5 ml) and add endothelial cell buffer as well as pre-warmed collagenase II in a 1:1:1 ratio. Pipette gently up and down using a 5 ml pipette.
Incubate the enzyme mixture in a 37 °C water bath for 50 min and thoroughly shake the tube after 25 and 50 min of incubation to homogenize the suspension until no white clumps are visible.
Stop the enzymatic digestion (~4.5 ml volume) by adding endothelial cell buffer to 15 ml and mix suspension by thoroughly pipetting up and down.
Centrifuge cell suspension for 7 min at 300 × g at 4 °C and aspirate the supernatant using a vacuum pump.
-
Myelin removal and erythrocyte depletion
Add 3 ml 25% BSA and transfer to a new 15 ml tube. Rinse the original tube one more time with 3 ml 25% BSA and then top off to 15 ml, thoroughly mixing the suspension with a 10 ml pipette.
Centrifuge for 30 min at 1,000 × g at 4 °C in order to separate the myelin (top) and to enrich for capillary fragments (bottom) (Figure 1D).
Aspirate the myelin layer with a vacuum pump. Before removing the clear BSA supernatant, switch to a new tip to minimize residual myelin in the cell pellet.
To deplete erythrocytes, incubate the pellet in 2 ml Red Blood Cell Lysis Buffer for 90 s at room temperature with occasional shaking. Add 1 ml Red Blood Cell Lysis Buffer with a P1000 pipette, transfer suspension to a new 15 ml Falcon tube, and rinse the tube one more time with Red Blood Cell Lysis Buffer 1 ml and combine.
Inhibit cell lysis by adding 13 ml endothelial cell buffer and put sample back on ice.
Centrifuge cell suspension for 7 min at 300 × g at 4 °C and aspirate the supernatant using a vacuum pump and leave ~2 ml in the tube. Use a 1 ml pipette to carefully remove the remaining supernatant.
-
Secondary digestion–single cell suspension
Resuspend the cell pellet in 2 ml endothelial cell buffer and transfer cell suspension to a new 15 ml Falcon tube.
To digest the microvessel fragments into a single cell suspension, add 1 mg/ml Collagenase/Dispase and incubate the mixture in a 37 °C water bath for 13 min.
Notice the formation of endothelial microvessel fragment aggregates clustered by DNA. Add 1 μg/ml DNase I, pipette up and down a few times until the microvessels are dissociated using a P1000 pipette, and incubate for an additional 2 min in the 37 °C water bath.
To quench the digestion reaction, add 13 ml endothelial cell buffer and mix by gently inverting the tube (do not pipette up and down).
Centrifuge cell suspension for 10 min at 300 × g at 4 °C.
Aspirate the supernatant using a vacuum pump and leave ~2 ml in the tube. Use a 1 ml pipette to carefully remove the remaining supernatant and store cell pellet on ice.
Resuspend the pelleted cells in FACS buffer (see Recipes). The total resuspension volume should be 200 μl per antibody cocktail FACS sample plus 50 µl for the unstained control (for additional controls see Notes section below).
Distribute the resuspended cells into appropriately labeled 1.5 ml tubes for the antibody cocktail FACS sample (200 µl) and the unstained control (50 µl) and store on ice.
Notes:- Important controls for FACS parameter setup include single-color positive controls, to compensate for channel spillover. Since the sample cell numbers are a limiting factor, we recommend using compensation beads in combination with an unstained cell sample control to set up forward and side scatter.
- We further recommend to establish proper gating using FMO (fluorescence minus one) controls, in which cells are stained with all antibodies except one. This is particularly important for initial FACS setup and should be repeated when using new antibody batches, as signal intensity may vary from batch to batch. For detailed information see Tung et al. (2007).
- Plan to run an experiment just for establishing these settings on your FACS device. In our hands, the procedure is highly reproducible and saved experimental settings with minor adjustments may be used for subsequent experiments, maximizing the available cell sample for sorting.
-
Immunostaining for FACS
Label 5 ml FACS test tubes for each fluorochrome used in the experiment plus one for unstained control and add 150 µl FACS buffer per tube (compensation controls).
Mix UltraComp eBeads by vigorously inverting at least 10 times and add three drops to a 1.5 ml tube and add 100 µl FACS buffer.
Apply 50 µl of the diluted compensation bead dilution to each compensation control tube.
Add antibodies at the same dilution (see Recipes) used for FACS sample staining (chose one antibody for PerCP-Cy5.5).
Mix well by flicking the tube and incubate on ice for 20 min in the dark.
Add all antibodies as indicated to the antibody cocktail FACS sample (see Recipes) and carefully mix by pipetting up and down using a P100 pipette (avoid bubbles).
Incubate on ice for 30 min in the dark.
-
During the incubation time, prepare collection tubes for desired downstream application:
RNA extraction: prepare two 1.5 ml screw cap tubes with 750 µl RLT Plus lysis buffer each (see manufacturer guidelines). Ensure that the volume is not significantly lower than 750 µl as a ratio lower than 1:1 of lysis buffer to sorted sample may result in reduced RNA quality. Keep collection tubes at room temperature.
Intact cells: prepare two 1.5 ml screw cap tubes with 500 µl FBS each and chill on ice.
Wash off excess antibodies by adding 1 ml FACS buffer to each tube (FACS sample as well as compensation beads) and centrifuge for 5 min at 300 × g at 4 °C.
Decant supernatant of compensation beads tubes.
Carefully aspirate FACS sample supernatant using a P1000 and then P200 pipette.
Resuspend FACS sample pellet in 300 µl and compensation beads in 200 µl FACS buffer.
Keep compensation controls on ice and in the dark until FACS.
Add 150 µl FACS buffer to the unstained cell sample kept on ice and mix well as above.
Ensure to dissociate potential sample cell aggregates using a P200 pipette before transferring the FACS samples onto the blue filter cap of a 5 ml Test Tube with Cell Strainer.
Spin cells through the filter for 2 min at 300 × g at 4 °C and store samples on ice in the dark.
Transport samples and a 1:10 propidium iodide (PI) pre-dilution (in FACS buffer) to the FACS facility. Keep cells on ice and in the dark at all times.
Notes:
This protocol does not cover detailed instructions for FACS parameter setup. For assistance, please contact your FACS core facility manager or FACS machine vendor.
Use a 100 µm nozzle.
While sorting, keep the threshold rate around or below 100x the value of the set frequency (typically fewer than 2,800 events per second).
Estimated sort time is 15-20 min per stained sample.
10%-25% of total events are CD31/MECA-99+ brain endothelial cells; sorted brain endothelial cell count ranges from 1-5 x 105 cells; average count from four adult mice: 3 x 105 cells.
-
FACS and sample collection
Run unstained cells to set up forward (FSC-A) and side scatter (SSC-A) (Figure 4).
Run single-color controls to set FACS parameters and compensate for channel spillover.
Add 1:3,000 (1:300 from 1:10 pre-dilution; equals 0.33 µg/ml) PI for live/dead staining right before running the individual sample, as it affects cell viability.
Run FMO controls for new batches of antibody, to determine positive cell populations and set up gates accordingly (Figure 4). For more detailed information see Tung et al. (2007) .
-
Run stained FACS sample
Gate cells on forward (FSC-A = size) and side scatter (SSC-A = internal structure) to exclude cell debris and residual myelin (Figure 4A).
Plot FSC-A against FSC-W to discriminate single cells from cell doublets/aggregates (Figure 4B). For more detailed information see Tung et al. (2007) .
Exclude PI positive (dead) cells (Figure 4C).
Exclude erythrocytes, monocytes/macrophages, and microglia by gating on CD45, CD11a/b, and Ter-119 negative cells (Figure 4D).
Exclude pericytes (CD13+) and astrocytes (ACSA-2+) by gating on the double negative population (Figure 4E) (see Notes).
Gate on CD31/MECA-99 double positive cells and collect cells from this gate to obtain a pure brain endothelial cell population (Figure 4F). Record 1 million events.
Collect cells in 1.5 ml screw-cap microcentrifuge tubes containing (a) RLT Plus lysis buffer, for direct cell lysis and bulk RNA downstream applications or (b) FBS, for downstream applications requiring intact/single cells.
-
Selection of optional downstream applications
RNA microarray (Sealfon and Chu, 2011) (Figure 5).
RNA-Seq, bulk (Kukurba and Montgomery, 2015).
Single cell RNA-Seq ( Haque et al., 2017 ).
Assay for Transposase-Accessible Chromatin using sequencing (ATAC-Seq) ( Buenrostro et al., 2013 and 2015).
We do not recommend cultivation of sorted endothelial cells as the procedure causes stress to the cells that impacts expression profiles. Cells may attach to extracellular matrix-coated surfaces but won’t proliferate sufficiently.
Figure 2. Brain tissue harvest.
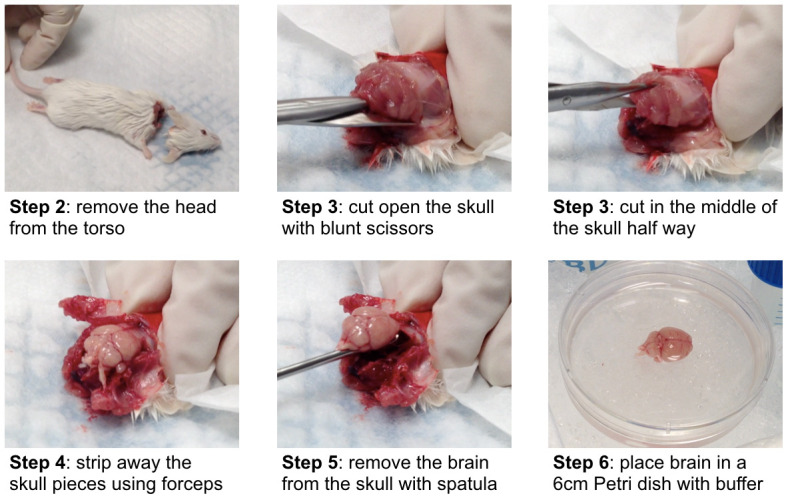
Illustration of the brain tissue harvest procedure described in Procedure A (adult wild-type Balb/c mouse). Step 1 is animal sacrifice.
Figure 3. Selected steps of the cortex/hippocampus dissection described in Procedure B.

Bottom right image illustrates the location of the hippocampus indicated by the green dotted line.
Figure 4. FACS gating strategy for purification of single cells endothelial cells.

A. Cells are gated on forward (FSC-A = size) and side scatter (SSC-A = internal structure) to exclude cell debris and residual myelin. B. FSC-A and FSC-W plotting was used to discriminate single cells from cell doublets/aggregates. C. PI uptake indicates dead cells, which are excluded. D. CD45, CD11a/b, and Ter-119 negative cells are gated to exclude erythrocytes, monocytes/macrophages, and microglia. E. CD13 and ACSA-2 staining is applied to exclude pericytes (orange) and astrocytes (red), respectively. The CD13 positive population consists of pericytes and pericyte/endothelial cell doublets (orange) and may be further gated on both endothelial markers CD31/MECA-99 (not shown), resulting in the double negative population to represent pericytes. F. CD31/MECA-99 double positive cells are defined as the brain endothelial cell population. The blue numbers indicate the event percentage of the parental gate. Dot plots show 50k recoded events.
Figure 5. Quality control of FACS sorted cell populations from the murine brain.
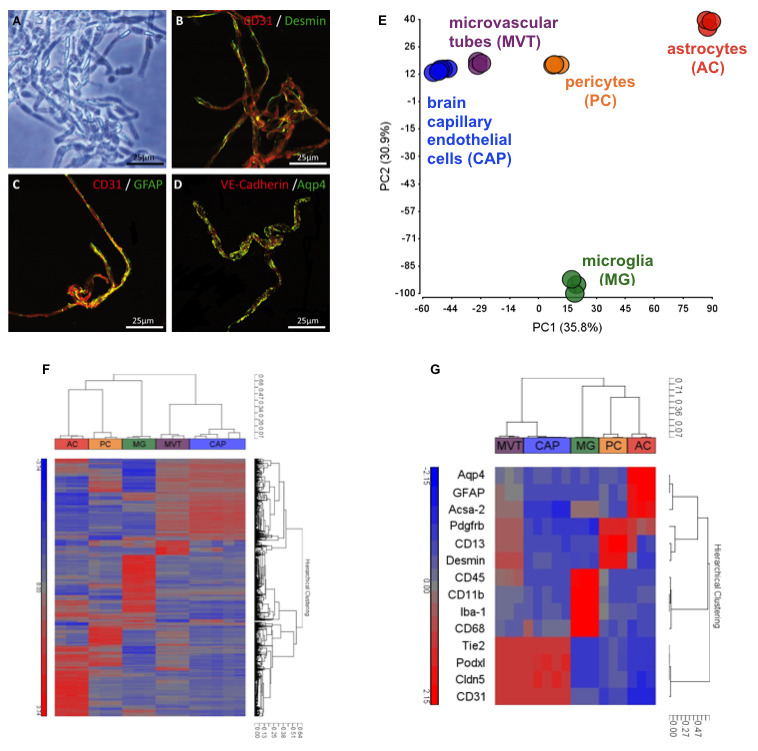
. A-D. Microvascular tubes (MVT) were isolated ( Fisher et al., 2007 ) and seeded on fibronectin-coated glass slides for double immunohistochemistry staining of brain endothelial marker CD31, pericyte marker Desmin, and astrocytic markers GFAP and Aqp4 indicating insufficient cell separation using mechanical tissue dissociation. Black scale 50 µm and white scales 25 µm. MVTs served as mixed cell population control for RNA microarray studies (E-G). E. Principal-component analysis (PCA) of the transcriptomes of brain capillary endothelial cell (CAP), pericyte (PC), astrocyte (AC), and microglia (MG) single cell populations isolated using flow cytometry. For purification of CAP and PC populations, the outlined protocol was applied, while ACs and MGs were isolated using an unpublished method. PCA was calculated for normalized EVs with a difference of at least two-fold between any pair of samples (P < 0.05 [one-way ANOVA]) and with raw expression (EV) of >140 in 100% of replicates of at least one sample population. Numbers in parentheses indicate the proportion of total variability calculated for each principal component (PC). Each data point represents a biological replicate of cells sorted from tissues pooled from 2-8 adult C57/Bl6 mice. Analysis implicates well-defined cell populations clustering together based on their cell type-specific transcriptomes. F. Hierarchical clustering by correlation of samples MVT, CAP, PC, AC, and MG with the gene list defined in E based on log2 normalized EVs. Each terminal branch represents results from a single microarray analysis of an independent biological replicate of sorted cells as described in E. G. Hierarchical clustering by correlation of selected cell-type defining gene markers: Aqp4, GFAP, and Acsa-2 (AC); Pdgfrb, CD13, and Desmin (PC); CD45, CD11b, Iba-1, and CD68 (MG); Tie2, Podxl, Cldn5, and CD31 (CAP). Individual cell types cluster together and express high levels of known cell type-specific genes indicating highly pure cell populations. As a control, arrays of MVT show the presence of pericyte, endothelial, MG and astrocyte genes.
Data analysis
The data analysis described below refers to purification quality control transcriptome analysis using RNA microarray technology shown in Figure 5. For purification of brain endothelial and pericyte populations the outlined protocol was applied, while microvascular tubes, astrocytes, and microglia were isolated using different techniques ( Fisher et al., 2007 and Fisher et al., unpublished).
RNA sample quality and quantity were determined by Agilent Bioanalyzer Total RNA Pico Euk chip readings at the Stanford University PAN facility.
Three to fifty nanogram of total RNA, with a minimum RNA integrity number (RIN) of 8, was used for amplification, labeling, and hybridization performed by Expression Analysis (Q2Solutions). Samples were hybridized on Affymetrix Genechip Mouse Gene 1.0 ST arrays.
GeneSpring GX sample preprocessing and default normalization (RMA-16) were applied. Dynamic ranges of all RNA samples were at least 40.
GeneSpring GX and Partek Genomic Suite software were used for processing and analysis of the transcriptome data. Principal-component analysis (PCA) was calculated for normalized EVs with a difference of at least two-fold between any pair of samples (P < 0.05 [one-way ANOVA], permeative 100 multiple testing, Benjamini-Hochberg correction) and with raw expression (EV) of >140 in 100% of replicates of at least one sample population.
Each PCA data point represents a biological replicate of cells sorted from tissues pooled from 2-8 adult C57/Bl6 mice (ages: 6-14 weeks [cells] and 4 months [MVTs]).
Hierarchical clustering was performed by correlation of all samples with the gene list defined in the PCA analysis based on log2 normalized EVs (Figure 5F). Each terminal branch represents results from a single microarray analysis of an independent biological replicate of sorted cells.
Hierarchical clustering was performed by correlation of selected cell-type defining gene markers: Aqp4, GFAP, and Acsa-2 (astrocytes); Pdgfrb, CD13, and Desmin (pericytes); CD45, CD11b, Iba-1, and CD68 (microglia); Tie2, Podxl, Cldn5, and CD31 (brain capillary endothelial cells).
Notes
This protocol may be applied to brain tissue derived from adult mice of older ages as well. Be advised that the yield may decline with increasing age or may potentially be affected by transgene expression.
This protocol may also be applied to individual brains. Use 5 ml instead of 15 ml tubes and 1.5 ml conical tubes as applicable and scale down the buffer and enzyme solution volumes (e.g., for one brain, use a quarter of the volumes indicated; incubation times remain the same).
Cut Whatman filter paper sheet to an appropriate size (e.g., 18 x 12 cm) and autoclave wrapped in aluminum foil or autoclavable container.
Ensure that the brain tissue stays moistened while removing meninges and during tissue dissection.
Due to high fat content, brain tissue pellets are rather unstable and viscous. Use conical tubes at all times and make sure to use a swing bucket centrifuge to collect cells on the bottom of the tube. Never decant supernatant, but carefully aspirate using a vacuum pump for big volumes and a handheld pipettor for small volumes.
Antibody concentrations are recommendations and should be tested since staining intensity might vary from batch to batch.
The CD13+ pericyte population consists of individual pericytes as well as of pericyte/endothelial cell doublets. CD13+ cells may be further gated on both endothelial markers (not shown). The resulting CD31/MECA-99 double negative population represent pericytes. It is of note that the yield of pure pericytes is rather low (1-1.5 x 104) and requires more starting material, typically up to eight mice.
MECA-99 antibody was produced and conjugated to DL488 in-house. You may choose a different conjugate if you wish to extend the fluorochrome panel by an additional color. E.g., in one of our studies, we injected VCAM-1-DL488 in vivo and stained cells with MECA-99-DL405 ( Yousef et al., 2018a ).
RNA samples should not be put on ice but instead be processed right after sorting is finished. Alternatively, freeze RNA samples in RLP Plus buffer at -80 °C for short-term storage (up to four weeks).
Recipes
-
Stock solutions (in dH2O)
2 M NaOH (50 ml)
5 M NaCl (100 ml)
1 M KCl (50 ml)
1 M CaCl2 (50 ml)
1 M MgCl2 (50 ml)
1 M Na2HPO4 (50 ml)
1 M KH2PO4 (50 ml)
Notes:- Stock solutions b-g are for making endothelial cell buffer and PBS for BSA solution.
- Aliquot in 50 ml Falcon tubes and store at room temperature (check for precipitate before use and prepare fresh as required).
-
PBS for BSA (combine all components in a 1,000 ml cylinder)
140 mM NaCl: 28.4 ml/5 M solution
0.2 mM CaCl2: 0.2 ml/1 M solution
0.2 mM MgCl2: 0.2 ml/1 M solution
10 mM Na2HPO4: 10 ml/1 M solution
10 mM KH2PO4: 10 ml/1 M solution
Fill the column up to 800 ml with dH2O and add magnet bar
Adjust with NaOH to pH 7.4 using a pH meter
Filter sterilize with a 0.22 µm bottle-top filter under sterile conditions
Use one filter for 1,000 ml buffer, filter 500 ml each in two sterile disposable bottles
Store at 4 °C for up to four weeks (check before use, if cloudy prepare fresh)
-
Endothelial cell buffer (combine all components in a 1,000 ml cylinder)
153 mM NaCl: 30.6 ml/5 M solution
5.6 mM KCl: 5.6 ml/1 M solution
1.7 mM CaCl2: 1.7 ml/1 M solution
1.2 mM MgCl2: 1.2 ml/1 M solution
15 mM HEPES: 15 ml/1 M solution
Fill the column up to 800 ml with dH2O and add magnet bar
Weigh 10 g BSA and layer on top on the surface
Place the cylinder at 4 °C (fridge or cold room) and cover with aluminum foil
When BSA has completely dissolved (~30 min), put the column on a magnet stirrer
Adjust with NaOH to pH 7.4 using a pH meter
Filter sterilize with a 0.22 µm bottle-top filter under sterile conditions
Use one filter for 1,000 ml buffer, filter 500 ml each in two sterile disposable bottles
Store at 4 °C for up to four weeks (check before use, if cloudy prepare fresh)
-
Collagenase II solution
Prepare 2,400 U/ml collagenase II endothelial cell buffer
Filter sterilize with a 0.22 µm bottle-top filter under sterile conditions
Aliquot 2 ml into 15 ml tubes and store at -20 °C up to a year
25% BSA
Fill a 2,000 ml Erlenmeyer flask with 400 ml PBS for BSA
Weigh 100 g BSA and evenly layer on top on the surface and do not mix
Place the flask in a -20 °C freezer overnight and cover with aluminum foil. The freezing process will allow the BSA to go into solution. We do not recommend using a magnetic stirrer for this step
Thaw BSA in a 37 °C water bath. Put a weight ring on top of the flask to immerse completely in water. Swirl flask intermittently to facilitate the thawing process
As soon as the BSA has completely thawed, add a magnet bar and let the solution stir until all residues stuck to the glass wall have been dissolved
Place the funnel with the Whatman Grade 1573-1/2 filter paper in the 1,000 ml glass beaker and filter
Under sterile conditions, filter BSA through a 0.45 µm bottle-top filter
Aliquot (14 ml suggested) into 15 ml Falcon tubes and store at -20 °C up to a year
-
Collagenase/Dispase solution
Prepare a 100 mg/ml solution in molecular grade water
Filter sterile into sterile 1.5 ml tube using a 1 ml syringe and the Millex-GV Filter 0.22 µm under sterile conditions
Aliquot (45 µl suggested) into sterile 1.5 ml tubes and store at -20 °C up to a year
-
DNase I solution
Prepare a 1 mg/ml solution in molecular grade water. Do not vortex, mix gently
Filter sterile into sterile 1.5 ml tube using a 1 ml syringe and the Millex-GV Filter 0.22 µm under sterile conditions
Aliquot (5 µl suggested) into sterile 1.5 ml tubes and store at -20 °C up to a year
-
FACS Buffer
Prepare 10% FBS (endotoxin-low) in HBSS (50 ml)
Store at 4 °C for up to two weeks (check for precipitate before use)
-
Antibody dilutions (Table 1)
*MECA-99 is a homemade antibody that was labeled with DyLightTM 488 Antibody Labeling Kit (Thermo Fisher Scientific). Antibody staining might vary from batch to batch. Determine optimal dilution for FACS if detected signals are too high or low.
Table 1. List of conjugated antibodies for flow cytometry.
| Antibody | Clone | Conjugate | Dilution (vol/vol) |
|---|---|---|---|
| CD31 (PECAM-1) | 390 | PE-Cy7 | 1:100 |
| MECA-99 | MECA-99 | DL488* | 1:1,000 |
| Aminopeptidase N/CD13 | ER-BMDM1 | APC | 1:50 |
| ACSA-2 | ACSA-2 | PE | 1:100 |
| Anti-Mouse CD45 | 30-F11 | PerCP-Cy5.5 | 1:1,000 |
| CD11a/CD18 (LFA-1) | H155-78 | PerCP-Cy5.5 | 1:100 |
| CD11b | M1/70 | PerCP-Cy5.5 | 1:100 |
| Ter-119 | TER-119 | PerCP-Cy5.5 | 1:100 |
Acknowledgments
This work was funded by the Edinger Institute, Frankfurt, Germany, the Stanford Institute for Immunity, Transplantation and Infection (ITI), Stanford, USA (C.J.C.), and by R01-GM37734 and R01-AI130471 from the National Institutes of Health (NIH) and an award from the Department of Veterans Affairs to E.C.B.
We thank Stefan Liebner from the Edinger Institute, Frankfurt, Germany for providing the procedure of brain endothelial cell isolation for cultivation, which served as a starting protocol for this further developed and modified protocol.
Competing interests
The authors declare that there are no conflicts of interest.
Ethics
The mice used in this protocol were bred and aged in-house and lived under a 12 h light-dark cycle in pathogenic-free conditions, in accordance with the Guide for Care and Use of Laboratory Animals of the National Institutes of Health.
Citation
Readers should cite both the Bio-protocol article and the original research article where this protocol was used.
References
- 1. Abbott N. J. and Friedman A.(2012). Overview and introduction: the blood-brain barrier in health and disease. Epilepsia 6: 1–6.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Abbott N. J., Patabendige A. A., Dolman D. E., Yusof S. R. and Begley D. J.(2010). Structure and function of the blood-brain barrier. Neurobiol Dis 37(1): 13-25. [DOI] [PubMed] [Google Scholar]
- 3. Buenrostro J. D., Giresi P. G., Zaba L. C., Chang H. Y. and Greenleaf W. J.(2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10(12): 1213-1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Buenrostro J. D., Wu B., Chang H. Y. and Greenleaf W. J.(2015). ATAC-seq: A method for assaying chromatin accessibility genome-wide. Curr Protoc Mol Biol 109: 21..29.1-21.29.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Czupalla C. J., Liebner S. and Devraj K.(2014). In vitro models of the blood-brain barrier . Methods Mol Biol 1135: 415-437. [DOI] [PubMed] [Google Scholar]
- 6. Daneman R., Zhou L., Agalliu D., Cahoy J. D., Kaushal A. and Barres B. A.(2010). The mouse blood-brain barrier transcriptome: a new resource for understanding the development and function of brain endothelial cells. PLoS One 5(10): e13741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fisher J., Devraj K., Ingram J., Slagle-Webb B., Madhankumar A. B., Liu X., Klinger M., Simpson I. A. and Connor J. R.(2007). Ferritin: a novel mechanism for delivery of iron to the brain and other organs. Am J Physiol Cell Physiol 293(2): C641-C649. [DOI] [PubMed] [Google Scholar]
- 8. Haque A., Engel J., Teichmann S. A. and Lonnberg T.(2017). A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications. Genome Med 9(1): 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kruse A., Hallmann R. and Butcher E. C.(1999). Specialized patterns of vascular differentiation antigens in the pregnant mouse uterus and the placenta. Biol Reprod 61(6): 1393-1401. [DOI] [PubMed] [Google Scholar]
- 10. Kukurba K. R. and Montgomery S. B.(2015). RNA sequencing and analysis. Cold Spring Harb Protoc 2015(11): 951-969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee M., Kiefel H., LaJevic M. D., Macauley M. S., Kawashima H., O'Hara E., Pan J., Paulson J. C. and Butcher E. C.(2014). Transcriptional programs of lymphoid tissue capillary and high endothelium reveal control mechanisms for lymphocyte homing. Nat Immunol 15(10): 982-995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liebner S., Corada M., Bangsow T., Babbage J., Taddei A., Czupalla C. J., Reis M., Felici A., Wolburg H., Fruttiger M., Taketo M. M., von Melchner H., Plate K. H., Gerhardt H. and Dejana E.(2008). Wnt/β-catenin signaling controls development of the blood-brain barrier. J Cell Biol 183(3): 409-417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liebner S., Czupalla C. J. and Wolburg H.(2011). Current concepts of blood-brain barrier development. Int J Dev Biol 55(4-5): 467-476. [DOI] [PubMed] [Google Scholar]
- 14. Liebner S., Dijkhuizen R. M., Reiss Y., Plate K. H., Agalliu D. and Constantin G.(2018). Functional morphology of the blood-brain barrier in health and disease. Acta Neuropathol 135(3): 311-336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Marchi N., Granata T., Ghosh C. and Janigro D.(2012). Blood-brain barrier dysfunction and epilepsy: pathophysiologic role and therapeutic approaches. Epilepsia 53(11): 1877-1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sealfon S. C. and Chu T. T.(2011). RNA and DNA microarrays. Methods Mol Biol 671: 3-34. [DOI] [PubMed] [Google Scholar]
- 17. Tung J. W., Heydari K., Tirouvanziam R., Sahaf B., Parks D. R., Herzenberg L. A. and Herzenberg L. A.(2007). Modern flow cytometry: a practical approach. Clin Lab Med 27(3): 453–468., v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vanlandewijck M., He L., Mae M. A., Andrae J., Ando K., Del Gaudio F., Nahar K., Lebouvier T., Lavina B., Gouveia L., Sun Y., Raschperger E., Rasanen M., Zarb Y., Mochizuki N., Keller A., Lendahl U. and Betsholtz C.(2018). A molecular atlas of cell types and zonation in the brain vasculature. Nature 554(7693): 475-480. [DOI] [PubMed] [Google Scholar]
- 19. Wolburg H., Noell S., Fallier-Becker P., Mack A. F. and Wolburg-Buchholz K.(2012). The disturbed blood-brain barrier in human glioblastoma. Mol Aspects Med 33(5-6): 579-589. [DOI] [PubMed] [Google Scholar]
- 20. Yousef H., Czupalla C. J., Lee D., Burke A., Chen M., Zandstra J., Berber E., Lehallier B., Mathur V., Nair R. V., Bonanno L., Merkel T., Schwaninger M., Quake S., Butcher E. C. and Wyss-Coray T.(2018). Aged blood inhibits hippocampal neurogenesis and activates microglia through VCAM1 at the blood-brain barrier. bioRxiv. doi: 10.1101/242198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yousef H., Czupalla C. J., Lee D., Butcher E. C. and Wyss-Coray T.(2018). Papain-based single cell isolation of primary murine brain endothelial cells using flow cytometry. Bio-protocol 8(22): e3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zlokovic B. V.(2008). The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 57(2): 178-201. [DOI] [PubMed] [Google Scholar]