

Abstract
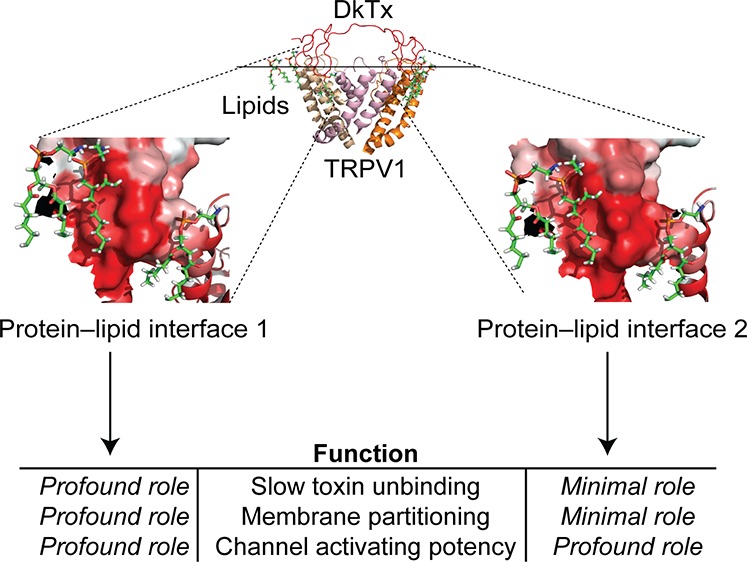
The roles of surrounding membrane lipids in the functions of transmembrane and peripheral membrane proteins are largely unknown. Herein, we utilize the recently reported structures of the TRPV1 ion channel protein bound to its potent protein agonist, the double-knot toxin (DkTx), as a model system to investigate the roles of toxin–lipid interfaces in TRPV1 activation by characterizing a series of DkTx variants electrophysiologically. Together with membrane partitioning experiments, these studies reveal that toxin–lipid interfaces play an overwhelmingly dominant role in channel activation as compared to lipid-devoid toxin–channel interfaces. Additionally, we find that whereas the membrane interfaces formed by one of the knots of the toxin endow it with its low channel-dissociation rate, those formed by other knot contribute primarily to its potency. These studies establish that protein–lipid interfaces play nuanced yet profound roles in the function of protein–protein complexes within membranes.
Membrane lipids have long been considered passive entities that serve merely to anchor membrane proteins, allowing them to execute complicated processes such as the transport of ions and small molecules across the membrane bilayer, catalysis of biochemical reactions, and signal transduction. This classical view has been challenged by recent advances in structural biology1−6 and mass spectroscopy7−10 that reveal that lipids engage in intimate and specific interactions with a diverse range of membrane proteins including ion channels, transporters, and membrane enzymes, suggesting that protein–lipid interfaces may play important roles in protein function.11−19 A detailed understanding of the functional roles of these protein–lipid interfaces, therefore, is imperative to fully understand the function of both integral and peripheral membrane proteins.
Recent structural work on the complex of the homotetrameric rat TRPV1 ion channel protein and its potent agonist, the double-knot spider toxin (DkTx;1,2Figure 1a) provides one of the best available snapshots of protein–protein complexes in a native membrane-like milieu. These studies have demonstrated that each of the two lobes (cystine knots K1 and K2) of the toxin interacts with the channel by inserting two of their loops (loops 2 and 4) into the membrane to form a toxin–lipid–channel tripartite complex (Figure 1a and b). DkTx20 belongs to the family of the inhibitory cystine-knot (ICK) toxins21−23 known to target several types of ion channels and is the only member of this family of toxins that contains two ICK motifs (K1 and K2 separated by a seven-amino-acids-long linker). Another unique aspect of DkTx is that it is the only pore-binding toxin that is known to partition into the membrane.2 Indeed, other membrane-partitioning toxins such as Hanatoxin24−26 inhibit voltage-activated ion channels by targeting their voltage sensors that are far removed from yet tightly coupled to the pore. In contrast to the DkTx–TRPV1 complex, no structural information is available currently on the toxin–channel complexes formed by these voltage sensor-binding toxins, thereby obviating molecular-level investigations into the role of toxin–lipid interfaces in channel modulation.
Figure 1.

DkTx–TRPV1 complex. (a) The DkTx–lipid–TRPV1 tripartite complex viewed sideways from within the membrane, PDB: 5irx,1 and a zoomed-in view of the complex depicting the pore domains of three channel monomers (right panel; the fourth monomer is not depicted to enhance clarity). The solid horizontal lines represent the membrane boundaries (the region above the top line is extracellular, and the region below the bottom line is intracellular). (b) Structure of DkTx depicting residues that interact exclusively with TRPV1 residues and not with lipids in green and those that interact with lipids in blue. Only the lipid-interacting residues are labeled. The same color code is used to denote the sequence of the K1 and K2 knots of DkTx at the bottom. The residues of loops 2 and 4 of each knot are boxed. (c) A two-electrode voltage clamp electrophysiology recording of TRPV1 depicting activation by capsaicin (Caps, 5 μM) and subsequently by DkTx (3.3 μM), followed by wash-off for 30 min, and ultimately complete channel block by ruthenium red (RR, 7 μM). The dotted line represents zero current.
A notable feature of DkTx is that it demonstrates extremely slow wash-off of the toxin after channel activation20,27 (Figure 1c)—an intriguing observation considering that the TRPV1–DkTx complex structures1,2 do not reveal any electrostatic,28 cation–pi,29 or pi stacking30 interactions between the residues of the toxin and those of the channel that commonly underlie tight complex formation between proteins.31 In contrast to this relatively sparse protein–protein interaction landscape, the structures depict two intimate protein–lipid interfaces, one formed by the K1 knot of DkTx and the other one formed by its K2 knot (Figure 1a, right panel). This observation engenders the hypothesis that protein–lipid interfaces play a major role in the binding and potency of the toxin. Taken together, these structural and functional attributes of the DkTx–TRPV1 complex render it an ideal model system for mechanistic investigations into the poorly understood roles of protein–lipid interfaces in the function of protein–protein complexes present within membranes.
Results and Discussion
Results
DkTx–TRPV1 Structures Enable Generation of Protein–Lipid and Protein–Protein Interaction Maps
To compare and contrast the roles of protein–protein interactions with protein–lipid interactions in DkTx-mediated activation of TRPV1, we performed systematic site-directed mutagenesis to generate DkTx variants and characterized their TRPV1 activation properties electrophysiologically. Our selection of toxin residues for substitution was guided by the recent cryo-EM structure of the rat TRPV1–DkTx complex solved in a lipidic environment1 and by another recent study2 that utilized molecular dynamics simulations to predict lipid-binding sites on the toxin and also reported a molecular model of the DkTx–TRPV1 complex structure. We classified the toxin residues lying within 4.4 Å of the atoms of the nearby channel residues/lipid molecules in either of these structural frameworks as “interacting.” The interaction maps depicting all contact points obtained from these analyses are tabulated in Table S1, Supporting Information. These maps enabled us to pinpoint 27 toxin residues that interact with the channel or/and lipids, 13 of which were observed to be exclusively channel-interacting (no lipid contacts) and 14 of which were identified as lipid-interacting, depicted in green and blue, respectively, in Figure 1b. Among the lipid-interacting residues, eight (E7, V8, I28, and H30 of the K1 knot of the toxin and E49, V50, I68, and Y70 of the K2 knot) were observed to directly interact only with lipids and not with the channel, whereas six (W11, E26, and F27 of K1 and W53, A66, and F67 of K2) were identified as interacting both with lipids and with channel residues.
DkTx–Lipid Interfaces Play Crucial Roles in the Efficacy of the Toxin for TRPV1 Activation
After identifying channel or/and lipid-interacting DkTx residues, we generated alanine variants of each of these residues, except for A66, which was substituted to serine. Wild-type DkTx and its variants were expressed recombinantly in E. coli inclusion bodies and refolded and purified to homogeneity by HPLC (representative protein purification data are depicted in Figure S1, Supporting Information; HPLC traces for purity evaluation of each of the variants except for E49A that did not refold under the conditions tested are depicted in Figure S2; and the MALDI mass characterization data of each variant are tabulated in Table S2). Subsequently, the toxin variants were characterized by performing two-electrode voltage clamp experiments on rat TRPV1 expressed in Xenopus laevis oocytes to yield dose–response curves that are depicted in Figure 2a (representative electrophysiological recordings obtained for each of the toxin variants are depicted in Figure S3, Supporting Information). These dose–response curves clearly show that the variants of DkTx residues that interact with channel residues independent of lipids (Figure 2a, top panel) demonstrate, at best, a moderate effect on the potency of the toxin. Indeed, except for the alanine variants of the G12 and G54 residues of DkTx present at similar positions of the K1 and K2 knots (sequence alignment of K1 and K2 is provided at the bottom of Figure 1b) that yielded an 8- and 9-fold increase in the EC50 values, respectively, as compared to the wild-type toxin (Table S2), the other 11 exclusively channel-interacting variants gave EC50 values that were within 2–4 fold of that of the wild-type toxin (bars in green in Figure 2b and Table S2). In contrast, variants of several of the lipid-interacting toxin residues demonstrated a profound abrogation of toxin potency (bottom panel of Figure 2a and blue bars in Figure 2b). In particular, the W11A and W53A DkTx variants of the K1 and K2 knot respectively demonstrated a substantially reduced potency—whereas the W11A variant yielded an 80-fold higher EC50 value (Table S2) than the wild-type toxin, the W53A variant was such a poor TRPV1 agonist that we were unable to generate its complete dose response curve due to a lack of solubility of the toxin at the high concentrations required to achieve full channel activation (Figure 2a), demonstrating that the EC50 value of this variant was well above 100-fold higher than that of the wild-type toxin. Our interaction maps (Table S1) show that these tryptophan residues interact intimately with both the channel residues and also with lipid molecules. To investigate the importance of the W11 and W53 residues further, we generated their leucine and tyrosine variants and characterized their activity. Similar to the alanine variants of these residues, their leucine variants also demonstrated poor TRPV1 activation properties, whereas their tyrosine variants demonstrated significant rescue of toxin activity (EC50 values obtained for the W11Y and W53Y variants were ∼10-fold higher than that of the wild-type toxin; Figures 2a,b), suggesting that an aromatic side chain is important for toxin activity at these positions. The alanine variant of another DkTx residue that constitutes the toxin–lipid interface, V8, located in loop 1 of the K1 knot, demonstrated an appreciably reduced potency (∼16-fold higher EC50 value) as compared to the wild-type toxin (Figure 2 and Table S2). The alanine substituent of the corresponding residue of the K2 knot, V50, also demonstrated a similarly significant loss of toxin potency (Figure 2b). Interestingly, neither of our two interaction maps (Table S1) depicts these two valine residues in the proximity of any channel residues and, instead, shows both of them as exclusively lipid-interacting. Strikingly, except for the lipid-interacting residue E7 of loop 1 of the K1 knot whose alanine substituent demonstrated an 11-fold higher EC50 value as compared to the wild-type toxin (Figure 2 and Table S2), not a single charged or polar residue of the toxin was observed to play an important role in the toxin’s activity. Indeed, the residues that were found to be most important for toxin activity were hydrophobic—whereas some of them were aliphatic (V8, I28, V50, and I68), the others were aromatic (W11, F27, W53, and F67).
Figure 2.

Effects on the potency of DkTx for TRPV1 activation upon alteration of protein–protein and protein–lipid interfaces. (a) Dose–response plots of the variants of exclusively channel-interacting (top) and lipid-interacting (bottom) residues of DkTx. The K1 knot variants of DkTx are depicted on the left and K2 variants on the right. (b) EC50 values of all DkTx variants. The bars in green correspond to variants of exclusively channel-interacting DkTx residues, and the ones in blue are of those that interact with lipids. The EC50 values for the W53A and W53L variants could not be obtained owing to their extremely low potency (this is denoted by the arrows on the top of the bars corresponding to these variants). “WT” is an abbreviation for “wild-type”. Each data point is an average of three to five recordings, and the error bars correspond to standard deviation values.
Protein–Lipid Interfaces Formed by the K1 Knot of DkTx Are Responsible for Slow Toxin Wash-Off
A prominent observation from our electrophysiological recordings was that several of our DkTx variants demonstrate enhanced wash-off rates as compared to the wild-type toxin (Figure 3a). Interestingly, all the DkTx residues whose variants demonstrated fast wash-off (W11, F27, I28, and V8) are lipid-interacting residues of the K1 knot. Moreover, none of the variants of the exclusively channel-interacting DkTx residues, except G12 (another K1 residue), and none of the K2 knot DkTx variants demonstrated this phenotype (Figure S3). The fastest wash-off rates were observed for the W11A variant that washed off completely within ∼3 min (Figure 3a), in contrast with the wild-type toxin that does not wash-off completely even after 30 min of buffer perfusion (Figure 1c).
Figure 3.

Toxin wash-off studies. (a) Electrophysiological recordings obtained for fast washing-off DkTx variants. Capsaicin was applied at a concentration of 5 μM, and the toxins were applied at 6.6 μM, except for V8A, which was tested at 13.2 μM. (b) Wash-off studies on wild-type DkTx showing fast wash-off when applied at a low concentration (top panel) and concentration dependence of wash-off kinetics (middle and bottom panels). The y axis of the plot on the bottom panel depicts % reduction in current after 3 min of buffer perfusion post wild-type DkTx-mediated channel activation. The blue line shown was generated by fitting a linear equation to the data. (c) Averaged wash-off current traces obtained for wild-type DkTx and fast washing-off lipid-interacting K1 variants of DkTx (left panel) and those of their corresponding K2 variants (right panel). (d) Bar graph depicting the percentage reduction in current after 3 min of buffer perfusion post-toxin-mediated channel activation for all variants (bars corresponding to the variants of lipid-independent channel-interacting residues are depicted in green and those for lipid-interacting residues are depicted in blue). The experiments (in both c and d) were performed at saturation concentrations of the respective toxins denoted within parentheses in Figure 3d. Each current trace/data point is an average of three to five recordings, and the error bars correspond to standard deviation values.
The large differences in the wash-off kinetics demonstrated by DkTx variants motivated us to study toxin wash-off in more detail. We began our studies by first determining whether toxin wash-off rates were dependent on the concentration of the applied toxin. We observed that when a low concentration (0.06 μM) of wild-type DkTx was employed to activate TRPV1, the toxin washed-off completely in 12 min (Figure 3b, top panel), in contrast to the much slower wash-off observed with 3.3 μM of the toxin (Figure 1c). Wash-off studies with a range of concentrations of DkTx revealed that wash-off kinetics were indeed concentration dependent—when 0.01 μM of wild-type DkTx was employed to activate TRPV1, the wash-off was complete within 1 min, in stark contrast to the negligible wash-off in 3 min observed upon channel activation with 2.2 μM toxin (Figures 3b, middle panel). These studies established an inverse correlation between the wash-off rates and the concentration of the toxin used in the experiment (Figure 3b, bottom panel; Pearson’s r = −0.90). The observation that wash-off kinetics depends on the concentration of the toxin used for channel activation suggests the possibility that wash-off kinetics vary as a function of channel occupancy. Consequently, we reasoned that a valid comparison of wash-off kinetics between the toxin variants can only be performed at toxin concentrations where the channel occupancy is the same in each case, for example at the top of each variant’s dose response curve, where all channels would be toxin-bound resulting in 100% channel occupancy before wash-off. All subsequent wash-off experiments were therefore performed with saturating concentrations of each of the toxin variants.
The wash-off current traces obtained over a 5 min buffer perfusion time-period subsequent to channel activation with saturating concentrations of our fast washing-off DkTx variants, W11A, W11L, W11Y, V8A, F27A, and I28A, are depicted in Figure 3c (left panel). Notably, none of the DkTx variants of the K2 knot demonstrated fast wash-off (see Figure S3 for representative traces). Indeed, the wash-off kinetics obtained with saturation concentrations of W53Y, F67A, and I68A and the V50A variants overlapped with that of the wild-type toxin (right panel of Figure 3c). Toxin variants W53A and W53L were not amenable to these experiments as their poor potency obviated preparation of toxin solutions at concentrations required to attain saturation of their dose–response curves (Figure 2). Our entire wash-off data set for both the channel-interacting and lipid-interacting residues of the DkTx depicted in Figure 3d shows that none of the residues of the toxin that interact with the channel residues exclusively via protein–protein interactions (green bars), except for G12A, demonstrated significantly faster wash-off rates as compared to that of the wild-type toxin.
K1 Knot-Driven Efficient Membrane Partitioning of DkTx Underlies Its Slow Wash-Off
The observation that the fast washing-off toxin variants were generated by substituting lipid-interacting residues suggests that the wash-off kinetics are linked to toxin–membrane lipid interactions. To compare the intrinsic membrane lipid-interacting properties of our toxin variants, we subjected them to tryptophan fluorescence-based membrane partitioning experiments on POPC–POPG (1:1) large unilamellar vesicles (LUVs) by employing approaches similar to the ones previously reported.2,32−34 Consistent with the results of a previous study,2 our experiments with wild-type DkTx revealed that it partitions well into vesicles. Indeed, in the presence of LUVs generated from 0.1 mM lipid concentration, a robust increase in fluorescence emission with a concomitant blue shift of 13 nm was observed (black traces in Figure 4a), characteristic of proteins that interact favorably with membrane lipids and partition into the membrane. In comparison, the W11A variant demonstrated merely a 3 nm blue shift and a modest increase in emission intensity in the presence of vesicles generated with the same concentration of lipids (red traces in Figure 4a). Consistent with these observations, normalized relative fluorescence versus lipid concentration plots (Figure 4b) revealed that in contrast to the wild-type toxin that partitions so well that the curve saturates at ∼0.25 mM lipid concentration, the W11A variant partitions poorly, not saturating even at a 1.25 mM lipid concentration. Other fast washing-off K1 knot variants of DkTx, W11L, and F27A also demonstrated greatly abrogated membrane partitioning as compared to the wild-type toxin (Figure 4b). The variants of analogous K2 knot residues of DkTx, W53A, and F67A, on the other hand, yielded curves that overlapped with that of the wild-type toxin demonstrating that they partition very well into membranes (Figure 4b). We generated similar partitioning plots for our other fast washing-off DkTx variants and their analogous K2 knot variants that yielded slow wash-off and obtained their Kx values (see Figure S4 of the Supporting Information for each variant’s partitioning plots and fluorescence emission spectra, and Table S2 for their Kx values). Plotting the Kx values of the wild-type toxin and those of the K2 and fast washing-off K1 variants versus the % wash-off yielded the plot shown in Figure 4c, and fitting a linear equation to the plot for the fast washing-off K1 variants gave a good correlation (Pearson’s r = −0.90). The K2 variants of DkTx such as W53Y, I68A, and F67A that wash-off slowly demonstrated efficient membrane partitioning (open squares in Figure 4c and Figure S5) similar to wild-type DkTx. These results are consistent with the notion that the slow wash-off demonstrated by DkTx is a consequence of its high intrinsic affinity for the membrane that is orchestrated by the protein–lipid interactions of the K1 knot, and not the K2 knot.
Figure 4.

Toxin–membrane interaction studies on DkTx and its variants by employing tryptophan fluorescence (a–c) and oocyte depletion (d,e). (a) Tryptophan emission spectra of wild-type DkTx (black) and the W11A variant (red) in the presence of 1:1 POPC–POPG liposomes (total lipid concentration: 0.1 mM) have been depicted as solid curves, whereas the ones obtained in the absence of lipids are shown as dashed curves. (b) Plots of normalized relative fluorescence intensity (F/Fo) at 320 nm for the wild-type toxin and the variants of analogous phenylalanine and tryptophan residues of the K1 and K2 knots as a function of the available lipid concentration. (c) Plot of % wash-off of wild-type DkTx and K1 variants (solid squares), and those of K2 variants (open squares) after 3 min of buffer perfusion post TRPV1 activation by saturation concentrations of the toxins (obtained from the electrophysiology experiments) versus mol. fraction partitioning coefficients (Kx) values (obtained from the tryptophan fluorescence experiments). The blue line shown was generated by fitting a linear equation to the data for the K1 variants. (d) HPLC traces depicting toxin depletion upon incubation of DkTx and its variants with Xenopus laevis oocytes. Traces in black correspond to the controls wherein the toxins solubilized in buffer devoid of oocytes were subjected to HPLC analysis, whereas those in red were obtained when the supernatants of toxin solutions incubated with 100 oocytes were subjected to HPLC. (e) Plot of % wash-off of wild-type DkTx and K1 variants (solid squares) and K2 variants (open squares) after 3 min of buffer perfusion post TRPV1 activation by saturation concentrations of the toxins (obtained from the electrophysiology experiments) versus fractional depletion (obtained from HPLC peak areas as described in the Supporting Information section). The blue line shown was generated by fitting a linear equation to the data for the K1 variants. Each data point is an average of three to five recordings/assays, and the error bars correspond to standard deviation values.
To investigate the interactions of DkTx and its variants with more physiologically representative membranes, we characterized their binding to Xenopus laevis oocytes. These experiments were performed by following previously reported protocols developed by Swartz and co-workers for studying the membrane-interacting properties of toxins that target voltage-activated ion channels.33 Oocytes were incubated in toxin solutions, and the unbound toxin was quantitated by subjecting the supernatant to HPLC. The depletion in the concentration of DkTx and its variants due to partitioning into oocyte plasma membranes was calculated by using the areas under the HPLC peaks corresponding to the toxins as a measure of the amount of the unbound toxin (details in the Supporting Information). The results of these experiments (Figure 4d,e) revealed that, whereas the slow washing-off toxins (wild-type DkTx and the variants of the K2 knot residues W53, F67, and I68) were substantially depleted, the fast washing-off K1 variants (W11A, W11L, and F27A) demonstrated significantly lower depletion. The HPLC chromatograms for the depletion of the variants not depicted in Figure 4d are provided in Figure S6a (Supporting Information), and the results of a control experiment demonstrating enhanced DkTx depletion upon incubation with TRPV1-expressing oocytes is shown in Figure S6b. Plotting % wash-off vs fractional depletion for the fast washing-off K1 variants (akin to the analysis depicted in Figure 4c for LUVs) and fitting a linear equation to this plot gave a good correlation (Pearson’s r = −0.94). In contrast to their wash-off kinetics, the EC50 values of DkTx and its variants did not correlate well with their membrane partitioning parameters (Kx, Figure S7a; and fractional depletion, Figure S7b), suggesting that bulk membrane partitioning properties of DkTx do not contribute substantially to the toxin’s potency. Taken together, the results of these depletion experiments performed on physiological membranes are consistent with those of the fluorescence-based membrane partitioning experiments on LUVs and implicate the high membrane affinity of DkTx for its slow washing-off feature.
The conclusions drawn from the results described above are valid only if the toxin variants fold properly and retain the wild-type toxin’s overall structure. To answer this question, we performed CD experiments on wild-type DkTx and our most functionally disrupted DkTx variants (V8A, W11A, F27A, I28A, W53A, F67A, and I68A). The CD spectra of these variants were observed to be identical to that of the wild-type toxin (Figure S8, Supporting Information) demonstrating that the toxin variants fold properly.
Discussion
The overarching goal of this study was to elucidate the functional roles of protein–membrane lipid interfaces by leveraging the power of electrophysiology that enables the characterization of the function of ion channel membrane proteins under physiological conditions. By complementing these studies with fluorescence-based membrane partitioning and cellular depletion experiments, we have dissected the functional contributions of protein–lipid and protein–protein interactions orchestrated by the spider toxin, DkTx, to its TRPV1 ion channel activation properties.
An important insight that our results provide is that protein–lipid interactions do not merely subtly modulate but, rather, can drive protein function. To illustrate this point, the entire data set obtained from our electrophysiology experiments on DkTx is summarized in the potency (EC50 values) versus channel-dissociation rate (% wash-off) plots depicted in Figure 5, separately for the K1 and K2 knots (except for the lipid-interacting K2 knot variants, W53A and W53L, that could not be fully characterized owing to their low potency). The plot for the K1 knot variants depicts several data points (corresponding to the W11A, W11L, V8A, G12A, W11Y, I28A, and F27A variants) that are localized at its top-right portion, implying that substituting these K1 knot residues of the toxin resulted in a substantial increase in channel-dissociation rates of the toxin as well as appreciable ablation of the toxin’s potency. Except for G12, all of these residues are lipid-interacting. On the other hand, the majority of the data points for the K2 knot variants are clustered toward the left side of the plot, highlighting that these variants demonstrate slow wash-off similar to the wild-type toxin. Several K2 knot variants, however, demonstrate a considerably reduced potency for activating TRPV1 including the W53A, W53L, V50A, F67A, W53Y, G54A, and I68A variants, among which all except G54A are variants of lipid-interacting toxin residues (Figure 5b). This observation that the lipid-interacting K1 residues contribute profoundly to both the toxin’s potency and in endowing it with slow channel-dissociation rates, whereas those of the K2 knot contribute substantially to the former but negligibly to the latter, reveals the ability of protein–lipid interactions to fine-tune protein function in different ways. This functional dichotomy of the K1 and K2 lipid-interacting interfaces is particularly noteworthy considering that both map to similar regions of two structurally similar motifs (K1 and K2 knots) present within the same protein molecule.
Figure 5.
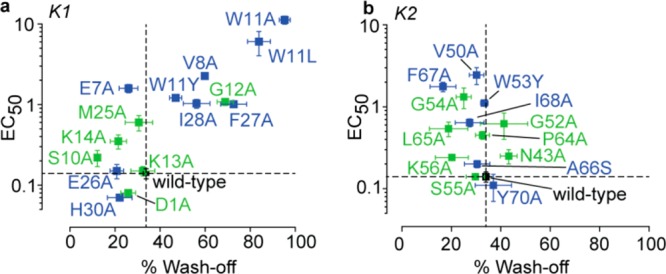
Plots of potency for TRPV1 activation (EC50 values) versus channel-dissociation rates (% wash-off), of DkTx variants. (a) DkTx variants of the K1 knot residues and (b) those of the K2 knot residues. Data points for the variants of lipid-interacting residues are depicted in blue, those of channel-interacting residues in green, and that for the wild-type toxin in black. Percentage wash-off depicts the % reduction in current after 3 min of buffer perfusion post channel activation with saturation concentrations of the toxin variants. The dotted lines represent the data for wild-type DkTx.
Mapping of the functionally critical toxin residues identified in this study to the DkTx–TRPV1 complex structure reveals that the toxin activates the channel by employing a unique mechanism that entails the formation of two hydrophobic interfaces comprising membrane lipids, hydrophobic residues of the toxin, and those of the channel (Figure 6). The high degree of hydrophobicity of both of these tripartite complexes (one formed by the K1 knot and the other by the K2 knot) is prominently seen in their surface renditions colored by employing the Eisenberg hydrophobicity scale35 (right panels of Figure 6a and b). Each complex lies at the interface of two adjacent channel subunits (labeled subunits 1 and 2 for the K1 knot complex and subunits 2 and 3 for the K2 knot complex in Figure 6) such that the entire DkTx–TRPV1 complex spans three subunits of the tetrameric channel. Whereas one of the TRPV1 subunits of each complex contributes the hydrophobic F655, A657, V658, and I661 residues of its S6 helix, the other contributes the Y631 residue of its pore helix. Consistent with our discovery that these toxin–lipid–channel interfaces play critical roles in toxin activation of the channel, variants of two of these channel residues, A657 and Y631, have been shown to disrupt the DkTx-sensitivity of the channel.20 The complexes contain three putative phosphatidylethanolamine lipid molecules each, one of which (lipid 1 of the K1 knot complex and lipid 4 of the K2 knot complex) is embedded deep in a hydrophobic pocket formed by the functionally critical toxin residues identified in this study—V8, W11, I28, and F27 of the K1 knot and V50, W53, I68, and F67 of the K2 knot and channel residues F655, A657, V658, and I661 (Figure 6). The tryptophan residues of the toxin form the floor of these hydrophobic pockets, whereas the walls of the pockets are formed by the hydrophobic side chains of the isoleucine and phenylalanine toxin residues on one side and those of the channel residues, F655, V658, and A657, on the other, whereas the channel residue I661 forms the outer edge of the pocket. The tails of each of the lipid molecules in the pdb file for the structure are truncated, and no structural information on their peripherally located CH2 groups is available. It is, however, tempting to speculate that the hydrophobic valleys formed by the phenylalanine residues of the toxin (F27 in case of the K1 knot complex and F67 in case of the K2 knot complex) and I661 of the channel serve as hydrophobic conduits for one of the hydrophobic tails of lipid 1 and lipid 4 to thread through, and that the other hydrophobic tail of each of these lipids threads through the hydrophobic valleys formed by the side chains of the V8/V50 and I28/I68 residues of the toxin (right panels of Figure 6a and b). In addition to binding to the toxin and channel residues, lipids 1 and 4 also engage in hydrophobic interactions with lipids 2 and 5 that are located nearby (Figure 6). The third lipid of each complex (lipid 3/lipid 6) forms a second hydrophobic patch adjacent to the one detailed above. Whereas lipid 1 and lipid 4 interact with loop 1 and loop 2 of the knots, lipid 3 and lipid 6 form hydrophobic patches with the I28/I68 and F27/F67 residues of loop 4 of the knots along with the Y631 residue of the pore helix of an adjacent TRPV1 subunit. Interestingly, one of the functionally important residues identified in this study, G12 (Figures 2b, 3d, and 5), does not make direct contact with any of the lipids in the structure (Table S1). Considering the unique property of glycine to confer conformational flexibility to protein structure,36,37 it is possible that G12 enables the adjacent W11 residue to reach down into the membrane and form the heart of the tripartite complex. A similar logic may explain the functionally important roles of the G52 and G54 residues of the K2 knot that flank its critical W53 residue.
Figure 6.

Functionally critical toxin residues of the (a) K1 knot and (b) K2 knot, identified in this study mapped onto the structure (pdb: 5irx(1)) along with the TRPV1 residues and lipid molecules they interact with. The left panels depict the toxin residues in blue, the lipids in the sticks representation, and the channel residues in the color employed to depict the channel subunit they belong to (the color scheme used to render the channel subunits is the same one that was used in Figure 1). The right panels depict the same interface shown on the left panel in the surface orientation rendered by employing the Eisenberg scale, wherein the intensity of the red color is proportional to the hydrophobicity.
Previous work has demonstrated that the individual knots of DkTx can activate TRPV1 albeit with a lower potency than that of the double knot and that the K1 knot’s potency is much lower than that of the K2 knot.20,27 If the contributions of the individual knots toward the potency of the double knot are in accordance with their potency as single knots, variants of the K1 knot residues of DkTx would be expected to cause minimal alteration of the toxin’s potency. In contrast to this expectation, we observed that several K1 knot variants dramatically disrupted TRPV1 activation properties. Indeed, among the 15 K1 knot variants we tested, eight (W11A, W11L, V8A, E7A, W11Y, G12A, I28A, and F27A in the order of increasing potency) demonstrated a >7-fold increase in the EC50 values as compared to that of the wild-type toxin (Figure 5a and Table S2). This dependence of the toxin’s potency on K1 knot residues despite its low intrinsic potency as a single knot suggests that the knots on their own activate the channel by employing a different mechanism as compared to that of the double knot. This notion is supported by the observation that, whereas introducing the L65A substitution into the K2 single knot substantially reduces its potency,2 this substitution has only a moderate effect in the context of the double knot (Figure 2). Obtaining structures of single knots bound to the channel in lipidic environments coupled with mutagenesis-based structure–function studies on the individual knots akin to the one reported in this work on the double knot can enable testing of this intriguing hypothesis.
One of the most striking results we obtained in this study is that in contrast to wild-type DkTx, several variants of lipid-interacting residues of the K1 knot of DkTx demonstrate rapid wash-off rates (Figures 3 and 5a). Interestingly, these wash-off rates are inversely correlated with the membrane partitioning ability of these toxin variants measured both on artificial lipid vesicles (Figure 4c) and also on cells (Figure 4e). Since the partitioning experiments were performed on lipid vesicles/cells not containing the TRPV1 channel, this observation suggests that the high intrinsic membrane affinity of the wild-type toxin underlies its slow wash-off. Efficacious membrane partitioning of the toxin will lead to its accumulation at high concentrations within the membrane, suggesting that the slow wash-off phenotype of the toxin may be due to its presence at high concentrations in the membrane. These results are consistent with a “toxin relay mechanism” depicted in Figure 7. When the toxin is applied, it accumulates in the membrane at high concentrations owing to its highly efficient partitioning ability, and a few of these molecules bind to TRPV1 channels resulting in channel activation. Upon subsequent initiation of wash-off by buffer perfusion, the channel-bound toxin molecules (the knots of which are depicted as filled ellipses) begin to get dislodged and are rapidly replaced by the surrounding membrane-partitioned toxin molecules that are present at a high concentration. Buffer perfusion also causes toxin molecules within the membrane that are not bound to TRPV1 (whose knots are depicted as open ellipses) to wash-off, resulting in depletion of the toxin in the vicinity of the channels, thereby slowing down the relay process, leading to an increase in the population of the toxin-free closed channels that manifests itself as a slowly decreasing TRPV1 current in the electrophysiology recording. This model convincingly explains our observation that the toxin variants that partition poorly wash-off rapidly (Figure 4c and e) and can also explain the concentration dependence of toxin wash-off kinetics (Figure 3b). The slow channel-dissociation rate of the toxin is, therefore, not merely due to a tight bimolecular binding event but also due to the high concentration of neighboring toxin molecules in the membrane that are available to replace channel-bound toxin molecules as they are lost due to buffer perfusion.
Figure 7.
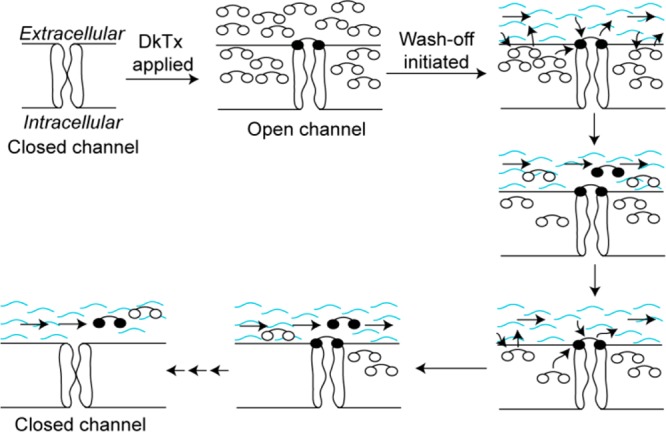
Our proposed “toxin relay” model that explains the slow wash-off feature of DkTx. DkTx is represented by a dumbbell-shaped object with its two ellipses denoting the two knots of the toxin. The knots of the toxin molecules that do not bind to the channel are depicted as empty ellipses and those that bind to the channel as filled ellipses. The wavy blue curves depict buffer flow.
Taken together, our studies on DkTx-activation of TRPV1 reveal that this process is driven primarily via protein–lipid interfaces, and minimally by lipid-independent protein–protein interactions. Furthermore, we report that DkTx activates the TRPV1 ion channel by employing a mechanism that entails the formation of hydrophobic toxin–lipid–channel interfaces, rather than by utilizing its charged amino acids to interact directly with the channel, as observed in the case of the potassium channel-targeting pore-blocking toxins such as charybdotoxin,38,39 and also for the acid-sensing ion channel (ASIC)-activating toxin psalmotoxin.40,41 It remains to be seen whether a similar mechanism of action is employed by the voltage sensor-targeting toxins, the channel complexes of which await structural characterization. Finally, our functional and membrane partitioning data support a mechanistic model that posits that the high propensity of DkTx for membrane partitioning in addition to its bivalent mode of binding underlies its slow channel-unbinding property.
Methods
Please see the Supporting Information for the interaction map table and detailed experimental procedures for the recombinant production of DkTx and its variants, electrophysiology, membrane partitioning, and CD experiments.
Acknowledgments
This work was supported by the Wellcome Trust/DBT India Alliance Fellowship [grant number IA/I/14/2/501551] awarded to J.K. and funds from IISER Pune. D.S. and Y.S. thank CSIR and IISER Pune, respectively, for providing fellowships. The contributions of rotation students, Y. Mathur, V. Pande, S. Sarkar, S. Singh, and N. Zacharia, towards production of DkTx variants are duly acknowledged.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.8b00644.
Detailed protocol for generating the interaction map of DkTx-lipids-TRPV1 complex, table depicting the map of interactions between DkTx and TRPV1/lipids, detailed procedure for the production of DkTx and its variants, representative DkTx purification data HPLC traces for purity evaluation of all our DkTx variants, procedure for electrophysiology experiments and data analysis, representative electrophysiological recordings of the DkTx variants, procedure for membrane partitioning experiments on LUVs and oocytes, tryptophan fluorescence data for the interaction of DkTx and its variants with LUVs, bar graph depicting mol fraction partition coefficients of DkTx variants of K2 knot residues, HPLC traces for oocyte-based toxin depletion assays performed with DkTx and its variants, table depicting consolidated data summary for all DkTx variants, potency (EC50) vs mol partitioning coefficient (Kx), and potency (EC50) vs fractional depletion plots, procedure and spectra for the CD-based structural characterization of DkTx and its variants (PDF)
Author Present Address
† Department of Biological Sciences, Indian Institute of Science Education and Research (IISER) Bhopal, Bhopal Bypass Road, Bhauri, Bhopal–462066, Madhya Pradesh, India
Author Contributions
D.S. and Y.S. performed the experiments. D.S., Y.S., and J.K. designed the experiments, analyzed the data, and wrote the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Gao Y.; Cao E.; Julius D.; Cheng Y. (2016) TRPV1 structures in nanodiscs reveal mechanisms of ligand and lipid action. Nature 534, 347–351. 10.1038/nature17964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae C.; Anselmi C.; Kalia J.; Jara-Oseguera A.; Schwieters C. D.; Krepkiy D.; Won Lee C.; Kim E. H.; Kim J. I.; Faraldo-Gomez J. D.; Swartz K. J. (2016) Structural insights into the mechanism of activation of the TRPV1 channel by a membrane-bound tarantula toxin. eLife 5, e11273,. 10.7554/eLife.11273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payandeh J.; Scheuer T.; Zheng N.; Catterall W. A. (2011) The crystal structure of a voltage-gated sodium channel. Nature 475, 353–358. 10.1038/nature10238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nury H.; Van Renterghem C.; Weng Y.; Tran A.; Baaden M.; Dufresne V.; Changeux J. P.; Sonner J. M.; Delarue M.; Corringer P. J. (2011) X-ray structures of general anaesthetics bound to a pentameric ligand-gated ion channel. Nature 469, 428–431. 10.1038/nature09647. [DOI] [PubMed] [Google Scholar]
- Long S. B.; Tao X.; Campbell E. B.; MacKinnon R. (2007) Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature 450, 376–382. 10.1038/nature06265. [DOI] [PubMed] [Google Scholar]
- Autzen H. E.; Myasnikov A. G.; Campbell M. G.; Asarnow D.; Julius D.; Cheng Y. (2018) Structure of the human TRPM4 ion channel in a lipid nanodisc. Science 359, 228–232. 10.1126/science.aar4510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laganowsky A.; Reading E.; Allison T. M.; Ulmschneider M. B.; Degiacomi M. T.; Baldwin A. J.; Robinson C. V. (2014) Membrane proteins bind lipids selectively to modulate their structure and function. Nature 510, 172–175. 10.1038/nature13419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong X.; Liu Y.; Liu W.; Liang X.; Laganowsky A. (2017) Allosteric modulation of protein-protein interactions by individual lipid binding events. Nat. Commun. 8, 2203. 10.1038/s41467-017-02397-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M.; Morgner N.; Barrera N. P.; Politis A.; Isaacson S. C.; Matak-Vinkovic D.; Murata T.; Bernal R. A.; Stock D.; Robinson C. V. (2011) Mass spectrometry of intact V-type ATPases reveals bound lipids and the effects of nucleotide binding. Science 334, 380–385. 10.1126/science.1210148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolla J. R.; Sauer J. B.; Wu D.; Mehmood S.; Allison T. M.; Robinson C. V. (2018) Direct observation of the influence of cardiolipin and antibiotics on lipid II binding to MurJ. Nat. Chem. 10, 363–371. 10.1038/nchem.2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A. G. (2011) Lipid-protein interactions. Biochem. Soc. Trans. 39, 761–766. 10.1042/BST0390761. [DOI] [PubMed] [Google Scholar]
- Phillips R.; Ursell T.; Wiggins P.; Sens P. (2009) Emerging roles for lipids in shaping membrane-protein function. Nature 459, 379–385. 10.1038/nature08147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunte C.; Richers S. (2008) Lipids and membrane protein structures. Curr. Opin. Struct. Biol. 18, 406–411. 10.1016/j.sbi.2008.03.008. [DOI] [PubMed] [Google Scholar]
- Polley A.; Orłowski A.; Danne R.; Gurtovenko A. A.; Bernardino de la Serna J.; Eggeling C.; Davis S. J.; Róg T.; Vattulainen I. (2017) Glycosylation and lipids working in concert direct CD2 ectodomain orientation and presentation. J. Phys. Chem. Lett. 8, 1060–1066. 10.1021/acs.jpclett.6b02824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coskun Ü.; Grzybek M.; Drechsel D.; Simons K. (2011) Regulation of human EGF receptor by lipids. Proc. Natl. Acad. Sci. U. S. A. 108, 9044–9048. 10.1073/pnas.1105666108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L.; Lee Y.-H.; Pappone P.; Palma A.; McNamee M. G. (1992) Site-specific mutations of nicotinic acetylcholine receptor at the lipid-protein interface dramatically alter ion channel gating. Biophys. J. 62, 61–63. 10.1016/S0006-3495(92)81779-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras F.-X.; Ernst A. M.; Haberkant P.; Björkholm P.; Lindahl E.; Gönen B.; Tischer C.; Elofsson A.; von Heijne G.; Thiele C.; Pepperkok R.; Wieland F.; Brügger B. (2012) Molecular recognition of a single sphingolipid species by a protein’s transmembrane domain. Nature 481, 525. 10.1038/nature10742. [DOI] [PubMed] [Google Scholar]
- Rissanen S.; Grzybek M.; Orłowski A.; Róg T.; Cramariuc O.; Levental I.; Eggeling C.; Sezgin E.; Vattulainen I. (2017) Phase Partitioning of GM1 and Its Bodipy-Labeled Analog Determine Their Different Binding to Cholera Toxin. Front. Physiol. 8, 252. 10.3389/fphys.2017.00252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blouin C. M.; Hamon Y.; Gonnord P.; Boularan C.; Kagan J.; de Lesegno C. V.; Ruez R.; Mailfert S.; Bertaux N.; Loew D.; et al. (2016) Glycosylation-dependent IFN-γR partitioning in lipid and actin nanodomains is critical for JAK activation. Cell 166, 920–934. 10.1016/j.cell.2016.07.003. [DOI] [PubMed] [Google Scholar]
- Bohlen C. J.; Priel A.; Zhou S.; King D.; Siemens J.; Julius D. (2010) A bivalent tarantula toxin activates the capsaicin receptor, TRPV1, by targeting the outer pore domain. Cell 141, 834–845. 10.1016/j.cell.2010.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalia J.; Milescu M.; Salvatierra J.; Wagner J.; Klint J. K.; King G. F.; Olivera B. M.; Bosmans F. (2015) From foe to friend: using animal toxins to investigate ion channel function. J. Mol. Biol. 427, 158–175. 10.1016/j.jmb.2014.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartz K. J. (2007) Tarantula toxins interacting with voltage sensors in potassium channels. Toxicon 49, 213–230. 10.1016/j.toxicon.2006.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohlen C. J.; Julius D. (2012) Receptor-targeting mechanisms of pain-causing toxins: How ow?. Toxicon 60, 254–264. 10.1016/j.toxicon.2012.04.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartz K. J.; MacKinnon R. (1995) An inhibitor of the Kv2.1 potassium channel isolated from the venom of a Chilean tarantula. Neuron 15, 941–949. 10.1016/0896-6273(95)90184-1. [DOI] [PubMed] [Google Scholar]
- Swartz K. J.; MacKinnon R. (1997) Hanatoxin modifies the gating of a voltage-dependent K+ channel through multiple binding sites. Neuron 18, 665–673. 10.1016/S0896-6273(00)80306-2. [DOI] [PubMed] [Google Scholar]
- Swartz K. J.; MacKinnon R. (1997) Mapping the receptor site for hanatoxin, a gating modifier of voltage-dependent K+ channels. Neuron 18, 675–682. 10.1016/S0896-6273(00)80307-4. [DOI] [PubMed] [Google Scholar]
- Bae C.; Kalia J.; Song I.; Yu J.; Kim H. H.; Swartz K. J.; Kim J. I. (2012) High yield production and refolding of the double-knot toxin, an activator of TRPV1 channels. PLoS One 7, e51516. 10.1371/journal.pone.0051516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheinerman F. B.; Norel R.; Honig B. (2000) Electrostatic aspects of protein-protein interactions. Curr. Opin. Struct. Biol. 10, 153–159. 10.1016/S0959-440X(00)00065-8. [DOI] [PubMed] [Google Scholar]
- Crowley P. B.; Golovin A. (2005) Cation-pi interactions in protein-protein interfaces. Proteins: Struct., Funct., Genet. 59, 231–239. 10.1002/prot.20417. [DOI] [PubMed] [Google Scholar]
- McGaughey G. B.; Gagne M.; Rappe A. K. (1998) pi-Stacking interactions. Alive and well in proteins. J. Biol. Chem. 273, 15458–15463. 10.1074/jbc.273.25.15458. [DOI] [PubMed] [Google Scholar]
- Stites W. E. (1997) Protein–Protein Interactions: Interface Structure, Binding Thermodynamics, and Mutational Analysis. Chem. Rev. 97, 1233–1250. 10.1021/cr960387h. [DOI] [PubMed] [Google Scholar]
- Lee S. Y.; MacKinnon R. (2004) A membrane-access mechanism of ion channel inhibition by voltage sensor toxins from spider venom. Nature 430, 232–235. 10.1038/nature02632. [DOI] [PubMed] [Google Scholar]
- Milescu M.; Vobecky J.; Roh S. H.; Kim S. H.; Jung H. J.; Kim J. I.; Swartz K. J. (2007) Tarantula toxins interact with voltage sensors within lipid membranes. J. Gen. Physiol. 130, 497–511. 10.1085/jgp.200709869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta K.; Zamanian M.; Bae C.; Milescu M.; Krepkiy D.; Tilley D. C.; Sack J. T.; Yarov-Yarovoy V.; Kim J. I.; Swartz K. J. (2015) Tarantula toxins use common surfaces for interacting with Kv and ASIC ion channels. eLife 4, e06774 10.7554/eLife.06774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg D.; Schwarz E.; Komaromy M.; Wall R. (1984) Analysis of membrane and surface protein sequences with the hydrophobic moment plot. J. Mol. Biol. 179, 125–142. 10.1016/0022-2836(84)90309-7. [DOI] [PubMed] [Google Scholar]
- Krieger F.; Moglich A.; Kiefhaber T. (2005) Effect of proline and glycine residues on dynamics and barriers of loop formation in polypeptide chains. J. Am. Chem. Soc. 127, 3346–3352. 10.1021/ja042798i. [DOI] [PubMed] [Google Scholar]
- Yan B. X.; Sun Y. Q. (1997) Glycine residues provide flexibility for enzyme active sites. J. Biol. Chem. 272, 3190–3194. 10.1074/jbc.272.6.3190. [DOI] [PubMed] [Google Scholar]
- Banerjee A.; Lee A.; Campbell E.; Mackinnon R. (2013) Structure of a pore-blocking toxin in complex with a eukaryotic voltage-dependent K(+) channel. eLife 2, e00594 10.7554/eLife.00594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein S. A.; Pheasant D. J.; Miller C. (1994) The charybdotoxin receptor of a Shaker K+ channel: peptide and channel residues mediating molecular recognition. Neuron 12, 1377–1388. 10.1016/0896-6273(94)90452-9. [DOI] [PubMed] [Google Scholar]
- Dawson R. J.; Benz J.; Stohler P.; Tetaz T.; Joseph C.; Huber S.; Schmid G.; Hugin D.; Pflimlin P.; Trube G.; Rudolph M. G.; Hennig M.; Ruf A. (2012) Structure of the acid-sensing ion channel 1 in complex with the gating modifier Psalmotoxin 1. Nat. Commun. 3, 936. 10.1038/ncomms1917. [DOI] [PubMed] [Google Scholar]
- Baconguis I.; Gouaux E. (2012) Structural plasticity and dynamic selectivity of acid-sensing ion channel-spider toxin complexes. Nature 489, 400–405. 10.1038/nature11375. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.