

Abstract
Radiotherapy has been used for over hundred years as a local tumor treatment. The occurrence of systemic anti-tumor effects manifesting as regression of tumors outside of the irradiated field (abscopal effect) was occasionally observed but deemed too rare and unpredictable to be a therapeutic goal. This has changed with the advent of immunotherapy. Remarkable systemic effects have been observed in patients receiving radiotherapy to control tumors that were progressing during immune checkpoint blockade, stimulating interest in using radiation to overcome primary and acquired cancer resistance to immunotherapy. Here we review the immunological mechanisms that are responsible for the ability of focal radiation to promote antitumor T cell responses that mediate tumor rejection and, in some cases, result in systemic effects.
Keywords: Abscopal effect, Anti-tumor immunity, DNA damage, Immunotherapy, Ionizing radiation, Tumor microenvironment
Interactions between radiotherapy and immune responses in cancer
Localized radiotherapy has a broad role in cancer treatment. It is used to reduce risk of recurrence after surgery, by treating the surgical cavity (e.g., in early breast cancer), as a curative treatment in some localized cancers (e.g., early prostate cancer) or as a palliative modality in metastatic disease, aiming at reducing the bulk of the tumor and relieve symptoms like pain or mechanical compression of organs. Depending on the clinical setting, the efficacy of ionizing radiation (IR), has been defined by its ability to either delay cancer growth or eliminate the irradiated tumor. Clinical observations of tumor responses outside the irradiated field, albeit extremely rare, are referred to as abscopal effect (from the Latin “ab” away from and “scopus”, target) [1]. A recent review of abscopal effects of radiotherapy reported between 1969 and 2014 the literature detected only 46 cases, despite millions of patients being treated worldwide [2]. Thus, it is not surprising that the abscopal effect remained a relatively obscure and largely ignored phenomenon until the advent of cancer immunotherapy.
The discovery that in some patients blocking the main immune checkpoints, CTLA-4 and/or PD-1, leads to immune-mediated tumor regression has reignited interest in cancer immunotherapy [3]. Experimental evidence in pre-clinical models has show that IR can enhance immune-mediated tumor recognition and rejection, acting in synergy with immune checkpoint blockade (ICB) [4, 5]. Observations of abscopal responses in patients receiving palliative radiotherapy due to tumor progression while on treatment with ICB [6, 7] have further contributed to the interest in deciphering the mechanisms underlying the immunogenicity of radiotherapy and its interaction with cancer immunotherapy. In this context, the abscopal effect from an anecdotal phenomenon has become a surrogate endpoint of efficacy in clinical trials of radiation and ICB [8]. The optimal application of radiotherapy in its new role as an immune-stimulator is under active investigation, and will be described in this review..
Cellular responses to ionizing radiation shape the immunogenicity of irradiated cancer cells
Whereas normal lymphocytes can undergo interphase apoptosis, most cancer cells that contain lethal damage die when attempting mitosis or sometimes after a few cycles of replication. Alternatively, they may become senescent, a metabolically active form of irreversible growth arrest that is often associated with a secretory phenotype [9]. It is becoming increasingly clear that the type of cellular response induced by IR is intimately connected to the immunogenicity of irradiated tumors. Here we will discuss the link between DNA damage repair (DDR) mechanisms (described in BOX 1) and the cytoplasmic nucleic acids sensing that activate interferon type I (IFN-I)-driven immune activation and inflammation.
BOX 1. Ionizing radiation-induced DNA damage and repair mechanisms.
The type of radiation most commonly used for cancer treatment is X-ray or γ-ray ionizing radiation (IR). As opposed to non-ionizing radiations (i.e. radio- or micro- waves, visible, infrared or UV light), IR causes DNA damage by tearing off electrons from atoms, which cause damage directly or indirectly via the production of reactive oxygen species (ROS) generated by interactions between free radicals and molecular oxygen (Figure I) [9]. The ensuing cellular response depends upon several factors, including the dose of IR applied, the type of DNA injury, the phase of the cell cycle, and the integrity of the DNA damage repair (DDR) machinery. DNA double-strand breaks (DSBs) represent the principal damage induced by IR. DNA DSBs can be simple or complex depending on the physical characteristics of the break, the chromatin architecture surrounding the break and the kinetics of break repair (Figure II) [80–82]. While simple DSBs are amenable to fast repair, complex DSBs require activation of coordinated DDR processes to prevent genomic instability and cell death. Cells use two main mechanisms to repair DSBs: homologous recombination (HR), and non-homologous end joining (NHEJ) [80]. NHEJ and HR contribute to the fast and slow component of DSB repair with half-lives ranging from 5 to 30 min and 2 to 5 hours respectively [80].
NHEJ pathway catalyzes simple rejoining reactions between DNA ends with no sequence homology, making it highly efficient but also more error-prone. NHEJ repairs the majority of direct two-ended radiation-induced DSBs and is the predominant repair mechanism in the G1 phase of the cell cycle. In addition to the classical NHEJ process, the alternative end joining (alt-EJ) mechanism can be triggered to either compensate for the lack of some NHEJ core proteins or for the failure of NHEJ or HR to complete repair. Alt-EJ uses the PARP1 polyribosylating enzyme and the XRCC1/DNA ligase III complex to repair DNA breaks [80].
The HR pathway is elicited to repair complex DNA DSBs. This mechanism is relatively slow but highly accurate because it uses an undamaged DNA guide strand to repair the DSBs. As a consequence, HR can only operate during the S and G2 phase of the cell cycle when sister chromatids are present to obtain homologous DNA sequences, while NHEJ is inhibited by the cell cycle regulator CYREN [83].
Defects in DDR pathways are known to promote carcinogenesis by allowing the generation of mutations. Recently it has become clear that dysfunction of DDR processes are both extremely frequent in cancer and critical for allowing the proliferation of cells carrying damaged DNA, thereby facilitating the persistence of genomic instability [10]. While downregulation of DDR processes underlies the enhanced sensitivity of cancer cells to DNA damaging treatments such as IR, which has been exploited therapeutically for a long time, the immunological implications of defective DDR are only beginning to be appreciated. Progression of cancer cells through mitosis following IR-induced unrepaired DNA DSBs leads to the formation of micronuclei. Micronuclei have defective membranes that easily break down exposing double-stranded DNA (dsDNA) to the cytosolic dsDNA sensor cyclic GMP–AMP synthase (cGAS) [11–13]. cGAS is a pattern recognition receptor that triggers IFN-I production via the downstream adaptor STING and is critical for activation of immune responses to viruses [14]. Thus, the accumulation of dsDNA in the cytosol of irradiated cancer cells activates canonical defense pathways that are hard-wired in the immune system, and chiefly rely on CD8 T cell activation to clear viral infections. Likewise, successful anti-tumor immunity elicited by IR is mediated by cGAS and STING pathways in the irradiated tumor, and relies on activation of anti-tumor CD8 T cells [11, 15, 16].
These recent results emphasize the importance of dsDNA as a danger signal or damage-associated molecular pattern (DAMP) that defines IR-induced immunogenic cell death (ICD) [17]. Like other IR-generated DAMPs, including plasma membrane-exposed calreticulin, secreted HMGB1, and ATP [18, 19], DNA derived from the cancer cells is also sensed by tumor-infiltrating dendritic cells (DCs) via the cGAS/STING pathway, promoting DC activation and effective cross-presentation of tumor-derived antigens to T cells [16] (Figure 1).
Figure 1: Radiation-induced secretion of IFN-I is critical for abscopal responses.
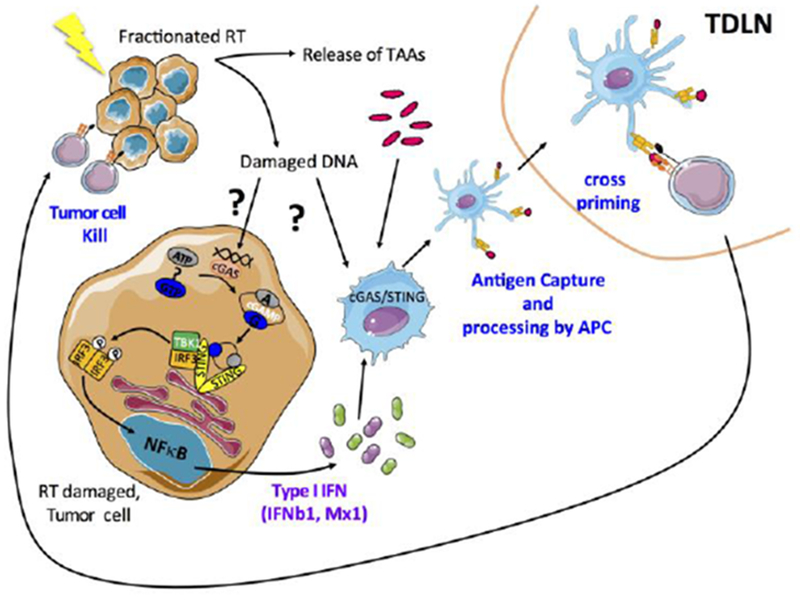
Ionizing radiation leads to the release of tumor associated antigens (TAA) and damaged DNA that can stimulate the production of type-I interferon via the cGAS/STING pathway in tumor cells. The subsequent secretion of IFN-I and interferon-stimulated genes (including CXCL10) promote the recruitment and activation of BATF3-DCs. Once in the tumor, BATF3-DCs take up the TAA and tumor-derived DNA to further stimulate the production of IFN-I. Activated BATF3-DCs then migrate to the tumor draining lymph node where they can prime CD8+ T cells to initiate cytotoxic T-cell responses. Once activated, cytotoxic T lymphocytes (CTLs) migrate to the irradiated tumor and eliminate residual cancer cells, and to distant metastatic sites leading to systemic tumor regression (abscopal effect).
Poly-(adenosine diphosphate-ribose)-polymerase (PARP) proteins play multiple roles in DDR and their inhibition hinders DNA repair and enhances cancer sensitivity to IR [20]. Interestingly, improved tumor control in mice treated with the combination of a PARP inhibitor and IR was found to depend on the induction of senescence in the cancer cells, associated with secretion of multiple pro-inflammatory cytokines and chemokines that stimulated T cell-mediated tumor rejection [21]. Although senescent cells do not proliferate and cannot generate micronuclei, it has been recently demonstrated that they accumulate dsDNA in the cytosol that is sensed by cGAS. The DNA is transferred to the cytosol as chromatin fragments that pinch off from the nuclei, a process that occurs in conjunction with loss of the nuclear lamina protein lamin B1 [22]. In senescent cells, however, cGAS/STING pathway activation resulted in a NFkB pathway-regulated pro-inflammatory cytokine production whereas STING-mediated IFN-I induction was inhibited by p38 MAP-kinase activation [22]. These data provide further support for a role of cytosolic DNA as a regulator of immune signaling following cancer cells irradiation, and suggest that multiple factors can modulate the type of signals generated.
DNA is not the only nucleic acid that functions as an IR-induced DAMP. Small endogenous non-coding RNAs, such as U1 and U2, translocate to the cytoplasm after IR where they bind to the dsRNA sensor DExD/H-box helicase 58 (DDX58, also known as retinoic acid-inducible gene-1, RIG-I), leading to downstream activation of mitochondrial antiviral signaling protein (MAVS) and IFN-I production [23].
Overall, the relative contribution to anti-tumor immunity of each of the members of this growing list of IR-induced DAMPs remains to be determined, but it is likely to vary depending on the intrinsic immunogenicity of a given tumor [24], the type of radiation dose and fractionation regimen and the immunotherapy agent used, as discussed below.
In context: Effects of ionizing radiation in the tumor microenvironment
The tumor microenvironment (TME) is dominated by immunosuppression, with multiple mechanisms that allow tumor escape from immune-mediated control that have been linked to tumor responsiveness to immunotherapy [25]. Here we will discuss the mechanisms that have been shown to influence IR effects and/or that are modulated by IR and can hinder or promote anti-tumor immune responses.
Mechanisms that limit anti-tumor immunity
Hypoxia is a common feature of the TME, especially prevalent in aggressive and rapidly growing tumors. Hypoxia drives many processes that favor invasion and metastasis, and contribute to establishing an immunosuppressive TME [26]. The hypoxia-inducible factor-1α (HIF-1α) upregulates genes that control angiogenesis, metabolism and metastasis [27]. In addition, HIF-1α drives the expression of programmed death-ligand 1 (PD-L1) on both cancer cells and myeloid-derived suppressor cells (MDSCs) [26, 28], and the production of vascular endothelial growth factor A (VEGFA) by cancer cells. VEGFA promotes the recruitment of regulatory T cells and MDSCs to the tumor and suppresses the maturation of DCs [29]. Moreover, HIF-1α increases shedding of the NKG2D ligand MICA from the surface of tumor cells reducing their killing by NK cells [30]. However, HIF-1α and VEGFA are required for CD8 T cells adaptation to the hypoxic TME and maintenance of effector function [31], indicating that therapeutic targeting of these factors could impair tumor rejection.
The interplay between IR and tumor hypoxia is complex. On the one hand, hypoxic tumors have reduced radiosensitivity, since indirect DNA damage induced by IR depends on the formation of ROS (BOX 1). On the other hand, radiation leads to re-oxygenation of hypoxic tumors, an effect attributed to increased perfusion and decreased oxygen consumption [27]. However, this is paradoxically associated with an increase in levels of HIF-1α and HIF-regulated proteins, due, in part, to ROS-mediated induction of HIF-1 [32]. Importantly, macrophages that infiltrated the tumor and produced NO were shown to be responsible for elevated levels of HIF-1α in normoxic tumors a few days after irradiation due to NO-mediated S-nitrosylation and stabilization of HIF-1α [33]. Thus, HIF-1α-induced effects play an important role in shaping the TME after IR.
Cancer-associated fibroblasts (CAFs) are relatively resistant to IR but undergo stress-induced senescence, which is associated with a secretory phenotype known as senescence-associated secretory phenotype (SASP). Secreted cytokines, chemokines, pro-angiogenic and growth factors promote chronic inflammation and a pro-tumorigenic TME [34]. A recent report provides evidence for a role of IR-activated CAFs in fostering a metabolic switch in colorectal cancer that increases glutamine consumption via activation of insulin-like growth factor-1 receptor (IGF-1R). As a consequence, the paracrine IGF-1/IGF-1R signaling initiated by IR-activated CAFs enhanced progression in a colorectal cancer model [35]. While this study did not investigate the immune effect of the IR-induced IGF-1/IGF-1R pathway, there is evidence that its stimulation in the myeloid compartment promotes M2-like macrophages polarization [36].
Transforming growth factor-beta (TGF-β) signaling has effects on the tumor cell itself and on host-tumor cell interactions. Tumor-cell-autonomous TGFβ effects can paradoxically result in tumor suppression as well as tumor promotion [37]. TGFβ crosstalk with the TME shapes cell type-dependent and context-dependent regulatory effects. Activation of TGFβ occurs in the irradiated tumor via a ROS-induced conformational change of the latency-associated peptide/TGFβ complex releasing active TGFβ [38]. Activated TGFβ promotes DNA repair thus contributing to tumor radio-resistance [37]. Most importantly, TGFβ drives immunosuppressive effects which limit IR-induced immune activation. In two mouse tumor models of breast cancer neutralization of TGFβ was required for IR-induced priming of CD8 T cells to multiple tumor antigens, and for T cell-mediated inhibition of non-irradiated metastases [39]. These data highlight the importance of TGFβ-mediated immunosuppression in the context of the irradiated tumor.
The recruitment of myeloid cells with immunosuppressive function to irradiated tumors can also hinder the development of anti-tumor immunity. IR was shown to increase the expression of colony-stimulating factor-1 (CSF-1) by mouse and human prostate cancer cells in vitro by inducing binding of the DNA damage-induced kinase ABL1 to the CSF1 gene promoter. In vivo, IR-induced CSF-1 enhanced the recruitment of both, monocytic (CD11bhily6Clo) and granulocytic (CD11bloly6Chi) MDSC to mouse tumors, reducing therapeutic response. Elevated CSF-1 levels in the serum of prostate cancer patients were seen after radiotherapy, suggesting clinical relevance of this pathway [40]. The chemokine stromal cell-derived factor 1 (SDF-1, also known as CXCL12) has also been implicated in IR-induced myeloid cell recruitment to the tumor. Blockade of SDF-1 interaction with its receptor C-X-C chemokine receptor 4 (CXCR4) prevented IR-induced recruitment of tumor associated macrophages (TAM) and inhibited revascularization in intracranial human GBM xenografts [41].
In a mouse tumor model of pancreatic adenocarcinoma, the C-C ligand motif 2 (CCL2) chemokine was upregulated by IR in the cancer cells, resulting in vivo in increased tumor infiltration by inflammatory macrophages expressing C-C chemokine receptor type 2 (CCR2) [42]. The mobilization of inflammatory monocytes via CCL2/CCR2 axis has been described as a negative prognosticator in several cancers, including pancreatic cancer [43], suggesting that IR-induced upregulation of CCL2 is detrimental.
Upregulation of PD-L1-following IR has been reported in several preclinical studies, and seems to reflect an acquired immune resistance response to interferon-γ (IFNγ)-producing effector T cells induced by IR [5, 39, 44]. Thus, PD-1/PD-L1 axis may represent a major obstacle to RT-induced tumor rejection, a hypothesis currently being tested in several clinical studies [8].
Mechanisms that promote anti-tumor immunity
IR has multiple immune-modulatory effects on the TME that favor tumor rejection. As discussed above, cytosolic dsDNA generated by IR leads to production of IFNβ by irradiated cancer cells, as well as by the DCs infiltrating the tumor, promoting the cross-presentation of tumor antigens to CD8 T cells [15, 16]. Moreover, IFN-stimulated genes (ISGs) that encode the chemokines CCL5, CXCL16 (C-X-C motif chemokine ligand 16), and CXCL10, are upregulated and drive the primed effector T cells to the tumor [15, 45, 46]. The upregulation of adhesion molecules on the tumor vascular endothelium following IR can also contribute to improved T cell infiltration [47, 48].
Surviving cancer cells after radiation exposure can become targets for elimination by NK and effector CD8 T cells. This effect is in part due to upregulation of NKG2D ligands, a direct consequence of DDR response activation [49–51]. Increased surface MHC class I and Fas/CD95 were also shown to increase rejection of the irradiated tumor by adoptively transferred T cells [52, 53]. There is at least some evidence that the massive alterations in the post-radiation transcriptome results in changes in the antigenic repertoire presented by the cancer cells on MHC class I [53]. However, it is not clear if the presentation of a more diverse peptide pool underlies the broadening of the TCR repertoire of infiltrating T cells that has been observed after IR [54, 55].
Finally, by promoting transfer of tumor antigens to the infiltrating myeloid cells IR induces their killing by anti-tumor T cells. The latter effect resulted in tumor regression in an experimental situation where the cancer cells themselves could not be recognized by T cells [56]. It remains to be determined if such mechanisms can contribute to overcoming the resistance to immunotherapy attributed to cancer cell loss of β2-microglobulin or other components of the MHC class I antigen presentation machinery [25].
Overall, the interaction of IR with the TME is complex. The degree of pre-existing hypoxia and immunosuppression determine the threshold of positive changes that radiation must induce to shift the balance towards immune activation, possibly explaining the rarity of abscopal responses in the absence of immunotherapy. Additionally, radiotherapy’s ability to induce activating versus suppressive effects may depend on the radiation dose and fractionation and how they interact with a given tumor, as further discussed below.
In situ vaccination by radiotherapy: from local to abscopal
The demonstration in multiple pre-clinical studies that IR induces anti-tumor T cells when applied to an in vivo growing tumor has fostered enthusiasm for its use as a tool to convert a tumor into an in situ vaccine in patients [57, 58]. However, while T cell contribution to regression of the irradiated tumor is well-proven [59], the generation of T cell responses that are effective against non-irradiated metastases has remained a challenge. It is not surprising that IR alone is capable to elicit abscopal responses only in a very small portion of patients, considering that most other strategies for therapeutic vaccination in cancer have failed due to the now well-recognized immunosuppressive barrier [2, 60]. Encouragingly, pilot studies have shown abscopal responses in about a third of the patients when IR was combined with stimulation of the immune system by GM-CSF or TLR agonists [57, 61]. The most striking abscopal responses have been seen when IR was used with immune checkpoint blockade (ICB), [6, 62]. An example is a patient with metastatic non small cell lung cancer (NSCLC) refractory to chemotherapy who was treated with radiotherapy to a single liver metastasis and anti-CTLA-4 and showed complete regression of multiple liver, lung, bone and lymph node metastases [62]. What is especially remarkable is that this response is durable, lasting now for more than 4 years in a disease that has shown minimal response to anti-CTLA-4 alone or combined with chemotherapy [63, 64]. However, as discussed in the next session, a significant induction of abscopal responses by the combination of IR and ICB has yet to be confirmed in prospective studies [65]. Two strategies are currently being pursued in pre-clinical studies to improve the results: (1) combinations of multiple immunotherapy agents to overcome suppressive pathways and/or to increase T cell activation, and (2) optimization of radiation delivery to achieve the best ratio of immunea-ctivating to immune-suppressive signals.
Preclinical studies investigating combinations of multiple immunotherapy agents.
Several strategies are in development to enhance systemic responses to radiotherapy and ICB, and some have shown benefits in preclinical studies, as detailed below.
Abscopal responses were seen in 17% of mice bearing bilateral B16/F10 melanoma treated with 20 GyXl to one tumor and anti-CTLA-4. The modest response was attributed to tumor upregulation of PD-L1 and T-cell exhaustion. Dual immune checkpoint blockade by anti-CTLA-4 and anti-PD-Ll or anti-PD-1 improved responses and long-term survival of the mice. Radiotherapy broadened the T cell repertoire, while anti-CTLA-4 depleted intratumoral regulatory T cells, and anti-PD-Ll reinvigorated the exhausted T cells [53].
In one study using two poorly immunogenic breast carcinomas (TSA and 4T1), concurrent blockade of TGFβ with radiation (6GyX5) induced CD8 T cell responses to multiple tumor antigens and regression of irradiated tumors and non-irradiated lung metastases or synchronous tumors. However, PD-L1 was upregulated in the tumor, and all tumors recurred. Addition of anti-PD-1 to the combination of radiation and TGFβ blockade delayed tumor recurrence and extended mice survival [39]. In another study, simultaneous targeting of TGFβ and PD-L1 was achieved using a bifunctional fusion protein (M7824). Treatment of MCA-38-tumor bearing mice with M7824 and 5 Gy X 5 radiation led to an increase of tumor-specific CD8 T cells and rejection of irradiated and abscopal tumors, while each treatment alone had modest effects [65].
Abscopal responses were reported in mice bearing breast, melanoma and colorectal bilateral tumors following local radiation to one tumor with an agonistic anti-CD137 (4-IBB) and a neutralizing PD-1 antibody. Tumor regression was improved when the three treatments were combined, and was associated with increased T cell tumor infiltration. The response was abrogated in mice deficient in BATF-3-dependent dendritic cells, indicating that these cells were required for cross-priming of tumor-specific CD8 T-cells [64].
An oligonucleotide aptamer that simultaneously targets VEGF and 4-IBB was used to activate T cells via 4-1BB/CD137 only in the tumor and avoid the toxicity seen with systemic anti-4-1BB antibody. As monotherapy VEGF-targeted 4-1BB failed to induce responses in VEGFlow 4T07 tumors. Tumor-targeted radiation (12GyX1) upregulated VEGF and enhanced tumor responses to the aptamer. In mice with two 4T1 tumors, which express high levels of VEGF, abscopal responses were achieved when mice received radiation to one tumor together with VEGF-targeted 4-1BB [66].
Preclinical studies investigating the optimization of radiation delivery
The second strategy was initially explored by Dewan et al. [66], in a study comparing side by side three IR regimens for the ability to induce abscopal effects in combination with anti-CTLA-4 in mouse models of breast and colorectal cancer. Two regimens consisting of 8GyX3 and 6GyX5 were effective, whereas a single 20Gy dose was not. This result was puzzling since induction of anti-tumor T cells was observed in pre-clinical studies with the use of single IR doses as high as 30 Gy in some tumor models, however without evidence of generating abscopal responses [67]. Many of the IR-induced effects described above result in rejection of the irradiated tumor, but the bar is higher at the abscopal sites, explaining the need for more robust T cell responses. Recent data have provided new insights into the requirements for achieving abscopal responses. In the setting of combinations of radiation and ICB the induction of abscopal responses relies on the activation of IFN-I via cGAS and STING pathway in the irradiated cancer cells. This burst of IFN-I jump-starts the immune response by attracting BATF3-dependent DCs (best known as DC1, ref. [68]) to poorly immunogenic tumors that are intrinsically inept at recruiting these DCs [15]. As mentioned above, dsDNA that accumulates in the cytosol of the irradiated cancer cells is the trigger of this response, which is under the control of the DNA exonuclease TREX1. TREX1 is expressed at low levels in all cells and clears cytosolic DNA to prevent autoimmunity driven by improper IFN-I activation [69]. In cancer cells TREX1 levels are upregulated in an IR dose-dependent fashion: after a certain dose threshold TREX1 is induced in quantity sufficient to digest the accumulated cytosolic DNA, resulting in abrogation of IR immunogenicity [15]. In the mouse breast and colorectal carcinoma cells tested by Dewan et al. [66], 6 and 8 Gy doses enhanced dsDNA without increasing TREX1, but 20 Gy induced a marked upregulation of TREX1 [15]. The threshold for the induction of TREX1 upregulation is dictated by the size of the single IR dose, not by the total dose delivered.
In fact, repeating daily doses of 8Gy three times enhanced the upregulation of IFN-I. For most carcinoma cells the window of maximal cytosolic dsDNA accumulation ranges between 6 to 12 Gy, but can vary considerably among different cancer cell lines. If the importance of this mechanisms is confirmed in the clinic, careful consideration and possibly ex vivo testing may be needed to identify the optimal IR dose before treatment with the combination of IR and ICB [70].
Partnership of radiotherapy and immunotherapy
Since many ICBs have entered mainstream oncology in the last few years, their use in patients that were treated with IR has become common. Many retrospective studies that analyzed the interaction between IR and ICBs found at least some evidence of abscopal responses, but in cancers responsive to ICBs it cannot be ruled out that these responses reflect a later activity of the immunotherapy itself, which can manifest slowly [71]. Two retrospective studies are of particular interest in supporting the hypothesis that IR can increase responses to ICB: one study analyzed 98 NSCLC patients treated in a pembrolizumab trial and found that progression-free survival was significantly longer in patients who previously received radiotherapy than in patients who did not [72]. In another study analyzing 101 patients with advanced melanoma it was found that patients who had received concurrent radiotherapy while on ipilimumab showed a higher rate of complete response (25.7% vs. 6.5% for ipilimumab alone; p=0.04) and increased median overall survival of 19 months vs. 10 months for ipilimumab alone (p = 0.01) [73]. These intriguing data await confirmation in prospective randomized trials. Many studies that combine IR and ICBs are ongoing (reviewed in [8]). Among the few studies designed to assess abscopal responses two early phase trials have shown early positive but limited effects [74, 75] while others failed to demonstrate a clear benefit of IR [55, 76]. Possible factors that might have influenced the results of these trials have been discussed elsewhere [65, 77]. The achievement of local tumor control by IR and ipilimumab was shown to correlate with abscopal responses in a retrospective trial [7], suggesting that it may reflect the induction of effective anti-tumor immunity. A better understanding of the mechanisms underlying the immunogenicity of IR will help patients selection and design of new clinical studies testing the benefits of IR with immunotherapy.
One setting with enormous potential is early stage disease, where the in situ vaccination by IR could induce T cell responses that are able to control micro-metastatic/low-burden disease and reduce recurrence. This notion is supported by the recently reported results of a trial in 713 patients with locally advanced non-resectable NSCLC. Responders to initial chemo-radiation were randomly assigned to either anti-PDL-1 antibody durvalumab or placebo. At an interim analysis patients randomized to durvalumab demonstrated a median progression-free survival of 16.8 months versus 5.6 months with placebo [78].
Concluding Remarks
Until recently, immunology and radiation biology have evolved in parallel, with limited interactions between the two disciplines. It is now clear that the two fields of investigation are converging, in the context of oncology. The tumor response to IR is clearly dependent on its stimulatory or suppressive effects on the immune system [79]. On the other end, the ability of IR to harness canonical immune responses to viral infection can be exploited to elicit anti-tumor CD8 T cell responses. The hope, supported by pre-clinical data and clinical observations, is that such responses can be elicited in patients with “cold” tumors that do not engage the immune system, thus enabling them to better respond to ICB [24]. Progress in establishing the value of IR and its place in the context of cancer immunotherapy requires an improved knowledge of the molecular and cellular mechanisms that define the immunogenicity of IR, and their application in the context of IR doses and fractionation. IR in the clinic is used in a large range of doses delivered in a single, a few, or many fractions, based on the tumor type and size, its location, and the therapeutic goal. Use of IR to generate an in situ tumor vaccine requires optimization of the dose and fractionation, and may result in paradigm shifts when compared with standard radiotherapy. It also depends on the choice of immunotherapy agents and route of administration (systemic versus intra-lesional) to combine with radiation to assure the best likelihood of clinical response.
Figure I:
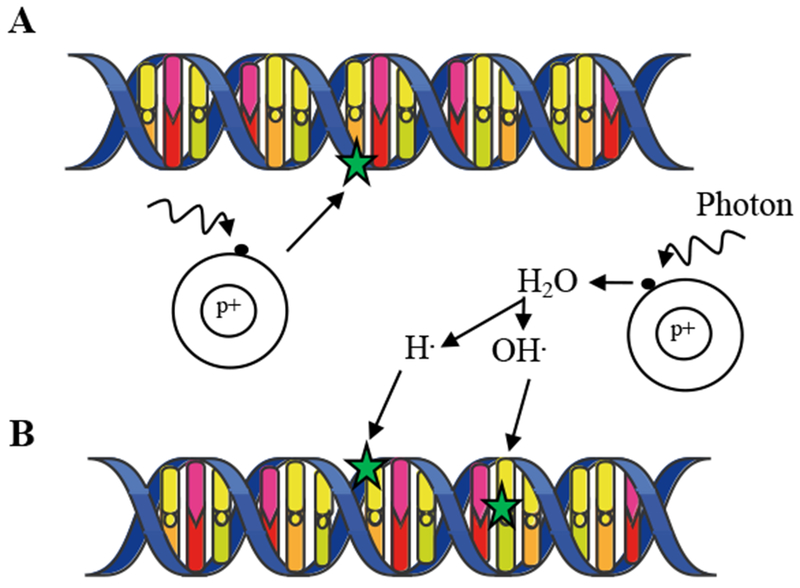
A- Direct effect. Following X-ray, an electron is ejected and directly damages DNA. B- Indirect effect. The incident photon ejects an electron, which creates a free radical. The diffusing free radical then causes damage to DNA.
Figure II:
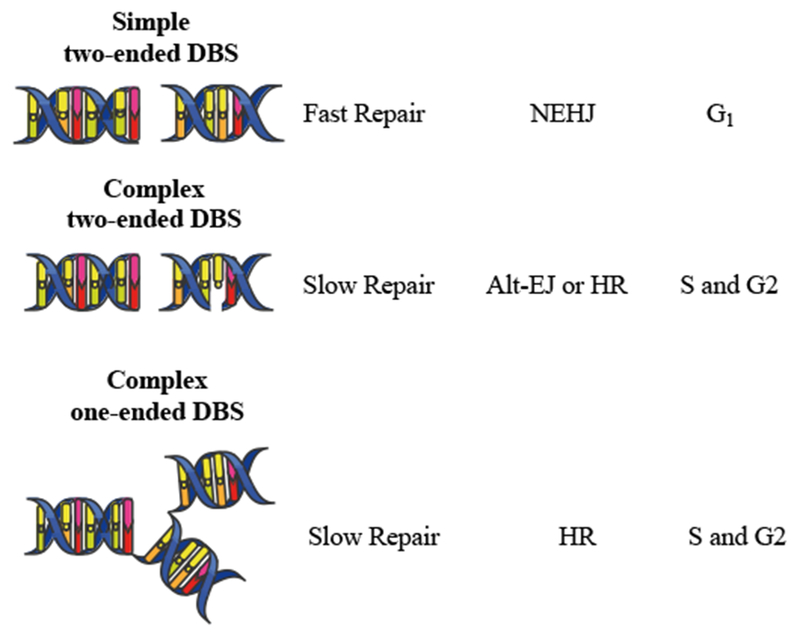
Major type of DBS breaks. Simple DSBs involve two broken DNA ends (i.e. two-ended DSB) in close proximity. Complex DSBs consist in two broken DNA ends in proximity to additional DNA damage (i.e., cross-links, single stranded breaks, etc...) or a DSB within a replication fork (one-ended DSB).
HIGHLIGHTS.
Tumor-targeted radiation occasionally elicits immune-mediated systemic tumor regression.
Evidence of synergy between radiotherapy and immune checkpoint blockade (ICB) supports the concept of in situ vaccination by radiation, and ICB combinations together with an optimization of the radiation dose and fractionation offer paths to improved responses.
Radiation alters the balance between immune-activating and suppressive signals in the tumor microenvironment.
Pathways involved in autoimmunity and microbial immunity are responsible for regulating the induction of type I interferon via cGAS/STING in irradiated tumors, and are stimulated upon tumor cell irradiation and activation of the DNA damage response.
OUTSTANDING QUESTIONS BOX.
Besides micronuclei formation, what are the other mechanisms responsible for accumulation of dsDNA in the cysosol of irradiated cancer cells?
What mechanisms regulate radiation-induced TREX1 upregulation?
Is the DNA that stimulates cGAS in irradiated cancer cells of nuclear or mitochondrial origin?
Can pharmacological TREX1 inhibition be used to increase responses to radiation and immune checkpoint blockade?
Can intratumoral delivery of TLR or STING agonists that induce IFN-I achieve the same efficacy as radiotherapy in enhancing local and systemic responses to immune checkpoint blockade?
Can standard multi-fraction radiation regimens induce a cumulative cytosolic dsDNA accumulation and activate the IFN-I pathway?
Are similar mechanisms critical for the immunogenicity of chemo-radiation?
Can radiation of cancer cells expose antigenic mutations to the immune system?
Is the paucity of BATF3 DCs in non-irradiated tumors a barrier to achieving abscopal effects with immune checkpoint blockade?
Acknowledgements:
This work was supported by NIH grants to S.D. (R01 CA201246 and R01 CA198533) and to S.C.F. (S10 RR027619). Additional funding was provided by the Breast Cancer Research Foundation (to S.C.F. and S.D; BCRF-16-054) and the Chemotherapy Foundation (to S.D.). C.V-B. is supported in part by the 2017-Kellen Junior Faculty Fellowship from the Anna-Maria and Stephen Kellen Foundation.
GLOSSARY
- Abscopal effect:
from the Latin ab scopus (away from the target), this term indicates tumor regression seen outside of the field of radiation.
- BATF3-dependent DCs (also known as DC1):
Key antigen-presenting cells for CD8 T cell activation that are dependent for ontogeny on the transcription factors IRF-8 and BATF-3.
- Cyclic GMP-AMP synthase (cGAS):
a cytosolic DNA sensor that binds to microbial DNA as well as self DNA and catalyzes cGAMP synthesis.
- Damage-associated molecular pattern (DAMPs):
Endogenous molecules that functions as endogenous adjuvant once released by stressed or dying cells.
- Homologous recombination (HR):
High-fidelity DNA damage repair mechanism that occurs in S and G2 phases of the cell cycle using a sister chromatid as a template.
- Hypoxia:
Refers to a condition in which there is a lower-than-normal concentration of oxygen at the tissue level.
- Immune checkpoint blockade (ICB):
Therapeutic strategy based on the inhibition of immune checkpoint pathways that are in place to maintain self-tolerance and are co-opted by cancer to evade immune rejection. Currently approved drugs (antibodies) block immune checkpoint receptor cytotoxic T lymphocyte antigen 4 (CTLA-4) and Programmed Death 1 (PD-1) and its major ligand PD-L1.
- Immunogenic cell death (ICD):
A form of cell death that is associated with the generation and release of DAMPs and cytokines that activate innate immune cells and promote the cross-presentation of antigens derived from the dying cells to T cells.
- Non-Homologous End Joining (NHEJ):
Error-prone repair of double-stranded DNA lesions that occurs in G1 phase of the cell cycle to attach the free DNA ends without a homologous template.
- Radiation dose:
Energy deposited by ionizing radiation per unit mass, measured in gray (Gy): 1 Gy = 1 J/kg.
- Stimulator of interferon genes (STING):
a protein that resides on the outer leaflet of the endoplasmic reticulum and the ER-Golgi intermediate compartment and is activated by cGAMP produced by cGAS and by other cyclic dinucleotides of bacterial origin. STING activates the type I IFN and NFkB pathways.
- Three-prime repair exonuclease 1 (TREX1):
DNA exonuclease that degrades cytosolic single-stranded and double-stranded DNA.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Formenti SC and Demaria S (2009) Systemic effects of local radiotherapy. Lancet Oncol 10 (7), 718–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abuodeh Y et al. (2016) Systematic review of case reports on the abscopal effect. Curr Probl Cancer 40 (1), 25–37. [DOI] [PubMed] [Google Scholar]
- 3.Sharma P and Allison JP (2015) Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell 161 (2), 205–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Demaria S et al. (2005) Immune-mediated inhibition of metastases after treatment with local radiation and CTLA-4 blockade in a mouse model of breast cancer. Clin Cancer Res 11 (2 Pt 1), 728–34. [PubMed] [Google Scholar]
- 5.Deng L et al. (2014) Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J Clin Invest 124 (2), 687–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Postow MA et al. (2012) Immunologic correlates of the abscopal effect in a patient with melanoma. N Engl J Med 366 (10), 925–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grimaldi AM et al. (2014) Abscopal effects of radiotherapy on advanced melanoma patients who progressed after ipilimumab immunotherapy. Oncoimmunology 3, e28780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kang J et al. (2016) Current clinical trials testing the combination of immunotherapy with radiotherapy. J Immunother Cancer 4, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Willers H and Held KD (2006) Introduction to clinical radiation biology. Hematol Oncol Clin North Am 20 (1), 1–24. [DOI] [PubMed] [Google Scholar]
- 10.Jeggo PA et al. (2016) DNA repair, genome stability and cancer: a historical perspective. Nat Rev Cancer 16 (1), 35–42. [DOI] [PubMed] [Google Scholar]
- 11.Harding SM et al. (2017) Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 548 (7668), 466–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mackenzie KJ et al. (2017) cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 548 (7668), 461–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bartsch K et al. (2017) Absence of RNase H2 triggers generation of immunogenic micronuclei removed by autophagy. Hum Mol Genet 26 (20), 3960–3972. [DOI] [PubMed] [Google Scholar]
- 14.Cai X et al. (2014) The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol Cell 54 (2), 289–96. [DOI] [PubMed] [Google Scholar]
- 15.Vanpouille-Box C et al. (2017) DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat Commun 8, 15618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deng L et al. (2014) STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 41 (5), 843–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Galluzzi L et al. (2017) Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol 17 (2), 97–111. [DOI] [PubMed] [Google Scholar]
- 18.Apetoh L et al. (2007) Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med 13 (9), 1050–9. [DOI] [PubMed] [Google Scholar]
- 19.Obeid M et al. (2007) Calreticulin exposure is required for the immunogenicity of gamma-irradiation and UVC light-induced apoptosis. Cell Death Differ 14 (10), 1848–50. [DOI] [PubMed] [Google Scholar]
- 20.Shrivastav M et al. (2008) Regulation of DNA double-strand break repair pathway choice. Cell Res 18 (1), 134–47. [DOI] [PubMed] [Google Scholar]
- 21.Meng Y et al. (2012) Radiation-inducible immunotherapy for cancer: senescent tumor cells as a cancer vaccine. Mol Ther 20 (5), 1046–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dou Z et al. (2017) Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 550 (7676), 402–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ranoa DR et al. (2016) Cancer therapies activate RIG-I-like receptor pathway through endogenous non-coding RNAs. Oncotarget 7 (18), 26496–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sharma P and Allison JP (2015) The future of immune checkpoint therapy. Science 348 (6230), 56–61. [DOI] [PubMed] [Google Scholar]
- 25.Sharma P et al. (2017) Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 168 (4), 707–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barsoum IB et al. (2014) A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res 74 (3), 665–74. [DOI] [PubMed] [Google Scholar]
- 27.Dewhirst MW et al. (2008) Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat Rev Cancer 8 (6), 425–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Noman MZ et al. (2014) PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med 211 (5), 781–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Voron T et al. (2014) Control of the immune response by pro-angiogenic factors. Front Oncol 4, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barsoum IB et al. (2011) Hypoxia induces escape from innate immunity in cancer cells via increased expression of ADAM10: role of nitric oxide. Cancer Res 71 (24), 7433–41. [DOI] [PubMed] [Google Scholar]
- 31.Palazon A et al. (2017) An HIF-1alpha/VEGF-A Axis in Cytotoxic T Cells Regulates Tumor Progression. Cancer Cell 32 (5), 669–683e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moeller BJ et al. (2004) Radiation activates HIF-1 to regulate vascular radiosensitivity in tumors: role of reoxygenation, free radicals, and stress granules. Cancer Cell 5 (5), 429–41. [DOI] [PubMed] [Google Scholar]
- 33.Li F et al. (2007) Regulation of HIF-lalpha stability through S-nitrosylation. Mol Cell 26 (1), 63–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hellevik T and Martinez-Zubiaurre I (2014) Radiotherapy and the tumor stroma: the importance of dose and fractionation. Front Oncol 4, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tommelein J et al. (2018) Radiotherapy-Activated Cancer-Associated Fibroblasts Promote Tumor Progression through Paracrine IGF1R Activation. Cancer Res 78 (3), 659–670. [DOI] [PubMed] [Google Scholar]
- 36.Spadaro O et al. (2017) IGF1 Shapes Macrophage Activation in Response to Immunometabolic Challenge. Cell Rep 19 (2), 225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barcellos-Hoff MH and Cucinotta FA (2014) New tricks for an old fox: impact of TGFbeta on the DNA damage response and genomic stability. Sci Signal 7 (341), re5. [DOI] [PubMed] [Google Scholar]
- 38.Jobling MF et al. (2006) Isoform-specific activation of latent transforming growth factor beta (LTGF-beta) by reactive oxygen species. Radiat Res 166 (6), 839–48. [DOI] [PubMed] [Google Scholar]
- 39.Vanpouille-Box C et al. (2015) TGFbeta Is a Master Regulator of Radiation Therapy-Induced Antitumor Immunity. Cancer Res 75 (11), 2232–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu J et al. (2013) CSF1R signaling blockade stanches tumor-infiltrating myeloid cells and improves the efficacy of radiotherapy in prostate cancer. Cancer Res 73 (9), 2782–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kioi M et al. (2010) Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J Clin Invest 120 (3), 694–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kalbasi A et al. (2017) Tumor-Derived CCL2 Mediates Resistance to Radiotherapy in Pancreatic Ductal Adenocarcinoma. Clin Cancer Res 23 (1), 137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sanford DE et al. (2013) Inflammatory monocyte mobilization decreases patient survival in pancreatic cancer: a role for targeting the CCL2/CCR2 axis. Clin Cancer Res 19 (13), 3404–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dovedi SJ et al. (2014) Acquired Resistance to Fractionated Radiotherapy Can Be Overcome by Concurrent PD-L1 Blockade. Cancer Research 74 (19), 5458–5468. [DOI] [PubMed] [Google Scholar]
- 45.Matsumura S et al. (2008) Radiation-induced CXCL16 release by breast cancer cells attracts effector T cells. J Immunol 181 (5), 3099–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Burnette BC et al. (2011) The efficacy of radiotherapy relies upon induction of type i interferon-dependent innate and adaptive immunity. Cancer Res 71 (7), 2488–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lugade AA et al. (2005) Local radiation therapy of B16 melanoma tumors increases the generation of tumor antigen-specific effector cells that traffic to the tumor. J Immunol 174 (12), 7516–23. [DOI] [PubMed] [Google Scholar]
- 48.Klug F et al. (2013) Low-dose irradiation programs macrophage differentiation to an iNOS(+)/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell 24 (5), 589–602. [DOI] [PubMed] [Google Scholar]
- 49.Gasser S et al. (2005) The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature 436 (7054), 1186–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lam AR et al. (2014) RAE1 ligands for the NKG2D receptor are regulated by STING-dependent DNA sensor pathways in lymphoma. Cancer Res 74 (8), 2193–2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ruocco MG et al. (2012) Suppressing T cell motility induced by anti-CTLA-4 monotherapy improves antitumor effects. J Clin Invest 122 (10), 3718–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chakraborty M et al. (2003) Irradiation of tumor cells up-regulates Fas and enhances CTL lytic activity and CTL adoptive immunotherapy. J Immunol 170 (12), 6338–47. [DOI] [PubMed] [Google Scholar]
- 53.Reits EA et al. (2006) Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J Exp Med 203 (5), 1259–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rudqvist NP et al. (2018) Radiotherapy and CTLA-4 Blockade Shape the TCR Repertoire of Tumor-Infiltrating T Cells. Cancer Immunol Res 6 (2), 139–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Twyman-Saint Victor C et al. (2015) Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 520 (7547), 373–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang B et al. (2007) Induced sensitization of tumor stroma leads to eradication of established cancer by T cells. J Exp Med 204 (1), 49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brody JD et al. (2010) In situ vaccination with a TLR9 agonist induces systemic lymphoma regression: a phase I/II study. J Clin Oncol 28 (28), 4324–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Formenti SC and Demaria S (2012) Radiation therapy to convert the tumor into an in situ vaccine. Int J Radiat Oncol Biol Phys 84 (4), 879–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee Y et al. (2009) Therapeutic effects of ablative radiation on local tumor require CD8+ T cells: changing strategies for cancer treatment. Blood 114 (3), 589–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Finn OJ (2017) The dawn of vaccines for cancer prevention. Nat Rev Immunol. [DOI] [PubMed] [Google Scholar]
- 61.Golden EB et al. (2015) Local radiotherapy and granulocyte-macrophage colony-stimulating factor to generate abscopal responses in patients with metastatic solid tumours: a proof-of-principle trial. Lancet Oncol 16 (7), 795–803. [DOI] [PubMed] [Google Scholar]
- 62.Golden EB et al. (2013) An abscopal response to radiation and ipilimumab in a patient with metastatic non-small cell lung cancer. Cancer Immunol Res 1 (6), 365–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zatloukal P et al. (2009) Randomized phase II clinical trial comparing tremelimumab (CP-675, 206) with best supportive care (BSC) following first-line platinum-based therapy in patients (pts) with advanced non-small cell lung cancer (NSCLC). J Clin Oncol 27 (15S), 8071. [Google Scholar]
- 64.Lynch TJ et al. (2012) Ipilimumab in combination with paclitaxel and carboplatin as first-line treatment in stage IIIB/IV non-small-cell lung cancer: results from a randomized, double-blind, multicenter phase II study. J Clin Oncol 30 (17), 2046–54. [DOI] [PubMed] [Google Scholar]
- 65.Demaria S et al. (2016) Radiotherapy: Changing the Game in Immunotherapy. Trends Cancer 2 (6), 286–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dewan MZ et al. (2009) Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin Cancer Res 15 (17), 5379–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Filatenkov A et al. (2015) Ablative Tumor Radiation Can Change the Tumor Immune Cell Microenvironment to Induce Durable Complete Remissions. Clin Cancer Res 21 (16), 3727–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sanchez-Paulete AR et al. (2017) Antigen cross-presentation and T-cell cross-priming in cancer immunology and immunotherapy. Ann Oncol 28 (suppl_12), xii44–xii55. [DOI] [PubMed] [Google Scholar]
- 69.Stetson DB et al. (2008) Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell 134 (4), 587–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vanpouille-Box C et al. (2018) Toward Precision Radiotherapy for Use with Immune Checkpoint Blockers. Clin Cancer Res 24 (2), 259–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Menis J et al. (2016) The European Organization for Research and Treatment of Cancer perspective on designing clinical trials with immune therapeutics. Ann Transl Med 4 (14), 267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shaverdian N et al. (2017) Previous radiotherapy and the clinical activity and toxicity of pembrolizumab in the treatment of non-small-cell lung cancer: a secondary analysis of the KEYNOTE-001 phase 1 trial. Lancet Oncol 18 (7), 895–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Koller KM et al. (2017) Improved survival and complete response rates in patients with advanced melanoma treated with concurrent ipilimumab and radiotherapy versus ipilimumab alone. Cancer Biol Ther 18 (1), 36–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tang C et al. (2017) Ipilimumab with Stereotactic Ablative Radiation Therapy: Phase I Results and Immunologic Correlates from Peripheral T Cells. Clin Cancer Res 23 (6), 1388–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hiniker SM et al. (2016) A Prospective Clinical Trial Combining Radiation Therapy With Systemic Immunotherapy in Metastatic Melanoma. Int J Radiat Oncol Biol Phys 96 (3), 578–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kwon ED et al. (2014) Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184–043): a multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol 15 (7), 700–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Golden EB and Formenti SC (2015) Radiation therapy and immunotherapy: growing pains. Int J Radiat Oncol Biol Phys 91 (2), 252–4. [DOI] [PubMed] [Google Scholar]
- 78.Antonia SJ et al. (2017) Durvalumab after Chemoradiotherapy in Stage III Non-Small-Cell Lung Cancer. N Engl J Med 377 (20), 1919–1929. [DOI] [PubMed] [Google Scholar]
- 79.Golden EB and Formenti SC (2014) Is tumor (R)ejection by the immune system the “5th R” of radiobiology? Oncoimmunology 3 (1), e28133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ceccaldi R et al. (2016) Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol 26 (1), 52–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Goodarzi AA et al. (2010) The influence of heterochromatin on DNA double strand break repair: Getting the strong, silent type to relax. DNA Repair (Amst) 9 (12), 1273–82. [DOI] [PubMed] [Google Scholar]
- 82.Shibata A et al. (2014) DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Mol Cell 53 (1), 7–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Arnoult N et al. (2017) Regulation of DNA repair pathway choice in S and G2 phases by the NHEJ inhibitor CYREN. Nature 549 (7673), 548–552. [DOI] [PMC free article] [PubMed] [Google Scholar]