

Abstract
Recent advances in molecular design have displayed striking examples of dynamic chirality transfer between various elements of chirality, e.g., from central to either helical or axial chirality and vice versa. While considerable progress in atroposelective synthesis has been made, it is intriguing to design chiral molecular switches able to provide selective and dynamic control of axial chirality with an external stimulus to modulate stereochemical functions. Here, we report the synthesis and characterization of a photoresponsive bis(2-phenol)-substituted molecular switch 1. The unique design exhibits a dynamic hybrid central-helical-axial transfer of chirality. The change of preferential axial chirality in the biaryl motif is coupled to the reversible switching of helicity of the overcrowded alkene core, dictated by the fixed stereogenic center. The potential for dynamic control of axial chirality was demonstrated by using (R)-1 as switchable catalyst to direct the stereochemical outcome of the catalytic enantioselective addition of diethylzinc to aromatic aldehydes, with successful reversal of enantioselectivity for several substrates.
Introduction
Chirality plays a fundamental role in a myriad of biological processes, including information storage and transmission, gene expression, energy production, and cellular motion.1−4 For instance, life has developed on Earth by optimizing its biological functions using l-amino acids as polypeptide building blocks and d-glucose as chemical energy source. The chirality of d-deoxyribose is amplified to the (almost) exclusively right handed helices of DNA.5 The supreme control of directional movement showcased by biological machine structures like ATP synthase,6 proteasomes,7 ribosomes,8 myosin,9 kinesin,10 and bacterial flagella11 are astonishing demonstrations of how transfer of chiral information leads to accurate control of metabolic functions and motion in cells. None of these processes could take place without precise propagation, amplification and coupling of movement, from the very bottom scale of single molecular chiral entities to the fine interplay of large protein subunits. While early research on stereochemistry mainly focused on point chirality, other motifs that feature axial chirality,12,13 helical chirality,14 and planar chirality15,16 have been extensively investigated for their potential use in synthesis, in asymmetric catalysis, and as chiral dopants. Compared with molecules that feature fixed central chirality (i.e., point chirality), axially chiral compounds may not comprise stereogenic center(s) yet exist as enantiomers.17,18 Atropisomers belong to the class of axially chiral compounds: in this case the enantiomers exist due to the restricted rotation around a single bond. The phenomenon of equilibration of stereoisomers about a rotational axis—atropisomerization19—has become a main topic of investigation in organic,20 materials,21 and medicinal chemistry.22 Despite a number of responsive molecular devices based on reversible cis–trans isomerization of double bonds,23−25 cyclizations,26 redox cycles27 and rotation around single bonds,21,28 only scattered examples of stimuli responsive systems featuring elements of axial chirality have been reported.25,29−31 Focused efforts have produced elegant systems displaying unidirectional aryl–aryl bond rotation of biaryl structures via sequential addition of chemical stimuli, overcoming the atropisomerization energy barrier inherently featured by the open structures via more flexible macrocyclic or tricyclic intermediates.21,28,32−35 Solvent-dependent atropisomerism of a flexible 2,2′-biphenol core bridged tricyclic structure was reported by Reichert and Breit.36 However, a catalytic application in which the biphenol acts as a responsive ligand was not shown. Therefore, the major challenge remains to design chiral based molecular switches able to selectively and dynamically generate and harness biaryl axial chirality with an external stimulus.
Combining dynamic chirality, chirality transfer, and photoswitches, our group has previously achieved control of activity and stereoselectivity by switchable catalysts29,37−39 based on first generation molecular motors.40−44 These responsive catalysts harness the intra- and intermolecular transfer of chirality to ultimately control the stereochemical outcome of a catalytic transformation via photochemical E–Z isomerization and thermal helix inversions of a functionalized unidirectional four-stage rotary motor based on an overcrowded alkene core. By combining the fixed point chirality originating from the stereocenters in first generation molecular motors with the dynamic alkene configuration and helical chirality, the enantiomeric excess and configuration of the chiral product can be reversibly controlled. However, out of the four possible diastereoisomeric forms, only the two pseudoenantiomeric cisoid isomers exert significant asymmetric induction via internal cooperative catalysis, while an exclusive light-driven switchable catalyst based on these chiral motor remains elusive.
We anticipated that the development of new bis(2-phenol)-functionalized switchable catalysts that harness the pairing of hybrid helical-axial chiralities within chiroptical switchable units could provide unprecedented levels of dual stereoselectivity with noninvasive control and high spatiotemporal resolution. Here we report the photochemical control of axial biaryl chirality in a light-responsive BINOL-type catalyst based on a chiral molecular switch, which displays dual stereocontrol in an asymmetric addition of organozinc reagents to aromatic aldehydes. The bis(2-phenol) unit operates as a chiral flexible bifunctional catalytic unit, the conformation of which is dictated by the dynamic stereochemistry of the central photoswitchable scaffold via internal dynamic transfer of chirality. Moreover, the catalyst described here benefits from a highly thermally stable switch core unit that can be reversibly photoisomerized exclusively between two pseudoenantiomeric forms.
Results and Discussion
Design
Molecular motors of the second generation are helical-shaped overcrowded alkenes consisting of a symmetric tricyclic lower half and an asymmetric upper half that features a single stereocenter (Scheme 1).45−47 Utilizing the hybrid chirality generated by the stereogenic center and the helical structure during the photochemical E–Z isomerization (PEZI) and thermal helix inversion (THI) of the central alkene bond allows unidirectional rotary motion controlled by a light- and heat-driven four-stage cycle (Scheme 1). The combinations of an upper half containing a six-membered ring and a lower half featuring a five membered ring are characterized by a high activation energy for the thermal relaxation process and have been recently reported as a new class of bistable photoswitches.46−48 Due to the long half-life at room temperature, i.e., high thermal stability, of their photogenerated metastable isomers,47 they allow for the design of systems capable of displaying dual stereocontrol while retaining the desired configuration for extended time intervals at elevated temperatures. This property, combined with their unique dynamic helical chirality, is highly desirable in the field of switchable asymmetric catalysis.
Scheme 1. Isomerization Processes Leading to Unidirectional Rotation in Second Generation Molecular Motor.

Four-stage cycle with only two distinct stereoisomers in case of symmetrically substituted lower half (here R = R′). S = stable isomer, MS = metastable isomer.
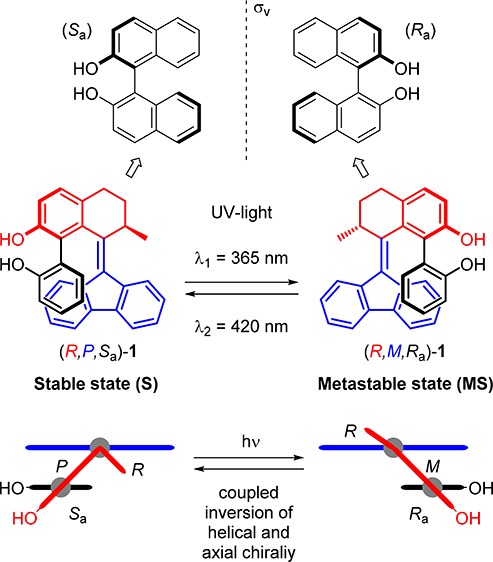
We envisioned that merging a flexible 2,2′-biphenol core with the rotor of a rigid second generation overcrowded alkene scaffold would result in transfer of chirality from the helical core of the overcrowded alkene to the biphenyl unit by steric interactions (Scheme 2). We expected the lower phenol group to remain parallel to the fluorene half, as a consequence of the system’s tendency to reduce steric hindrance between these two moieties. In this way the distinctive dynamic helicity of the switch unit and the conformational versatility of the substituted biaryl motif are combined. The system described herein features three stereochemical elements (Scheme 2). The first element is the stereogenic center of the switch (highlighted in red), which can exist with either the R or S configuration. The second element is the helicity of the overcrowded alkene (highlighted in blue), which is controlled by the configuration at the stereogenic center but can be inverted upon photoisomerization.
Scheme 2. Design of Photoswitchable 2,2′-Biphenol-Substituted Overcrowded Alkene 1.
The assigned descriptors are based on the structure of compound (R)-1 (for explanation of the chiral descriptors, vide infra). Axial helicity and chirality (green) of the 2,2′-biphenol core are coupled to axial helicity (blue) and point chirality (red) of the molecular switch scaffold. Two diastereomers with opposite coupled helicity can be selectively addressed by irradiation with UV light: (R,P,Sa)-1 (S); (R,M,Ra)-1 (MS).
More precisely, the more stable diastereoisomer (stable isomer, S) of the R enantiomer will adopt a P helicity, while the photogenerated diastereoisomer with higher energy (metastable isomer, MS) will adopt an M helicity. The axial chirality of the biaryl unit (highlighted in green) is the third stereochemical element that can be assigned either Ra or Sa according to the CIP rules.49,50 For biphenyls with an average dihedral angle of 90°, such as ortho substituted biphenyls, these stereochemistry descriptors are interchangeably used with M and P, respectively. Depending on the size of the groups and substitution pattern at the ortho positions, the dihedral angle can be smaller than 90°. Each rotamer with either Ra or Sa absolute configuration possesses two conformational helical geometries, also assigned as right-handed (P) or left-handed (M) according to the CIP rules.18
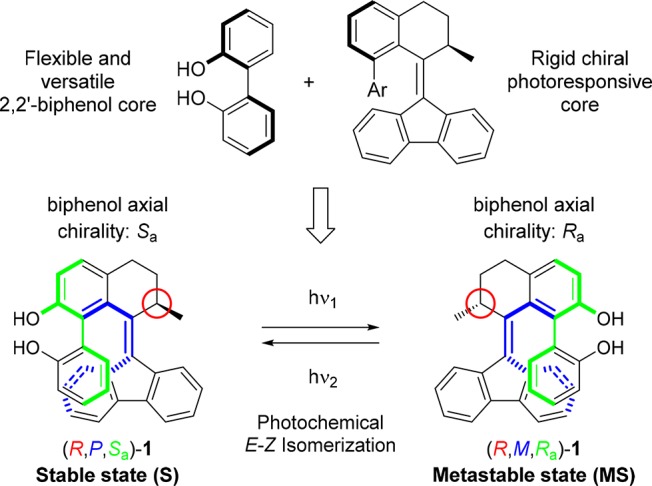
Recently our group reported a study on the tidal locking of an aryl moiety in a molecular motor, showing that among the four theoretically possible conformations of a biaryl unit, only conformations in which the nonannulated aryl group was parallel to the fluorenyl lower half were adopted.51 The other conformations with the aryl orientated perpendicular with respect to the lower half are expected to induce significant steric strain. With such a diastereotopic constraint, the true helicity (Pa/Ma) of the biaryl is inextricably connected to the helicity (P=/M=) of the overcrowded alkene chromophore, and is identical to it in each isomer (Figure 1b). Therefore, three stereodescriptors (R/S, P/M, and Ra/Sa) will be sufficient for the assignment of any expected isomer reported in this work. So for isomer (R,P=,Pa,Sa)-1: R = configuration of stereogenic center, P= = helicity of alkene, Pa = helicity of biaryl, Sa = axial chirality of biaryl (Figure 1a).
Figure 1.
(a) Example of top-down schematic view and front structural view of (R,P=,Pa,Sa)-1. Upper half ring (red, methyl substituent omitted); fluorenyl lower half (blue); biaryl moiety (black). Assigned stereodescriptors based on the structure of compound (R)-1 (see main text for details). (b) Depiction of four conformations of the biaryl moiety as viewed from the top along the central double bond and biaryl single bond. (c) Hydrogen-bonding assisted biaryl rotation of 2,2′-biphenol with inversion of stereochemistry. (d) Schematic energy vs biaryl torsional angle profile upon clockwise rotation of lower phenol group around the biaryl single bond in (R)-1.
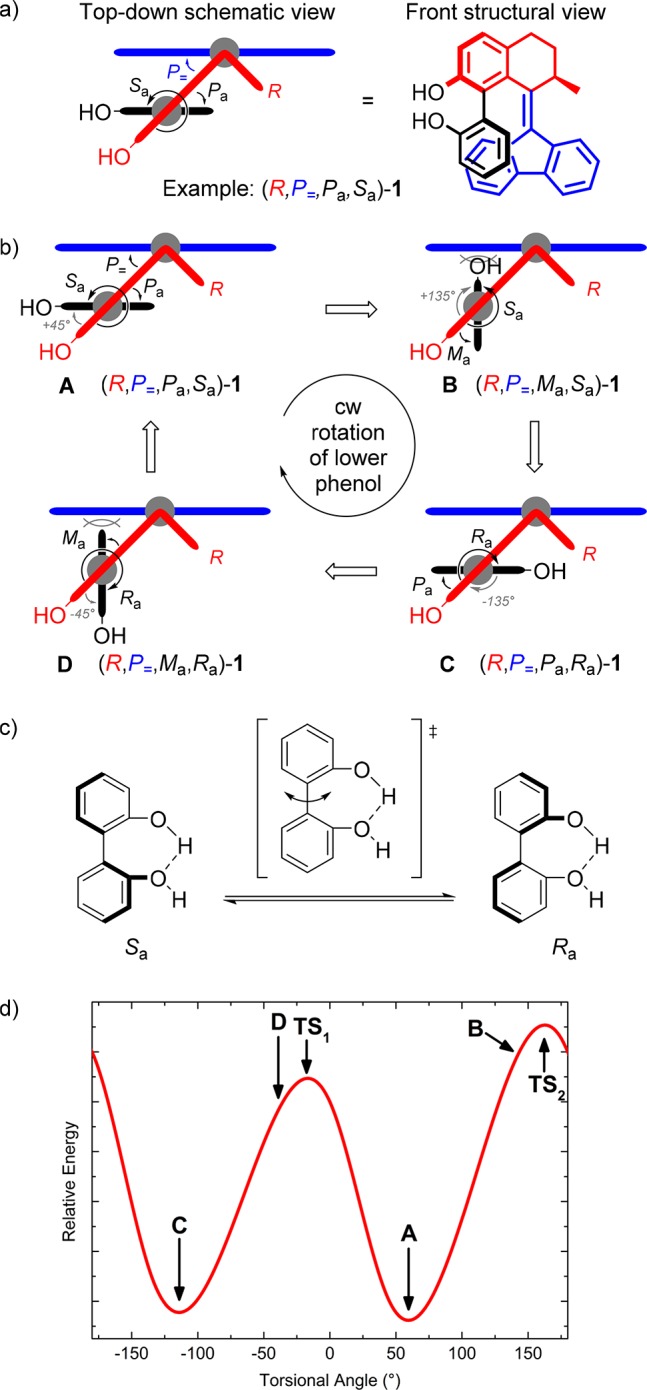
The asterisks at the stereodescriptors throughout the text denote a racemic mixture of isomers with identical relative stereochemistry (e.g., R*,P*,Sa* means a mixture of R,P,Sa and S,M,Ra). The doubly expressed axial stereodescriptor (Ra/Sa) throughout the text denotes a mixture of rotamers with identical absolute stereochemistry at the stereocenter and configurational helicity but opposite axial chirality (e.g., R,P,Sa/Ra means a mixture of atropisomers R,P,Sa and R,P,Ra). The inversion of axial chirality in 2,2′-biphenols is likely to take place via a coplanar transition state along the syn-periplanar conformation of the phenol rings taking advantage of the intramolecular hydrogen bonds between the hydroxyl groups (Δ‡G° = 48.1 kJ mol–1, T = 298.15 K; Figure 1c), based on a density functional theory (DFT) study by Fujimura and co-workers.52 These calculations support the proposal of reversible axial chirality when applied to our system, as we expected the syn- and anti-conformers (hydroxyl groups in proximity or pointing away from each other, respectively) to be in equilibrium in solution in the absence of metals or other coordinating species. Recently our group has shown how the behavior of a related biphenol-functionalized molecular motor can be controlled via reversible covalent and noncovalent modifications.53 A schematic representation of four possible conformations of 1 upon rotation of the aryl–aryl bond is presented in Figure 1b. We expect conformations with matching helicities of biaryl and overcrowded alkene units to be highly favored (A and C), while the two conformers with the aryl perpendicular to the lower half to experience steric hindrance (B and D), as shown in the relative energy vs torsional angle profile plot (see Figure 1d) based on DFT calculations (vide infra). Scheme 3 illustrates the delicate interplay of dynamic stereochemical elements and the switching process between the stable and metastable isomers of (R)-1. The rotamers of the stable state (R,P,Ra)-1 and (R,P,Sa)-1 interchange via atropisomerization (Scheme 3, left equilibrium) presumably facilitated by internal hydrogen bonding between the two phenolic moieties.52 We envisioned that upon irradiation with UV light of a mixture of (R,P,Ra)-1 and (R,P,Sa)-1 brings about isomerization into the corresponding conformers of the metastable state (R,M,Ra)-1 and (R,M,Sa)-1 isomers, respectively, during which the upper half containing the biaryl motif rotates with respect to the fluorenyl lower half yielding isomers with opposite helicity (P → M). Notably, the metastable isomer was also expected to display atropisomerization (Scheme 3, right equilibrium). We undertook a theoretical study a priori to verify the design as shown in Figure 1, with particular attention to the barrier for biaryl rotation and the relative energy of the four accessible conformers upon reversible irradiation. The structures of the four ground states were computed via DFT method calculations (see Supporting Information for details), which suggested an energetic preference in both biaryl rotation equilibria for the conformers (R,P,Sa)-1 and (R,M,Ra)-1, respectively (Scheme 3). These conformers are characterized by having the lower hydroxyl substituent pointing away from the central overcrowded alkene in a syn conformation with the upper phenol group.
Scheme 3. Switching Process between the Rotamers of Stable and Metastable Isomers of (R)-1.
Proposed ground states of rotamers and transition states (middle) of atropisomerization processes, as viewed from the top along the axis given by the double bond. Top and bottom left: rotamers of stable isomer (R,P,Sa/Ra)-1; top and bottom right: metastable isomer (R,M,Ra/Sa)-1.
In summary, our design is based on the following elements: (a) the selective and reversible photoisomerization of the overcrowded alkene scaffold between only two states; (b) the unique change in helicity of the chiroptical switch governed by the fixed configuration of the stereogenic center; (c) the coupled change in axial chirality of the biaryl core achieved via a central-to-helical-to-axial transfer of chirality; (d) the introduction of the switchable chiral biphenol functionality with the potential of various applications in catalytic enantioselective transformations.
Synthesis
Key steps in the synthesis of 1 are the Barton–Kellogg coupling of thioketone 7 and diazo-derivative 6, followed by deprotection of the biphenol moiety and chiral resolution of the target molecule 1 as illustrated in Scheme 4. Commercially available 7-methoxy-1-tetralone was brominated with N-bromosuccinimide in acetonitrile to yield 2 (93%),54 followed by Suzuki–Miyaura cross-coupling catalyzed by Pd2 dba3 and SPhos to provide the dimethoxy-biaryl motif in ketone 3 (94%). α-Methylation provided ketone 4 (86%), which was converted to the corresponding hydrazone 5 (75%) via condensation with hydrazine monohydrate using Sc(OTf)3 as a catalyst. The diazo coupling partner 6 was accessed via in situ oxidation with [bis(trifluoroacetoxy)iodo]-benzene at low temperature. Fluorene-9-thione 7, freshly synthesized by thionation of 9-fluorenone with Lawesson’s reagent, was subsequently added to yield a variable mixture of episulfide 8 and overcrowded alkene 9.
Scheme 4. Synthesis and Chiral Resolution of 2,2′-Biphenol Molecular Switch 1.

Note on resolution of 1: (i) result from first resolution; (ii) (S,M,Ra/Sa)-1 obtained by second resolution of the solid fraction: (8S,9R)-(−)-N-benzylcinchonidinium chloride 10 (0.9 equiv), 79% yield, > 99% ee (solid); (R,P,Sa/Ra)-1 obtained by second resolution of the residue from solution: 10 (0.3 equiv), 81% ee (residue from solution), followed by recrystallization from EtOH/H2O = 1:1 of the residue from solution, 15% yield, 96% ee.
After separation, the remaining episulfide was desulfurized by treatment with HMPT at elevated temperature to provide 9 (85%, for the 3-step sequence). The use of boron tribromide, widely applied for the deprotection of methoxy-substituents, resulted in partial decomposition of the overcrowded alkene and in an inseparable mixture of target compound and side-products. Successful deprotection was accomplished using methyl magnesium iodide at 165 °C55 to afford racemic (R*,P*,Sa/Ra)-1 (86%) as a mixture of two atropisomers in their thermodynamic equilibrium ratio of 60:40 (in CDCl3) according to 1H NMR analysis. Optical resolution of 1 was accomplished by two-step resolution with (8S,9R)-(−)-N-benzylcinchonidinium chloride (10) in ethyl acetate.55 Both enantiomeric mixtures of conformers were obtained in high optical purity: (R,P,Sa/Ra)-1 (96% ee, 15%); (S,M,Ra/Sa)-1 (>99% ee, 31%). The structure of 1 was unequivocally proven by NMR spectroscopy (vide infra), HRMS, as well as by single-crystal X-ray structure analysis. By means of a high-brilliance Cu IμS microfocus source (Cu Kα radiation wavelength = 1.54178 Å), the absolute configuration of enantiomerically pure (R)-1 was determined despite the absence of atoms that show significant anomalous scattering.56−58 The reconstructed unit cell of the lattice was shown to contain only the syn-conformer (R,P,Sa)-1 (see Figure 2). The experimental data confirmed the proposed model of coupled helical-to-axial transfer of helicity, demonstrating the most favored conformation of the lower aryl substituent to be parallel to the fluorenyl lower half of the switch core (synclinal) in the crystal lattice. The dihedral angle over the biaryl motif determined from the X-ray structure in the solid state was found to be +55.7° (+52.7° by calculation, vide infra).
Figure 2.
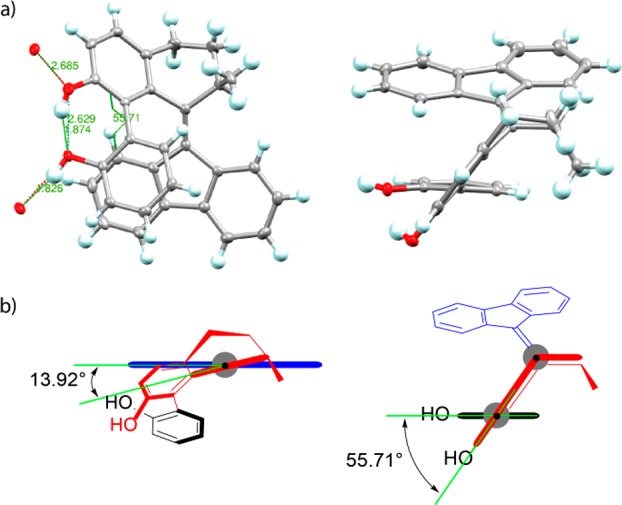
(a) X-ray structure of (R,P,Sa)-1. Left: front view; right: top view. Ellipsoids set at 50% probability. Hydrogen bond lengths (intra: H101–O1 1.874 Å, inter: H100–O1′ 1.826 Å) and oxygen–oxygen distances (intra: O1–O2 2.629 Å, inter: O1–O2′ 2.685 Å). (b) Newman projections. Left: top view through overcrowded alkene bond. Right: top view through aryl–aryl bond of biaryl unit. Torsional angles of alkene unit (13.92°) and biaryl unit (55.71°) are shown.
The angles found by calculation agree with those found by X-ray, and the small deviation between the two can be explained by a packing effect in the crystal structure that is expected to bring about small distortions with respect to a free molecule in solution or gas phase.
Atropisomerization Process
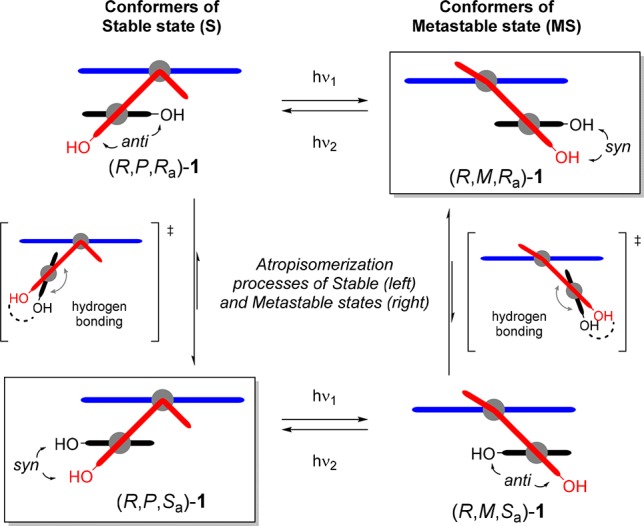
The chiral resolution and initial characterization of 1 by 1H NMR (Figure 3b) disclosed a very interesting yet initially unexpected phenomenon. Compound 1 was obtained from synthesis as pure stable isomer and could be resolved in two enantiomerically pure fractions, which by chiral HPLC analysis appeared as single eluted fractions with sharp symmetric peaks (see Supporting Information for details). However, both racemic and enantiopure fractions comprised two inseparable species, as displayed by 1H NMR spectroscopy analysis. We observed two sets of absorptions in the 1H NMR spectra recorded with a solution of stable (R)-1 in toluene-d8 (Figure 3b): the relative integration of the best resolved peaks of the atropisomers revealed a ratio of A:B = 67:33 [methyl protons: δ 1.34 ppm (A), 1.22 ppm (B); proton H1 at the stereogenic center: δ 3.95 ppm (A), 3.86 ppm (B); proton H2 of fluorenyl substituent: δ 7.78 ppm (A), 7.70 ppm (B)]. On the basis of calculated 1H NMR spectra (see Table S3–S4 and Figure S30 in Supporting Information), we assigned the experimental sets of absorptions A and B to the atropisomers (R,P,Sa)-1 and (R,P,Ra)-1, respectively (Figure 3a, see Supporting Information for atropisomer assignment study). Similar behavior with minor variation in the ratio was observed in other deuterated solvents (see Supporting Information for details). On the basis of our design, we assumed the syn and anti atropisomers to equilibrate via biaryl rotation (Scheme 3). Initial attempts to determine the rate of the atropisomerization process via dynamic NMR focused on the coalescence of the aforementioned diagnostic absorption peaks in the aromatic region (see Figure 3b).59−611H NMR spectra (400 MHz) of a sample of stable isomer (R,P,Sa/Ra)-1 in toluene-d8, were recorded at temperatures ranging from 50 to 100 °C.62 No coalescence of the aforementioned diagnostic absorption peaks was observed, suggesting the activation barrier for the biaryl rotation process to be higher than typical exchange processes usually determined via Dynamic NMR (see Supporting Information for details).63 Dynamic HPLC (DHPLC) analysis was also considered, as it was previously reported to allow for the successful determination of rotational barriers for other substituted biphenyl atropisomers.64−67 Despite the screening of temperatures down to 0 °C, no splitting of the elution peaks was observed, indicative of a relatively fast equilibration process even at lower temperatures. The rotational process was eventually demonstrated and studied by one-dimensional exchange spectroscopy (EXSY, 1H–1H nuclear Overhauser enhancement spectra). The measurements were conducted in the temperature range of 39.2–60.9 °C, consisting of an arrayed cluster of multiple mixing times per temperature (see Supporting Information for details). Exponential curve fitting of the EXSY traces provided the exchange constant (k) and associated standard error (σk) for each temperature. Eyring analysis of rate versus temperature afforded the Gibbs free energy of activation for the isomerization of the major into the minor atropisomer (Δ‡GBI at rt = 78.2 ± 1.1 kJ mol–1). The equilibration time at room temperature (20 °C) is extrapolated to be in order of minutes (t1/2 = 1.2 ± 0.4 min), while the temperature at which t1/2 equals 1 h is calculated to be equal to −50.5 ± 0.5 °C. This analysis explains why the isolation of atropisomers was not successful, as well as the lack of coalescence in the 1H NMR spectrum even at higher temperatures and the unresolved elution profile in the analytical HPLC chromatograms. Notably, when the isolated metastable isomer (R,M,Ra/Sa)-1 (via preparative HPLC, vide infra) was subjected to the same EXSY experiments, no exchange was observed (measurement temperatures up to 60 °C). This observation is in accordance with the larger elution band of the metastable isomers fraction compared with the elution band of the stable isomers observed in the analytical HPLC analysis (see Supporting Information for details), which suggests a lower atropisomerization rate, hence a slightly higher activation barrier for the biaryl rotation process in the metastable isomers. As observed in the X-ray structure analysis and based on the model investigated by Fujimura and co-workers,51 we propose a thermodynamically favored cyclic seven-membered ring conformation generated upon internal coordination via hydrogen bonding of the two hydroxyl substituents (see Scheme 3). Experimental evidence and calculation data suggest that such a conformation provides access to a transition state with a relatively low barrier for atropisomerization, allowing for a fast exchange of two conformers in solution at room temperature. In these two calculated transition states (TSBI-(R,P,Syn)-1‡ and TSBI-(R,M,Syn)-1‡, see Figures S25-S26 in Supporting Information), the hydrogen bond between the two phenol moieties is shorter than it is in any other conformation, suggesting additional stabilization of the transition state with respect to its corresponding minima explaining the relatively low barrier for atropisomerization. Moreover, the barrier for biaryl rotation is sufficiently low to allow the desired syn atropisomer to act as a thermodynamic sink upon its depletion in a reaction selective for it, for instance, by bidentate coordination of the biphenol to a metal center (vide infra, Scheme 5a). Such a complexation would require a syn conformation of the biaryl motif and concordant alkene and biaryl helicity, due to the system’s tendency to reduce steric hindrance between the lower phenol moiety and the fluorenyl lower half (Figure 1; vide infra, Scheme 5b, see Supporting Information for details).
Figure 3.
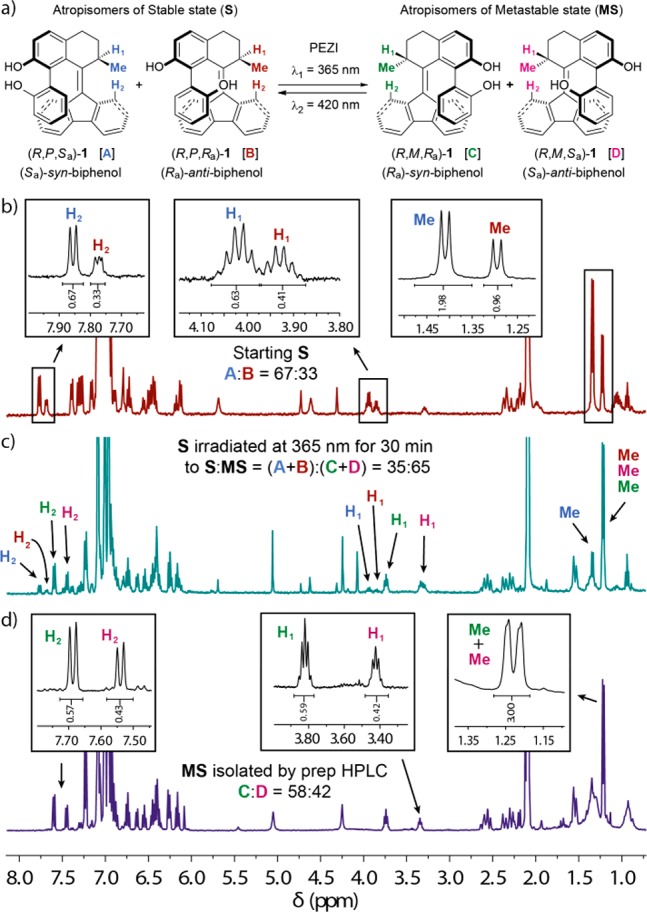
(a) Schematic representation of the photochemical E–Z isomerization of stable atropisomers (R,P,Sa/Ra)-1 to metastable atropisomers (R,M,Ra/Sa)-1. 1H NMR spectra of (R)-1 (∼5.0 mg, toluene-d8 (0.7 mL), 25 °C): (b) stable state (R,P,Sa/Ra)-1 (A:B = 67:33); (c) after irradiation with UV light (365 nm) of (R)-1 to the metastable state (R,M,Ra/Sa)-1 (∼65% of MS); (d) metastable state (R,M,Ra/Sa)-1 (C:D = 58:42) isolated by preparative HPLC from the irradiated mixture (see Supporting Information for details).
Scheme 5. Mono- and Bidentate Coordination Equilibrium upon Reaction of (R)-1 with Organozinc Reagents.
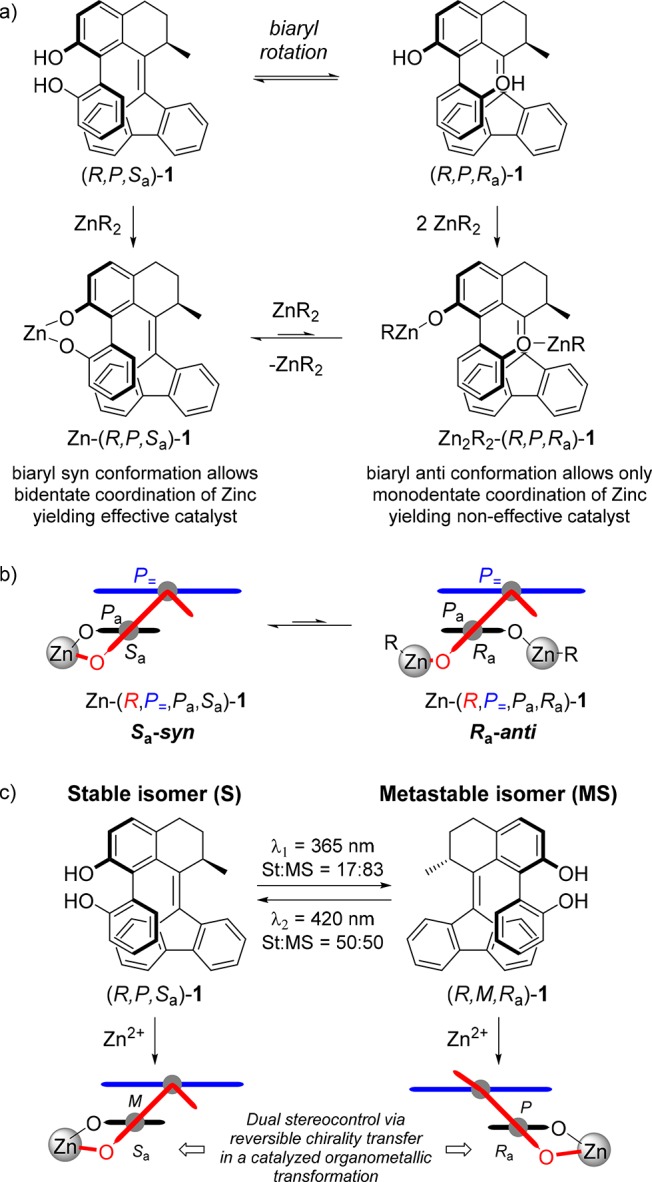
(a) Depiction of the possible mono- and bidentate coordination species upon reaction of stable isomers of 1 with ZnR2. (b) Only the isomers with a syn conformation (torsion angle = 0° to ±90°) were expected to efficiently bind a metal center and successfully transfer the chirality within a catalytically active complex. (c) Light-assisted dual stereocontrol could be achieved in a catalyzed organometallic reaction upon photoisomerization of (R)-1 and internal transfer of chirality to the coordinated metal site.
NMR Spectroscopy
In order to investigate the photochemical behavior of 1 in more detail, an NMR sample of stable isomer (R,P,Sa/Ra)-1 in toluene-d8 was irradiated with UV light (365 nm) for 30 min at room temperature. 1H NMR spectra were taken before (Figure 3b) and after exposure (Figure 3c). Upon irradiation two new sets of absorptions, C and D, with intensities increasing over time were obtained [methyl protons: δ 1.22 ppm (C + D, peaks not resolved); proton H1at the stereogenic center: δ 3.75 ppm (C), 3.35 ppm (D); proton H2 of fluorenyl substituent: δ 7.61 ppm (C), 7.47 ppm (D)]. This is indicative of the photoinduced isomerization to the metastable isomer (R,M,Ra/Sa)-1 comprising of two distinct atropisomeric species, (R,M,Ra)-1 (C) and (R,M,Sa)-1 (D), respectively. The relative integration revealed a final ratio in toluene-d8 of (R,P,Sa/Ra)-1 (A + B):(R,M,Ra/Sa)-1 (C + D) = 35:65, upon irradiation over 30 min. It should be specified that the photostationary state (PSS) was not reached in order to avoid degradation upon prolonged exposure. Due to the high thermal stability of the metastable isomers [(R,M,Ra/Sa)-1], their isolation from a crude mixture of an irradiated solution of (R)-1 was achieved by preparative HPLC (see Supporting Information for details). Analysis by 1H NMR revealed the metastable isomer to comprise a mixture of atropisomers (R,M,Ra)-1 (C) and (R,M,Sa)-1 (D) in a ratio of C:D = 58:42 (Figure 3d).
Photochemical Isomerization
The switching properties of (R)-1 were monitored by UV–vis absorption and circular dichroism (CD) spectroscopy (Figure 4). A schematic representation of the reversible photochemical E–Z isomerization process of (R)-1 is shown in Figure 4a. A solution of stable (R,P,Sa/Ra)-1 (toluene, 4.5 × 10–5 M) in quartz cuvettes was purged with argon and irradiated at room temperature toward either the metastable isomer using UV light (365 nm, Figure 4b, black to red gradient) or the stable isomer using visible light (420 nm, Figure 4c, red to blue gradient). The reversible photochemical E–Z isomerization was found to be characterized by a clear isosbestic point at 368 nm, indicating the absence of side reactions. A bathochromic shift of the major absorption band (π → π*) of about 40 nm was observed, indicative of an increase in alkene strain and consistent with other second generation motors and switches, as is expected for the metastable form (R,M,Ra/Sa)-1.48 The sample was subsequently subjected to irradiation cycles (see Figure S6 in Supporting Information), displaying nonperfect switching fatigue resistance with minor decomposition, as opposed to the highly resistant unfunctionalized parent compounds recently studied.48 However, upon addition of the radical scavenger TEMPO (10–5 M) to a solution of (R)-1 (toluene, 4.0 × 10–5 M), we observed no evidence of degradation after six irradiation cycles (Figure 4d). This observation suggests that radicals may be involved in the decomposition process.68 Lastly, a solution of stable (R,P,Sa/Ra)-1 (toluene, 4.5 × 10–5 M) was subjected to CD spectroscopy in order to perform a qualitative analysis of the change in its helical structure (Figure 4e). The CD spectrum displayed a strong Cotton effect in the area of 320–370 nm. Upon irradiation with 365 nm light, an inversion of the absorption band was observed, which is indicative of an inversion in helicity and shows that the photochemical isomerization of the stable isomers (R,P,Sa/Ra)-1 to the metastable isomers (R,M,Ra/Sa)-1 has occurred. Upon irradiation with 420 nm light, the original absorption band could be partially recovered. Chiral HPLC analysis confirmed the presence of the metastable species and allowed determining the S:MS ratio at the PSS of the irradiated mixtures upon selection of detector wavelength at the isosbestic point (368 nm, see Supporting Information for details). An efficient photoswitching process was observed upon irradiation with 365 nm light, with a high ratio toward the metastable diastereoisomer (S:MS = 17:83) at the PSS365. However, the reverse process achieved upon irradiation at 420 nm light was found to be less selective, affording an equimolar mixture of stable and metastable isomers (S:MS = 50:50) at the PSS420.
Figure 4.

(a) Schematic representation of photochemical E–Z isomerization of stable isomer (R,P,Sa/Ra)-1 to metastable isomer (R,M,Ra/Sa)-1. (b) Experimental UV–vis absorption spectra of stable (R,P,Sa/Ra)-1 (toluene, 4.5 × 10–5 M, black) and upon irradiation with UV light (365 nm) of (R,P,Sa/Ra)-1 toward the metastable isomer affording a PSS365 mixture (S/MS = 17:83, red) with an isosbestic point at 368 nm. (c) Experimental UV–vis absorption spectra after irradiation of the PSS365 sample using visible light (420 nm), resulting in reversed E–Z isomerization toward the stable isomer affording a new PSS420 mixture (S/MS = 50:50). (d) Irradiation cycles of (R)-1 (toluene, ∼4.0 × 10–5 M) in the presence of TEMPO (∼10–5 M) toward opposite PSS mixtures (red: 365 nm, 4 min; blue: 420 nm, 15 min). (e) Experimental and calculated CD spectra of (R)-1 (toluene, 5.0 × 10–1 M): black, starting stable isomer (R,P,Sa/Ra)-1; red: CD spectra of PSS365 mixture; blue: CD spectra of PSS420 mixture; cyan: metastable isomer (R,M,Ra/Sa)-1. Note: PSS ratios determined by HPLC analysis of the irradiated solutions via quantitative analysis with PDA detector wavelength set at the isosbestic point (368 nm).
Switchable Asymmetric Catalysis
Having established the reversible switching process between (R,P,Sa/Ra)-1 and (R,M,Ra/Sa)-1, we investigated their abilities for dual stereocontrol in a model asymmetric catalysis reaction.37
As a proof of principle, we envisioned to use compound (R)-1 as a switchable bidentate ligand, which could coordinate a metal center and eventually be applied to an asymmetric transformation acting as a tunable stereoselective catalyst (vide infra, Scheme 5). We anticipated the isomers of 1 having an anti conformation of the biphenol unit (torsion angle = ±90° to 180°; hydroxyl groups pointing away from each other) to be poor bidentate ligands. Therefore, only the isomers with syn conformation (torsion angle = 0° to ±90°; hydroxyl groups in proximity) were expected to efficiently bind metal ions in a bidentate fashions common to binaphthols (BINOL’s) and successfully transfer the chirality within a catalytically active complex (Scheme 5a,b). Hence we proposed that the tunable helicity (P or M) of the switch core in turn would dictate the preferential axial configuration (Ra or Sa) of the desirable syn conformation of the biaryl moiety and eventually, for instance, the configuration (R or S) of a newly formed stereogenic center when applied to an enantioselective catalytic event (Scheme 5c). Zn-BINOL-derived complexes have previously been reported to successfully mediate the catalytic asymmetric aldol69−71 and hetero-Diels–Alder72 reactions. We decided to use compound (R)-1 as a switchable bidentate ligand in 1,2-additions of diethylzinc to benzaldehydes. Numerous efforts have been devoted in the past decades to develop new effective chiral ligands for asymmetric addition of diethylzinc to benzaldehyde.73−77 However, only few cases have been reported in which dual stereocontrol was achieved by tuning the reaction conditions. The switching of enantioselectivity in the catalytic addition of diethylzinc to aldehydes was obtained by changes in the reaction conditions (e.g., solvent, temperature) while using the same chiral additive.78−81 Alternatively, complementary catalytic systems were developed by the use of distinct structural derivatives from a common chiral catalyst scaffold to access both enantiomers of the desired products.82−85 In the representative reaction (see scheme in Table 1), benzaldehyde 11a was added to a mixture of ligand (R)-1 and a solution of diethylzinc in toluene, yielding secondary alcohol 12a and in an amount of the side-product benzyl alcohol 13a. The latter is the product of the aldehyde reduction, a known process occurring in the case of a slow addition process and proposed to derive from the β-hydride elimination of organozinc species and subsequent reduction of the substrate in case of poorly activated zinc complexes.86,87 As we anticipated, photoinduced switching of ligand (R)-1 allowed successful reversal of stereoselectivity in the 1,2-addition of diethylzinc to benzaldehyde.
Table 1. Dynamic Enantioselective Addition of Organozinc to Aromatic Aldehydes with (R)-1.


General reaction conditions: 0.0125 mmol of (R,P,Sa/Ra)-1 in 0.5 mL of dry toluene at 0 °C; 0.375 mmol of R2Zn (Et2Zn, 1.0 M in hexane; i-Pr2Zn, 1.0 M in toluene) added dropwise and stirred over 10 min; 0.125 mmol of 11 added to the mixture. Reaction mixture stirred for 7 d at 0 °C. Reaction with irradiated mixture of (R)-1: 0.00125 mmol of (R,P,Sa/Ra)-1 in 15 mL of dry, degassed Et2O, irradiated with UV light (365 nm) for 30 min until the PSS was reached (S:MS = 17:83). PSS ratio determined by chiral HPLC analysis. Reaction procedure follows as described above.
Determined by 1H NMR analysis of crude.
Determined by chiral GC or chiral HPLC analysis of isolated product.
Isolated yield. Abbreviations: N.A., Not Applicable.
The results of the catalysis experiments are presented in Table 1. 1H NMR analysis allowed determining the conversion and selectivity of organozinc addition versus aldehyde reduction to benzylic alcohol. The enantiomeric excess (ee) of chiral secondary alcohols 12a–g was determined by chiral HPLC or GC analysis. In addition to benzaldehyde, several para- and ortho-substituted aromatic aldehydes bearing electron-withdrawing or electron-donating groups were tested as substrates. In all cases, when the stable form (R,P,Sa/Ra)-1 was used as a catalyst, the preferred formation of the (R)-enantiomer of secondary alcohols 12 was observed (Table 1, odd-number entries), with ee’s up to 68% (entry 1).88 In sharp contrast, upon use of the irradiated mixture of catalyst (R,P,Sa/Ra)-1 (365 nm light, PSS ratio S:MS = 17:83), the addition proceeded with reversed enantioselectivity under the same reaction conditions. Preferred formation of the (S)-enantiomer of secondary alcohols 12 was also observed in all cases after irradiation (even-number entries), with ee’s up to 55% (entry 8). The difference in enantioselectivity (Δee) between nonirradiated and irradiated catalyst solution was up to 113% (from 68% (R) to 45% (S), comparison of entries 1–2). Notably, use of diisopropylzinc led to no enantioselectivity in either case (entries 13–14).75 A control experiment was conducted by performing the addition of diethylzinc in absence of (R)-1, which resulted in a marked decrease in conversion, as well as lower addition vs reduction selectivity and isolated yield of 12a (entry 15).89 Addition of tetrabutylammonium bromide did not improve the catalytic activity, as otherwise observed in previously reported systems.90 It should be emphasized that no decomposition, racemization or significant thermal relaxation of the recovered catalyst (90% average catalyst recovery) was observed, as determined by 1H NMR and chiral HPLC analysis (see Supporting Information for details). Moreover, in all cases the catalyst was recovered after an experiment conducted using a nonirradiated catalyst solution that was recycled to perform a subsequent experiment with the same substrate after irradiation of catalyst solution without notable loss of catalytic performance (see Supporting Information for further details). Point chirality of 1 governs the dynamic helical chirality, which in turn is coupled to the axial chirality resulting in a syn conformation in the catalytically active ligand species. The inversion of enantioselectivity is an indication of the reversed local chirality around the alkyl-transferring zinc center and the coordinated aldehyde, achieved by using a ligand with opposite chiral induction.82−84 In this system we propose that upon irradiation and subsequent inversion of the biaryl axial chirality, the metastable isomer (R,M,Ra/Sa)-1 resembles the enantiomer of the stable isomer (R,P,Sa/Ra)-1 (Scheme 5c). As the flexible biphenol unit is the chiral ligand for zinc, tunable chiral induction can be achieved in the proximity of the zinc-aldehyde substrate complex, providing a novel approach for light-assisted dual stereocontrol in catalysis.
Conclusions
The design, synthesis, and resolution of a photoresponsive molecular switch featuring a versatile 2,2′-biphenol motif in which chirality is transferred across three stereochemical elements is described.
The comparison of experimental and computational data confirmed the proposed model of coupled central-to-helical-to-axial transfer of chirality, demonstrating the most favored conformation of the lower aryl substituent of the biaryl unit to be parallel to the fluorenyl lower half of the switch core. Compared with previously reported molecular motor based systems, the reduction from four to two isomerization stages featured by the biaryl-functionalized design described herein provides a simple, reusable, and efficient dynamic responsive chiral core. Extensive studies based on chiral HPLC analysis, 1H NMR, UV–vis absorption, and CD spectroscopy proved the reversible photoswitching of 1, with no fatigue over multiple cycles in the presence of substoichiometric amount of TEMPO. The chirality transfer was successfully applied to creation of another stereogenic element as demonstrated via dynamic central-to-helical-to-axial-to-central transfer of chirality by using (R)-1 as switchable catalyst in the enantioselective addition of diethylzinc to various benzaldehydes. Reversal of enantioselectivity was accomplished for each substrate, with ee values for 12 up to 68%, Δee’s up to 113% and yields up to 87%. These results achieved in switchable asymmetric catalysis highlight the proof-of principle of a two-stage dynamically responsive chiral biaryl-functionalized switch scaffold. The future development of analogous biaryl-switch structures combined with the well-established role of biaryls as privileged structures in asymmetric catalysis may lead to the construction of a variety of unprecedented switchable chiral catalysts that could perform multiple enantioselective transformation in a sequential manner. In addition, this chiral switch system has considerable potential as responsive chirality selector for a range of other applications beyond the field of asymmetric catalysis, such as control of supramolecular architecture, host–guest interaction, and polymer or liquid crystal morphology.
Acknowledgments
Financial support from The Netherlands Organization for Scientific Research (NWO–CW), Foundation for Fundamental Research on Matter (FOM, a subsidiary of NWO), the Zernike Institute for Advanced Materials, The Royal Netherlands Academy of Arts and Sciences (KNAW), The European Research Council (Advanced Investigator Grant No. 694345 to BLF), Ministry of Education, Culture and Science (Gravitation Program 024.001.035), and the University of Groningen are acknowledged. The authors would like to thank Ing. P. van der Meulen for the technical support during the EXSY experiments.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b10816.
The authors declare no competing financial interest.
Supplementary Material
References
- Eliel E. L.; Wilen S. H.. Stereochemistry of Organic Compounds; Wiley: New York, 1994. [Google Scholar]
- Yamamoto H.; Carreira E. M.. Comprehensive Chirality; Elsevier Science: Oxford, 2012. [Google Scholar]
- Blackmond D. G. Cold Spring Harbor Perspect. Biol. 2010, 2 (5), a002147 10.1101/cshperspect.a002147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley R. In Encyclopedia of Molecular Cell Biology and Molecular Medicine; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2006. [Google Scholar]
- Timsit Y.; Youri DNA Self-Assembly: From chirality to evolution. Int. J. Mol. Sci. 2013, 14 (4), 8252–8270. 10.3390/ijms14048252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer P. D. Molecular motors: What makes ATP synthase spin?. Nature 1999, 402 (6759), 247–249. 10.1038/46193. [DOI] [PubMed] [Google Scholar]
- Murata S.; Yashiroda H.; Tanaka K. Molecular mechanisms of proteasome assembly. Nat. Rev. Mol. Cell Biol. 2009, 10 (2), 104–115. 10.1038/nrm2630. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan V. Ribosome structure and the mechanism of translation. Cell 2002, 108 (4), 557–572. 10.1016/S0092-8674(02)00619-0. [DOI] [PubMed] [Google Scholar]
- Lodish H.; Berk A.; Zipursky S. L.; Matsudaira P.; Baltimore D.; Darnell J. In Mol. Cell. Biol., 4th ed.; W. H. Freeman: New York, 2000; Section 18.3. [Google Scholar]
- Berg J.; Tymoczko J.; Stryer L. In Biochemistry, 5th ed.; W. H. Freeman: New York, 2002; Section 34.3. [Google Scholar]
- Berg H. C.; Anderson R. A. Bacteria swim by rotating their flagellar filaments. Nature 1973, 245 (5425), 380–382. 10.1038/245380a0. [DOI] [PubMed] [Google Scholar]
- Bringmann G.; Price Mortimer A. J.; Keller P. A.; Gresser M. J.; Garner J.; Breuning M. Atroposelective synthesis of axially chiral biaryl compounds. Angew. Chem., Int. Ed. 2005, 44 (34), 5384–5427. 10.1002/anie.200462661. [DOI] [PubMed] [Google Scholar]
- Bringmann G.; Gulder T.; Gulder T. A.M.; Breuning M. Atroposelective total synthesis of axially chiral biaryl natural products. Chem. Rev. 2010, 111 (2), 563–639. 10.1021/cr100155e. [DOI] [PubMed] [Google Scholar]
- Rickhaus M.; Mayor M.; Juríček M. Strain-induced helical chirality in polyaromatic systems. Chem. Soc. Rev. 2016, 45 (6), 1542–1556. 10.1039/C5CS00620A. [DOI] [PubMed] [Google Scholar]
- Bolm C.; Muñiz K. Asymmetric Claisen rearrangement. Chem. Soc. Rev. 1999, 28 (1), 51–59. 10.1039/a801291a. [DOI] [Google Scholar]
- Sokolov V. I.Chirality and Optical Activity in Organometallic Compounds; Gordon and Breach Science Publishers: New York, 1990. [Google Scholar]
- Mislow K.Introduction to Stereochemistry; Dover Publications: Dover, 2003. [Google Scholar]
- Nguyen T. Giving atropisomers another chance. Chem. Eng. News 2018, 96, 33. [Google Scholar]
- Oki M. Recent Advances in atropisomerism. Top. Stereochem. 2007, 14, 1–81. 10.1002/9780470147238.ch1. [DOI] [Google Scholar]
- Leroux F. Atropisomerism, biphenyls, and fluorine: a comparison of rotational barriers and twist angles. ChemBioChem 2004, 5 (5), 644–649. 10.1002/cbic.200300906. [DOI] [PubMed] [Google Scholar]
- Collins B. S.L.; Kistemaker J. C.M.; Otten E.; Feringa B. L. A chemically powered unidirectional rotary molecular motor based on a palladium redox cycle. Nat. Chem. 2016, 8 (9), 860–866. 10.1038/nchem.2543. [DOI] [Google Scholar]
- LaPlante S. R.; Fader L. D.; Fandrick K. R.; Fandrick D. R.; Hucke O.; Kemper R.; Miller S. P.F.; Edwards P. J. Assessing atropisomer axial chirality in drug discovery and development. J. Med. Chem. 2011, 54 (20), 7005–7022. 10.1021/jm200584g. [DOI] [PubMed] [Google Scholar]
- Bandara H. M.D.; Burdette S. C. Photoisomerization in different classes of azobenzene. Chem. Soc. Rev. 2012, 41 (5), 1809–1825. 10.1039/C1CS15179G. [DOI] [PubMed] [Google Scholar]
- Feringa B. L.; van Delden R. A.; Koumura N.; Geertsema E. M. Chiroptical molecular switches. Chem. Rev. 2000, 100 (5), 1789–1816. 10.1021/cr9900228. [DOI] [PubMed] [Google Scholar]
- Erbas-Cakmak S.; Leigh D. A.; McTernan C. T.; Nussbaumer A. L. Artificial molecular machines. Chem. Rev. 2015, 115 (18), 10081–10206. 10.1021/acs.chemrev.5b00146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irie M.; Fukaminato T.; Matsuda K.; Kobatake S. Photochromism of diarylethene molecules and crystals: memories, switches, and actuators. Chem. Rev. 2014, 114 (24), 12174–12277. 10.1021/cr500249p. [DOI] [PubMed] [Google Scholar]
- Canary J. W. Redox-triggered chiroptical molecular switches. Chem. Soc. Rev. 2009, 38 (3), 747–756. 10.1039/b800412a. [DOI] [PubMed] [Google Scholar]
- Kelly T. R.; De Silva H.; Silva R. A. Unidirectional rotary motion in a molecular system. Nature 1999, 401 (6749), 150–152. 10.1038/43639. [DOI] [PubMed] [Google Scholar]
- Blanco V.; Leigh D. A.; Marcos V. Artificial switchable catalysts. Chem. Soc. Rev. 2015, 44 (15), 5341–5370. 10.1039/C5CS00096C. [DOI] [PubMed] [Google Scholar]
- Kottas G. S.; Clarke L. I.; Horinek D.; Michl J. Artificial molecular rotors. Chem. Rev. 2005, 105 (4), 1281–1376. 10.1021/cr0300993. [DOI] [PubMed] [Google Scholar]
- De Bo G.; Leigh D. A.; McTernan C. T.; Wang S. A complementary pair of enantioselective switchable organocatalysts. Chem. Sci. 2017, 8 (10), 7077–7081. 10.1039/C7SC02462B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher S. P.; Dumur F.; Pollard M. M.; Feringa B. L. A reversible, unidirectional molecular rotary motor driven by chemical energy. Science 2005, 310 (5745), 80–82. 10.1126/science.1117090. [DOI] [PubMed] [Google Scholar]
- Lin Y.; Dahl B. J.; Branchaud B. P. Net directed 180° aryl–aryl bond rotation in a prototypical achiral biaryl lactone synthetic molecular motor. Tetrahedron Lett. 2005, 46 (48), 8359–8362. 10.1016/j.tetlet.2005.09.151. [DOI] [Google Scholar]
- Dahl B. J.; Branchaud B. P. 180° Unidirectional bond rotation in a biaryl lactone artificial molecular motor prototype. Org. Lett. 2006, 8 (25), 5841–5844. 10.1021/ol062428u. [DOI] [PubMed] [Google Scholar]
- Anyika M.; Gholami H.; Ashtekar K. D.; Acho R.; Borhan B. Point-to-axial chirality transfer—a new probe for “sensing” the absolute configurations of monoamines. J. Am. Chem. Soc. 2014, 136 (2), 550–553. 10.1021/ja408317b. [DOI] [PubMed] [Google Scholar]
- Reichert S.; Breit B. Development of an axial chirality switch. Org. Lett. 2007, 9 (5), 899–902. 10.1021/ol0700660. [DOI] [PubMed] [Google Scholar]
- Escorihuela J.; Burguete M. I.; Luis S. V. New advances in dual stereocontrol for asymmetric reactions. Chem. Soc. Rev. 2013, 42 (12), 5595–5617. 10.1039/c3cs60068h. [DOI] [PubMed] [Google Scholar]
- Vlatković M.; Collins B. S.L.; Feringa B. L. Dynamic responsive systems for catalytic function. Chem. - Eur. J. 2016, 22 (48), 17080–17111. 10.1002/chem.201602453. [DOI] [PubMed] [Google Scholar]
- Romanazzi G.; Degennaro L.; Mastrorilli P.; Luisi R. Chiral switchable catalysts for dynamic control of enantioselectivity. ACS Catal. 2017, 7 (6), 4100–4114. 10.1021/acscatal.7b00827. [DOI] [Google Scholar]
- Wang J.; Feringa B. L. Dynamic control of chiral space in a catalytic asymmetric reaction using a molecular motor. Science 2011, 331 (6023), 1429–1432. 10.1126/science.1199844. [DOI] [PubMed] [Google Scholar]
- Vlatković M.; Bernardi L.; Otten E.; Feringa B. L. Dual stereocontrol over the Henry reaction using a light- and heat-triggered organocatalyst. Chem. Commun. 2014, 50 (58), 7773–7775. 10.1039/c4cc00794h. [DOI] [PubMed] [Google Scholar]
- Zhao D.; Neubauer T. M.; Feringa B. L. Dynamic control of chirality in phosphine ligands for enantioselective catalysis. Nat. Commun. 2015, 6, 6652. 10.1038/ncomms7652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao D.; van Leeuwen T.; Cheng J.; Feringa B. L. Dynamic control of chirality and self-assembly of double-stranded helicates with light. Nat. Chem. 2017, 9 (3), 250–256. 10.1038/nchem.2668. [DOI] [PubMed] [Google Scholar]
- Vlatković M.; Volarić J.; Collins B. S.L.; Bernardi L.; Feringa B. L. Org. Biomol. Chem. 2017, 15 (39), 8285–8294. 10.1039/C7OB01851G. [DOI] [PubMed] [Google Scholar]
- Koumura N.; Geertsema E. M.; van Gelder M. B.; Meetsma A.; Feringa B. L. Second generation light-driven molecular motors. unidirectional rotation controlled by a single stereogenic center with near-perfect photoequilibria and acceleration of the speed of rotation by structural modification. J. Am. Chem. Soc. 2002, 124 (18), 5037–5051. 10.1021/ja012499i. [DOI] [PubMed] [Google Scholar]
- Klok M.; Walko M.; Geertsema E. M.; Ruangsupapichat N.; Kistemaker J. C.M.; Meetsma A.; Feringa B. L. New mechanistic insight in the thermal helix inversion of second-generation molecular motors. Chem. - Eur. J. 2008, 14 (35), 11183–11193. 10.1002/chem.200800969. [DOI] [PubMed] [Google Scholar]
- Vicario J.; Walko M.; Meetsma A.; Feringa B. L. Fine tuning of the rotary motion by structural modification in light-driven unidirectional molecular motors. J. Am. Chem. Soc. 2006, 128 (15), 5127–5135. 10.1021/ja058303m. [DOI] [PubMed] [Google Scholar]
- Kistemaker J. C.M.; Pizzolato S. F.; van Leeuwen T.; Pijper T. C.; Feringa B. L. Spectroscopic and theoretical identification of two thermal isomerization pathways for bistable chiral overcrowded alkenes. Chem. - Eur. J. 2016, 22 (38), 13478–13487. 10.1002/chem.201602276. [DOI] [PubMed] [Google Scholar]
- Cahn R. S.; Ingold C.; Prelog V. Specification of molecular chirality. Angew. Chem., Int. Ed. Engl. 1966, 5 (4), 385–415. 10.1002/anie.196603851. [DOI] [Google Scholar]
- Prelog V.; Helmchen G. Basic principles of the CIP-System and proposals for a revision. Angew. Chem., Int. Ed. Engl. 1982, 21 (8), 567–583. 10.1002/anie.198205671. [DOI] [Google Scholar]
- Štacko P.; Kistemaker J. C.M.; van Leeuwen T.; Chang M.-C.; Otten E.; Feringa B. L. Locked synchronous rotor motion in a molecular motor. Science 2017, 356 (6341), 964–968. 10.1126/science.aam8808. [DOI] [PubMed] [Google Scholar]
- Sahnoun R.; Koseki S.; Fujimura Y. Density functional theoretical study on enantiomerization of 2,2‘-Biphenol. J. Phys. Chem. A 2006, 110 (7), 2440–2447. 10.1021/jp0554137. [DOI] [PubMed] [Google Scholar]
- van Leeuwen T.; Danowski W.; Pizzolato S. F.; Štacko P.; Wezenberg S. J.; Feringa B. L. Braking of a light-driven molecular rotary motor by chemical stimuli. Chem. - Eur. J. 2018, 24, 81–84. 10.1002/chem.201704747. [DOI] [PubMed] [Google Scholar]
- Cabrera E. V.; Sanchez J. L.; Banerjee A. K. A practical synthesis of 7,8-dimethoxy-2-tetralone. Org. Prep. Proced. Int. 2011, 43 (4), 364–367. 10.1080/00304948.2011.594008. [DOI] [Google Scholar]
- van Leeuwen T.; Gan J.; Kistemaker J. C.M.; Pizzolato S. F.; Chang M.-C.; Feringa B. L. Enantiopure functional molecular motors obtained by a switchable chiral-resolution process. Chem. - Eur. J. 2016, 22 (21), 7054–7058. 10.1002/chem.201600628. [DOI] [PubMed] [Google Scholar]
- Sheldrick G. M. Acta Crystallogr., Sect. A: Found. Adv. 2015, 71 (Pt 1), 3–8. 10.1107/S2053273314026370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldrick G. M. Acta Crystallogr., Sect. A: Found. Crystallogr. 2008, 64 (Pt 1), 112–122. 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- Hooft R. W.W.; Straver L. H.; Spek A. L. Using the t-distribution to improve the absolute structure assignment with likelihood calculations. J. Appl. Crystallogr. 2010, 43 (4), 665–668. 10.1107/S0021889810018601. [DOI] [Google Scholar]
- Lunazzi L.; Mancinelli M.; Mazzanti A. Correct values of the rotation barriers of 1,8-ditolylanthracenes. J. Org. Chem. 2007, 72 (26), 10045–10050. 10.1021/jo701917v. [DOI] [PubMed] [Google Scholar]
- Mazzanti A.; Lunazzi L.; Minzoni M.; Anderson J. E. Rotation in biphenyls with a single ortho-substituent. J. Org. Chem. 2006, 71 (15), 5474–5481. 10.1021/jo060205b. [DOI] [PubMed] [Google Scholar]
- Casarini D.; Lunazzi L.; Mancinelli M.; Mazzanti A.; Rosini C. Structure, conformation, stereodynamics, dimer formation, and absolute configuration of axially chiral atropisomers of hindered biphenyl carbinols. J. Org. Chem. 2007, 72 (20), 7667–7676. 10.1021/jo701481m. [DOI] [PubMed] [Google Scholar]
- Claridge T. D. W.High-Resolution NMR Techniques in Organic Chemistry; Newnes, 2008; Vol. 19. [Google Scholar]
- It should be emphasized that compared with compound 1, the methoxy-protected precursor 9 could be obtained and isolated as variable mixtures of nonequilibrating atropisomers (see Supporting Information for data). Notably, no isomerization or coalescence of sets of absorption was detected by 1H NMR spectroscopy even after prolonged heating of a sample of 9 (toluene solution, 110 °C, 12 h), confirming the important role of the hydroxyl groups and internal hydrogen-bonding in the atropisomerization process.
- Ceccacci F.; Mancini G.; Mencarelli P.; Villani C. Determination of the rotational barrier of a chiral biphenyl: Comparison of theoretical and experimental data. Tetrahedron: Asymmetry 2003, 14 (20), 3117–3122. 10.1016/S0957-4166(03)00544-5. [DOI] [Google Scholar]
- Trapp O.; Trapp G.; Schurig V. Direct calculation and computer simulation of the enantiomerization barrier of oxazepam in dynamic HPLC experiments—a comparative study. J. Biochem. Biophys. Methods 2002, 54 (1), 301–313. 10.1016/S0165-022X(02)00118-5. [DOI] [PubMed] [Google Scholar]
- Cabrera K.; Jung M.; Fluck M.; Schurig V. Determination of enantiomerization barriers by computer simulation of experimental elution profiles obtained by high-performance liquid chromatography on a chiral stationary phase. J. Chromatogr. A 1996, 731 (1–2), 315–321. 10.1016/0021-9673(95)01230-3. [DOI] [Google Scholar]
- Andreani R.; Bombelli C.; Borocci S.; Lah J.; Mancini G.; Mencarelli P.; Vesnaver G.; Villani C. New biphenylic derivatives: synthesis, characterisation and enantiodiscrimination in chiral aggregates. Tetrahedron: Asymmetry 2004, 15 (6), 987–994. 10.1016/j.tetasy.2004.01.038. [DOI] [Google Scholar]
- Degradation of compound 1 was observed in presence of strong bases and radical species, which led to addition of lower phenolic hydroxyl group to overcrowded alkene motif and loss of switching properties.
- Yoshikawa N.; Kumagai N.; Matsunaga S.; Moll G.; Ohshima T.; Suzuki T.; Shibasaki M. Direct catalytic asymmetric aldol reaction: synthesis of either syn- or anti-α,β-dihydroxy ketones. J. Am. Chem. Soc. 2001, 123 (10), 2466–2467. 10.1021/ja015580u. [DOI] [PubMed] [Google Scholar]
- Kumagai N.; Matsunaga S.; Yoshikawa N.; Ohshima T.; Shibasaki M. Direct catalytic enantio- and diastereoselective aldol reaction using a Zn–Zn-Linked-BINOL complex: A practical synthesis of syn-1,2-Diols. Org. Lett. 2001, 3 (10), 1539–1542. 10.1021/ol015878p. [DOI] [PubMed] [Google Scholar]
- Kumagai N.; Matsunaga S.; Kinoshita T.; Harada S.; Okada S.; Sakamoto S.; Yamaguchi K.; Shibasaki M. Direct catalytic asymmetric aldol reaction of hydroxyketones: asymmetric Zn catalysis with a Et2Zn/linked-BINOL complex. J. Am. Chem. Soc. 2003, 125 (8), 2169–2178. 10.1021/ja028926p. [DOI] [PubMed] [Google Scholar]
- Du H.; Long J.; Hu J.; Li X.; Ding K. 3,3‘-Br2-BINOL-Zn complex: a highly efficient catalyst for the enantioselective hetero-Diels–Alder reaction. Org. Lett. 2002, 4 (24), 4349–4352. 10.1021/ol027034r. [DOI] [PubMed] [Google Scholar]
- Noyori R.Asymmetric Catalysis in Organic Synthesis; Wiley: Chichester, 1994. [Google Scholar]
- Pu L.; Yu H.-B. Catalytic asymmetric organozinc additions to carbonyl compounds. Chem. Rev. 2001, 101 (3), 757–824. 10.1021/cr000411y. [DOI] [PubMed] [Google Scholar]
- Soai K.; Niwa S. Enantioselective addition of organozinc reagents to aldehydes. Chem. Rev. 1992, 92 (5), 833–856. 10.1021/cr00013a004. [DOI] [Google Scholar]
- Noyori R.; Kitamura M. Enantioselective addition of organometallic reagents to carbonyl compounds: chirality transfer, multiplication, and amplification. Angew. Chem., Int. Ed. Engl. 1991, 30 (1), 49–69. 10.1002/anie.199100491. [DOI] [Google Scholar]
- Hirose T.; Kodama K. In Comprehensive Organic Synthesis II; 2014; pp 204–266. [Google Scholar]
- Schmidt B.; Seebach D. Catalytic and stoichiometric enantioselective addition of diethylzinc to aldehydes using a novel chiral spirotitanate. Angew. Chem., Int. Ed. Engl. 1991, 30 (1), 99–101. 10.1002/anie.199100991. [DOI] [Google Scholar]
- Song T.; Zheng L.-S.; Ye F.; Deng W.-H.; Wei Y.-L.; Jiang K.-Z.; Xu L.-W. Modular synthesis of Ar-BINMOL-Phos for catalytic asymmetric alkynylation of aromatic aldehydes with unexpected reversal of enantioselectivity. Adv. Synth. Catal. 2014, 356 (8), 1708–1718. 10.1002/adsc.201301128. [DOI] [Google Scholar]
- Dean M. A.; Hitchcock S. R. Divergent enantioselective pathways in the catalytic asymmetric addition of diethylzinc to aldehydes in the presence and absence of titanium tetraisopropoxide. Tetrahedron: Asymmetry 2008, 19 (22), 2563–2567. 10.1016/j.tetasy.2008.10.033. [DOI] [Google Scholar]
- Shibata T.; Tarumi H.; Kawasaki T.; Soai K. Reversal in enantioselectivity induced by achiral alcohols in asymmetric autocatalysis in the presence of a chiral diol as a chiral initiator. Tetrahedron: Asymmetry 2012, 23 (14), 1023–1027. 10.1016/j.tetasy.2012.07.010. [DOI] [Google Scholar]
- Hirose T.; Sugawara K.; Kodama K. Switching of enantioselectivity in the catalytic addition of diethylzinc to aldehydes by regioisomeric chiral 1, 3-amino sulfonamide ligands. J. Org. Chem. 2011, 76 (13), 5413–5428. 10.1021/jo200834n. [DOI] [PubMed] [Google Scholar]
- Yue H.; Huang H.; Bian G.; Zong H.; Li F.; Song L. Enantioselectivity switch controlled by N,N′-di- or N,N,N′,N′-tetra-substituted chiral thiophosphorodiamide ligands, structural relatives of thioureas, in catalytic additions of diethylzinc to aldehydes. Tetrahedron: Asymmetry 2014, 25 (2), 170–180. 10.1016/j.tetasy.2013.12.014. [DOI] [Google Scholar]
- Mao J.; Wan B.; Zhang Z.; Wang R.; Wu F.; Lu S. Reversal of enantioselectivity by adding Ti (OiPr) 4 novel sulfamide-amine alcohol ligands for the catalytic asymmetric addition of diethylzinc to aldehydes. J. Mol. Catal. A: Chem. 2005, 225 (1), 33–37. 10.1016/j.molcata.2004.08.027. [DOI] [Google Scholar]
- Costa A. M.; Jimeno C.; Gavenonis J.; Carroll P. J.; Walsh P. J. Optimization of catalyst enantioselectivity and activity using achiral and meso ligands. J. Am. Chem. Soc. 2002, 124 (24), 6929–6941. 10.1021/ja0166601. [DOI] [PubMed] [Google Scholar]
- Erdik E. In Organozinc Reagents in Organic Synthesis; CRC Press: Boca Raton, FL, 1996; pp 215–216. [Google Scholar]
- Weber B.; Seebach D. Ti-TADDOLate-catalyzed, highly enantioselective addition of alkyl-and aryl-titanum derivatives to aldehydes. Tetrahedron 1994, 50 (25), 7473–7484. 10.1016/S0040-4020(01)90475-2. [DOI] [Google Scholar]
- The same reaction catalyzed by (P)-3,3′-diphenyl BINOL was reported to yield the product in 44% ee (see ref (82)).
- It should be noted that the quality of the commercial diethylzinc solution had a large impact on the results of the catalytic reactions, affording lower yields and selectivities as degradation of the reagent progressed.
- Zong H.; Huang H.; Bian G.; Song L. Synergistic effect of achiral quaternary ammonium salt on asymmetric additions of diethylzinc to aldehydes. Tetrahedron Lett. 2013, 54 (21), 2722–2725. 10.1016/j.tetlet.2013.03.066. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.