

A self-illuminating nanoparticle can function as an effective nanomedicine for inflammation imaging and cancer therapy.
Abstract
Nanoparticles have been extensively used for inflammation imaging and photodynamic therapy of cancer. However, the major translational barriers to most nanoparticle-based imaging and therapy applications are the limited depth of tissue penetration, inevitable requirement of external irradiation, and poor biocompatibility of the nanoparticles. To overcome these critical limitations, we synthesized a sensitive, specific, biodegradable luminescent nanoparticle that is self-assembled from an amphiphilic polymeric conjugate with a luminescent donor (luminol) and a fluorescent acceptor [chlorin e6 (Ce6)] for in vivo luminescence imaging and photodynamic therapy in deep tissues. Mechanistically, reactive oxygen species (ROS) and myeloperoxidase generated in inflammatory sites or the tumor microenvironment trigger bioluminescence resonance energy transfer and the production of singlet oxygen (1O2) from the nanoparticle, enabling in vivo imaging and cancer therapy, respectively. This self-illuminating nanoparticle shows an excellent in vivo imaging capability with suitable tissue penetration and resolution in diverse animal models of inflammation. It is also proven to be a selective, potent, and safe antitumor nanomedicine that specifically kills cancer cells via in situ 1O2 produced in the tumor microenvironment, which contains a high level of ROS.
INTRODUCTION
Nanoparticles have been used to detect and treat many harmful diseases ranging from cancer (1), obesity (2), diabetes (3), and inflammatory disorders (4) to cardiovascular diseases (5). As well documented, inflammation is closely associated with the pathogenesis of many devastating diseases (6–8). Over the past few decades, different nanoprobes have been developed and applied to molecular imaging of inflammation by computed tomography (9), magnetic resonance imaging (10), optical methods (11), positron emission tomography (12), and multimodal approaches (13). These nanomedicine strategies have greatly improved our understanding of molecular and cellular processes underlying various disease etiologies, thereby facilitating diagnosis, stratification, therapeutic evaluation, and drug discovery. Among these techniques, luminescence imaging is a facile, low-cost, rapid, straightforward, and highly sensitive approach that enables real-time detection of a wide variety of cellular, subcellular, or molecular events during an inflammatory response in unperturbed environments (14–16). To increase the spatiotemporal resolution, different luminescent nanoprobes, such as polymeric nanoparticles (17), inorganic nanoparticles (18), and rare earth-based nanophosphors (19), have been developed. Although these nanoparticles are sensitive luminescent probes for in vitro detection, their effectiveness for in vivo imaging is limited in superficial tissues (14, 16, 20). The inadequate in vivo application of existing nanoparticles in deep tissues is mainly attributed to the poor penetration of nanoprobes in animal tissues (14, 21, 22). In addition, because a large number of luminescent nanoprobes are derived from nondegradable conjugated polymers and inorganic nanoparticles, their in vivo applications in inflammation imaging are compromised by their poor degradability and possible proinflammatory effects, although they have shown promising results in tumor imaging (14, 23–26).
In addition to molecular imaging, nanoparticles have been extensively investigated in targeted cancer therapy (1, 27, 28). Increasing evidence has demonstrated that nanoparticles can significantly improve the in vivo efficacies of small-molecule photosensitizers conventionally used for photodynamic therapy (PDT) of cancer by enhancing their intracellular delivery and targeting specificity (29). To date, diverse arrays of nanoparticles have been developed for PDT (27, 30). More recently, two-photon excitation nanoparticles (31), upconversion nanoparticles (32), and near-infrared photosensitizers (33) were used to improve tissue penetration for the effective treatment of large and deep-seated solid tumors. Despite their great success in animal studies, clinical translation of most existing photosensitizers is largely hindered by the limited penetration depth of external light required for their activation (27, 32). To overcome this barrier, self-illuminated PDT has been proposed for tumor therapy in the absence of external excitation (32, 34–36). In this modality, the photosensitizer is excited by internal chemiluminescence or bioluminescence triggered by the tumor microenvironment, producing cytotoxins such as singlet oxygen (1O2) to kill cancer cells. In general, quantum dots and nanoparticles based on conjugated polymers are combined with luminescent proteins (such as luciferase and horseradish peroxidase) to produce self-illuminating systems (20, 26, 35–39). However, only very few self-illuminating platforms have been investigated for tumor therapy (35, 36). Moreover, the use of either quantum dots or conjugated polymer–derived nanoparticles raises in vivo safety concerns, especially for long-term therapy (40).
To overcome the abovementioned critical barriers to nanoparticle-based inflammation imaging and cancer therapy, we devised a nanoparticle capable of bioluminescence resonance energy transfer (BRET). This self-illuminating nanoparticle not only showed excellent imaging quality in detecting inflammation in diverse animal models but also demonstrated selective and potent antitumor efficacy in vivo with a desirable safety profile.
RESULTS
Design and synthesis of a nanoparticle for imaging and therapy
We synthesized a nanoparticle using a Ce6-luminol-PEG (CLP) conjugate capable of BRET for sensitive in vivo inflammation imaging and cancer therapy. This CLP conjugate is composed of chlorin e6 (Ce6, a commonly used photosensitizer), luminol, and polyethylene glycol (PEG) (Fig. 1A) and can self-assemble into a core-shell structured nanoparticle (Fig. 1B). Under oxidative conditions with excess reactive oxygen species (ROS) and myeloperoxidase (MPO), the in situ bioluminescence of luminol excites the photosensitizer Ce6 to generate fluorescence and 1O2 via BRET (Fig. 1B), enabling direct inflammation detection and tumor PDT.
Fig. 1. Engineering of a self-illuminating nanoparticle with luminescence, fluorescence, and intrinsic singlet oxygen generation capabilities.
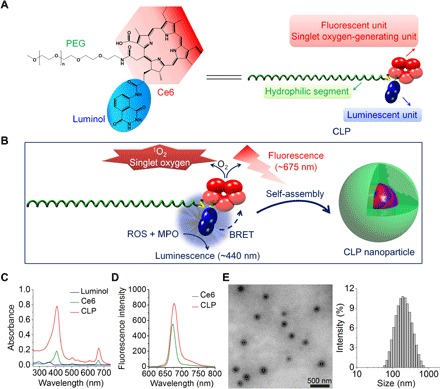
(A) Chemical structure and schematic of a designed amphiphile of a CLP conjugate. (B) Sketch of self-assembly of the CLP conjugate into a core-shell structured nanoparticle. Through BRET between the luminol and Ce6 units, luminescence originating from the energy donor luminol upon triggering by inflammatory mediators of ROS and MPO may excite the acceptor Ce6 to produce fluorescence and singlet oxygen (1O2). (C and D) UV-visible (C) and fluorescence (D) spectroscopic characterization of CLP. (E) Transmission electron microscopy (TEM) image (left) and size distribution profile in deionized water (right) of assembled CLP nanoparticles.
The CLP conjugate was synthesized by sequential coupling reactions of Ce6, luminol, and PEG (fig. S1A). Infrared spectroscopy showed characteristic absorption bands of Ce6, luminol, and PEG in the obtained product (fig. S1B). 1H nuclear magnetic resonance (NMR) spectroscopy indicated that the proton signals corresponding to the vinyl group of Ce6 [at ~6.2 to 6.4 and 8.2 to 8.3 parts per million (ppm)] and the phenyl group of luminol (at ~7.3 to 7.6 ppm) could be observed, while the ethylene proton signals of PEG appeared at ~3.2 to 3.6 ppm, in a typical spectrum of CLP in DMSO (dimethyl sulfoxide)–D6 (fig. S1C). These measurements demonstrated the simultaneous conjugation of luminol and PEG onto Ce6. Successful synthesis of the CLP conjugate was also confirmed by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (fig. S1, D and E). The 1H NMR spectrum revealed that the molar ratio of Ce6, luminol, and PEG in the synthesized CLP conjugate was approximately 2:1:1. In addition, as expected, the CLP conjugate displayed the characteristic UV (ultraviolet)–visible absorption (peaked at ~400 and ~660 nm) and fluorescent emission of Ce6 (Fig. 1, C and D).
Different from Ce6, which is hydrophobic and soluble only in highly polar solvents such as DMSO and dimethylformamide, the CLP conjugate is very soluble in water and common organic solvents such as methanol, chloroform, and tetrahydrofuran. Because of its amphiphilicity, the CLP conjugate can self-assemble into core-shell structured nanoparticles in aqueous solution, as demonstrated by transmission electron microscopy (TEM) (Fig. 1E, left panel), with a mean hydrodynamic diameter of 171 nm (polydispersity index, 0.29 ± 0.02) and a zeta-potential of −19 ± 2 mV (Fig. 1E, right panel). Comparable size distribution profiles were detected for CLP nanoparticles in phosphate-buffered saline (PBS), cell culture medium, fetal bovine serum (FBS), and rat serum (fig. S2, A to D). Significant aggregation of CLP nanoparticles occurred only when they were incubated in PBS and culture medium for a relatively long period of time, which was different from that in deionized water and FBS (fig. S2E). In addition, no significant decrease in the mean diameter was detected upon dilution of CLP nanoparticles in deionized water, although the scattering intensity was notably reduced (fig. S2F). This suggested that CLP nanoparticles have good kinetic stability, which is advantageous for in vivo applications. In line with the hydrophobic force–mediated self-assembly in aqueous solution, both the UV-visible absorption and fluorescence intensity corresponding to Ce6 of CLP in water were notably reduced compared to those in organic solvents (fig. S2, G and H). Accordingly, the emission intensity of CLP in water initially increased with its concentration, peaked at a critical association concentration (CAC) of ~0.06 mg/ml or 13 μM, and then started to decrease as its concentration increased further (fig. S2I), showing an inverted V-shape relationship between the concentration and the fluorescence (fig. S2J). This decrease in fluorescence at high CLP concentrations was attributed to fluorescence quenching, resulting from aggregation of the hydrophobic Ce6 units. At a CLP concentration above the CAC, the proton signals of Ce6 and the luminol unit in the 1H NMR spectrum in D2O almost disappeared, which was different from the spectrum in DMSO-D6 (fig. S2K). This is due to the severe attenuation of proton signals of the lipophilic moieties (i.e., Ce6 and luminol) as a result of hydrophobic interaction–dominated aggregation in aqueous solution (41).
Luminescence properties of CLP
We examined the luminescence properties of CLP under different conditions. In the presence of H2O2 and ClO−, the luminescence spectrum of CLP showed two emission peaks: one at 450 nm, corresponding to the luminescence of luminol, and the other at 675 nm, belonging to the fluorescence emission of Ce6 (Fig. 2A). The second emission peak was the result of BRET between the donor luminol and the acceptor Ce6 in the CLP conjugate (Fig. 1B), as there is considerable overlap between the luminescence spectrum of luminol (λmax = 440 nm) and the absorption spectrum of Ce6 (λmax = 401 nm) (Fig. 2B). The BRET efficiency was ~0.56, as calculated based on a previously established method (39). By contrast, luminol itself displayed only its characteristic luminescent emission, while the Ce6-PEG conjugate (Ce6-PEG) did not show any luminescence in the absence of the BRET donor luminol (Fig. 2A). Consequently, these results substantiated our hypothesis that luminol can function as an effective luminescent donor to excite an acceptor moiety Ce6 under oxidative conditions to generate fluorescence through a BRET process.
Fig. 2. In vitro characterization of BRET and luminescent properties of the CLP conjugate.
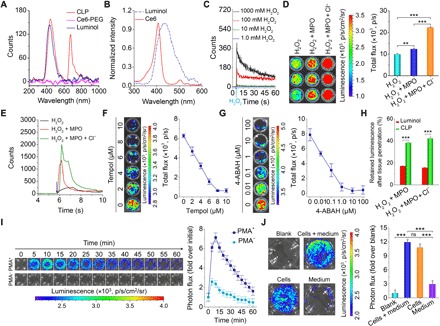
(A) Luminescent spectra of a CLP conjugate, a Ce6-PEG conjugate, and luminol in the presence of 100 mM H2O2 and 100 mM ClO−. (B) The luminescence spectrum of luminol in PBS and the absorption spectrum of Ce6 in DMSO. (C) Time-dependent luminescence of CLP (0.5 mg/ml) upon incubation with various concentrations of H2O2. (D and E) Luminescence at 1.0 mg/ml CLP (D) and time-resolved luminescent signals of CLP at 0.5 mg/ml (E) in the presence of 50 μM H2O2 with or without 15 mU MPO and 50 mM Cl−. (F and G) Attenuation of the luminescence at 0.5 mg/ml CLP and in the presence of 50 μM H2O2 and 15 mU MPO by a ROS scavenger, Tempol (F), and an MPO inhibitor, 4-aminobenzoic hydrazide (4-ABAH) (G). (H) In vitro tissue penetration capacity of luminol and CLP. After a black 96-well plate containing CLP or luminol at the same dose of 0.5 mg/ml was covered with 3 mm of porcine muscle tissue, the penetrating luminescent signals were determined. (I) Time-resolved luminescence imaging of neutrophils. Immediately after 5.4 μM CLP (i.e., 25 μg/ml) was incubated with neutrophils (5 × 105 cells per well) with (PMA+) or without (PMA−) stimulation with phorbol 12-myristate 13-acetate (PMA; 100 ng/ml ) for 1 hour, images were acquired at predetermined time points. (J) Intracellular and extracellular luminescent signals. The Blank group indicates neutrophils stimulated with PMA and without treatment with CLP nanoparticles. After PMA-stimulated neutrophils were incubated with 5.4 μM CLP for 0.5 hours, luminescence images were acquired and indicated as the Cells + medium group. Immediately after this imaging, cells and the culture medium containing PMA were separated, and images were separately collected for the Cells and Medium groups. For all luminescence imaging results (D, F, G, I, and J) measured with an IVIS Spectrum imaging system [exposure time = 5 min, focal length/stop (f/stop) = 1, binning = 8, field of view (FOV) = 12.8 cm], the left panels are luminescent images, while the right panels show quantitative data. Data in (D) and (F) to (J) are means ± SEM. (n = 3). One-way analysis of variance (ANOVA) was used for statistical analysis. **P < 0.01, ***P < 0.001; ns, no significance.
We next tested the time course of luminescence of CLP at different H2O2 concentrations using an ultraweak luminescence analyzer. At a CLP concentration of 0.5 mg/ml, there was no detectable luminescence when the H2O2 concentration was below 10 mM, and at higher H2O2 concentrations (>100 mM), the luminescence exponentially decreased with time (Fig. 2C). Further analysis via an In Vivo Imaging System (IVIS) Spectrum imaging system revealed a linear increase in luminescence as a function of the H2O2 concentration (fig. S3A). We found that the luminescence of CLP at 675 nm was substantially enhanced by adding ClO− (fig. S3B), which is a highly oxidative anion that is produced in vivo by the H2O2-MPO-Cl− antibacterial system (42). Accordingly, we further confirmed that adding MPO or MPO/Cl− significantly enhanced CLP luminescence (Fig. 2, D and E), resulting from the generation of more oxidative ClO− by the H2O2-MPO-Cl− system, which accelerated oxidation of the luminol unit. As well demonstrated, ClO− is able to directly or indirectly oxidize luminol to produce the blue glow (43). Consistently, among different reactive species of ROS, the luminescence of CLP displayed the highest sensitivity to ClO− at either a low or a high level (fig. S3, C and D).
Consistent with the fluorescence results in water (fig. S2, I and J), the luminescent signals of CLP in the presence of H2O2 initially increased, peaked at 0.5 mg/ml, and then decreased with the increase in CLP concentration (fig. S3, E and F). This observation was the consequence of the quenching of BRET-derived luminescence at high CLP concentrations, since hydrophobic aggregation of CLP conjugates may occur in these cases. After adding a micelle destabilizing SDS, to disrupt the assembled micelle-like nanoparticle, the luminescence of CLP significantly increased (fig. S3G). Disruption of the core-shell structure exposed the luminol units, which might also contribute to the increased luminescence due to luminol, at least to a certain degree. In addition, the luminescence of CLP showed a sustainable profile during the examined period of time (fig. S3, H and I), which is beneficial for practical applications.
In line with H2O2- and MPO-triggered luminescence profiles of CLP, adding a ROS scavenger, Tempol, or an MPO inhibitor, 4-aminobenzoic hydrazide (4-ABAH), abolished the luminescence (Fig. 2, F and G), highlighting the oxidation- and MPO-dependent illumination. In addition, the tissue penetration capability of the CLP conjugate and luminol was examined. In this case, they were separately mixed with aqueous solutions of H2O2/MPO with or without Cl− in a black 96-well plate that was covered by a 3-mm-thick porcine muscle tissue, and immediately, the bioluminescent signals were detected. The CLP conjugate showed ~2-fold more tissue penetration than luminol alone (Fig. 2H). The reason is that the wavelength (~675 nm) of the BRET-derived bioluminescence of CLP is much longer than that of the luminescence (~440 nm) of luminol. It is well established that photons with wavelengths between 650 and 950 nm are more penetrative through animal tissues than short-wavelength photons, because of reduced absorption and scattering (21, 22). Consequently, the CLP conjugate displayed deeper penetration than luminol. Further examination revealed a threshold thickness of 5 mm for high-resolution ex vivo imaging under the tested conditions (fig. S3J). Together, these results substantiated that CLP, with its expected BRET-derived luminescence, displayed desirable luminescence performance under varied oxidative conditions.
In vitro luminescence imaging of inflammation
Neutrophils play an important role in both acute and chronic inflammatory responses, which produce ROS and MPO upon activation at inflammatory sites (44). Therefore, we aimed to use our newly engineered CLP nanoparticle to detect inflammation by targeting the neutrophils. Neutrophils were isolated from peritoneal exudates of mice to conduct a series of in vitro experiments. First, we tested the cellular uptake of CLP nanoparticles by neutrophils at different time points. We found that neutrophils rapidly and effectively took up the CLP nanoparticles, as demonstrated by flow cytometric and confocal microscopic analysis (fig. S4, A to C). This is consistent with the previous finding that neutrophils can internalize different nanoparticles by endocytosis (45, 46). We also confirmed that CLP exhibited negligible cytotoxicity to neutrophils at working concentrations (<270 μM; fig. S4D), and the neutrophils still showed ~70% viability even at a high concentration of 1080 μM. In addition, both time- and dose-dependent cellular uptake of CLP nanoparticles was observed in macrophages (fig. S4, E and F).
Then, in vitro imaging in neutrophils was examined. Our data showed that, in the presence of CLP nanoparticles, neutrophils prestimulated with phorbol 12-myristate 13-acetate (PMA) emitted luminescence, whereas unstimulated neutrophils did not (Fig. 2I). The decrease in luminescence after 15 min in stimulated neutrophils indicated depletion of intracellular ROS or luminol units in CLP nanoparticles (Fig. 2I). The luminescence was predominantly from intracellular CLP nanoparticles (Fig. 2J), which was consistent with its effective internalization in neutrophils (fig. S4, A to C). In addition, the luminescence of CLP nanoparticles in neutrophils was positively correlated with the nanoparticle dose and neutrophil count (fig. S4, G and H). These findings demonstrated that the CLP nanoparticle can serve as an effective luminescent probe for imaging inflammation by self-illuminating in activated neutrophils.
In vivo luminescence imaging of inflammation
We then tested the application of the CLP nanoparticle as a probe for inflammation in mouse models. We first examined it in a mouse peritonitis model, which is a well-characterized model of acute inflammation extensively used in mechanistic and drug screening studies (47). After intraperitoneal administration of 5 mg of CLP nanoparticles (i.e., 1.08 μmol CLP) or free luminol at the same dose of the luminol unit to each mouse with zymosan-induced peritonitis, we found that the luminescent signal from the nanoparticles is much stronger than that from the luminol molecules (Fig. 3A). Flow cytometric analysis indicated that the locally administered CLP nanoparticles were mainly illuminated in neutrophils (CD11b+Ly-6G+) of the peritoneal exudate cells from peritonitis mice (fig. S5A), due to their abundance in the peritoneal cavity in the acute phase of peritonitis and their high expression of MPO (47, 48). Therefore, the CLP nanoparticle can function as an effective nanoprobe for luminescence imaging of peritonitis via local administration. After receiving intravenous injections, both CLP nanoparticle–treated healthy mice and luminol-treated peritonitis mice showed no notable luminescence (Fig. 3B, the left and middle mice in the left panel). By contrast, strong luminescence was observed at the peritoneal sites of mice intravenously administered with CLP nanoparticles (Fig. 3B, the right mouse in the left panel). In this case, CLP nanoparticles yielded 31-fold higher luminescent intensity than luminol molecules (Fig. 3B, right panel), highlighting the sensitivity and specificity of CLP nanoparticles for inflammation imaging.
Fig. 3. In vivo imaging in mouse models of inflammation.
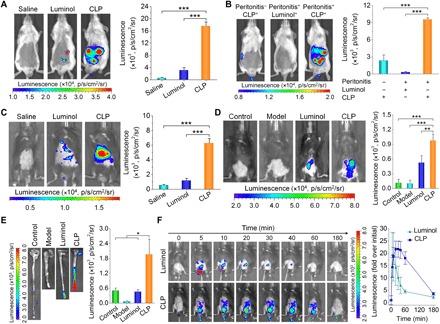
(A) Luminescent imaging of mice with peritonitis immediately after intraperitoneal administration of different formulations. To induce peritonitis, a saline solution containing 1 mg of zymosan was intraperitoneally injected into each mouse at 6 hours before imaging. Mice in the saline group were treated with saline, while those in the CLP and luminol groups were each administered with 5 mg of CLP nanoparticles or free luminol at the same dose of the luminol unit, respectively. (B) Luminescence imaging of peritonitis in mice at 0.5 hours after intravenous injection of different probes. In the Peritonitis−CLP+ group, healthy mice were treated with CLP nanoparticles. Peritonitis mice in the Peritonitis+Luminol+ and Peritonitis+CLP+ groups were administered with free luminol and CLP nanoparticles (5 mg in each mouse) at the same dose of the luminol unit, respectively. (C) Imaging of acute liver injury in mice at 24 hours after challenging via intraperitoneal injection of acetaminophen at 300 mg/kg. Mice in the saline group were intravenously injected with saline, while mice in the luminol and CLP groups were separately intravenously administered with free luminol and CLP nanoparticles (5 mg in each mouse) at the same dose of the luminol unit. After 0.5 hours, imaging was conducted. (D) In vivo luminescence imaging of mice with colitis induced by drinking water containing 3% (w/v) dextran sulfate sodium (DSS) for 7 days. (E) Ex vivo luminescent images of colonic tissues isolated immediately after in vivo imaging. In both cases, the control group represents healthy mice treated with CLP nanoparticles, while the model group denotes diseased mice administered with saline. In the luminol and CLP groups, mice with colitis were treated with free luminol and CLP nanoparticles (5 mg in each mouse) at the same dose of the luminol unit per mouse, respectively. At 15 min after different treatments via enema, luminescent images were acquired. Then, the mice were euthanized, and their colonic tissues were excised for ex vivo imaging. (F) Time-lapse in vivo luminescence imaging of mice with DSS-induced colitis after local administration of 5 mg of CLP nanoparticles or free luminol at the same dose of the luminol unit in each animal. In all images, the left panels show representative luminescent images, while the right panels illustrate quantified intensities. Data are means ± SEM (A to C and F, n = 4; D and E, n = 5). One-way ANOVA was used for statistical analysis. *P < 0.05, **P < 0.01, ***P < 0.001.
In an acute liver injury mouse model that is related to inflammation and the hepatic infiltration of neutrophils, intravenous injection of CLP nanoparticles in mice also resulted in significantly higher luminescence than treatment of mice with luminol molecules (Fig. 3C), consistent with the ex vivo luminescence imaging results of excised hepatic tissues (fig. S5B). These results demonstrated that the CLP nanoparticle can be used as a suitable probe for high-resolution luminescence imaging of acute liver injury, owing to its passive targeting effect, as illustrated by the accumulation of CLP in the liver (fig. S5C). By contrast, currently available small-molecule luminescent probes such as luminol showed poor resolution due to their limited tissue penetration capability and systemic distribution after intravenous administration.
We then tested the in vivo imaging capability of CLP nanoparticles in ulcerative colitis, which is a common inflammatory bowel disease (49). Dextran sulfate sodium (DSS)–induced colitis in mice was established as another inflammation model in the deep tissue of the colon. Compared to the saline group, the group locally administered with luminol molecules showed luminescence at the colonic site of mice bearing colitis (Fig. 3D, left panel), and the group treated with CLP nanoparticles exhibited significantly stronger luminescence. Quantitative analysis showed that there was a significant difference between luminol molecules and CLP nanoparticles (Fig. 3D, right panel). Consistent results were obtained using ex vivo imaging of isolated colonic tissues (Fig. 3E). CLP nanoparticles showed much higher luminescence than luminol molecules in both experiments (Fig. 3, D and E). Generally, diseased mice with ulcerative colitis had shorter colons than healthy mice (49). Moreover, the CLP nanoparticle could also be used for fluorescence imaging via external excitation by taking advantage of the fluorescence of the Ce6 component (Fig. 1A and fig. S5, D and E). Diseased mice displayed significantly higher fluorescence intensity than normal mice treated with the same dose of CLP nanoparticles. This can be attributed to the enhanced distribution of the CLP nanoparticle in diseased colonic tissues with injured epithelial cells and mucosal barrier (50). Both the BRET-based luminescence and regular fluorescence measurements consistently proved precise targeting of inflammation by the CLP nanoparticle. Our BRET nanoparticle can prevent false-positive signals with this double sensing effect. We further tested the dynamics of CLP-based luminescence in inflammation. After enema administration of 5 mg of CLP nanoparticles or free luminol at the same dose of the luminol unit in each mouse, real-time imaging revealed substantially strong and sustainable luminescence in CLP nanoparticle–treated mice (Fig. 3F). This sustained luminescence of the CLP nanoparticle was attributed to its accumulation and gradual hydrolysis in inflamed colonic tissues (50), as a considerable amount of CLP remained even at 3 hours after administration (fig. S6). By contrast, small-molecule luminol was rapidly absorbed and distributed through molecular diffusion (fig. S6A).
We further used the CLP nanoparticle to monitor the development of colitis in mice at different stages of pathogenesis in vivo. At days 1, 3, 5, and 7 after stimulation with DSS, the luminescence of CLP nanoparticles gradually increased (Fig. 4, A and B). In a separate study without CLP nanoparticle treatment to avoid its possible effects on colitis development, the disease activity index (DAI), the levels of MPO and ROS, and the count of neutrophils were determined at varied time points corresponding to real-time luminescence imaging (Fig. 4, C to G). We found that the luminescence intensity was well correlated with DAI (Fig. 4H), which is a common parameter for evaluating the degree of colitis (49). Similar positive correlations were observed with the MPO and H2O2 levels, as well as the neutrophil count, using sigmoidal fitting. The increased luminescence was a collective effect of enhanced MPO, H2O2, and neutrophils, and therefore, it showed a more substantial increase as separately compared to MPO, H2O2, and neutrophils. These data are consistent with histological analyses of hematoxylin and eosin (H&E)–stained sections of colonic tissues (Fig. 4I). In this aspect, normal microstructure was observed at days 0 and 1, with the straight luminal surface, tightly packed tubules closely approximating the muscularis mucosae and numerous goblet cells. At day 3, epithelial erosion and crypt damage occurred, which were more serious at day 5. Nearly complete loss of crypts, significant injury of mucous membrane, and a high level of inflammatory cell infiltration were found at day 7. Furthermore, the histological activity index (HAI) was evaluated based on the epithelium and infiltration (51), which showed notably increased scores after 3 days of DSS challenge (Fig. 4J).
Fig. 4. In vivo luminescence imaging of the development of colitis in mice.
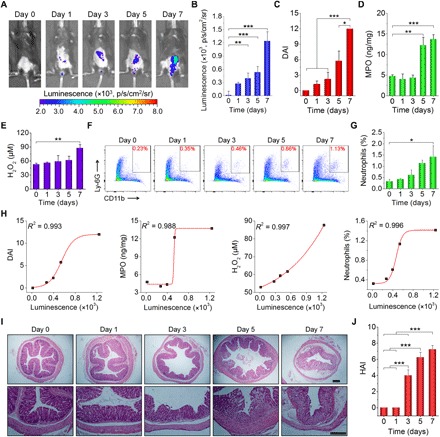
(A and B) Luminescent images (A) and quantitative analysis (B) of mice with colitis induced by drinking water containing 3% (w/v) DSS for different periods of time. For each time point, images were acquired at 15 min after enema administration of 5 mg of CLP nanoparticles in each mouse. (C to E) Changes in DAI (C) and the levels of MPO (D) and H2O2 (E) during colitis development. DAI is defined as the summation of the stool consistency index, fecal bleeding index, and weight loss index. Colonic tissues were excised from mice subjected to drinking water containing DSS at specific time points to quantify MPO and H2O2 levels. (F and G) Representative flow cytometric profiles (F) and quantitative data (G) of neutrophil counts. At different time points after mice were treated with drinking water containing DSS, they were euthanized, and cells in colonic tissues were isolated for analysis. (H) Correlation analyses of luminescent intensities with DAI, MPO, and H2O2 levels and neutrophil count. All data were fitted with a sigmoid growth function. (I and J) Microscopic images (I) and HAI scores (J) of H&E-stained sections of colonic tissues isolated from mice stimulated with DSS for different periods of time. Scale bars, 200 μm. Data in (B) to (E), (G), and (J) are means ± SEM (n = 4). One-way ANOVA was used for statistical analysis. *P < 0.05, **P < 0.01, ***P < 0.001.
Overall, the results described above demonstrate that the CLP nanoparticle is an effective nanoprobe for in vivo luminescence imaging of different inflammatory disorders that are associated with high MPO and ROS expression.
In vitro antitumor activity of the CLP nanoparticle
Oxidative stress can lead to inflammation and cancer (52). In addition to using the CLP nanoparticle as a nanoprobe for the detection of inflammation, we further explored its antitumor applications. We examined whether the CLP nanoparticle was able to produce 1O2, a high-energy form of oxygen, under oxidative conditions. To monitor the generation of 1O2, we used 9,10-diphenylanthracene (DPA) to chemically trap 1O2. In the presence of 1O2, the absorption of DPA at 355 nm will be irreversibly attenuated (53). After incubating different concentrations of CLP nanoparticles with 100 mM H2O2 for 6 hours, we detected reduced DPA absorbance at 355 nm, and the decrease was proportional to the concentration of CLP nanoparticles (fig. S7A and Fig. 5A). At a constant CLP nanoparticle dose of 20 μM, the absorbance of DPA at 355 nm decreased with increasing H2O2 concentration (fig. S7B and Fig. 5B). These results demonstrated the ROS-induced generation of 1O2 by CLP nanoparticles under oxidative conditions.
Fig. 5. In vitro and in vivo antitumor activity of CLP nanoparticles.
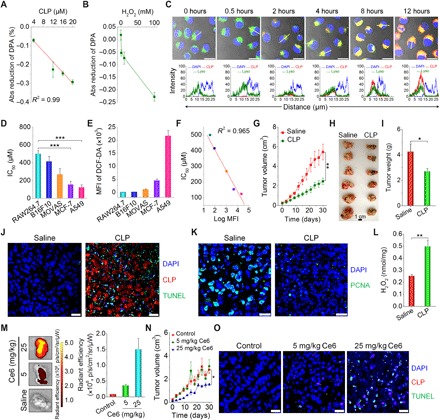
(A and B) The effects of CLP dose (A) and H2O2 concentration (B) on the generation of 1O2, as quantified using DPA as a probe. In both cases, the images show the reduction in DPA absorbance at 355 nm using that at 0 hours as the baseline. For the dose effect, quantification was performed after CLP nanoparticles were incubated with 100 mM H2O2 for 6 hours. To examine the influence of the H2O2 concentration, absorbance was measured after 6 hours of incubation with 20 μM CLP nanoparticles. (C) Confocal microscopy images illustrating time-dependent endocytosis of CLP nanoparticles in A549 lung carcinoma cells. Images in the lower panel display quantitative analysis of fluorescent intensities corresponding to indicated yellow lines in single cells of the upper fluorescence images. After A549 cells were cultured with 10.8 μM CLP nanoparticles for predetermined time periods, nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI), while endolysosomes were labeled with Lyso. Scale bars, 20 μm. (D) The half-maximal inhibitory concentration (IC50) values of different cells [including RAW264.7 murine macrophages, B16F10 mouse melanoma cells, MOVAS (mouse vascular aortic smooth muscle cell), MCF-7 human breast cancer cells, and A549 cells] after incubation with various doses of CLP nanoparticles for 24 hours. (E) Quantification of intracellular ROS levels in different cells by flow cytometry. After cells were incubated with 10 μM DCF-DA for 20 min and washed with PBS, measurements were performed. The ROS level is proportional to the mean fluorescence intensity (MFI) of a fluorescent probe DCF-DA. (F) Correlation between IC50 values and ROS levels. (G) Changes in tumor volume during treatment by intratumoral administration of saline or CLP nanoparticles at 3.25 mg/kg of Ce6 every 3 days in nude mice bearing A549 xenografts. (H and I) Digital photos (H) and quantified tumor weight (I) of excised tumors at day 30 after different treatments. (J) DAPI and TUNEL (terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling) double-labeling assay of cell apoptosis in paraffin sections. (K) Immunofluorescence analysis of proliferating cell nuclear antigen (PCNA) in tumor sections. (L) The ROS level in tumor tissues after treatment via intratumoral injection of saline or CLP nanoparticles. (M) Ex vivo fluorescence images (left) and quantitative data (right) of the distribution of CLP nanoparticles in A549 xenografts. At day 7 after the last intravenous injection of CLP nanoparticles at 5 or 25 mg/kg of Ce6, tumors were excised for imaging. (N) Tumor volume changes during treatment with intravenously administered saline or CLP nanoparticles at 5 or 25 mg/kg of Ce6. The control group was treated with saline. (O) DAPI/TUNEL double-labeling assay of apoptotic cells in tumor sections after 30 days of treatment with saline or CLP nanoparticles. Scale bars, 20 μm. Data are means ± SEM (A, B, D, E, and L to N, n = 4; G and I, n = 6). *P < 0.05, **P < 0.01, ***P < 0.001. (Photo credit: Huijie An, Third Military Medical University).
As extensively demonstrated, 1O2, a highly reactive product generated from a photochemical reaction during PDT, causes rapid cytotoxicity, leading to cell death via apoptosis or necrosis (54). Accordingly, we hypothesized that the CLP nanoparticle can be used for cancer treatment via PDT in the absence of external light. Before using the self-illuminating CLP nanoparticles for PDT in cancer, we examined the endocytosis of CLP nanoparticles using a human lung carcinoma cell line, A549 (Fig. 5C). In addition to a time-dependent increase in red fluorescence from CLP nanoparticles in A549 cells, we observed their localization in late endosomes and lysosomes by probing with LysoTracker Green DND-26 (Lyso). This indicated that the internalized CLP nanoparticles were mainly transported via the endolysosomal pathway. Then, we tested the in vitro antitumor activity of CLP nanoparticles in cancer cell lines, including mouse melanoma cells (B16F10), human breast cancer cells (MCF-7), and A549 cells, while the RAW264.7 murine macrophage and MOVAS (mouse aortic vascular smooth muscle cell) lines were used as normal control cells. We found that the cell viability in all examined cells was dose dependent (fig. S7C) and that the half-maximal inhibitory concentration (IC50) was 498.8, 414.2, 267.9, 153.0, and 123.7 μM for RAW264.7, B16F10, MOVAS, MCF-7, and A549 cells, respectively (Fig. 5D). Flow cytometric quantification using a ROS-sensitive fluorescent probe, 2′,7′-dichlorofluorescin diacetate (DCF-DA), to stain the cells, showed different ROS levels for different cell types (fig. S7D and Fig. 5E). The intracellular ROS expression was well correlated with the antitumor activity of the CLP nanoparticles in cancer cells (Fig. 5F), with the most notable activity in A549 cells with the highest ROS level. Low cytotoxicity was found for B16F10 cells because of their low intracellular ROS level (Fig. 5E), which is consistent with the low cytotoxicity detected in RAW264.7 and MOVAS cells (fig. S7, C and E, and Fig. 5D). For A549 cells with relatively high intracellular ROS, treatment with CLP nanoparticles could further increase the ROS level in cells (fig. S7, F and G). This agrees with the ROS-induced generation of 1O2 (Fig. 5, A and B).
Together, our results demonstrate that CLP nanoparticles generate 1O2 through intramolecular BRET in an oxidative cellular environment. CLP nanoparticles exhibited significant cytotoxicity to cancer cell lines with high intracellular ROS levels.
In vivo antitumor studies of the CLP nanoparticle
We then examined the in vivo antitumor efficacy of CLP nanoparticles in mice bearing A549 xenografts. Immunofluorescence analysis showed the infiltration of neutrophils and production of MPO in tumors (fig. S8A). Further quantification revealed the presence of a considerable amount of MPO and H2O2 in tumor tissues (fig. S8B). After intratumoral administration of the CLP nanoparticles at 3.25 mg/kg of Ce6 (fig. S8C), tumor growth was significantly suppressed for a treatment period of 30 days (Fig. 5G). At day 30, the CLP nanoparticle–treated group exhibited significantly reduced tumor size and weight compared to the saline control group (Fig. 5, H and I). H&E-stained histological sections of isolated tumors showed marked fragmentation of the nucleus and notable cell death in the CLP nanoparticle–treated group but not in the saline control group (fig. S8D). Using the TUNEL (terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling) assay, we detected considerable cell apoptosis in the sections of tumor tissues from CLP nanoparticle–treated mice. By contrast, negligible TUNEL-positive cells appeared in the saline control (Fig. 5J and fig. S8E). Immunofluorescence analysis also showed that treatment with CLP resulted in markedly decreased proliferating cell nuclear antigen (PCNA), a marker of cell proliferation in various cancers (Fig. 5K). In line with these results, we detected significantly enhanced ROS levels in tumors for the CLP nanoparticle–treated animals compared to those administered with saline (Fig. 5L). For intratumoral administration, we mainly detected the fluorescent signals of CLP nanoparticles in the tumor tissue but not in other major organs of the mice (fig. S8, F and G), suggesting that the CLP nanoparticles were maintained in the tumor and that their diffusion into other tissues was negligible.
Subsequently, we examined the antitumor efficacy after intravenous administration of CLP nanoparticles. First, an in vivo pharmacokinetic study showed that the circulation half-life of CLP nanoparticles was approximately 0.9 ± 0.1 hours after intravenous injection in mice (fig. S9A), and no CLP was detected after 72 hours. For the examined major organs, the CLP contents were considerably reduced after 24 hours (fig. S9, B to F), which revealed that most of the nanoparticles could be eliminated within 1 day. The gradually decreased accumulation in the kidneys suggested that the disassembled CLP molecules were mainly eliminated via glomerular filtration due to their low molecular weight. Correspondingly, considerable urinary excretion was detected, particularly at day 1 after administration (fig. S9G), with a total elimination of 39.0% at day 7. Nevertheless, relatively low levels of CLP were found in feces (fig. S9H), exhibiting a cumulative excretion of 2.7%. Consequently, CLP is mainly eliminated through urinary excretion. Whether it can be eliminated in the form of its metabolites remains to be addressed, since the cumulative urinary and fecal excretion of CLP was less than 50%. On the other hand, these results indicated that CLP might be accumulated in different tissues and organs.
Then, ex vivo fluorescence imaging was conducted to examine the biodistribution profile after intravenous administration of CLP nanoparticles containing 5 or 25 mg/kg of Ce6 units. We found intratumoral accumulation of CLP in mice bearing A549 xenografts (Fig. 5M), which was due to the enhanced permeability and retention effect (55). In addition to tumor sites, CLP nanoparticles (as indicated by fluorescence) were mainly distributed in the liver, lung, and kidney (fig. S10), consistent with the quantified biodistribution data in different organs (fig. S9). Although intravenous injection of CLP nanoparticles at 5 mg/kg of Ce6 every 6 days did not reduce tumor volumes (fig. S11A and Fig. 5N), therapy with CLP nanoparticles at 25 mg/kg of Ce6 effectively attenuated tumor growth during 30 days of treatment. Histological analysis showed substantial fragmentation of the nuclei in tumor sections of CLP nanoparticle–treated mice (fig. S11B). In addition, notable cell apoptosis was observed in tumor sections from mice treated with CLP nanoparticles containing 25 mg/kg of Ce6 (Fig. 5O and fig. S11C). It should be emphasized that the relatively weak fluorescence of CLP in the immunofluorescence image might be due to fluorescence quenching by in vivo biomolecules or metal ions since the tumors were isolated at day 7 after the last injection of CLP nanoparticles.
In a separate study, we compared in vivo efficacy of CLP nanoparticles with a typical chemotherapy drug cisplatin (CDDP) in mice with A549 tumors. After treatment by intravenous administration, CLP nanoparticles at a dose equivalent to 25 mg/kg of Ce6 showed antitumor activity comparable to CDDP at 6 mg/kg (CDDP-H), with respect to delaying tumor growth and reducing tumor weight (fig. S12, A to C). However, no significant effects were found at 1 mg/kg CDDP (CDDP-L). Treatment with CDDP caused significant side effects, as indicated by abnormal changes in the body weight, organ index, and number of platelets (fig. S12, D to F), particularly for mice treated with CDDP (6 mg/kg). Moreover, increased plasma levels of aspartate aminotransferase and urea were detected for CDDP groups (fig. S12, G and H). These results revealed that CDDP treatment led to varied degrees of toxic effects on the hemopoietic system, liver, and kidney. By contrast, no severe side effects were observed for the CLP group.
Consequently, these preliminary results demonstrate that the CLP nanoparticle may serve as a potentially effective and safe nanomedicine for PDT via the intravenous route in the absence of external excitation, although its efficacy remains to be enhanced by different strategies such as increasing targeting efficiency.
In vivo safety tests in mice
We further tested the safety profile of CLP nanoparticles administered to mice. After intratumoral injection of CLP nanoparticles containing 3.25 mg/kg of Ce6, we observed a moderate increase in the body weight of the mice treated with the CLP nanoparticles compared to that of the untreated mice (fig. S13A). At day 30 after treatment, the CLP nanoparticle–treated mice showed comparable organ index values and plasma levels of typical biochemical markers relevant to liver and kidney functions to the control animals (fig. S13, B to D). Histological analyses indicated no notable injuries or inflammatory responses in H&E sections of different organs (fig. S13E). These data suggest that the CLP nanoparticle is safe for local administration. In addition, during treatment with CLP nanoparticles by intravenous administration at 5 or 25 mg/kg of Ce6, all mice displayed comparable body weight changes (fig. S14A). In addition, the CLP nanoparticle–treated groups showed no abnormal variations in the organ index values and H&E sections of typical major organs (fig. S14, B and C).
Moreover, preliminary in vivo studies were conducted to evaluate the acute toxicity of CLP nanoparticles in healthy mice. After intravenous injection of CLP nanoparticles in mice at either 65 or 130 mg/kg of Ce6, all mice behaved normally, without notable side effects. In addition, all mice showed gradually increasing body weight (fig. S15A). Excluding the reversible change in the liver index at the high dose, no significant differences in organ index were detected for the other examined organs between the saline control and CLP nanoparticle–treated groups (fig. S15B). Quantification of typical biomarkers associated with liver (alanine aminotransferase and aspartate aminotransferase) and kidney (urea and creatinine) functions showed no abnormally increased levels for the CLP nanoparticle–treated animals in comparison to those of the saline group (fig. S15, C and D); notably increased markers generally reveal liver and kidney injuries. In addition, inspection of H&E sections revealed indiscernible pathological changes in various major organs (fig. S15E). Consequently, these data showed that CLP nanoparticles exhibited a good safety profile for intravenous administration, even at a single dose as high as 1000 mg/kg of CLP nanoparticles (i.e., 130 mg/kg of Ce6). Nevertheless, in combination with the excretion and tissue distribution results (figs. S9 and S10), the possible chronic toxicity to CLP-accumulated tissues (such as the liver) needs to be examined by in-depth studies, after short-term and long-term treatment.
Mechanism of CLP nanoparticle–mediated antitumor activity
We further probed the possible mechanism of CLP nanoparticle–mediated antitumor activity in A549 cells. We found that the apoptosis of A549 cells increased with the dose of CLP nanoparticles (fig. S16, A and B), indicating that apoptosis might be the major cause of cell death. Our data further revealed that CLP nanoparticles targeted the mitochondria of A549 cells. As shown by confocal microscopy, the accumulation of CLP nanoparticles was observed even after 0.5 hours of incubation, and the amount continuously increased with time (Fig. 6A). In addition, CLP nanoparticles induced mitochondrial membrane depolarization in a dose- and time-dependent manner (fig. S16, C and D, and Fig. 6, B and C). Since the loss of mitochondrial membrane potential is an early event in mitochondria-triggered apoptosis, our results suggest that CLP-induced apoptosis is mediated by the mitochondrial pathway of apoptosis. To test this hypothesis, we performed Western blotting analysis, and our results revealed a significant increase in cleaved caspase-3 level after treating A549 cells with CLP nanoparticles (Fig. 6D). The level of cleaved caspase-3 was linearly correlated with the dose of CLP nanoparticles (Fig. 6E) and the percentage of apoptotic cells (Fig. 6F). Similarly, reduction in total caspase-8 and caspase-9 (particularly caspase-9) and considerable increase in cleaved caspase-9 were detected after treatment with CLP nanoparticles (Fig. 6, G and H). In the caspase-mediated apoptosis, caspase-8 and caspase-9 are molecules responsible for the initiation of apoptosis, while caspase-3 is a downstream execution molecule. The activation of these molecules collectively revealed that CLP nanoparticles induced cell death through the apoptotic pathway. Together, these data demonstrate that CLP nanoparticle–induced cell apoptosis is largely mediated via the intrinsic mitochondrial pathway (Fig. 6I).
Fig. 6. In vitro mechanistic studies of the antitumor activity of CLP nanoparticles in A549 cells.
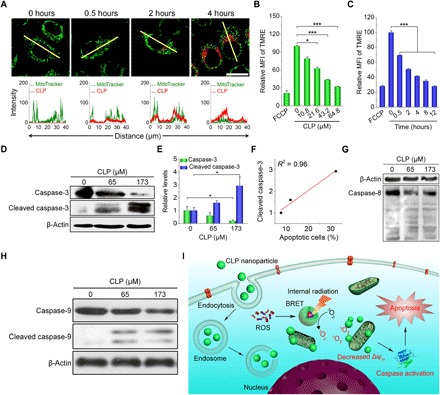
(A) Confocal microscopy images showing the mitochondrial localization of CLP nanoparticles. Images in the lower panel display quantitative analysis of the fluorescent intensities in the single cells as highlighted by the yellow lines in the upper fluorescence images. After cells were incubated with 10.8 μM CLP nanoparticles for different periods of time, mitochondria were stained with MitoTracker Green FM (MitoTracker). Scale bar, 20 μm. (B and C) The effects of the CLP nanoparticle dose (B) and incubation time (C) on mitochondrial membrane potential as analyzed by flow cytometry using a fluorescent probe tetramethylrhodamine ethyl ester (TMRE). For the dose-response experiments, cells were incubated with different doses of CLP nanoparticles for 12 hours, while they were cultured with 64.8 μM CLP to examine the time effect. FCCP (carbonyl cyanide p-trifluoromethoxy phenylhydrazone) was used as a positive control that can effectively depolarize the mitochondrial membrane potential. (D and E) Representative Western blot bands (D) and quantitative analysis (E) of total and cleaved caspase-3 in A549 cells after treatment with different doses of CLP nanoparticles for 24 hours. (F) Correlation between the percentage of apoptotic cells and the level of cleaved caspase-3. (G and H) Western blot analysis of total and cleaved caspase-8 (G) and caspase-9 (H) in A549 cells after 24 hours of incubation with CLP nanoparticles. (I) Schematic illustration of the antitumor mechanisms of CLP nanoparticles. Data are means ± SEM (n = 3). *P < 0.05, ***P < 0.001.
DISCUSSION
Real-time, noninvasive luminescence imaging holds promise in the future for the detection, diagnosis, and monitoring of a plethora of inflammation-associated diseases (4, 11, 24). However, effective, sensitive, safe, and reliable luminescent agents remain to be developed. Herein, we engineered a luminescent material, CLP, with near-infrared light emission based on intramolecular BRET between a donor, luminol, and a fluorescent acceptor, Ce6. Because of their amphiphilicity, CLP molecules can self-assemble into a core-shell structured micelle-like nanoparticle in aqueous solution. The luminescence of CLP nanoparticles is positively related to the levels of environmental ROS and MPO, two proinflammatory mediators commonly detected in both acute and chronic inflammatory conditions. After being internalized into neutrophils, CLP nanoparticles self-illuminated the activated neutrophils that produced ROS and MPO, showing a CLP dose- and cell number–dependent luminescence profile. In mouse models of inflammatory diseases, including acute peritonitis, acute liver injury, and ulcerative colitis, CLP nanoparticles delivered via local or intravenous administration showed significantly higher sensitivity than luminol, a frequently used small-molecule luminescent probe. Furthermore, our newly engineered CLP nanoparticles can be used to track the initiation and progression of colitis and possibly other inflammation-related diseases.
Compared to luminol molecules, our CLP nanoparticles showed a markedly enhanced luminescent signal, which should be attributed to their increased vascular permeability and high accumulation rate at inflammatory sites (4, 50). In addition, translocation via inflammatory cells (such as monocytes/macrophages and neutrophils) also contributed to the accumulation of nanoparticles administered via different routes at inflamed sites. This has been demonstrated by previous studies in atherosclerosis (56–58), a chronic inflammatory disease. By contrast, small-molecule probes are systemically absorbed, indiscriminately distributed, and rapidly excreted after delivery, leading to an undesirable signal-to-noise ratio and poor imaging quality in diseased sites (16). BRET contributes to the enhanced luminescence of the CLP nanoparticles by increasing tissue penetration through the long-wavelength, red-shifted emission (14, 17, 25, 26). Most existing luminescent agents capable of BRET or chemiluminescence resonance energy transfer, however, are mainly limited to in vitro biosensing applications (59–61), and their effectiveness and safety for in vivo applications remain to be validated. In comparison, our CLP nanoparticle offers high detection sensitivity, excellent specificity, superior imaging quality, and satisfactory safety for in vitro and in vivo applications across a broad range of cell types and mouse models, representing a major advance in this promising field.
In addition to producing bright and sustainable luminescence for in vivo real-time imaging and tracking, our CLP nanoparticle can also serve as a fluorescent probe for inflammation using flow cytometry and fluorescence microscopy. Compared with many other luminescent nanoprobes derived from conjugated polymers and inorganic materials (14, 20, 26), our CLP nanoparticle is highly biocompatible and safe for basic research and therapeutic assessments of inflammation-associated diseases. It was easily hydrolyzed, quickly disassembled, and completely excreted from the body. No significant side effects were found during and after local or intravenous administration of CLP nanoparticles to mice. The desirable in vivo biocompatibility and safety profile of CLP nanoparticles provide additional advantages over most currently developed self-illuminating systems, which contain toxic, inorganic nanomaterials (20, 36, 37), for both imaging and therapeutic applications. Although the distribution of our CLP nanoparticle in inflamed tissues is largely dominated by passive accumulation, its targeting efficiency and specificity can be further improved by adding functional moieties such as antibodies to specifically target molecules and/or cells involved in inflammation to realize precision imaging and therapy. In combination with aggregation-induced emission luminogens (62–64), it should be able to considerably improve the fluorescence efficiency of this type of self-illuminating nanoparticles.
As extensively demonstrated, oxidative stress, inflammation, and cancer are closely linked (52). Whereas cancer cells generally show enhanced ROS defense systems by their increased expression of free-radical scavengers that contribute to tumor radioresistance and drug resistance (65), significantly increasing the ROS level by targeting the oxidative stress-response pathway or PDT represents an effective strategy for cancer treatment (54). Unfortunately, clinical applications of numerous nanotherapies for the PDT of solid tumors have been considerably limited in spite of their promising efficacies in preclinical studies (27, 29, 30). The requirement of exogenous radiation is one of the major translational barriers for PDT, particularly for deep-seated solid tumors (32). By contrast, our CLP nanoparticle can generate 1O2 in the absence of external excitation, which is achieved by in situ excitation of the photosensitizer Ce6 via luminescence of the luminol unit under oxidative conditions in the tumor microenvironment. The CLP nanoparticle showed high in vitro antitumor activity in cancer cells expressing high ROS, such as A549 cells. Both intratumoral and intravenous injections of CLP nanoparticles significantly inhibited tumor growth in nude mice bearing A549 xenografts. In vitro mechanistic studies and in vivo examinations indicated that the anticancer activity of CLP nanoparticles is largely mediated by a mitochondria-mediated caspase activation pathway-triggered cell apoptosis. As compared to typical chemotherapeutic agents, the CLP nanoparticle showed a more desirable in vivo safety profile.
Treatment with the CLP nanoparticle via either intratumoral or intravenous injection cannot completely abrogate tumor growth, and its in vivo efficacies are not as highly effective as expected. This can be largely attributed to the low level of 1O2 generated at tumor sites, resulting from the relatively low content of luminol in the CLP conjugate that causes low BRET efficiency. The low ROS in the tumor microenvironment and limited targeting efficiency (in the case of intravenous injection) also contribute to undesirable therapeutic effects. Nevertheless, different strategies can be applied to further potentiate in vivo efficacies of this PDT approach. First, the number of luminol and Ce6 units incorporated in this type of self-illuminating nanoparticles should be markedly increased to enhance the 1O2 production efficiency, which can be achieved by using macromolecules containing much more reactive groups (such as graft or multiblock copolymers and dendrimers) as scaffolds. Second, the local ROS level may be intentionally amplified by simultaneously using ROS-generating agents that can be easily loaded into the core of CLP nanoparticles (30, 66). Third, it is possible to enhance targeting efficiency by decoration with active targeting moieties or decreasing particle size. Moreover, our self-illuminating nanoparticles can be used in combination with chemotherapeutic drugs to achieve synergistic effects.
In summary, we developed an amphiphilic luminescent polymer that can self-assemble into a nanoparticle for inflammation imaging and cancer therapy. This self-illuminating nanoparticle can be used for the stratification of inflammation-associated diseases and high-content screening of anti-inflammatory therapeutics. The nanoparticle can simultaneously function as an effective photodynamic nanotherapy for solid tumors expressing high ROS levels without the need for external excitation and achieve combination therapy by loading it with different anticancer drugs.
MATERIALS AND METHODS
Synthesis of a CLP conjugate
An amphiphilic conjugate based on Ce6, luminol, and PEG (abbreviated as CLP) was synthesized by conjugating both luminol and PEG with Ce6 using a standard N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC)/N-hydroxysuccinimide (NHS)–activated condensation reaction. Briefly, 50 mg (0.08 mmol) of Ce6 was dissolved in 10 ml of anhydrous DMSO, into which EDC [236 mg (1.23 mmol)] and NHS [144 mg (1.25 mmol)] were sequentially added. The reaction mixture was stirred at 50°C in the dark for 17 hours. Then, luminol [28 mg (0.16 mmol)] was added to the activated solution. After 3 days, monomethyl-terminated PEG monoamine with a molecular weight of 2000 [320 mg (0.16 mmol)] was added. Reaction was conducted at 50°C in the dark for 4 days. Last, the reaction mixture was dialyzed (molecular weight cutoff, 3500) against deionized water to remove unreacted reagents and by-products. After filtration through a 0.22-μm syringe filter, the obtained aqueous solution was lyophilized to give rise to a dark green powder.
Luminescence properties of the CLP conjugate
Luminescent curves were directly acquired with a fiber optic spectrometer (AvaSpec-HS, Avantes Inc., The Netherlands) in the presence of H2O2 and ClO−. The photon counts and time-dependent changes of luminescent signals of CLP were quantified by a BPCL-2-KGC ultraweak luminescence analyzer under different conditions.
In addition, luminescent signals of CLP in different solutions were examined using an IVIS Spectrum imaging system (PerkinElmer, USA). To this end, CLP at 0.22 mM (i.e., 1.0 mg/ml) was mixed with H2O2 at different concentrations in a black 96-well plate. Alternatively, various doses of CLP were mixed with 50 μM H2O2 in a black 96-well plate. After thorough mixing, the plate was immediately put into the imaging system to acquire luminescent signals, with an exposure time of 5 min [focal length/stop (f/stop) = 1, binning = 8, field of view (FOV) = 12.8 cm, no optical filter]. Similarly, luminescent signals in the existence of H2O2/MPO or H2O2/MPO/Cl− were detected. In another experiment, the attenuated luminescence of CLP was quantified after incubation with a ROS scavenger, Tempol, or an MPO inhibitor, 4-ABAH, in the presence of H2O2/MPO.
To examine the sensitivity to different components of ROS, CLP nanoparticles at 0.5 mg/ml were separately incubated with different ROS at 50 or 500 μM in a black 96-well plate. Aqueous solutions containing either H2O2 or OCl− were prepared based on commercially available agents. Peroxynitrite (ONOO−) was obtained by the reaction of H2O2 and nitrite. Hydroxyl radical (•OH) was generated by the Fenton reaction between ferrous acetate and H2O2, while superoxide anion was produced by the xanthine-xanthine oxidase system. Luminescence imaging was immediately performed as aforementioned to determine the luminescent intensities.
Tissue penetration efficiency study
To demonstrate the capability of CLP nanoparticles for deep-tissue imaging, the relative bioluminescence intensity of CLP nanoparticles and luminol was compared. Briefly, CLP nanoparticles (0.5 mg/ml) or luminol in aqueous solution was mixed with 50 μM H2O2 and 15 mU MPO with or without 50 mM Cl− in a black 96-well plate, which was covered with porcine muscle tissue with a thickness of 3 mm. The same samples without covering with muscle tissue were used as controls. In another study, the in vitro luminescence penetration threshold of CLP nanoparticles was examined. To this end, CLP nanoparticles (1 mg/ml) in aqueous solution was mixed with 100 μM H2O2, 30 mU MPO, and 100 mM Cl− in a black 96-well plate, which was covered with muscle tissues with different thicknesses. In all cases, the bioluminescent signals were quantified by an IVIS Spectrum imaging system (exposure time = 5 min, f/stop = 1, binning = 8, FOV = 6.6 cm).
Detection of the generation of singlet oxygen
The generation of singlet oxygen (1O2) was characterized by chemical trapping 1O2 with DPA (67). The decrease in UV absorbance between 300 and 450 nm of the corresponding thermostable endoperoxide DPAO2 was detected. The decreased absorbance at 355 nm was quantified to analyze the amount of produced 1O2. Specifically, DPA was dissolved in a solvent mixture of acetonitrile and deuterium oxide [1:1 (v/v)] to obtain a saturated solution. The dose-dependent changes in the presence of 100 mM H2O2 before and after 6 hours of incubation were recorded. Subsequently, 20 μM CLP was incubated with various concentrations of H2O2 and a DPA stock solution. After 6 hours, absorption spectra were recorded on a UV-visible spectrometer.
Studies on intracellular uptake of CLP nanoparticles
To observe cellular internalization behaviors by confocal laser scanning microscopy (CLSM), A549 human lung carcinoma cells were incubated with CLP nanoparticles at 50 μg/ml (i.e., 10.8 μM) for different time periods (0, 0.5, 2, 4, 8, or 12 hours). Then, cells were washed with PBS, stained with 75 nM Lyso for 1 hour, rinsed with PBS, fixed, and counterstained with 4′,6-diamidino-2-phenylindole (DAPI). CLSM observation was performed using a confocal microscope (LSM 800, Zeiss, Germany).
Observation of mitochondrial localization of CLP nanoparticles
A549 cells were incubated with CLP nanoparticles at 50 μg/ml (i.e., 10.8 μM) for 0, 0.5, 2, or 4 hours at 37°C, respectively. Cells were then treated with MitoTracker Green FM (MitoTracker) at 100 nM for 30 min. After the cells were washed three times with PBS, they were observed by CLSM immediately.
In vitro cytotoxicity evaluation
Mouse melanoma cell B16F10, human breast cancer cell MCF-7, A549, mouse vascular aortic smooth muscle cell MOVAS, and RAW264.7 murine macrophage cells were seeded in a 96-well plate at 1 × 104 cells per well and incubated for 12 hours, respectively. Then, cells were treated with the medium containing CLP nanoparticles at various doses for 24 hours. Cell viability was quantified by the thiazolyl blue tetrazolium bromide (MTT) assay using a microplate reader (EMax Plus Microplate Reader, Molecular Devices, USA). The relative viability of the untreated controls was normalized to be 100%, while the medium served as the background control. According to the improved Karber’s method, IC50 values were calculated.
Detection of intracellular ROS levels
B16F10, MCF-7, A549, MOVAS, and RAW264.7 cells were seeded in a 12-well plate at 1 × 105 cells per well in 1 ml of growth medium. After 12 hours, 10 μM DCF-DA was added and incubated for 20 min. Then, cells were washed with PBS, digested with trypsin, collected by centrifugation, and resuspended in PBS for detection by flow cytometry (BD Accuri C6, Becton Dickinson, USA). The corresponding cells without staining with DCF-DA were used as controls.
Western blot analysis of activation of caspases in A549 cells
A549 cells were seeded into a six-well plate at 3 × 105 cells per well. After 12 hours, cells were treated with CLP nanoparticles at doses of 0, 65, or 173 μM for 24 hours. Then, cells were washed three times with ice-cold PBS and lysed in ice-cold radioimmunoprecipitation assay buffer containing protease inhibitors. The supernatant was collected by centrifugation at 12,000g for 5 min at 4°C. The concentration of total protein was quantified by the bicinchoninic acid (BCA) method. The protein samples were loaded into SDS–polyacrylamide gel electrophoresis. The separated proteins were transferred to polyvinylidene fluoride membranes, followed by blocking in tris-buffered saline–Tween 20 (TBST) containing 5% nonfat dry milk at room temperature for 2 hours. Subsequently, each membrane was incubated at 4°C overnight with respective primary antibodies. Membranes were washed three times with TBST, followed by incubation with appropriate horseradish peroxidase–labeled antibodies at room temperature for 1 hour. Last, membranes were washed and then detected using chemiluminescence reagents.
Measurement of mitochondrial membrane potential
Tetramethylrhodamine ethyl ester (TMRE) was used to analyze changes of mitochondrial membrane potential. Specifically, A549 cells were seeded into a 12-well plate and treated with CLP nanoparticles at various concentrations for 12 hours or at 65 μM for predetermined time periods. Then, cells were incubated with 100 nM TMRE for 30 min. The incubated cells were washed twice with PBS, collected, and subsequently assayed by flow cytometry, with FCCP (carbonyl cyanide p-trifluoromethoxy phenylhydrazone) used as a positive control.
Apoptosis assays
A549 cells were seeded in a 12-well plate at 1 × 105 cells per well for 12 hours. After incubation with CLP nanoparticles at various doses for 24 hours, cells were collected, washed with PBS, and stained with annexin V–fluorescein isothiocyanate (FITC) and propidium iodide. Cells were further incubated for 15 min at room temperature in the dark. The percentage of apoptotic cells was quantified by flow cytometry.
Animals
All animal experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals proposed by the National Institutes of Health. All procedures and protocols were approved by the Animal Ethics Committee at Third Military Medical University (Chongqing, China). Male Kuming, C57BL/6, BALB/c, and BALB/c nude mice were obtained from the Animal Center at the Third Military Medical University. Before further experiments, all mice were acclimatized for at least 7 days.
Studies on intracellular uptake of CLP nanoparticles in neutrophils
Neutrophils were isolated from the peritoneal exudates of BALB/c mice after 4 hours of stimulation with thioglycollate (3.0 weight %). Cellular uptake profiles of CLP nanoparticles in neutrophils were analyzed by the fluorescence-activated cell sorting technique. Specifically, neutrophils were seeded in a 12-well plate at 5 × 105 cells per well in 1 ml of sterile Hanks’ balanced salt solution (HBSS) and incubated for 10 min. After washing with fresh HBSS, cells were incubated with 1 ml of growth medium containing 21.6 μM CLP nanoparticles for predetermined time periods. In another study, cells were incubated with various concentrations of CLP nanoparticles for 2 hours. After cells were washed three times with PBS, blew down, and collected by centrifugation, they were resuspended in PBS for analysis by flow cytometry. To observe cellular internalization behaviors by CLSM, neutrophils were treated with CLP nanoparticles at 21.6 μM for different time intervals. After washing three times with PBS, cells were fixed and stained with DAPI. CLSM observation was performed by a fluorescence microscope.
Ex vivo bioluminescence imaging of neutrophils
Neutrophils were seeded in a 12-well plate at 5 × 105 cells per well in 1 ml of HBSS and washed with fresh HBSS after 10 min. After incubation in 900 μl of HBSS with or without PMA (100 ng/ml) for 1 hour, 100 μl of aqueous solution containing 54 μM CLP nanoparticles was added, followed by luminescence imaging at predetermined time points. To separate intracellular and extracellular bioluminescence, 100 μl of aqueous solution containing 54 μM CLP nanoparticles was added and incubated for 30 min with neutrophils that were stimulated with PMA (100 ng/ml). Immediately after luminescence imaging, the extracellular medium was collected, and additional imaging was performed for the separated cells and culture medium containing PMA. Neutrophils stimulated with PMA and without treatment with CLP nanoparticles served as the Blank group. In another experiment, various concentrations of CLP nanoparticles were incubated with neutrophils prestimulated with PMA to examine the dose-response luminescence. Similarly, the effect of neutrophil count was investigated. In all cases, the plates were imaged using an IVIS Spectrum imaging system (exposure time = 5 min, f/stop = 1, binning = 8, FOV = 12.8 cm). In all cases, the quantitative analysis was performed using the Image Analysis Software provided by the manufacturer. For all data obtained with the Image Analysis Software, the corresponding background values were deducted.
In vivo imaging of peritonitis in mice
To induce peritonitis, male BALB/c mice (6 to 8 weeks old) were injected intraperitoneally with 0.5 ml of sterile zymosan solution (2 mg/ml). At 5.5 hours after challenge, each mouse was intravenously injected with 0.1 ml of saline containing CLP nanoparticles [50 mg/ml (i.e., 10.8 mM)] or luminol at the same dose of the luminol unit. For CLP nanoparticles, the dose was equivalent to 250 mg/kg. The luminescence images were acquired after 30 min (exposure time = 5 min, f/stop = 1, binning = 8, FOV = 21.8 cm). In separate experiments, after 6 hours of stimulation with zymosan, mice were intraperitoneally injected with 0.1 ml of saline containing CLP nanoparticles (50 mg/ml) or luminol at the same dose of the luminol unit. Immediately, bioluminescence imaging was performed. The quantitative analysis was conducted as aforementioned.
To verify cellular distribution of CLP nanoparticles in the abdominal cavity, mice were euthanized and injected with 5 ml of ice-cold PBS. Cells in the peritoneal exudates were separated, stained with anti-mouse V450-CD11b and PE-Ly-6G antibodies, and resuspended in PBS for detection by flow cytometry.
In vivo imaging of acute liver injury in mice
Acute liver injury in mice was established by intraperitoneal injection of acetaminophen at 300 mg/kg. After 24 hours, mice were randomly divided into three groups. Mice in the saline group were intravenously administered with 0.1 ml of saline. The CLP group was treated with intravenous injection of 0.1 ml of saline containing CLP nanoparticles (50 mg/ml) in each mouse, while the luminol group was administered with the same dose of the luminol unit. Immediately after injection, in vivo bioluminescence imaging was performed. In addition, ex vivo images of isolated liver tissues were acquired.
In vivo imaging of ulcerative colitis
Ulcerative colitis in C57BL/6 mice was induced by free feeding of drinking water containing 3% (w/v) DSS for 7 days (49). At predetermined time periods, mice were administered with 0.1 ml of saline containing CLP nanoparticles (50 mg/ml) or luminol at the same dose of the luminol unit via enema after they were anesthetized with isoflurane. After mice were held in a vertical position for 1 min, in vivo imaging was performed after 15 min. Then, mice were euthanized and colons were isolated for ex vivo imaging. In addition, in vivo and ex vivo fluorescence images were collected by excitation at 430 nm, with emission at 680 nm.
Evaluation on the degree of colitis
Animals were monitored every day for observation of water consumption, piloerection, stool consistency, the presence of gross blood in feces and near the anus, and morbidity. DAI was defined according to a previously established method (68). According to the calculated DAI values, the degree of colitis can range from 0 (unaffected) to 12 (severe colitis). At predetermined time periods, animals were euthanized, and the entire colon was quickly isolated to measure colonic length. In addition, segments of colon tissues were fixed in formalin and embedded in paraffin, and H&E-stained sections were prepared for histological analysis.
In vivo antitumor therapy with CLP nanoparticles after intratumoral delivery
A549 xenografts in BALB/c nude mice were established as aforementioned. When the tumor volume reached ~100 mm3, mice were randomly divided into two groups (n = 6). The saline group was treated with 0.1 ml of saline, while animals in the CLP group were treated with 0.1 ml of saline containing CLP nanoparticles at 3.25 mg/kg of Ce6 by intratumoral injection every 3 days. The first day of intratumoral injection was considered as day 1. After administration, body weight and tumor size were monitored every 3 days for a total of 30 days. Tumor volume was estimated by the formula a × b2/2, where a and b refer to the major and minor axes of the tumor measured by a caliper, respectively. After treatment, mice were euthanized to collect blood for serum biochemistry assays. Tumor tissues and major organs were also harvested. Histological sections were prepared for H&E staining and detection of apoptosis by TUNEL assay. Immunofluorescence analysis was also performed by staining sections with FITC-labeled PCNA antibody and DAPI.
In vivo pharmacokinetic, biodistribution, and elimination profiles of CLP after intravenous injection
BALB/c mice (6 to 8 weeks) were intravenously injected with 0.1 ml of saline containing CLP nanoparticles at 25 mg/kg. At predetermined time periods, whole blood and major organs were collected after mice were euthanized. The plasma was obtained by centrifugation at 2000g for 10 min. After the proteins were precipitated via methanol, the plasma CLP concentrations were quantified by high-performance liquid chromatography (HPLC; Prominence-i LC-2030C, Shimadzu), using a Shim-pack GIST C18 column (250 mm by 4.6 mm; particle size, 5 μm) and an RF-20A fluorescence detector. The excitation wavelength was set at 401 nm, while the emission wavelength was set at 660 nm. The mobile phase consisted of water and methanol at a volume ratio of 10:90, with a flow rate of 1.0 ml/min. The column temperature was 40°C. For different organ tissues, they were homogenized in PBS and centrifuged, and the supernatant was collected. The CLP concentrations were also determined by HPLC.
In another study, excretion of CLP was examined in BALB/c mice that were housed individually in metabolic cages. Briefly, after CLP nanoparticles were administered via intravenous injection at 25 mg/kg, urine and feces were separately collected at predefined time points. Urine samples were precipitated via methanol, while feces were extracted with methanol. The CLP concentrations were quantified by HPLC.
Tumor targeting after intravenous injection in mice bearing A549 xenografts
CLP nanoparticles at 5 or 25 mg/kg of Ce6 were intravenously injected in mice with subcutaneous A549 xenografts for 30 days. At day 7 after the last administration, tumors and major organs were isolated and rinsed with cold PBS. Ex vivo imaging was carried out via an IVIS Spectrum imaging system. Quantitative analyses were performed to determine the distribution of CLP nanoparticles in different tissues.
In vivo antitumor efficacy of CLP nanoparticles after intravenous delivery
Mice with subcutaneous A549 xenografts were randomly divided into three groups (n = 4). The control group was treated with saline by intravenous injection every 6 days, while the other two groups were separately administered with CLP nanoparticles at 5 or 25 mg/kg of Ce6. The tumor volume and body weight were monitored every 3 days. After 30 days, mice were euthanized. Tumor tissues and major organs were collected for further analyses.
In a separate study, in vivo antitumor efficacy and safety of CLP nanoparticles were compared with a typical chemotherapy drug, CDDP. A549 xenograft–bearing mice were randomly assigned into four groups (n = 5). The control group was treated with 0.1 ml of saline. The dose of CLP nanoparticles was equivalent to 25 mg/kg of Ce6. Mice in two CDDP groups were administered with CDDP at 1 or 6 mg/kg, which were labeled as CDDP-L and CDDP-H, respectively. All treatments were performed once every 3 days by intravenous injection. Tumor volume and body weight were measured every 3 days. After 14 days, mice were euthanized for additional analyses.
Statistical analysis
All data are presented as means ± SEM. Student’s t test or one-way ANOVA was used to determine significance with Statistical Product and Service Solutions (SPSS) 19.0. Statistical significance was considered at P < 0.05.
Supplementary Material
Acknowledgments
Funding: This study was supported by the National Natural Science Foundation of China (nos. 81701832 and 81471774), the Science and Technology Innovation Program of Southwest Hospital (no. SWH2016LHYS-05), the Graduate Student Research Innovation Project of Chongqing (to X.X.), and the Program for Distinguished Young Scholars of TMMU. Author contributions: J.Z. conceived the project. J.Z., X.X., H.A., D.Z., H.T., Y.D., and J.H. designed the experiments. X.X., H.A., D.Z., H.T., and Y.D. performed all the experiments. J.Z., X.X., H.A., X.L., and J.H. analyzed the data and composed the manuscript. All authors discussed the results and reviewed the manuscript. Competing interests: X.X., H.A., D.Z., Y.D., and J.Z. are inventors in a pending patent filed by the National Intellectual Property Administration of the PRC (No. 2018070600581930, 6 July 2018) related to CLP, but the rights belong to Third Military Medical University. All other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/1/eaat2953/DC1
Supplementary Materials and Methods
Fig. S1. Synthesis and characterization of a CLP conjugate.
Fig. S2. Characterization of the CLP conjugate and its nanoparticles.
Fig. S3. Luminescence properties of the CLP conjugate under different conditions.
Fig. S4. Intracellular uptake of CLP nanoparticles and in vitro luminescence imaging in neutrophils.
Fig. S5. In vivo imaging in different mouse models of inflammation.
Fig. S6. The retention of CLP and luminol in the colon after enema administration in colitis mice.
Fig. S7. In vitro antitumor activity of CLP nanoparticles.
Fig. S8. Tumor therapy after intratumoral administration of CLP nanoparticles in mice bearing A549 xenografts.
Fig. S9. In vivo pharmacokinetic, tissue distribution, and elimination profiles of CLP after intravenous administration.
Fig. S10. Tissue distribution of CLP nanoparticles after intravenous administration in mice bearing A549 xenografts.
Fig. S11. Tumor therapy after intravenous administration of CLP nanoparticles in mice bearing A549 xenografts.
Fig. S12. Comparison of in vivo efficacy and safety of CLP nanoparticles with CDDP.
Fig. S13. Safety study after intratumoral administration of CLP nanoparticles in A549 xenograft–bearing mice.
Fig. S14. Safety profiles of CLP nanoparticles in A549 xenograft–bearing mice during and after 30 days of intravenous injection.
Fig. S15. Acute toxicity evaluation of CLP nanoparticles after a single intravenous injection in healthy mice.
Fig. S16. Mechanistic studies of in vitro antitumor activity of CLP nanoparticles in A549 cells.
REFERENCES AND NOTES
- 1.Shi J., Kantoff P. W., Wooster R., Farokhzad O. C., Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 17, 20–37 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xue Y., Xu X., Zhang X.-Q., Farokhzad O. C., Langer R., Preventing diet-induced obesity in mice by adipose tissue transformation and angiogenesis using targeted nanoparticles. Proc. Natl. Acad. Sci. U.S.A. 113, 5552–5557 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Veiseh O., Tang B. C., Whitehead K. A., Anderson D. G., Langer R., Managing diabetes with nanomedicine: Challenges and opportunities. Nat. Rev. Drug Discov. 14, 45–57 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weissleder R., Nahrendorf M., Pittet M. J., Imaging macrophages with nanoparticles. Nat. Mater. 13, 125–138 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Mahmoudi M., Yu M., Serpooshan V., Wu J. C., Langer R., Lee R. T., Karp J. M., Farokhzad O. C., Multiscale technologies for treatment of ischemic cardiomyopathy. Nat. Nanotechnol. 12, 845–855 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hotamisligil G. S., Inflammation and metabolic disorders. Nature 444, 860–867 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Elinav E., Nowarski R., Thaiss C. A., Hu B., Jin C., Flavell R. A., Inflammation-induced cancer: Crosstalk between tumours, immune cells and microorganisms. Nat. Rev. Cancer 13, 759–771 (2013). [DOI] [PubMed] [Google Scholar]
- 8.Swirski F. K., Nahrendorf M., Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science 339, 161–166 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hammoud D. A., Molecular imaging of inflammation: Current status. J. Nucl. Med. 57, 1161–1165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neuwelt A., Sidhu N., Hu C.-A. A., Mlady G., Eberhardt S. C., Sillerud L. O., Iron-based superparamagnetic nanoparticle contrast agents for MRI of infection and inflammation. Am. J. Roentgenol. 204, W302–W313 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hong G., Antaris A. L., Dai H., Near-infrared fluorophores for biomedical imaging. Nat. Biomed. Eng. 1, 0010 (2017). [Google Scholar]
- 12.Tarkin J. M., Joshi F. R., Rudd J. H. F., Pet imaging of inflammation in atherosclerosis. Nat. Rev. Cardiol. 11, 443–457 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Tarkin J. M., Dweck M. R., Evans N. R., Takx R. A. P., Brown A. J., Tawakol A., Fayad Z. A., Rudd J. H. F., Imaging atherosclerosis. Circ. Res. 118, 750–769 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang N., Francis K. P., Prakash A., Ansaldi D., Enhanced detection of myeloperoxidase activity in deep tissues through luminescent excitation of near-infrared nanoparticles. Nat. Med. 19, 500–505 (2013). [DOI] [PubMed] [Google Scholar]
- 15.Shuhendler A. J., Pu K., Cui L., Uetrecht J. P., Rao J., Real-time imaging of oxidative and nitrosative stress in the liver of live animals for drug-toxicity testing. Nat. Biotechnol. 32, 373–380 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guo J., Tao H., Dou Y., Li L., Xu X., Zhang Q., Cheng J., Han S., Huang J., Li X., Li X., Zhang J., A myeloperoxidase-responsive and biodegradable luminescent material for real-time imaging of inflammatory diseases. Mater. Today 20, 493–500 (2017). [Google Scholar]
- 17.Lee D., Khaja S., Velasquez-Castano J. C., Dasari M., Sun C., Petros J., Taylor W. R., Murthy N., In vivo imaging of hydrogen peroxide with chemiluminescent nanoparticles. Nat. Mater. 6, 765–769 (2007). [DOI] [PubMed] [Google Scholar]
- 18.Park J.-H., Gu L., von Maltzahn G., Ruoslahti E., Bhatia S. N., Sailor M. J., Biodegradable luminescent porous silicon nanoparticles for in vivo applications. Nat. Mater. 8, 331–336 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang G., Peng Q., Li Y., Lanthanide-doped nanocrystals: Synthesis, optical-magnetic properties, and applications. Acc. Chem. Res. 44, 322–332 (2011). [DOI] [PubMed] [Google Scholar]
- 20.So M.-K., Xu C., Loening A. M., Gambhir S. S., Rao J., Self-illuminating quantum dot conjugates for in vivo imaging. Nat. Biotechnol. 24, 339–343 (2006). [DOI] [PubMed] [Google Scholar]
- 21.Frangioni J. V., In vivo near-infrared fluorescence imaging. Curr. Opin. Chem. Biol. 7, 626–634 (2003). [DOI] [PubMed] [Google Scholar]
- 22.Weissleder R., A clearer vision for in vivo imaging. Nat. Biotechnol. 19, 316–317 (2001). [DOI] [PubMed] [Google Scholar]
- 23.Wang J., Ma Q., Hu X.-X., Liu H., Zheng W., Chen X., Yuan Q., Tan W., Autofluorescence-free targeted tumor imaging based on luminous nanoparticles with composition-dependent size and persistent luminescence. ACS Nano 11, 8010–8017 (2017). [DOI] [PubMed] [Google Scholar]
- 24.Pelaz B., Alexiou C., Alvarez-Puebla R. A., Alves F., Andrews A. M., Ashraf S., Balogh L. P., Ballerini L., Bestetti A., Brendel C., Bosi S., Carril M., Chan W. C. W., Chen C., Chen X., Chen X., Cheng Z., Cui D., Du J., Dullin C., Escudero A., Feliu N., Gao M., George M., Gogotsi Y., Grünweller A., Gu Z., Halas N. J., Hampp N., Hartmann R. K., Hersam M. C., Hunziker P., Jian J., Jiang X., Jungebluth P., Kadhiresan P., Kataoka K., Khademhosseini A., Kopeček J., Kotov N. A., Krug H. F., Lee D. S., Lehr C.-M., Leong K. W., Liang X.-J., Ling Lim M., Liz-Marzán L. M., Ma X., Macchiarini P., Meng H., Möhwald H., Mulvaney P., Nel A. E., Nie S., Nordlander P., Okano T., Oliveira J., Park T. H., Penner R. M., Prato M., Puntes V., Rotello V. M., Samarakoon A., Schaak R. E., Shen Y., Sjöqvist S., Skirtach A. G., Soliman M. G., Stevens M. M., Sung H.-W., Tang B. Z., Tietze R., Udugama B. N., VanEpps J. S., Weil T., Weiss P. S., Willner I., Wu Y., Yang L., Yue Z., Zhang Q., Zhang Q., Zhang X.-E., Zhao Y., Zhou X., Parak W. J., Diverse applications of nanomedicine. ACS Nano 11, 2313–2381 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li P., Liu L., Xiao H., Zhang W., Wang L., Tang B., A new polymer nanoprobe based on chemiluminescence resonance energy transfer for ultrasensitive imaging of intrinsic superoxide anion in mice. J. Am. Chem. Soc. 138, 2893–2896 (2016). [DOI] [PubMed] [Google Scholar]
- 26.Xiong L., Shuhendler A. J., Rao J., Self-luminescing BRET-FRET near-infrared dots for in vivo lymph-node mapping and tumour imaging. Nat. Commun. 3, 1193 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen H., Zhang W., Zhu G., Xie J., Chen X., Rethinking cancer nanotheranostics. Nat. Rev. Mater. 2, 17024 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wilhelm S., Tavares A. J., Dai Q., Ohta S., Audet J., Dvorak H. F., Chan W. C. W., Analysis of nanoparticle delivery to tumours. Nat. Rev. Mater. 1, 16014 (2016). [Google Scholar]
- 29.Lucky S. S., Soo K. C., Zhang Y., Nanoparticles in photodynamic therapy. Chem. Rev. 115, 1990–2042 (2015). [DOI] [PubMed] [Google Scholar]
- 30.Master A., Livingston M., Sen Gupta A., Photodynamic nanomedicine in the treatment of solid tumors: Perspectives and challenges. J. Control. Release 168, 88–102 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shen Y., Shuhendler A. J., Ye D., Xu J.-J., Chen H.-Y., Two-photon excitation nanoparticles for photodynamic therapy. Chem. Soc. Rev. 45, 6725–6741 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Idris N. M., Gnanasammandhan M. K., Zhang J., Ho P. C., Mahendran R., Zhang Y., In vivo photodynamic therapy using upconversion nanoparticles as remote-controlled nanotransducers. Nat. Med. 18, 1580–1585 (2012). [DOI] [PubMed] [Google Scholar]
- 33.He J., Wang Y., Missinato M. A., Onuoha E., Perkins L. A., Watkins S. C., St. Croix C. M., Tsang M., Bruchez M. P., A genetically targetable near-infrared photosensitizer. Nat. Methods 13, 263–268 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yuan H., Chong H., Wang B., Zhu C., Liu L., Yang Q., Lv F., Wang S., Chemical molecule-induced light-activated system for anticancer and antifungal activities. J. Am. Chem. Soc. 134, 13184–13187 (2012). [DOI] [PubMed] [Google Scholar]
- 35.Kim Y. R., Kim S., Choi J. W., Choi S. Y., Lee S.-H., Kim H., Hahn S. K., Koh G. Y., Yun S. H., Bioluminescence-activated deep-tissue photodynamic therapy of cancer. Theranostics 5, 805–817 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hsu C. Y., Chen C. W., Yu H. P., Lin Y. F., Lai P. S., Bioluminescence resonance energy transfer using luciferase-immobilized quantum dots for self-illuminated photodynamic therapy. Biomaterials 34, 1204–1212 (2013). [DOI] [PubMed] [Google Scholar]
- 37.Du J., Yu C., Pan D., Li J., Chen W., Yan M., Segura T., Lu Y., Quantum-dot-decorated robust transductable bioluminescent nanocapsules. J. Am. Chem. Soc. 132, 12780–12781 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yao H., Zhang Y., Xiao F., Xia Z., Rao J., Quantum dot/bioluminescence resonance energy transfer based highly sensitive detection of proteases. Angew. Chem. Int. Ed. 46, 4346–4349 (2007). [DOI] [PubMed] [Google Scholar]
- 39.So M.-K., Loening A. M., Gambhir S. S., Rao J., Creating self-illuminating quantum dot conjugates. Nat. Protoc. 1, 1160–1164 (2006). [DOI] [PubMed] [Google Scholar]
- 40.Tsoi K. M., Dai Q., Alman B. A., Chan W. C. W., Are quantum dots toxic? Exploring the discrepancy between cell culture and animal studies. Acc. Chem. Res. 46, 662–671 (2013). [DOI] [PubMed] [Google Scholar]
- 41.Kwon G., Naito M., Yokoyama M., Okano T., Sakurai Y., Kataoka K., Micelles based on Ab block copolymers of poly(ethylene oxide) and poly(beta-benzyl L-aspartate). Langmuir 9, 945–949 (1993). [Google Scholar]
- 42.Klebanoff S. J., Myeloperoxidase-halide-hydrogen peroxide antibacterial system. J. Bacteriol. 95, 2131–2138 (1968). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gross S., Gammon S. T., Moss B. L., Rauch D., Harding J., Heinecke J. W., Ratner L., Piwnica-Worms D., Bioluminescence imaging of myeloperoxidase activity in vivo. Nat. Med. 15, 455–461 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kolaczkowska E., Kubes P., Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 13, 159–175 (2013). [DOI] [PubMed] [Google Scholar]
- 45.Wang Z., Li J., Cho J., Malik A. B., Prevention of vascular inflammation by nanoparticle targeting of adherent neutrophils. Nat. Nanotechnol. 9, 204–210 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xue J., Zhao Z., Zhang L., Xue L., Shen S., Wen Y., Wei Z., Wang L., Kong L., Sun H., Ping Q., Mo R., Zhang C., Neutrophil-mediated anticancer drug delivery for suppression of postoperative malignant glioma recurrence. Nat. Nanotechnol. 12, 692–700 (2017). [DOI] [PubMed] [Google Scholar]
- 47.Schwab J. M., Chiang N., Arita M., Serhan C. N., Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature 447, 869–874 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nauseef W. M., Myeloperoxidase in human neutrophil host defence. Cell. Microbiol. 16, 1146–1155 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wirtz S., Popp V., Kindermann M., Gerlach K., Weigmann B., Fichtner-Feigl S., Neurath M. F., Chemically induced mouse models of acute and chronic intestinal inflammation. Nat. Protoc. 12, 1295–1309 (2017). [DOI] [PubMed] [Google Scholar]
- 50.Lamprecht A., Nanomedicines in gastroenterology and hepatology. Nat. Rev. Gastroenterol. Hepatol. 12, 195–204 (2015). [DOI] [PubMed] [Google Scholar]
- 51.Zhang Q., Tao H., Lin Y., Hu Y., An H., Zhang D., Feng S., Hu H., Wang R., Li X., Zhang J., A superoxide dismutase/catalase mimetic nanomedicine for targeted therapy of inflammatory bowel disease. Biomaterials 105, 206–221 (2016). [DOI] [PubMed] [Google Scholar]
- 52.Reuter S., Gupta S. C., Chaturvedi M. M., Aggarwal B. B., Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 49, 1603–1616 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lindig B. A., Rodgers M. A. J., Schaap A. P., Determination of the lifetime of singlet oxygen in water-d2 using 9,10-anthracenedipropionic acid, a water-soluble probe. J. Am. Chem. Soc. 102, 5590–5593 (1980). [Google Scholar]
- 54.Agostinis P., Berg K., Cengel K. A., Foster T. H., Girotti A. W., Gollnick S. O., Hahn S. M., Hamblin M. R., Juzeniene A., Kessel D., Korbelik M., Moan J., Mroz P., Nowis D., Piette J., Wilson B. C., Golab J., Photodynamic therapy of cancer: An update. CA Cancer J. Clin. 61, 250–281 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fang J., Nakamura H., Maeda H., The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv. Drug Deliv. Rev. 63, 136–151 (2011). [DOI] [PubMed] [Google Scholar]
- 56.Lobatto M. E., Fuster V., Fayad Z. A., Mulder W. J. M., Perspectives and opportunities for nanomedicine in the management of atherosclerosis. Nat. Rev. Drug Discov. 10, 835–852 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dou Y., Chen Y., Zhang X., Xu X., Chen Y., Guo J., Zhang D., Wang R., Li X., Zhang J., Non-proinflammatory and responsive nanoplatforms for targeted treatment of atherosclerosis. Biomaterials 143, 93–108 (2017). [DOI] [PubMed] [Google Scholar]
- 58.Katsuki S., Matoba T., Nakashiro S., Sato K., Koga J., Nakano K., Nakano Y., Egusa S., Sunagawa K., Egashira K., Nanoparticle-mediated delivery of pitavastatin inhibits atherosclerotic plaque destabilization/rupture in mice by regulating the recruitment of inflammatory monocytes. Circulation 129, 896–906 (2014). [DOI] [PubMed] [Google Scholar]
- 59.Freeman R., Liu X., Willner I., Chemiluminescent and chemiluminescence resonance energy transfer (CRET) detection of DNA, metal ions, and aptamer-substrate complexes using hemin/G-quadruplexes and CdSe/ZnS quantum dots. J. Am. Chem. Soc. 133, 11597–11604 (2011). [DOI] [PubMed] [Google Scholar]
- 60.Lee J. S., Joung H.-A., Kim M.-G., Park C. B., Graphene-based chemiluminescence resonance energy transfer for homogeneous immunoassay. ACS Nano 6, 2978–2983 (2012). [DOI] [PubMed] [Google Scholar]
- 61.Wang Q., Yin B. C., Ye B. C., A novel polydopamine-based chemiluminescence resonance energy transfer method for microrna detection coupling duplex-specific nuclease-aided target recycling strategy. Biosens. Bioelectron. 80, 366–372 (2016). [DOI] [PubMed] [Google Scholar]
- 62.Li K., Zhu Z., Cai P., Liu R., Tomczak N., Ding D., Liu J., Qin W., Zhao Z., Hu Y., Chen X., Tang B. Z., Liu B., Organic dots with aggregation-induced emission (AIE dots) characteristics for dual-color cell tracing. Chem. Mater. 25, 4181–4187 (2013). [Google Scholar]
- 63.Qin W., Ding D., Liu J., Yuan W. Z., Hu Y., Liu B., Tang B. Z., Biocompatible nanoparticles with aggregation-induced emission characteristics as far-red/near-infrared fluorescent bioprobes for in vitro and in vivo imaging applications. Adv. Funct. Mater. 22, 771–779 (2012). [Google Scholar]
- 64.Hong Y., Lam J. W. Y., Tang B. Z., Aggregation-induced emission. Chem. Soc. Rev. 40, 5361–5388 (2011). [DOI] [PubMed] [Google Scholar]
- 65.Ge W., Zhao K., Wang X., Li H., Yu M., He M., Xue X., Zhu Y., Zhang C., Cheng Y., Jiang S., Hu Y., iASPP is an antioxidative factor and drives cancer growth and drug resistance by competing with Nrf2 for Keap1 binding. Cancer Cell 32, 561–573.e6 (2017). [DOI] [PubMed] [Google Scholar]
- 66.Mao D., Wu W., Ji S., Chen C., Hu F., Kong D., Ding D., Liu B., Chemiluminescence-guided cancer therapy using a chemiexcited photosensitizer. Chemistry 3, 991–1007 (2017). [Google Scholar]
- 67.Umezawa N., Tanaka K., Urano Y., Kikuchi K., Higuchi T., Nagano T., Novel fluorescent probes for singlet oxygen. Angew. Chem. Int. Ed. 38, 2899–2901 (1999). [DOI] [PubMed] [Google Scholar]
- 68.Alex P., Zachos N. C., Nguyen T., Gonzales L., Chen T. E., Conklin L. S., Centola M., Li X., Distinct cytokine patterns identified from multiplex profiles of murine dss and tnbs-induced colitis. Inflamm. Bowel Dis. 15, 341–352 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/1/eaat2953/DC1
Supplementary Materials and Methods
Fig. S1. Synthesis and characterization of a CLP conjugate.
Fig. S2. Characterization of the CLP conjugate and its nanoparticles.
Fig. S3. Luminescence properties of the CLP conjugate under different conditions.
Fig. S4. Intracellular uptake of CLP nanoparticles and in vitro luminescence imaging in neutrophils.
Fig. S5. In vivo imaging in different mouse models of inflammation.
Fig. S6. The retention of CLP and luminol in the colon after enema administration in colitis mice.
Fig. S7. In vitro antitumor activity of CLP nanoparticles.
Fig. S8. Tumor therapy after intratumoral administration of CLP nanoparticles in mice bearing A549 xenografts.
Fig. S9. In vivo pharmacokinetic, tissue distribution, and elimination profiles of CLP after intravenous administration.
Fig. S10. Tissue distribution of CLP nanoparticles after intravenous administration in mice bearing A549 xenografts.
Fig. S11. Tumor therapy after intravenous administration of CLP nanoparticles in mice bearing A549 xenografts.
Fig. S12. Comparison of in vivo efficacy and safety of CLP nanoparticles with CDDP.
Fig. S13. Safety study after intratumoral administration of CLP nanoparticles in A549 xenograft–bearing mice.
Fig. S14. Safety profiles of CLP nanoparticles in A549 xenograft–bearing mice during and after 30 days of intravenous injection.
Fig. S15. Acute toxicity evaluation of CLP nanoparticles after a single intravenous injection in healthy mice.
Fig. S16. Mechanistic studies of in vitro antitumor activity of CLP nanoparticles in A549 cells.