

Abstract
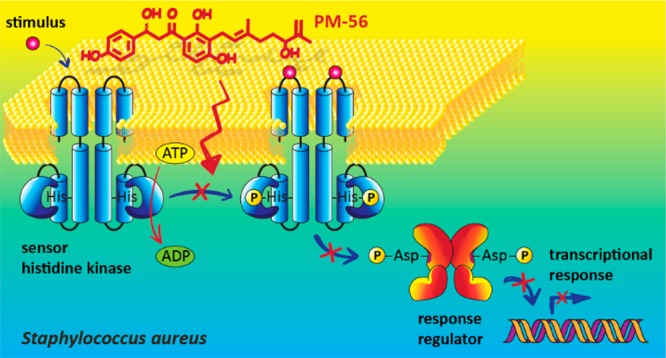
As an alternative strategy to fight antibiotic resistance, two-component systems (TCSs) have emerged as novel targets. Among TCSs, master virulence regulators that control the expression of multiple virulence factors are considered as excellent antivirulence targets. In Staphylococcus aureus, virulence factor expression is tightly regulated by a few master regulators, including the SaeRS TCS. In this study, we used a SaeRS GFP-reporter system to screen natural compound inhibitors of SaeRS, and identified xanthoangelol B 1, a prenylated chalcone from Angelica keiskei as a hit. We have synthesized 1 and its derivative PM-56 and shown that 1 and PM-56 both had excellent inhibitory potency against the SaeRS TCS, as demonstrated by various in vitro and in vivo experiments. As a mode of action, 1 and PM-56 were shown to bind directly to SaeS and inhibit its histidine kinase activity, which suggests a possibility of a broad spectrum inhibitor of histidine kinases.
Introduction
Antibiotic resistance has arisen as an extremely serious health problem in all parts of the world. New resistance mechanisms are emerging and spreading globally, threatening our ability to treat common infectious diseases. The list of resistant infections–such as pneumonia, tuberculosis, blood poisoning, gonorrhea, and foodborne diseases–is growing longer and sometimes these are impossible to treat as antibiotics become less effective.1,2 It is estimated that, by 2050, worldwide drug-resistant infections will claim 10 million deaths annually. One particularly problematic pathogen is methicillin-resistant Staphylococcus aureus (MRSA). MRSA is a nosocomial and communal menace that is resistant to many antibacterial drugs and antiseptics.3−5 The U.S. Centers for Disease Control and Prevention recently issued a report to various governments and organizations outlining the concern about MRSA. This report referenced the New Drugs for Bad Bugs (ND4BB) project initiated by the European Union, the United States military, and the World Health Organization (WHO).6,7 The Antimicrobial Resistance Global Report on Surveillance released by the WHO in 2014 outlined a global initiative to tackle the MRSA superbug and rekindle a campaign for new, effective, and safe drugs.8−10 Since they suppress bacterial growth, conventional antibiotics are confined to a limited lifespan. It is urgent to establish a new path forward for medicinal chemists to address successfully this problem.
Among many new approaches, the inhibition of virulence factors has shown promising outcomes. This strategy bypasses the route of killing pathogens and thereby has the potential to suppress the development of resistance. Many such concepts have been tested.11−13 Among these, two-component systems (TCSs) that control virulence factors have been of particular interest.14−17 Some TCSs control multiple virulence factors in pathogens and are thus called master virulence regulators.18 These TCSs are attractive targets for novel antivirulence agents. We identified one such TCS, the SaeRS system of S. aureus, as an excellent antivirulence target that is less likely to evolve resistance.19 We developed a GFP reporter system that can be applied to a high-throughput assay format. Using this assay, we discovered certain FDA-approved drugs that exert antivirulence effects on S. aureus.20 In continuing such studies, we identified natural compound inhibitors of the SaeRS TCS by screening plant-derived products21 using the same assay. Of the identified natural products that showed promising inhibitory activity, we chose xanthoangelol B for further study including the synthesis of complete xanthoangelol B, fragments, and a derivative, to test for antivirulence activities.
Xanthoangelol B is a prenylated chalcone isolated from Ashitaba (Angelica keiskei Koidzumi, Apiaceae). The Japanese herb Ashitaba is a perennial plant found along the Pacific coast of Japan. Ashitaba is enriched with numerous active compounds such as coumarins, flavanones, and chalcones.22−25 Xanthoangelol B is relatively scarce among these compounds, although it is found in all parts of the plant.26 It shows various therapeutic properties, such as antioxidative activity, antihypertensive activity, anti-inflammatory activity, and antiviral activity.27 However, although its related chalcones, xanthoangelol and 4-hydroxyderricin, were shown to have an antibacterial activity against Gram-positive bacteria,28 its antibacterial activity has not been established. Thus, given our intriguing screening results, we set out for an in-depth structure–activity study of xanthoangelol B. We were particularly interested in its effect on the virulence of S. aureus and its feasibility as an antivirulence agent.
Xanthoangelol B has a modular structure composed of chalcone and isoprene moieties. It was deemed important to analyze the biological activity of its fragments to establish any structure–activity relationships. Thus, we synthesized fragments of xanthoangelol B along with the parent compound and tested their activities. During the synthesis, we obtained a derivative of xanthoangelol B (compound PM-56) that had a comparable activity and tested its activities. Here, we report these processes and describe the antivirulence activities of the compounds that we synthesized.
Results and Discussion
Chemistry
The synthesis of biologically relevant geranylated flavanones and geranylated chalcones (such as (±)-prostratol F, (±)-6-geranyl-5,7-dihydroxy-3′,4′-dimethoxyflavanone, xanthoangelol, 3-geranyl-2,3′,4,4′-tetrahydroxychalcone, and (±)-lespeol) has been reported.29−31 With these precedents, we designed our syntheses using a retrosynthetic analysis, as outlined in Scheme 1. Xanthoangelol B 1 can be disconnected to two main fragments: fragment 3 and 4-hydroxybenzaldehyde 2. Fragment 3 can be obtained from 2′,4′-dihydroxyacetophenone 4, which is commercially available. There are two ways to synthesize this fragment 3 (Scheme 1). A synthetic route to 1 was also designed to generate various fragments that could be evaluated for their biological activity. The fragments shown in Scheme 1 can provide insight into the structure–activity relationship of 1 that may help to curtail or enhance its activity.
Scheme 1. Retrosynthetic Analysis of Xanthoangelol B 1.

Our synthetic design involved the limited use of protecting groups (Scheme 2). Our initial approach was to introduce 2,6-dimethylhepta-1,6-dien-3-ol at the end of the synthesis to reduce complications arising from the chiral aliphatic hydroxyl group. The 4′-hydroxyl position of 2′,4′-dihydroxyacetophenone was alkylated with allyl bromide that was heated at 220 °C in N,N-diethylaniline for 24 h. This allowed for Claisen rearrangement, giving a 3,3-sigmatropic-shifted major product 10 with a 76% yield. The minor product was 5-allyl-2,4-dihydroxy acetophenone. The regioselectivity of Claisen rearrangement giving a 3-allylated major product is well documented.32 Compound 10 was then subjected to aldol condensation by refluxing for 18 h in KF-alumina (40%), a solid-support catalyst in toluene. Compound 11 was obtained as the major product, with a yield of approximately 60%. This yield was slightly lower than that of most aldol condensation reactions owing to the two 2′,4′-OH groups. A reaction longer than 18 h resulted in the formation of a nonpolar mixture of spots observed by TLC analysis. The nature of the solvent also drastically influenced the yield of the reaction. 2,6-Dimethylhepta-1,6-dien-3-ol was introduced later by initially synthesizing 2,2-dimethyl-3-(3-methylbut-3-en-1-yl)oxirane 8 from citral by subjecting it to decarbonylation via heating at 130 °C in the presence of catalytic amounts of palladium acetate (10 mol %) in a sealed tube for 24 h. This produced 2,6-dimethylhepta-1,5-diene 12, with a yield of ca. 62%.33 Dimethylhepta-1,5-diene was subjected to selective oxidation using m-chloroperbenzoic acid (mCPBA) to obtain an epoxide 8 with a 45% yield.34 Compound 8 was then subjected to cross-olefin metathesis with 11, using a Hoveyda–Grubbs second-generation catalyst to yield 70% of an E-product 13.35 The epoxide ring was subsequently subjected to selective ring opening reactions or isomerization using in situ-generated diethylaluminum 2,2,6,6-tetramethyl-piperidide to yield the desired product xanthoangelol B 1 at a 69% yield.36
Scheme 2. Synthesis of Xanthoangelol B 1.

There were a few interesting observations during the synthesis of 1 (Scheme S1). As an alternative route to xanthoangelol B, the introduction of 2,6-dimethylhepta-1,6-dien-3-ol to the 3′-position of 2,4-dihydroxyacetophenone resulted in decreased reactivity toward condensation reactions. Use of a solid-support reagent (such as KF-alumina) or use of highly basic conditions yielded no product or a complex mixture of products. Heating the reaction in the presence of a high boiling-point solvent (either in highly basic or acidic conditions) yielded no desired product. Finally, the use of l-proline as an organic catalyst37,38 resulted in an aldol addition product 15. However, this product did not react further (to elimination) to yield 1 in any of the acidic or basic conditions attempted. Thus, 15 was subjected to selective epoxide isomerization, and the resulting compound PM-56 was tested for antivirulence activities as a derivative of 1.
Synthesized xanthoangelol B 1 was structurally identical to the natural product, except for the chiral center. Selective epoxidation and subsequent epoxide ring opening resulted in an enantiomeric mixture. We speculated that the number of required steps to synthesize 1 could be reduced by using a longer chain (i.e., geranyl bromide) and carrying out subsequent selective epoxidations, which could be reacted further to yield 1. This would provide us with an additional fragment for structure–activity tests and allow us to incorporate the metal-free chemistry. Thus, we devised a shorter synthetic route (without using metal catalysts) by minimizing the use of protecting groups. Specifically, 2,4-dihydroxy acetophenone was subjected to aldol condensation to give an intermediate that was reacted with 1,2-epoxy geranyl bromide. This generated 13, which underwent selective epoxy-isomerization to give 1 as an enantiomeric mixture (Scheme 3). Then, we attempted an optical resolution of synthesized compound 1. We chose a method of enantioselective organocatalyst oxidative kinetic resolution using the 7-Bn-3-n-Bu-4-oxa-5-azahomoadamantane/trichloroisocyanuric acid (TCCA) system to yield a resolved 1.39 The 1H and 13C NMR spectra of the resolved compound 1 were in agreement with spectra previously reported.21 The mass spectrum showed a peak at 409.19 corresponding to [M + H]+, consistent with the calculated mass of the desired product. In a direct comparison using HPLC, the synthetic and resolved 1 was shown to elute at exactly the same retention time as the natural product (Figure S1). The optical rotation for kinetically resolved 1 was αD = +13.5 (MeOH, c = 0.5). This is consistent with the data reported for naturally extracted 1,23 which indicates the authenticity of the kinetically resolved synthetic xanthoangelol B 1.
Scheme 3. Alternative Route for the Synthesis of Xanthoangelol B 1.

No metal catalysts involved.
Biological Evaluation of Xanthoangelol B, Its Fragments, and Its Derivative
Previously, we constructed a SaeRS GFP reporter strain based on the S. aureus USA300 isolate and found promising leads among FDA-approved drugs through a high throughput screening.20 The same SaeRS GFP reporter strain was constructed for screening a natural product library in this study. Briefly, as the α-hemolysin promoter (Phla) is a well-characterized target of theSaeRS TCS, we generated a minimal Phla, termed Phlam, which contains only the SaeR binding sites and the −10/–35 promoter sequences of the hemolysin gene (hla). Phlam was fused to a green fluorescence protein (gfp) gene instead of hla in the single-copy integration plasmid pCL55. The resulting plasmid (pCL-Phlam-gfp) was inserted into S. aureus USA300. Hence, if the expression of GFP in a sample was lower than the control sample, it meant that the SaeRS TCS was inhibited. As the complete inhibition of Phlam requires a complete shutdown of the SaeRS TCS (such as a deletion of saeS), Phlam will detect only potent inhibitors of the SaeRS reporter. Xanthoangelol B 1 was one of the hits among the natural products screened with this method. Thus, we assayed 1, its fragments, and a derivative (PM-56). The structures of all the assayed compounds and fragments are presented in Scheme 4 and Scheme S2.
Scheme 4. Derivatives of Xanthoangelol B.

Structure–Activity Studies of Xanthoangelol B and Its Inhibitory Effect on the SaeRS TCS
On initial screening, 1 and PM-56 showed an excellent inhibitory activity against SaeRS. These compounds repressed GFP expression while allowing bacterial growth, making them ideal antivirulence candidates. In contrast, all of the fragments (Scheme S2) failed to inhibit the SaeRS TCS, even at 20 μM. This indicates that the whole structure is needed for the inhibitory activity. Furthermore, xanthoangelol and xanthoangelol F (Scheme 4) did not show inhibitory activity at 20 μM.
These two analogs are also natural prenylated chalcones from Ashitaba, but they lack a hydroxyl group in the terminal isoprene unit. This hydroxyl group appears to be crucial for the antivirulence activity of this class of compounds. Xanthoangelol with an identical structure to that of 1, except for this hydroxyl group, did not have any activity (although methoxylation in the central phenol hydroxyl group of xanthoangelol F may still have an impact). IC50 values against the SaeRS GFP reporter were measured to be 2.1 μM for 1 and 4.3 μM for PM-56. In these experiments (at up to 8 and 16 μM), 1 and PM-56 did not disrupt bacterial growth (Figure 1A,C). To elaborate the inhibitory effect of both compounds on the growth of S. aureus, we measured the growth of S. aureus in the presence of 1 and PM-56 at higher concentrations up to 20 h. From 16 μM, 1 started showing growth inhibition, and at 64 μM of 1, the growth of S. aureus was severely inhibited (Figure S2). However, up to 64 μM of PM-56, S. aureus still showed comparable growth to the control (Figure S2). The trends shown at 8 h did not change up to 20 h. This indicates that the rigidity conferred to the chalcone structure by the conjugated double bond is not essential for anti-SaeRS activity but may lead to higher toxicity (which affects bacterial growth). Together, these data strongly indicate that the isoprene and chalcone moieties are necessary in their entirety for the inhibitory activity.
Figure 1.

Various concentrations of compounds were used to treat the USA300-Phlam GFP strain, and GFP expression was analyzed at 8 h. (A) Measurement of bacterial growth in the presence of xanthoangelol B 1; (B) measurement of fluorescence in the presence of xanthoangelol B 1; (C) measurement of bacterial growth in the presence of PM-56; (D) measurement of fluorescence in the presence of PM-56. All the experiments were performed in triplicates, and significance was compared with control (no treatment) using one-way ANOVA.
Effects of Inhibitors on Downstream Virulence Factors
Staphylococcal α-hemolysin, which causes lysis of erythrocytes, is the target virulence factor of the SaeRS TCS. Therefore, if the SaeRS TCS is inhibited, the expression of α-hemolysin should decrease to a certain degree. When 1 and PM-56 were incubated with erythrocytes in the presence of methicillin-resistant S. aureus USA300, notable protection from hemolysis was observed (Figure 2 and Figure S3). At only 4 μM, both compounds showed visible protection. At 8 μM, a comparable level of protection to that of saeS knockout mutants (SaeS::Tn551) was observed. The hemolysis activity of this saeS mutant was reduced to 82.6% in comparison to the wild type S. aureus. To confirm the relevance of the protection effect observed with 1 and PM-56 to the SaeRS pathway, we also tested 1 and PM-56 against the saeS knockout mutant. This experiment conducted at 8 μM of 1 and 15 μM of PM-56 showed the reduction in hemolysis by 86.9% and 87.4%, respectively, compared to hemolysis by the wild type (Figure S4). Although apparent further inhibition appeared to exist, the differences were not statistically significant, which thus suggests that the protection against hemolysis by 1 and PM-56 is highly likely attributed to inhibition of the SaeRS pathway. The IC50 values of 1 and PM-56 for the inhibition of erythrocyte hemolysis were then measured to be 4.6 and 4.0 μM, respectively. These values were consistent with the IC50 values of both compounds for the SaeRS TCS GFP reporter. Therefore, these strongly suggests that the protection of erythrocytes from hemolysis in the presence of MRSA can be attributed to inhibition of the SaeRS TCS.
Figure 2.

Percentage hemolysis values were determined after treating samples with compounds 1 and PM-56 after an 8 h incubation. (A) Compound 1: various concentrations were used to treat human RBCs, and percentage hemolysis values were calculated relative to the control sample. (B) Compound PM-56: various concentrations were used to treat human RBCs, and percentage hemolysis values were calculated relative to the control sample. All the experiments were performed in triplicates and the data calculated by one-way ANOVA. CT, positive control; SaeS::Tn551, saeS knockout.
We next analyzed the transcription of virulence genes in the presence of 1 and PM-56 (Figure 3). We used concentrations of 8 μM (1) and 10 μM (PM-56), corresponding to an IC90 value for the SaeRS GFP reporter. We observed significant transcriptional suppression of the following genes: α-hemolysin (hla), aureolysin (aur), γ-hemolysin, and staphylokinase. These four genes have a direct SaeR binding site in their promoter regions and are tightly regulated by the SaeRS TCS.40 While it has been reported that hla is regulated by an accessory gene regulator (agr) as well as by sarA, a direct connection between these regulatory systems and the expression of hla remains under debate.41 Their suppression likely reflects the inhibitory effects of both compounds on the SaeRS TCS. Then, we analyzed the transcription of a gene encoding Staphylococcal accessory regulator (SarA). SarA is responsible for the biofilm formation and not regulated by saeS. Thus, it was considered a good negative control, and its transcription analyzed post treatment of xanthoangelol B 1 and PM-56 (Figure S5). As anticipated, it can be seen that the level of sarA transcription was not significantly affected by the two compounds. These results imply that 1 and PM-56 indeed inhibited the SaeRS pathway with specificity and thus are promising candidates for future development of antivirulence agents or chemical probes specific for the SaeRS two-component system.
Figure 3.

Effects of 1 and PM-56 on the virulence gene expression were analyzed: (A) hla; (B) aureolysin (aur); (C) ϒ-hemolysin; (D) staphylokinase. 16S rRNA was used as a reference control gene. All the experiments were performed in triplicates and the data compared with no treatment control and calculated by one-way ANOVA.
Cytotoxicity
In order to explore the potential of xanthoangelol B 1 and PM-56 as initial leads for further translation, we have examined the cytotoxicity of both compounds on HeLa cells, quantitatively and qualitatively. First, LDH release assay was performed, in which we have treated HeLa cells with 1 and PM-56 at various concentrations (2–128 μM) for 72 h and measured the optical density at 450 nm. It was shown that IC50 values for HeLa cell survivals were 19.70 and 28.94 μM with 1 and PM-56, respectively (Figure 4A,B). These values were 10-fold and 7-fold higher than IC50 values for the GFP-reporter inhibition by 1 and PM-56, respectively. Hence, the toxicity/efficacy ratio is reasonable for the initial leads. Next, the live/dead imaging experiment has been performed. The HeLa cells were seeded in 96-well plates for 24 h, and after attachment of cells, the culture media (DMEM, 10% FBS, 1% penicillin/streptomycin) were changed, followed by treatment with 1 and PM-56 at various concentrations (from 2 to 64 μM) for 72 h. The HeLa cells stained with fluorescein diacetate dye (live cell staining, green color) and propidium iodide dye (dead cell staining, red color) were observed using the confocal microscopy (Figure 4 C,D).
Figure 4.

Cytotoxicity of xanthoangelol B and PM-56 on HeLa. (A) Xanthoangelol B and (B) PM-56): LDH release assay. HeLa cells were seeded in 5 × 105 cells and treated with xanthoangelol B 1 and PM-56 from 2 to 128 μM and incubated for 72 h. The LDH release was analyzed at OD450 nm. All the experiments were performed in three biological replicates, and data are presented as a mean ± SEM. (C) Xanthoangelol B and (D) PM-56: live/dead cell staining. HeLa cells were seeded 5 × 105 CFU in a 96-well plate and incubated 24 h for attachment. The various concentrations of 1 and PM-56 (2 to 64 μM) were tested for 72 h. Then, the cells were stained with FDA and PI and imaged under the confocal microscope. Green, live; red, dead, (i) vehicle control; (ii) 2 μM; (iii) 4 μM; (iv) 8 μM; (v) 16 μM; (vi) 32 μM; (vii) 64 μM. Images are based on n = 3 independent experiment.
Confocal images were in accordance with the quantitative results as mentioned earlier. Both compounds at their IC90 (approximately 8 μM) for the GFP-reporter did not produce substantially visible toxicity to the HeLa cells, while a reduction in viability was observed only when the cells were treated with concentrations higher than IC50 for cytotoxicity. Cytotoxicity tests demonstrated that 1 and PM-56 indeed had the promising traits as initial leads.
Validation of the Antivirulence Activity of Compounds 1 and PM-56 Using a Worm Infection Model
With the encouraging in vitro data, we further validated the antivirulence activity of 1 and PM-56 in a Galleria mellonella infection model. G. mellonella worms possess both a humoral and an innate immune system. The worms provide an attractive, simple, and easy-to-handle infection model with few biosafety or ethical issues. The G. mellonella infection model has previously been used with several human pathogens to test the in vivo efficacy of antibacterial and antivirulence drugs.42 Thus, G. mellonella larvae was challenged with 5 × 106 colony forming units (CFUs), a load designed to allow only 20% survival after 72 h. The larvae was post-treated with various doses of either 1 or PM-56 at 12 h intervals, up to 72 h. Larvae health indices were strictly followed and recorded. A dose-dependent protection from virulence by both compounds (1 and PM-56) was observed in the larvae (Figure 5). Compared to the vehicle control, 1 and PM-56 generated a 3.5-fold higher survival at doses as low as 2.0 and 2.1 mg/kg, respectively. Compound 1 showed 90% survival at a dose of 3.1 mg/kg and complete survival at 4.1 mg/kg. PM-56 showed 90% survival at 3.2 mg/kg and complete survival at 4.3 mg/kg. These survival rates were comparable to or slightly better than survival rates observed in worms infected with saeS knockout strains.
Figure 5.
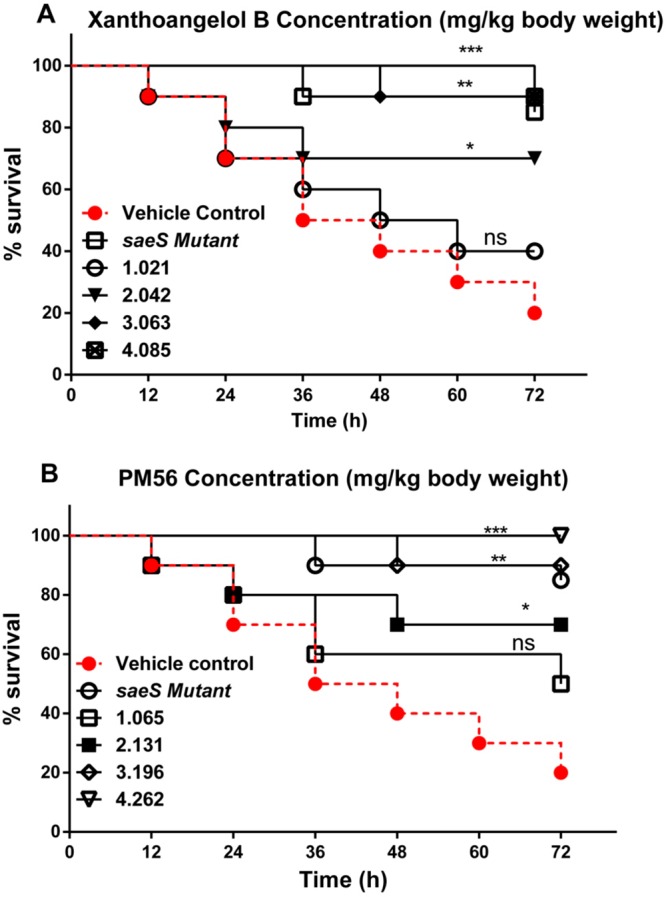
Ability of compounds 1 (A) and PM-56 (B) to protect G. mellonella from lethal S. aureus infection. Kaplan–Meier survival curves for G. mellonella challenged with 5 × 106 CFU S. aureus and treated with compounds 1 and PM-56 at 2 h postinfection. The compounds were injected every 12 h for 72 h. The experiments were performed using biological replicate samples (n = 10). ***P < 0.001, **P < 0.01, and *P < 0.05 by Student’s t-test.
Next, we measured bacterial burden in the host worms. As shown in Figure S4, the inactivation of saeS resulted in a decline in the bacterial burden in the host worms (up to 1.7 log10 CFU/worm). Treatment with 1 or PM-56 significantly lowered the bacterial burden in worms in a dose-dependent manner. Compared to the vehicle control, a dose of approximately 1 mg/kg allowed a sharp decrease in the bacterial burden. At concentrations of 4.1 and 4.3 mg/kg, respectively, 1 and PM-56 generated a decline of 1.72 log10 and 1.84 log10 CFU/worm (Figure S6). Both compounds again showed comparable efficacy to that of saeS knockout. These results are highly consistent with survival tests. Then, to evaluate whether the activities of 1 and PM-56 were based on the saeS-dependent pathway, we have measured the effects of both compounds on G. mellonella infected with the saeS knockout mutant under the same conditions as above. G. mellonella were challenged with 5 × 106 CFU of the wild type and the saeS knockout S. aureus, which were subsequently treated with 1 (2.042 mg/kg body weight) or PM-56 (2.131 mg/kg body weight) for every 12 h interval for 72 h. These concentrations were corresponding to IC90 for the SaeRS GFP reporter. The survival rates of G. mellonella infected with the knockout strain treated with 1 and PM-56 were 70% and 90%, respectively, while they were 20% for the wild type-infected larvae and 80% for the knockout strain-infected larvae without compounds (Figure S7A), which showed protection effects were comparable. Then, we counted CFU from G. mellonella of each group after a 72 h infection. It was shown that CFU was significantly decreased in the saeS knockout group as compared to the wild type infection group by up to 74.5%. Compounds 1 and PM-56 treated groups also showed comparable reduction in CFU by 76.8% and 83.3%, respectively (Figure S7B). Together, these results suggest that the protection effects initiated by the saeS knockout were not drastically improved by the treatment with 1 and PM-56, and subsequently, the activities of both compounds on G. mellonela were relevant to the saeS-dependent pathway although we cannot rule out an extra activity of PM-56 as the effects of PM-56 consistently showed a slight but nonsignificant improvement over the saeS knockout.
Overall, the superb protection given by 1 and PM-56 and the sharp decline in the bacterial burden following their use allowed us to draw comparisons with saeS knockout experiments. This hinted at a mode of action for these compounds. Thus, we next tested 1 and PM-56 for their ability to inhibit the histidine kinase or phosphotransferase activities of SaeS.
Effects of Compounds 1 and PM-56 on the Histidine Kinase and Phosphotransferase Activities of SaeS
The sensor histidine kinase (HK) of a bacterial two-component system represents a promising antibacterial target, as the catalytic and ATP-binding (CA) domain of the sensor HK is well conserved across bacterial species. Thus, an inhibitor binding this domain has the potential to be a broad-spectrum antibacterial agent.15 Indeed, many efforts have been made to discover such a class of inhibitors, with some degree of success.15,43,44 Compounds 1 and PM-56 displayed excellent SaeRS-related antivirulence activity in vitro and in vivo. These results were comparable to the effects seen in saeS knockout studies. Therefore, it was of great interest to determine whether these compounds inhibited the sensor HK SaeS. We tested the two compounds against the purified minimal kinase domain of SaeS. The minimal kinase domain contains a dimerization and histidine phosphotransfer (DHp) domain and a CA domain (amino acid residues 118–351). Measurements were conducted using a radioisotope method with SDS-PAGE autoradiography. Both compounds clearly inhibited the autophosphorylation of SaeS (Figure 6), with IC50 values of 220 and 160 μM for 1 and PM-56, respectively. This was generally consistent with their observed activities comparable to the saeS knockout. In contrast, when we measured the inhibition of phosphotransferase (PT) activity using radioactively phosphorylated SaeS in the presence of SaeR, no inhibition was observed (Figure S8). This implies that only the HK activity (and not the PT activity) of SaeS is relevant to the inhibitory activity of 1 and PM-56.
Figure 6.
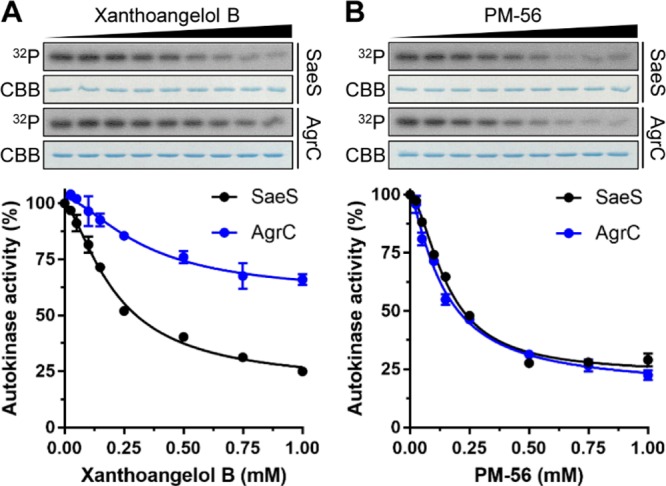
Inhibitory effects of xanthoangelol B 1 and PM-56 on the in vitro autokinase activities of SaeS and AgrC. Purified recombinant minimal kinase domains of SaeS and AgrC (5 μM each) were incubated at various concentrations (0–1 mM) of 1 (A) or PM-56 (B) for 15 min on ice before addition of ATP to initiate the autophosphorylation reactions. After SDS-PAGE analysis, total and 32P-labeled proteins were visualized by Coomassie-Brilliant Blue staining and autoradiography (32P), respectively. Each lane contains approximately 0.5 μg purified SaeS or AgrC. The 32P-labeled protein bands were quantified by densitometric analysis, and the autokinase activities were calculated as the percentages of 32P-labeled proteins in the samples treated with inhibitory chemicals compared to those in the control samples treated with 10% DMSO. Dose–response curves were generated by plotting the percent autokinase activity against the chemical concentration. The experiments were conducted in duplicate.
To probe a possibility of 1 and PM-56 as broader HK inhibitors, we also tested both compounds on AgrC, a component of another master virulence regulator and the most well characterized TCS in S. aureus.45 Thus, it was shown that 1 and PM-56 inhibited AgrC with IC50 values of 339 and 140 μM, respectively (Figure 6). PM-56 showed a comparable activity to that for SaeS, while 1 showed 1.5-fold lower activity compared to that for SaeS. This extra activity of PM-56 may explain slightly higher but nonsignificant improvement in the protection effect of PM-56 over the saeS knockout. Furthermore, PM-56 was 1.4-fold higher activity than 1 for SaeS, while this difference expanded to 2.4-fold for AgrC. Thus, this suggests a potential of the xanthoangelol B scaffold for further development of broad-spectrum or specific inhibitor of bacterial histidine kinases.
Next, we attempted to measure the direct binding of 1 and PM-56 with SaeS using the intrinsic fluorescence quenching assay. Since SaeS contains seven Tyr (and zero Trp) residues, the Tyr fluorescence at 306 nm was monitored before and after binding of 1 and PM-56. The reduction of fluorescence upon ligand binding was expressed as the percent fluorescence quenching, and the Kd value was calculated by nonlinear regression fitting (Figure S9). The quenching assay revealed that 1 and PM-56 bound to SaeS with the Kd values of 40 and 6.8 μM, respectively. These Kd values are smaller than IC50 values. It is not rare that IC50 values are measured to be much higher than Kd values,46−48 and the data are clear indication that both compounds directly bind to SaeS with consistent trends of stronger interaction of SaeS with PM-56 than with 1.
Since the binding of PM-56 to SaeS was stronger than that of 1 as revealed in the inhibition and fluorescence quenching studies, we selected PM-56 for further dissection of the inhibitor binding to SaeS. To identify the binding of PM-56 to SaeS, one-dimensional transverse relaxation filtered experiment with a Carr–Purcell–Meiboom–Gill pulse trains (1D CPMG) and 1D saturation transfer difference experiment (1D STD) were performed in the presence and absence of the minimal kinase domain of SaeS.49 The 1D CPMG experiment is able to detect the reduced T2-relaxation time of the PM-56 peaks caused by the interaction with SaeS, which results in the peak broadening and decreasing. The STD NMR spectra obtained with the minimal kinase domain of SaeS were virtually identical to those with the kinase domain of SaeS fused to maltose binding protein (MBP-SaeS) (Figure 7 and Figure S10), indicating that the presence of MBP does not influence the binding. Therefore, MBP-SaeS was used for interpretation of the 1D STD experiment to enhance the efficiency of saturation transfer from the protein to PM-56. The transferred saturation from the protein to the ligand is amplified by the binding exchange process and is efficient for target proteins with large molecular weight.50PM-56 clearly displayed the binding exchange process to SaeS. Simple 1D spectra also showed that the peaks of PM-56 were specifically broadened and shifted by SaeS, as compared to those of free PM-56 (Figure 7A). Although the SaeS and MBP-SaeS proteins were dialyzed in the NMR-buffer before the NMR experiments, the HEPES molecule included in the purified proteins was not completely removed. Nevertheless, the spectra of the 1D CPMG (Figure 7B) and 1D STD (Figure 5C) clearly show that PM-56 specifically binds to SaeS, but HEPES did not. Furthermore, the similar peak intensities between the 1D spectrum (Figure 7A, red spectrum) and the 1D STD spectrum (Figure 7C) showed putative interactions involving most parts of PM-56. Hence, the entire molecular structure appears to be needed to exert a meaningful inhibition, which is consistent with the test of fragments. The presence of binding exchange process accompanied by the clear peak broadening of PM-56 indicates that the binding affinity between SaeS and PM-56 is likely not too strong (i.e., nanomolar Kd) or too weak (i.e., millimolar Kd), which is in agreement with inhibition and binding measurements.
Figure 7.
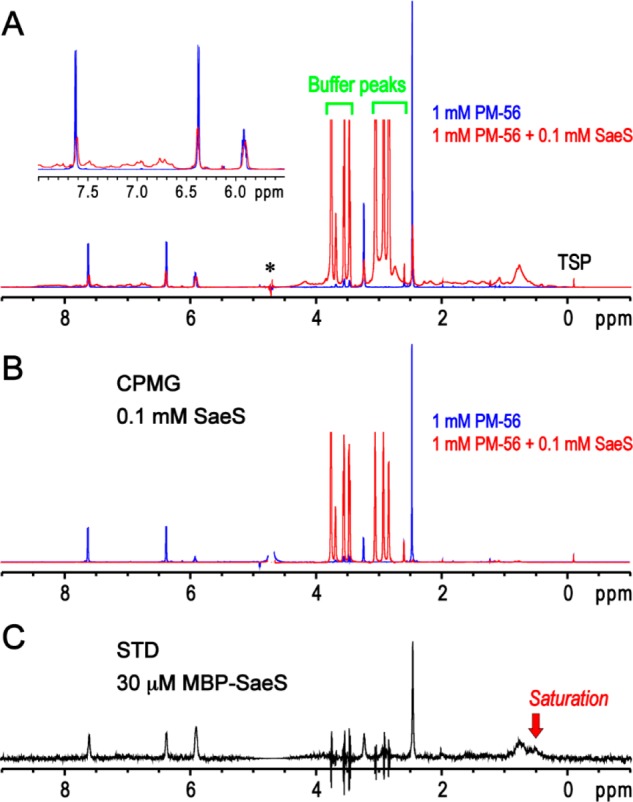
One-dimensional NMR analysis of the PM-56 binding to SaeS. (A) One-dimensional spectrum of PM-56 alone is used as a blank reference (blue). The several peaks of PM-56 are not shown in the spectrum due to an overlap with the huge water peak (marked with an asterisk). The presence of 0.1 mM of the minimal kinase domain of SaeS apparently decreases and broadens the peaks of PM-56 (red), and the peak shift of aromatic and olefin protons is also noted (inset). (B) Transverse relaxation filter using 200 ms CPMG pulse trains completely eliminated the peaks of PM-56 in the presence of 0.1 mM of the minimal kinase domain of SaeS (red). However, the reduction of the PM-56 peaks by the CPMG filter is not apparent in the absence of SaeS (blue). (C) Presence of a binding exchange process between PM-56 and MBP-SaeS is clearly confirmed by 1D STD experiment. The PM-56 peaks are specifically identified in 1D STD spectrum in the presence of 30 μM MBP-SaeS. Although the peak spikes of the remaining buffer (HEPES) are identified, their integrations are zero; slightly different peak shapes between on- and off-saturation at 0.5 and 30 ppm, respectively, could result in these peak spikes.
The IC50 values for HK activity were higher than expected considering the IC50 values for the SaeRS TCS GFP reporter in intact bacteria cells. Nevertheless, an observation of substantially lower activity for purified enzymes than enzymes in intact cells is not rare.51,52 This could be attributed to the different states of purified target proteins compared to proteins in intact cells. There are several factors that may affect protein activity in intact cells, such as the intracellular distribution of compounds, processing of target proteins, and presence of interacting proteins.52 In our case, the purified SaeS comprised the minimal kinase domain, which contains only the DHp and CA domains (amino acid residues 118–351). However, native SaeS is a modular protein containing transmembrane domains, a linker, and cytoplasmic domains. The presence of the linker has a significant impact on the kinase activity.53 Therefore, it is possible that integrity of the whole protein structure is crucial for its kinase activity. This would also affect its binding affinity to small molecules. Subsequently, the two inhibitors may appear to exert a lower inhibitory activity on the purified SaeS minimal kinase domain. Nevertheless, inhibition of the kinase activity of SaeS by 1 and PM-56, supported by STD NMR and the fluorescence quenching assay, is a significant indicator of their mechanism of action.
We cannot rule out possible contributions to inhibition by other mechanisms, such as SaeS sensing or SaeR binding to the promoter regions of virulence genes. Although a majority of the inhibitors of TCSs reported thus far54 have been HK inhibitors, a few have shown separate activities against the sensor16 or the response regulator.15 The evaluation of these possibilities is complicated, particularly for inhibitors of sensing activity because SaeS does not have a sensing domain and likely requires an unknown auxiliary protein.53 Nevertheless, these studies are currently underway and will be reported in due course. Given that 1 and PM-56 did not suppress bacterial growth at IC90 (while inhibiting the SaeRS TCS), these compounds are excellent candidates for specific action against the virulence of S. aureus. In light of this, the necessity of the whole xanthoangelol B molecule for optimal activity is an interesting finding. Neither the isoprenyl nor the chalcone unit alone displayed activity. Moreover, an overlapping fragment also failed to show activity. Compounds 1 and PM-56 are modular compounds composed of isoprene and chalcone. This result therefore deviates slightly from the logic of a fragment-based approach.55 This demonstrates that great caution is needed with fragment-based approaches (although these are still powerful methods of drug development). The major difference between 1 and PM-56 is the conversion of the α,β-unsaturated carbonyl in the chalcone moiety of 1 to the β-hydroxyl carbonyl in compound PM-56. This subtle change appears to confer a different activity profile. This will be valuable information in the further development of antivirulence agents. From the perspective of drug development, it may be beneficial to think about the route of administration. It was shown that orally administered xanthoangelol, a structurally related compound, was absorbed and distributed rapidly in mice in a single dose study,56 which allows speculation that 1 and PM-56 may behave similarly. However, in the same study, the absorption efficiency of the chalcone was evaluated to be low,56 potentially indicative of low oral bioavailability. The authors stated the limitation of a single dose study and the major difference between the results of bioavailability studies in animals and humans for possible reasons. Hence, this suggests that many aspects need to be taken into consideration, and it is considered that it is too early at the current stage to speculate the route of administration. Regardless, our data provide a strong starting point for further optimization.
Conclusion
In conclusion, we found a hit (xanthoangelol B 1) in a screening with inhibitory activity against the SaeRS TCS of S. aureus. A prenylated chalcone, xanthoangelol B 1, was synthesized in the racemic form and then resolved into an enantiomeric form. Synthesized xanthoangelol B 1 showed identical spectroscopic and optical properties to those of xanthoangelol B 1 extracted from a natural source. We also synthesized PM-56, a derivative of 1 with a hydrated chalcone double bond. The two compounds displayed similarly good inhibitory activities against the SaeRS TCS GFP reporter. They also inhibited the expression of downstream virulence factors, reducing hemolysis. PM-56 displayed a better tolerance for bacterial growth, which is desirable for antivirulence agents. Interestingly, fragments of the two compounds did not show any inhibitory activity. Thus, these compounds are required in their entirety for activity. Compounds 1 and PM-56 displayed excellent survival rates and low bacterial burdens in the G. mellonella infection model. These results were comparable to the effect of saeS knockout. The in vivo data demonstrated a good correlation with the in vitro data. Indeed, 1 and PM-56 showed substantial inhibition of the minimal kinase domain of SaeS, although measured IC50 values were higher than expected from the in vitro (bacteria) and in vivo data. However, the purified minimal kinase domain is not likely to have the same kinase activity as the full-length membrane protein SaeS. This might explain the lower potency of 1 and PM-56 against the purified minimal SaeS. Fluorescence quenching assay and NMR data support the direct binding of PM-56 with the minimal kinase domain of SaeS. This study demonstrates that natural products are still a valuable source of compounds for therapeutic agents. Efforts toward the partial or total chemical synthesis or modification of natural products can help diversify a candidate’s structure. With the expertise and insight gained from our synthesis of xanthoangelol B 1, we are now in a position to further develop inhibitors of the SaeRS TCS.
Experimental Section
General Methods
All reactions were carried out under open flask unless otherwise stated. THF, toluene, diethyl ether, and CH2Cl2 were procured from Sigma-Aldrich. Reagents were purified prior to use unless otherwise stated. Purification of reaction products was carried out by flash chromatography using Fisher silica gel (35–70 mesh). NMR spectra were recorded on Bruker DPX 400. 1H NMR spectra were measured at 400 and 500 MHz. 13C NMR spectra were measured at 126 and 150 MHz using CDCl3, acetone-d6, or DMSO-d6 as a solvent and internal reference. Coupling constants Js are given in Hz. Multiplicity as follows: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad signal. Mass spectrometric data were obtained on a Waters Acquity UPLC and UPC2 LC systems. Agilent 1260 infinity was used as an HPLC system to analyze and purify compounds. Synthesized xanthoangelol B 1 and PM-56 were screened for PAINS chemotypes using two online tools, PATTERNS (http://zinc15.docking.org/patterns/home) and FAFDrug4 (http://fafdrugs3.mti.univ-paris-diderot.fr/), and the results were negative or PASS.
Xanthoangelol B (1)
Diethylaluminum 2,2,6,6-tetramethylpiperidide (DATMP) was prepared in situ by reacting diethylaluminum chloride (6.79 mmol) and lithium 2,2,6,6-tetramethyl piperidide (6.79 mL) in dry toluene (10 mL) under argon atmosphere at 0 °C for 1 h. The DATMP prepared was used immediately.36 To a stirred mixture of DATMP (4 mmol) and dry toluene (10 mL), a solution of oxirane 13 (1 mmol) in toluene (3 mL) was added dropwise at 0 °C over a period of 5–10 min. The reaction mixture was stirred at 0 °C until the starting material was not detected by TLC. The reaction upon completion was quenched with ice-cold 1 M HCl, and the resulting organic phase was separated. The aqueous layer was extracted with diethyl ether (2 × 10 mL). The organic layer was combined, washed with brine, dried over anhydrous MgSO4, and concentrated in vacuo. The residue was purified by column chromatography (4/1 ethyl acetate/hexane, Rf = 0.4).36
Yield: 80%, yellow solid. 1H NMR (400 MHz, acetone-d6) δ ppm 1.62 (m, 2H), 1.74 (s, 3 H), 1.88 (s, 3 H), 2.03–2.29 (m, 3 H), 3.47 (br d, J = 6.8 Hz, 2 H), 3.97 (br t, J = 6 Hz, 1 H), 4.72–4.74 (m, 1 H), 4.87 (s, 1 H), 5.32–5.43 (m, 1 H), 6.50 (d, J = 9 Hz, 1 H), 6.93 (d, J = 8.5 Hz, 2 H), 7.73 (d, J = 15 Hz, 1 H), 7.73 (d, J = 8.5 Hz, 2 H), 7.78 (d, J = 9 Hz, 1 H), 7.86 (d, J = 15 Hz, 1 H). 13C NMR (125 MHz, CDCl3) 192.1, 163.8, 161.4, 157.9, 147.4, 143.9, 138.9, 130.5, 129.2, 127.8, 121.4, 118.6, 118.1, 115.9, 114.1, 111.5, 107.7, 75.6, 35.8, 32.9, 29.7, 18.1, 16.4. Chemical Formula: C25H28O5, calculated mass [M]: 408.19. Observed [M + H]+: 409.19. Data are consistent with those reported in ref (23).
Kinetic Resolution:39 Xanthoangelol B (1)
In a 25 mL round-bottomed flask, 1 (0.12 mmol), NaHCO3 (0.24 mmol), and 7-Bn-3-n-Bu-4-oxa-5-azahomoadamantane (2.4 μmol) were dissolved in CH2Cl2 (5 mL). After the reaction mixture was cooled to −40 °C, trichloroisocyanuric acid (TCCA, 0.024 mmol) was added, and the mixture was stirred at a fixed temperature (−40 °C) for 12 h. The mixture was diluted with i-PrOH (1 mL) followed by saturated aqueous NaHCO3 (10 mL) and warmed to room temperature. The aqueous layer was separated and extracted with CHCl3. The combined organic layers were dried over MgSO4 and concentrated under reduced pressure. The residue was purified by column chromatography (45/55 ethyl acetate/hexane, Rf = 0.3) to give the ketone (48%) and the desired chiral product (34%). Spectroscopic data were identical as above. [α]D20 = +13.5 (MeOH, c = 0.5).
Geraniolene Oxide (8)
A solution of geraniolene (8.85 mmol) in CHCl3 (30 mL) was cooled and stirred in an ice-bath to which a solution of 80% mCPBA (9.74 mmol) in ether (5 mL) was added dropwise over 30 min and stirred further for an additional 30 min. The reaction mixture was washed with 10% aqueous Na2CO3 (4 × 10 mL) and saturated NaCl (10 mL). The organic layer was dried (anh. Mg2SO4), and the solvents were removed on a rotary evaporator to yield a colorless liquid. The crude product was purified by short path distillation under vacuum (ca. 1 mmHg) at room temperature to yield 45% of geraniolene oxide
Yield 45%, pale yellow oil: 1H NMR (400 MHz, C6D6): 2.51 (dd, J = 4.6, 8.0 Hz, 1 H), 2.05 (m, 2 H), 1.76 (m, 1 H), 1.59 (s, 3 H), 1.49 (m, 1 H), 1.08 (s, 3 H), 1.04 (s, 3 H). Data are consistent with those reported in ref (57).
Allyl Aryl Ether (9)
2,4-Dihydroxy acetophenone (6.57 mmol) was dissolved in dry acetone (10 mL), and both anhydrous K2CO3 (9.85 mmol) and allyl bromide (7.23 mmol) were added. The mixture was refluxed and monitored by thin-layer chromatography (TLC). After completion of the reaction (8 h), the reaction mixture was filtered and acetone was removed under vacuum. The residue was purified by flash chromatography (9/1 ethyl acetate/hexane, Rf = 0.4) on silica gel to yield the desired ally aryl ether in an average yield of 76%.
1H NMR (400 MHz, CDCl3) δ = 2.57 (s, 3 H), 3.48 (d, J = 6.1 Hz, 2 H), 5.12 (d, J = 10.3 Hz, 1 H), 5.16 (d, J = 17.1 Hz, 1 H), 5.99 (ddt, J = 17.1 Hz, J = 10.3 Hz, J = 6.1 Hz, 1 H), 6.24 (s, OH, 1 H), 6.41 (d, J = 8.8 Hz, 1 H), 7.56 (d, J = 8.8 Hz, 1 H), 12.55 (s, OH, 1 H). Data are consistent with those reported in ref (32).
1-(3-Allyl-2,4-dihydroxyphenyl)ethan-1-one (10)
The allyl aryl ether (9) (10.41 mmol) was dissolved in 20 mL of N,N-diethylaniline and refluxed at 220 °C overnight. The reaction upon completion was cooled to room temperature and poured into 50 mL of precooled hexane. The separated solid was filtered and washed further with an excess amount of hexane. The product was purified by column chromatography (4/6 ethyl acetate/hexane, Rf = 0.5).
Yield 70%, Brown solid. 1H NMR (400 MHz, chloroform-d) δ ppm 2.57 (s, 3 H), 4.58 (dt, J = 5.38, 1.47 Hz, 3 H), 5.13–5.56 (m, 3 H), 5.91–6.23 (m, 1 H), 6.40–6.58 (m, 2 H), 7.65 (d, J = 8.80 Hz, 1 H), 12.74 (s, 1 H). 13C NMR (101 MHz, chloroform-d) δ ppm 26.22, 68.98, 101.69, 108.03, 114.02, 118.39, 132.32, 165.17, 202.58. Data are consistent with those reported in ref (32).
1-(3-Allyl-2,4-dihydroxyphenyl)-3-(4-hydroxyphenyl)prop-2-en-1-one (11)
Alumina (3 g, neutral alumina) was mixed with KF (2 g) in 20 mL of water, and the water was removed at 50–60 °C in a rotary evaporator. The impregnated alumina was then dried in a vacuum drying oven at 75 °C for several hours.58 To a solution of 10 (5.2 mmol) and 4-hydroxybenzaldehyde (5.7 mmol) in dry toluene (10 mL), the prepared KF-alumina 40% (15.6 mmol) was added. The reaction mixture was refluxed for 18 h until the reaction was completed. Upon completion of the reaction, KF-alumina was filtered off, the filtrate was evaporated, and the crude product was applied to column chromatography (1/1 ethyl acetate/hexane, Rf = 0.5) to give 60% of the pure product.
Yield 60%, yellow amorphous powder, 1H NMR (acetone-d6, 400 MHz): 3.39 (td, 2 H, J = 1.6, 6.0 Hz), 4.86 (qd, 1 H, J = 1.6, 10.0 Hz), 4.97 (qd, 1 H, J = 1.6, 16.8 Hz), 5.95 (m, 1 H), 6.51 (d, 1 H, J = 8.4 Hz), 6.90 (d, 2 H, J = 8.4 Hz), 7.71 (d, 2 H, J = 8.4 Hz), 7.72 (d, 1 H, J = 16.0 Hz), 7.81 (d, 1 H, J = 16.0 Hz), 7.97 (d, 1 H, J = 8.4 Hz), 8.98 (s, 1 H, OH), 9.31 (s, 1 H, OH), 13.95 (s, 1 H, OH). Data are consistent with those reported in ref (59).
2,6-Dimethylhepta-1,5-diene (12)
A sealed tube was charged with molecular sieves (4 Å, 15 g), citral (32.84 mmol), and palladium acetate (10 mol %). The tube was then purged with argon several times. Under a counter flow of argon, remaining liquid reagents were added, followed by cyclohexane (10 mL) by syringe. The tube was tightly closed by screw cap and placed in a preheated oil bath at required temperature. The reaction mixture was vigorously stirred for 24 h. The reaction mixture was cooled to room temperature and filtered through Celite. Reaction tube and residue were washed with ethyl acetate (20 mL). The filtrate was concentrated, and the resulting deformylated product was purified via column chromatography using silica gel (1/99 ethyl acetate/petroleum ether, Rf = 0.7).
Yield 62%, colorless oil. 1H NMR (400 MHz, CDCl3) δ 5.19–5.08 (t, J = 6.4 Hz, 1 H), 4.71 (d, J = 9.3 Hz, 2 H), 2.21–2.09 (m, 2 H), 2.04 (dd, J = 9.1, 6.2 Hz, 2 H), 1.74 (s, 3 H), 1.70 (s, 3 H), 1.63 (s, 3 H). 13C NMR (100 MHz, CDCl3) δ 145.8, 131.5, 124.1, 109.7, 37.8, 26.3, 25.6, 22.7, 22.4, 17.6. Data are consistent with those reported in ref (60).
1-(3-((E)-5-(3,3-Dimethyloxiran-2-yl)-3-methylpent-2-en-1-yl)-2,4-dihydroxyphenyl)-3-(4-hydroxyphenyl)prop-2-en-1-one (13)
Method 1
The Hoveyda–Grubbs second generation catalyst (10 mol %) was purged with argon to a mixture of 1-(3-allyl-2,4-dihydroxyphenyl)ethan-1-one (4.38 mmol) and 2,2-dimethyl-3-(3-methylbut-3-en-1-yl)oxirane (14.26 mmol) in DCM. The resulting solution was stirred at 42 °C for 3 h. Removal of the solvent in vacuo gave crude product, which was purified using flash chromatography (1/1 ethyl acetate/hexane, Rf = 0.4).61 Yield: 70%.
Method 2
To a solution of 17 (7.8 mmol) and 18 (8.58 mmol) in 30 mL of methanol was added KOH (46.8 mmol) at 0 °C. The reaction was warmed to room temperature and refluxed for 10 h. The reaction mixture was cooled with the pH adjusted to 6 using 1 N HCl, and then extracted with ethyl acetate. The organic layer was evaporated and purified by column chromatography (1/1 ethyl acetate/hexane, Rf = 0.4). Yield: 34%.
Dark yellow solid, 1H NMR (400 MHz, acetone-d6) δ ppm 1.11 (d, J = 11.00 Hz, 5 H), 1.03–1.17 (m, 1 H), 1.37–1.47 (m, 1 H), 1.80 (s, 3 H), 1.98 (s, 1 H), 2.10 (s, 1 H), 3.18 (d, J = 0.73 Hz, 2 H), 3.35–3.45 (m, 1 H), 4.71–4.76 (m, 2 H), 5.53 (br s, 1 H), 6.36–6.61 (m, 1 H), 6.39 (dd, J = 2.45, 1.22 Hz, 1 H), 6.94–7.00 (m, 1 H), 7.05 (d, J = 8.80 Hz, 1 H), 7.79–7.82 (m, 4 H), 8.19 (d, J = 8.80 Hz, 1 H).
(E)-1-(3-(5-(3,3-Dimethyloxiran-2-yl)-3-methylpent-2-en-1-yl)-2,4-dihydroxyphenyl)ethan-1-one (14)
The Hoveyda–Grubbs second generation catalyst (10 mol %) was purged with argon to a mixture of 1-(3-allyl-2,4-dihydroxyphenyl)ethan-1-one (5.2 mmol) and 2,2-dimethyl-3-(3-methylbut-3-en-1-yl)oxirane (10.92 mmol) in DCM. The resulting solution was stirred at 42 °C for 3 h. Removal of the solvent in vacuo gave the crude product, which was purified using flash chromatography (35/75 ethyl acetate/hexane, Rf = 0.5).61
1H NMR (400 MHz, CDCl3) δ 13.09 (1 H, s), 7.57 (d, J = 9.0 Hz, 1 H), 6.42 (d, J = 9.0 Hz, 1 H), 5.25(t, J = 6.6 Hz, 1 H,), 4.73 (t, J = 6.3 Hz, 1 H), 3.43 (d, J = 6.3 Hz, 2 H), 2.58 (3 H, s), 2.15–2.01 (4 H, m), 1.80 (3 H, s), 1.65 (3 H, s), 1.57 (3 H, s). 13C NMR (100 MHz, CDCl3) δ 202.8, 162.7, 161.8, 137.3, 131.4, 130.0, 124.0, 121.3, 114.7, 113.4, 107.6, 39.6, 26.4, 25.9, 25.5, 21.4, 17.5, 16.0. Chemical formula: C16H24O4, calculated mass [M]: 304.4. Observed [M + H]+: 305.37
(E)-1-(3-(5-(3,3-Dimethyloxiran-2-yl)-3-methylpent-2-en-1-yl)-2,4-dihydroxyphenyl)-3-hydroxy-3-(4-hydroxyphenyl)propan-1-one (15)
To a solution of 14(2.34 mmol) and 2 (2.8 mmol) in 20 mL of DMSO, L-proline (0.23 mmol) was added and reaction was stirred for 8 h. Upon completion of the reaction, the solvent was evaporated and column chromatography (1/99 methanol/DCM, Rf = 0.5) performed to yield the desired product. Yield: 67%.
1H NMR (400 MHz, acetone-d6) δ 1.10 (s, 3 H), 1.68 (s, 3 H), 1.13 (3 H, s, CH3), 2.08–2.13 (m, 4 H), 3.18 (d, 2 H, J = 8.6 Hz), 4.72 (m, 1 H), 5.52 (m, 1 H, m), 5.69 (m, 1 H), 6.39 (m, 1 H), 6.50 (d, 1 H, J = 6.9 Hz), 6.94 (d, 2 H, J = 6.6 Hz), 7.06 (d, 1 H, d, J = 15.4 Hz), 7.76 (d, 2 H, J = 8.6 Hz), 7.82 (d, 1 H, J = 8.9 Hz), 7.84 (d, 1 H, J = 15.4 Hz), 13.88 (1 H, s, OH). 13C NMR (100 MHz, CDCl3) δ 16.29, 17.72, 21.72, 25.68, 26.33, 39.72, 107.90, 113.95, 114.02, 115.60, 118.19, 121.00, 123.68, 127.92, 129.23, 130.54, 132.15, 139.92, 143.93, 157.83, 161.84, 163.85, 192.14 Chemical formula: C25H28O5, calculated mass [M]: 408.19. Observed [M + H]+: 409.2
1-(2,4-Dihydroxyphenyl)-3-(4-hydroxyphenyl)prop-2-en-1-one (17)
To a solution of 60% KOH in ethanol (10 mL), 2,4-dihydroxy acetophenone (6.57 mmol) was added followed by 4-hydroxy benzaldehyde (7.23 mmol). The reaction mixture was refluxed for about 24 h, and upon completion of reaction, the pH of the reaction was adjusted to 5. The mixture was then extracted with DCM (3 × 10 mL), washed with water, and dried over anhydrous MgSO4 and evaporated under vacuum to give crude product. The product was purified by column chromatography (6/4 ethyl acetate/hexane, Rf = 0.7) to give 80% of the yield.
Yield 80% yellow solid. 1H NMR (400 MHz, acetone-d6) δ 13.6 (s, 1 H), 12.7 (br s, 1 H), 9.9 (br s, 1 H), 8.13 (d, J = 8.8 Hz, 1 H), 7.78 (s, 2 H), 7.75 (d, J = 8.7 Hz, 2 H), 6.94 (d, J = 8.7 Hz, 2 H), 6.46 (dd, J = 8.8, 2.3 Hz, 1 H), 6.34 (d, J = 2.4 Hz, 1 H). 13C NMR (100 MHz, DMSO-d6) δ 191.9, 166.7, 165.2, 164.7, 144.2, 133.5, 130.9, 126.7, 117.3, 115.8, 107.8, 102.5. Data are consistent with those reported in ref (29).
mCPBA Oxidation of Geranyl Bromide (18)
Geranyl bromide (13.35 mmol) was dissolved in 30 mL of chloroform and cooled to 0 °C. mCPBA (14.02 mmol) was added in small portions over 30 min. The reaction mixture was stirred further for 30 min at 0 °C. After completion of the reaction, it was extracted five times with 1 M NaHCO3. Organic layer was washed with water, brine, dried over MgSO4, and concentrated to give the crude product, which was used further without purification.
1-(2,4-Dihydroxy-3-(6-hydroxy-3,7-dimethylocta-2,7-dien-1-yl)phenyl)-3-hydroxy-3-(4-hydroxyphenyl)propan-1-one (PM-56)
The same procedure of selective isomerization of oxiranes used for xanthoangelol B (1) was employed on 15. Upon completion of the reaction, the solvent was evaporated and the product purified by column chromatography (5/95, methanol/DCM, Rf = 0.4).
Yield 67%, dark yellow solid. 1H NMR (400 MHz, CDCl3/D2O) δ ppm 1.52–1.64 (m, 2 H), 1.68 (s, 2 H), 1.78 (s, 3H), 1.81 (s, 3H), 2.90 (m, 2H), 3.51 (br d, 2 H), 4.08 (br t, 1 H), 4.85 (s, 1 H), 4.95 (s, 1 H), 5.24–5.38 (m, 3 H), 6.09 (br s, 1 H), 6.43 (d, J = 9 Hz, 1 H), 6.90 (d, J = 12 Hz, 2 H), 7.59 (d, J = 12 Hz, 2 H), 7.75 (d, J = 9 Hz, 1 H). 13C NMR (100 MHz, acetone-d6) 199.6, 162.2, 161.6, 156.8, 145.8, 138.4, 137.8, 127.2, 122.8, 117.3, 116.8, 116.1, 112.2, 110.8, 108.5, 76.3, 70.6, 51.9, 35.5, 33.9, 21.8, 17.8, 17.4. Chemical formula: C25H30O6, calculated mass [M]: 426.2. Observed [M + H]+: 427.2
HPLC Analysis for Purity
Kinetically resolved xanthoangelol B 1 and PM-56 were analyzed for their purity using an HPLC. The used system was the Agilent Technologies 1260 Infinity system with a C18 Column (Kromasil 100–5-C18, 5 μm, 21.2 × 250 mm). The column was eluted with 20/80 acetonitrile/water and the elution monitored with a UV detector at 254 nm. The program was run for 42 min for 1 and 25 min for PM-56. HPLC showed a single peak for each product, and the purity is judged to be over 95% (Figure S11).
HPLC Analysis
For all other HPLC analyses, the same system was used under the same condition except the mobile phase (15/85 acetonitrile/water). The program running time was typically 25 min.
Acknowledgments
This study was funded by the Wellcome Trust Seed Award in Science to S.S.L. (107988/Z/15/Z) and by National Research Foundation of Korea grants to R.A. (2015R1C1A2A01056004) and K.K.K. (2017M3A9E4078553). P.M. thanks the Wellcome Trust for funding.
Glossary
ABBREVIATIONS
- ATP
adenosine triphosphate
- Bn
benzoyl
- Bu
butyl
- CA
catalytic and ATP-binding
- CFU
colony-forming unit
- CPMG
Carr–Purcell–Meiboom–Gill
- DCM
dichloromethane
- DHp
dimerization and histidine phosphotransfer
- DMEM
Dulbecco’s modified Eagle’s medium
- FBS
fetal bovine serum
- GFP
green fluorescent protein
- HEPES
(4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid)
- HK
histidine kinase
- HPLC
high performance chromatography
- LDH
lactate dehydrogenase
- MBP
maltose-binding protein
- mCPBA
m-choloroperoxybenzoic acid
- MRSA
methicillin resistant Staphylococcus aureus
- PT
phosphotransfer
- STD
saturation transfer difference
- TCS
two component system
- TLC
thin layer chromatography
- TSB
tryptic soy broth
- WHO
World Health Organization
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.8b01012.
Author Contributions
# These authors contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Omran A. R. The epidemiologic transition: a theory of the epidemiology of population change. Milbank Quarterly 2005, 83 (4), 731–757. 10.1111/j.1468-0009.2005.00398.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal D. R.; Segal M. W.. America’s Military Population; Citeseer: University Park, PA, 2004; Vol. 59. [Google Scholar]
- Chambers H. F. The changing epidemiology of Staphylococcus aureus?. Emerging Infect. Dis. 2001, 7 (2), 178–182. 10.3201/eid0702.010204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowy F. D. Staphylococcus aureus infections. N. Engl. J. Med. 1998, 339 (8), 520–532. 10.1056/NEJM199808203390806. [DOI] [PubMed] [Google Scholar]
- Frank A. L.; Marcinak J. F.; Mangat P. D.; Schreckenberger P. C. Increase in community-acquired methicillin-resistant Staphylococcus aureus in children. Clin. Infect. Dis. 1999, 29 (4), 935–936. 10.1086/520463. [DOI] [PubMed] [Google Scholar]
- De Kraker M. E.; Davey P. G.; Grundmann H. Mortality and hospital stay associated with resistant Staphylococcus aureus and Escherichia coli bacteremia: estimating the burden of antibiotic resistance in Europe. PLoS Med. 2011, 8 (10), e1001104 10.1371/journal.pmed.1001104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewardson A. J.; Allignol A.; Beyersmann J.; Graves N.; Schumacher M.; Meyer R.; Tacconelli E.; De Angelis G.; Farina C.; Pezzoli F. The health and economic burden of bloodstream infections caused by antimicrobial-susceptible and non-susceptible Enterobacteriaceae and Staphylococcus aureus in European hospitals, 2010 and 2011: a multicentre retrospective cohort study. Eurosurveillance 2016, 21 (33), 30319. 10.2807/1560-7917.ES.2016.21.33.30319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell K. M.; Hodgkinson J. T.; Sore H. F.; Welch M.; Salmond G. P.; Spring D. R. Combating multidrug-resistant bacteria: current strategies for the discovery of novel antibacterials. Angew. Chem., Int. Ed. 2013, 52 (41), 10706–10733. 10.1002/anie.201209979. [DOI] [PubMed] [Google Scholar]
- Shallcross L. J.; Howard S. J.; Fowler T.; Davies S. C. Tackling the threat of antimicrobial resistance: from policy to sustainable action. Philos. Trans. R. Soc., B 2015, 370 (1670), 20140082. 10.1098/rstb.2014.0082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rex J. H. ND4BB: addressing the antimicrobial resistance crisis. Nat. Rev. Microbiol. 2014, 12 (4), 231–232. 10.1038/nrmicro3245. [DOI] [Google Scholar]
- Walsh C. Molecular mechanisms that confer antibacterial drug resistance. Nature 2000, 406 (6797), 775–781. 10.1038/35021219. [DOI] [PubMed] [Google Scholar]
- Davies J.; Davies D. Origins and evolution of antibiotic resistance. Microbiol. Mol. Biol. Rev. 2010, 74 (3), 417–433. 10.1128/MMBR.00016-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worthington R. J.; Melander C. Combination approaches to combat multidrug-resistant bacteria. Trends Biotechnol. 2013, 31 (3), 177–184. 10.1016/j.tibtech.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotoh Y.; Eguchi Y.; Watanabe T.; Okamoto S.; Doi A.; Utsumi R. Two-component signal transduction as potential drug targets in pathogenic bacteria. Curr. Opin. Microbiol. 2010, 13 (2), 232–239. 10.1016/j.mib.2010.01.008. [DOI] [PubMed] [Google Scholar]
- Bem A. E.; Velikova N.; Pellicer M. T.; Baarlen P. v.; Marina A.; Wells J. M. Bacterial histidine kinases as novel antibacterial drug targets. ACS Chem. Biol. 2015, 10 (1), 213–224. 10.1021/cb5007135. [DOI] [PubMed] [Google Scholar]
- Rasko D. A.; Moreira C. G.; Li D. R.; Reading N. C.; Ritchie J. M.; Waldor M. K.; Williams N.; Taussig R.; Wei S.; Roth M.; Hughes D. T.; Huntley J. F.; Fina M. W.; Falck J. R.; Sperandio V. Targeting QseC signaling and virulence for antibiotic development. Science 2008, 321 (5892), 1078–1080. 10.1126/science.1160354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldry M.; Nielsen A.; Bojer M. S.; Zhao Y.; Friberg C.; Ifrah D.; Glasser Heede N.; Larsen T. O.; Frøkiær H.; Frees D.; Zhang L.; Dai H.; Ingmer H. Norlichexanthone reduces virulence gene expression and biofilm formation in Staphylococcus aureus. PLoS One 2016, 11 (12), e0168305 10.1371/journal.pone.0168305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bone M. A.; Wilk A. J.; Perault A. I.; Marlatt S. A.; Scheller E. V.; Anthouard R.; Chen Q.; Stibitz S.; Cotter P. A.; Julio S. M. Bordetella PlrSR regulatory system controls BvgAS activity and virulence in the lower respiratory tract. Proc. Natl. Acad. Sci. U. S. A. 2017, 114 (8), E1519–E1527. 10.1073/pnas.1609565114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun F.; Cho H.; Jeong D.-W.; Li C.; He C.; Bae T. Aureusimines in Staphylococcus aureus are not involved in virulence. PLoS One 2010, 5 (12), e15703 10.1371/journal.pone.0015703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo W.-S.; Arya R.; Kim K. K.; Jeong H.; Cho K. H.; Bae T. The FDA-approved anti-cancer drugs, streptozotocin and floxuridine, reduce the virulence of Staphylococcus aureus. Sci. Rep. 2018, 8 (1), 2521. 10.1038/s41598-018-20617-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D. W.; Curtis-Long M. J.; Yuk H. J.; Wang Y.; Song Y. H.; Jeong S. H.; Park K. H. Quantitative analysis of phenolic metabolites from different parts of Angelica keiskei by HPLC–ESI MS/MS and their xanthine oxidase inhibition. Food Chem. 2014, 153, 20–27. 10.1016/j.foodchem.2013.12.026. [DOI] [PubMed] [Google Scholar]
- Baba K.; Kido T.; Yoneda Y.; Taniguchi M.; Kozawa M. Chemical components of Angelica keiskei Koidzumi v. components of the fruits and comparison of coumarins and chalcones in the fruits roots and leaves. Shoyakugaku Zasshi 1990, 44, 235–239. [Google Scholar]
- Baba K.; Nakata K.; Taniguchi M.; Kido T.; Kozawa M. Chalcones from Angelica keiskei. Phytochemistry 1990, 29 (12), 3907–3910. 10.1016/0031-9422(90)85357-L. [DOI] [Google Scholar]
- Nakata K.; Taniguchi M.; Baba K. Three chalcones from Angelica keiskei. Shoyakugaku Zasshi 1999, 53 (6), 329–332. [Google Scholar]
- Okuyama T.; Takata M.; Takayasu J.; Hasegawa T.; Tokuda H.; Nishino A.; Nishino H.; Iwashima A. Anti-tumor-promotion by principles obtained from Angelica keiskei. Planta Med. 1991, 57 (03), 242–246. 10.1055/s-2006-960082. [DOI] [PubMed] [Google Scholar]
- Kil Y.-S.; Choi S.-K.; Lee Y.-S.; Jafari M.; Seo E.-K. Chalcones from Angelica keiskei evaluation of their heat shock protein inducing activities. J. Nat. Prod. 2015, 78 (10), 2481–2487. 10.1021/acs.jnatprod.5b00633. [DOI] [PubMed] [Google Scholar]
- Kil Y.-S.; Pham S. T.; Seo E. K.; Jafari M. Angelica keiskei an emerging medicinal herb with various bioactive constituents and biological activities. Arch. Pharmacal Res. 2017, 40 (6), 655–675. 10.1007/s12272-017-0892-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inamori Y.; Baba K.; Tsujibo H.; Taniguchi M.; Nakata K.; Kozawa M. Antibacterial activity of two chalcones, xanthoangelol and 4-hydroxyderricin, isolated from the root of Angelica keiskei Koidzumi. Chem. Pharm. Bull. 1991, 39 (6), 1604–1605. 10.1248/cpb.39.1604. [DOI] [PubMed] [Google Scholar]
- Kakati D.; Barua N. C. Total synthesis and assignment of the absolute stereochemistry of xanthoangelol J: development of a highly efficient method for Claisen–Schmidt condensation. Tetrahedron 2014, 70 (3), 637–642. 10.1016/j.tet.2013.11.106. [DOI] [Google Scholar]
- Sugamoto K.; Matsusita Y.-i.; Matsui K.; Kurogi C.; Matsui T. Synthesis and antibacterial activity of chalcones bearing prenyl or geranyl groups from Angelica keiskei. Tetrahedron 2011, 67 (29), 5346–5359. 10.1016/j.tet.2011.04.104. [DOI] [Google Scholar]
- Jung D. H.; Lee Y. R.; Kim S. H. New synthetic routes to biologically interesting geranylated flavanones and geranylated chalcones: first total synthesis of (±)-prostratol F, xanthoangelol, and (±)-lespeol. Helv. Chim. Acta 2010, 93 (4), 635–647. 10.1002/hlca.200900251. [DOI] [Google Scholar]
- Gozzo F. C.; Fernandes S. A.; Rodrigues D. C.; Eberlin M. N.; Marsaioli A. J. Regioselectivity in aromatic Claisen rearrangements. J. Org. Chem. 2003, 68 (14), 5493–5499. 10.1021/jo026385g. [DOI] [PubMed] [Google Scholar]
- Modak A.; Deb A.; Patra T.; Rana S.; Maity S.; Maiti D. A general and efficient aldehyde decarbonylation reaction by using a palladium catalyst. Chem. Commun. (Cambridge, U. K.) 2012, 48 (35), 4253–4255. 10.1039/c2cc31144e. [DOI] [PubMed] [Google Scholar]
- Lacey J. R.; Anzalone P. W.; Duncan C. M.; Hackert M. J.; Mohan R. S. A study of epoxyolefin cyclizations catalyzed by bismuth trifluoromethanesulfonate and other metal triflates. Tetrahedron Lett. 2005, 46 (49), 8507–8511. 10.1016/j.tetlet.2005.10.013. [DOI] [Google Scholar]
- Stewart I. C.; Douglas C. J.; Grubbs R. H. Increased efficiency in cross-metathesis reactions of sterically hindered olefins. Org. Lett. 2008, 10 (3), 441–444. 10.1021/ol702624n. [DOI] [PubMed] [Google Scholar]
- Yasuda A.; Tanaka S.; Yamamoto H.; Nozaki H. A highly stereospecific procedure for the transformation of allylic alcohols into 1, 3-dienes. Bull. Chem. Soc. Jpn. 1979, 52 (6), 1752–1756. 10.1246/bcsj.52.1752. [DOI] [Google Scholar]
- Palomo C.; Oiarbide M.; García J. M. Current progress in the asymmetric aldol addition reaction. Chem. Soc. Rev. 2004, 33 (2), 65–75. 10.1039/B202901D. [DOI] [PubMed] [Google Scholar]
- List B.; Lerner R. A.; Barbas C. F. Proline-catalyzed direct asymmetric aldol reactions. J. Am. Chem. Soc. 2000, 122 (10), 2395–2396. 10.1021/ja994280y. [DOI] [Google Scholar]
- Murakami K.; Sasano Y.; Tomizawa M.; Shibuya M.; Kwon E.; Iwabuchi Y. Highly enantioselective organocatalytic oxidative kinetic resolution of secondary alcohols using chiral alkoxyamines as precatalysts: catalyst structure, active species, and substrate scope. J. Am. Chem. Soc. 2014, 136 (50), 17591–17600. 10.1021/ja509766f. [DOI] [PubMed] [Google Scholar]
- Liu Q.; Yeo W. S.; Bae T. The SaeRS two-component system of Staphylococcus aureus. Genes 2016, 7 (10), 81. 10.3390/genes7100081. [DOI] [Google Scholar]
- Dunman P. á.; Murphy E.; Haney S.; Palacios D.; Tucker-Kellogg G.; Wu S.; Brown E.; Zagursky R.; Shlaes D.; Projan S. Transcription profiling-based identification of Staphylococcus aureus genes regulated by the agr and/or sarA loci. J. Bacteriol. 2001, 183 (24), 7341–7353. 10.1128/JB.183.24.7341-7353.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai C. J.-Y.; Loh J. M. S.; Proft T. Galleria mellonella infection models for the study of bacterial diseases and for antimicrobial drug testing. Virulence 2016, 7 (3), 214–229. 10.1080/21505594.2015.1135289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eguchi Y.; Kubo N.; Matsunaga H.; Igarashi M.; Utsumi R. Development of an antivirulence drug against Streptococcus mutans repression of biofilm formation, acid tolerance, and competence by a histidine kinase inhibitor, walkmycin C. Antimicrob. Agents Chemother. 2011, 55 (4), 1475–1484. 10.1128/AAC.01646-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velikova N.; Fulle S.; Manso A. S.; Mechkarska M.; Finn P.; Conlon J. M.; Oggioni M. R.; Wells J. M.; Marina A. Putative histidine kinase inhibitors with antibacterial effect against multi-drug resistant clinical isolates identified by in vitro and in silico screens. Sci. Rep. 2016, 6, 26085. 10.1038/srep26085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi S. D.; Malachowa N.; Whitney A. R.; Braughton K. R.; Gardner D. J.; Long D.; Wardenburg J. B.; Schneewind O.; Otto M.; DeLeo F. R. Comparative analysis of USA300 virulence determinants in a rabbit model of skin and soft tissue infection. J. Infect. Dis. 2011, 204 (6), 937–941. 10.1093/infdis/jir441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasgar A.; Furdas S. D.; Maloney D. J.; Jadhav A.; Jung M.; Simeonov A. High-throughput 1,536-well fluorescence polarization assays for α1-acid glycoprotein and human serum albumin binding. PLoS One 2012, 7 (9), e45594 10.1371/journal.pone.0045594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond L. A.; Krinks C. H. V.; Durham J.; Tomkins S. E.; Burnett R. D.; Jones E. L.; Chandraratna R. A. S.; Brown G. Antagonists of retinoic acid receptors (RARs) are potent growth inhibitors of prostate carcinoma cells. Br. J. Cancer 2001, 85, 453–462. 10.1054/bjoc.2001.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James J.; Ruggeri B.; Armstrong R. C.; Rowbottom M. W.; Jones-Bolin S.; Gunawardane R. N.; Dobrzanski P.; Gardner M. F.; Zhao H.; Cramer M. D.; Hunter K.; Nepomuceno R. R.; Cheng M.; Gitnick D.; Yazdanian M.; Insko D. E.; Ator M. A.; Apuy J. L.; Faraoni R.; Dorsey B. D.; Williams M.; Bhagwat S. S.; Holladay M. W. CEP-32496: A novel orally active BRAF V600E inhibitor with selective cellular and in vivo antitumor activity. Mol. Cancer Ther. 2012, 11 (4), 930–941. 10.1158/1535-7163.MCT-11-0645. [DOI] [PubMed] [Google Scholar]
- Stockman B. J.; Dalvit C. NMR screening techniques in drug discovery and drug design. Prog. Nucl. Magn. Reson. Spectrosc. 2002, 41 (3), 187–231. 10.1016/S0079-6565(02)00049-3. [DOI] [Google Scholar]
- Mayer M.; Meyer B. Group epitope mapping by saturation transfer difference NMR to identify segments of a ligand in direct contact with a protein receptor. J. Am. Chem. Soc. 2001, 123 (25), 6108–6117. 10.1021/ja0100120. [DOI] [PubMed] [Google Scholar]
- Mitchell J. A.; Akarasereenont P.; Thiemermann C.; Flower R. J.; Vane J. R. Selectivity of nonsteroidal antiinflammatory drugs as inhibitors of constitutive and inducible cyclooxygenase. Proc. Natl. Acad. Sci. U. S. A. 1993, 90 (24), 11693–11697. 10.1073/pnas.90.24.11693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuhl A. M.; Nolan C. E.; Brodney M. A.; Niessen S.; Atchison K.; Houle C.; Karanian D. A.; Ambroise C.; Brulet J. W.; Beck E. M.; Doran S. D.; O’Neill B. T.; am Ende C. W.; Chang C.; Geoghegan K. F.; West G. M.; Judkins J. C.; Hou X.; Riddell D. R.; Johnson D. S. Chemoproteomic profiling reveals that cathepsin D off-target activity drives ocular toxicity of β-secretase inhibitors. Nat. Commun. 2016, 7, 13042. 10.1038/ncomms13042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q.; Cho H.; Yeo W.-S.; Bae T. The extracytoplasmic linker peptide of the sensor protein SaeS tunes the kinase activity required for Staphylococcal virulence in response to host signals. PLoS Pathog. 2015, 11 (4), e1004799 10.1371/journal.ppat.1004799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari S.; Jamal S. B.; Hassan S. S.; Carvalho P. V. S. D.; Almeida S.; Barh D.; Ghosh P.; Silva A.; Castro T. L. P.; Azevedo V. Two-component signal transduction systems of pathogenic bacteria as targets for antimicrobial therapy: an overview. Front. Microbiol. 2017, 8, 1878. 10.3389/fmicb.2017.01878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babaoglu K.; Shoichet B. K. Deconstructing fragment-based inhibitor discovery. Nat. Chem. Biol. 2006, 2 (12), 720–723. 10.1038/nchembio831. [DOI] [PubMed] [Google Scholar]
- Nakamura T.; Tokushima T.; Kawabata K.; Yamamoto N.; Miyamoto M.; Ashida H. Absorption and metabolism of 4-hydroxyderricin and xanthoangelol after oral administration of Angelica keiskei (Ashitaba) extract in mice. Arch. Biochem. Biophys. 2012, 521 (1), 71–76. 10.1016/j.abb.2012.03.013. [DOI] [PubMed] [Google Scholar]
- Corey E.; Staas D. D. Demonstration of a common concerted mechanistic pathway for the acid-catalyzed cyclization of 5, 6-unsaturated oxiranes in chemical and enzymatic systems. J. Am. Chem. Soc. 1998, 120 (14), 3526–3527. 10.1021/ja980096l. [DOI] [Google Scholar]
- Yamawaki J.; Kawate T.; Ando T.; Hanafusa T. Potassium fluoride on alumina. An efficient solid base for elimination, addition, and condensation. Bull. Chem. Soc. Jpn. 1983, 56 (6), 1885–1886. 10.1246/bcsj.56.1885. [DOI] [Google Scholar]
- Dong X.; Liu T.; Yan J.; Wu P.; Chen J.; Hu Y. Synthesis, biological evaluation and quantitative structure-activities relationship of flavonoids as vasorelaxant agents. Bioorg. Med. Chem. 2009, 17 (2), 716–726. 10.1016/j.bmc.2008.11.052. [DOI] [PubMed] [Google Scholar]
- Cheng H.; Zhang Z.; Yao H.; Zhang W.; Yu J.; Tong R. Unified asymmetric total syntheses of (−)-alotaketals A–D and (−)-phorbaketal A. Angew. Chem., Int. Ed. 2017, 56 (31), 9096–9100. 10.1002/anie.201704628. [DOI] [PubMed] [Google Scholar]
- Eignerova B.; Dračínský M.; Kotora M. Perfluoroalkylation through cross-metathesis between alkenes and (perfluoroalkyl) propenes. Eur. J. Org. Chem. 2008, 2008 (26), 4493–4499. 10.1002/ejoc.200800230. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.