

VSIG4 controls inflammation through MS4A6D-mediated transcriptional inhibition of Nlrp3 and Il-1β in macrophages.
Abstract
Hyperactivation of the NLRP3 inflammasome contributes to the pathogenesis of multiple diseases, but the mechanisms underlying transcriptional regulation of Nlrp3 remain elusive. We demonstrate here that macrophages lacking V-set and immunoglobulin domain–containing 4 (Vsig4) exhibit significant increases in Nlrp3 and Il-1β transcription, caspase-1 activation, pyroptosis, and interleukin-1β (IL-1β) secretion in response to NLRP3 inflammasome stimuli. VSIG4 interacts with MS4A6D in the formation of a surface signaling complex. VSIG4 occupancy triggers Ser232 and Ser235 phosphorylation in MS4A6D, leading to activation of JAK2-STAT3-A20 cascades that further results in nuclear factor κB suppression and Nlrp3 and Il-1β repression. Exaggerated NLRP3 and IL-1β expression in Vsig4−/− mice is accountable for deleterious disease severity in experimental autoimmune encephalomyelitis (EAE) and resistance to dextran sulfate sodium (DSS)–induced colitis. The agonistic VSIG4 antibodies (VG11), acting through NLRP3 and IL-1β suppression, show significant therapeutic efficacy in mouse EAE. These findings highlight VSIG4 as a prospective target for treating NLRP3-associated inflammatory disorders.
INTRODUCTION
The proinflammatory cytokines interleukin-1β (IL-1β) and IL-18, mainly derived from macrophages, play important roles in host defense against infection and inflammatory diseases. Biological activation of IL-1β/IL-18 typically requires cleavage of the inactive precursors (proIL-1β and proIL-18) by the intracellular cysteine-aspartic protease caspase-1, with its activity regulated by divergent multimeric protein platforms called inflammasomes (1). The best characterized inflammasome is the NLRP3 (nucleotide-binding domain, leucine-rich–containing family, pyrin domain–containing–3, or Nod-like receptor protein 3) inflammasome, a protein-scaffolding complex consisting NLRP3, caspase-1, and the adaptor molecule ASC (apoptosis-associated peck-like protein with CARD domain, Pycard). Dysregulation of the NLRP3 inflammasome contributes to the pathogenesis of various inflammatory syndromes, including multiple sclerosis (MS), cardiovascular disorders, neurodegenerative diseases, and metabolic disorders such as gout, obesity, and insulin resistance (2). Therefore, a fine balance must be maintained between activation and inhibition of the NLRP3 inflammasome to avoid overinflammatory damage in the host.
A two-signal model has been proposed for NLRP3 inflammasome activation, in which the priming signal comes from stimulation of pathogen recognition receptors (PRRs) that activate the nuclear factor κB (NF-κB) and mitogen-activated protein kinase (MAPK) pathways for up-regulating transcription of NLRP3 inflammasome components, and the second signal (activation) is induced by various triggers such as adenosine 5′-triphosphate (ATP), pore formation, potassium (K+) efflux, lysosomal destabilization/rupture, and mitochondrial reactive oxygen species (mtROS) (1). Upon activation, caspase-1 cleaves proIL-1β/proIL-18, generating bioactive IL-1β and IL-18 that promote inflammation or induce inflammatory cell death known as “pyroptosis” (3). The protein levels of NLRP3 inflammasome components are considered to be rate limiting for inflammasome activation, for which multiple regulatory mechanisms appear to exert inhibitory roles, such as autophagy, T cell activation, expressions of type I interferon, nitric oxide (NO), tripartite-motif protein 30 (TRIM30), pyrin-only protein-1 (POP1), and miR-223 (4). Among these regulators, several molecules can directly inhibit NLRP3 protein expression, thus attenuating inflammasome activation. For example, the type 2 plasminogen activator decreases NLRP3 expression via increasing autophagic degradation of NLRP3 (5). A20 [also called alterations of tumor necrosis factor–α–induced protein 3 (TNFAIP3)] limits NLRP3 inflammasome activation through restricting proIL-1β and NLRP3 ubiquitination (6). Most of the previously identified regulators seem to work at the posttranscriptional and posttranslational levels; however, transcriptional regulation of NLRP3 inflammasome components is elusive.
V-set and immunoglobulin domain–containing 4 (VSIG4) is a complement receptor of the immunoglobulin superfamily (CRIg) that specifically expresses in resting tissue-resident macrophages (7, 8). By binding complement component C3b or inactivated C3b (iC3b), VSIG4 mediates clearance of C3b/iC3b-opsonized pathogens including Listeria monocytogenes and Staphylococcus aureus (7). VSIG4 can also suppress T cell proliferation and promote the differentiation of Foxp3+ regulatory T cells (Treg) by binding an unidentified receptor on T cells (9). The VSIG4-Fc fusion protein seems to protect against the development of experimental autoimmune arthritis, uveoretinitis, and hepatitis (10). Nevertheless, whether VSIG4 is capable of controlling the transcription of NLRP3 inflammasome components remains undefined.
We show here that Vsig4−/− mice present exaggerated severity in myelin oligodendrocyte glycoprotein peptide (MOG35~55)–induced experimental autoimmune encephalomyelitis (EAE); nevertheless, these mice are resistant to dextran sulfate sodium (DSS)–induced colitis, in both cases resulting from increases in synthesis of NLRP3 and IL-1β in vivo. Whereas, inhibition of NLRP3 activity by CY09 (11), abrogating IL-1β activity by IL-1 receptor antagonist (IL-1Rα) (12), or deletion of IL-1 receptor 1 (Il-1R1) and Nlrp3 in Vsig4−/− mice all seem to exert opposite phenotypes. By using yeast split-ubiquitin screening system, we identified the member 6D of the membrane-spanning 4-domains subfamily A (MS4A6D) that interacts with VSIG4 to form a surface inhibitory signaling complex (SISC), which suppresses the expression of NLRP3 and IL-1β in macrophages during inflammatory responses. These findings reveal that VSIG4 plays an essential role in negative regulation of macrophage-driven intracellular inflammation.
RESULTS
Enhanced Nlrp3 and Il-1β gene transcription in Vsig4−/− macrophages
To investigate the potential role of VSIG4 in regulating NLRP3 inflammasome signaling, we isolated peritoneal exudate macrophages (PEMs) from Vsig4−/− mice and wild-type (WT) littermates. Quantitative reverse transcription polymerase chain reaction (qRT-PCR) revealed that Vsig4−/− PEMs showed significant elevation in mRNA encoding for Nlrp3 and Il-1β; in contrast, other transcripts of inflammasome components such as Caspase-1, ASC, and Il-18 were not significantly different from WT controls (Fig. 1A). Augmented protein levels of NLRP3 and proIL-1β in Vsig4−/− PEMs were also confirmed by Western blot (Fig. 1B). Lipopolysaccharide (LPS) triggered further enhancement of NLRP3 and proIL-1β protein expression in Vsig4−/− PEMs compared with WT controls (Fig. 1B). To avoid cellular heterogeneity of conventional PEMs, we next chose RAW264.7 cells, a macrophage line lacking Vsig4 expression, to examine functional specificity of VSIG4. Lentiviral restoration of Vsig4 (Len-Vsig4) expression in RAW264.7 cells revitalized reduction in both the basal and LPS-induced NLRP3 and proIL-1β expression as compared with the control (Fig. 1C).
Fig. 1. VSIG4 inhibits Nlrp3 and Il-1β transcription in vitro.
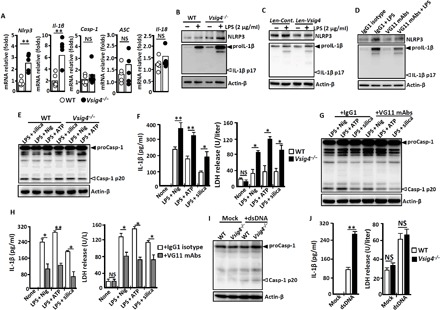
PEMs were isolated from Vsig4−/− mice and their C57BL/6 WT littermates. (A) qRT-PCR analysis of mRNA transcription encoding for NLRP3 inflammasome components (each circle represents an individual mouse). (B) PEMs were treatment without or with lipopolysaccharide (LPS; 2 μg/ml) for 6 hours, and cell extracts were immunoblotted for NLRP3 and IL-1β. (C) RAW264.7 cells were stably infected with Len-Cont. or Len-Vsig4 vectors, cells were further treated without or with LPS (2 μg/ml) for 6 hours, and cell extracts were immunoblotted for NLRP3 and IL-1β. (D) PEMs from WT mice were treated with the VG11 mAbs (50 μg/ml) or isotype IgG1 (50 μg/ml) for 6 hours, cells were further activated by LPS (2 μg/ml) for 6 hours, and cell extracts were immunoblotted for NLRP3 and IL-1β. PEMs were stimulated with LPS (2 μg/ml) for 3 hours and then treated with 1.5 μM ATP or 5 μM nigericin for 45 min, or added with SiO2 (500 μg/ml) for 4 hours. (E) Cell extracts were immunoblotted for caspase-1. (F) ELISA analysis of IL-1β and LDH release in cultured supernatants. PEMs were treated with NLRP3 inflammasome stimuli in the presence of isotype IgG1 mAbs (50 μg/ml) or VG11 mAbs (50 μg/ml). (G) Cell extracts were immunoblotted for caspase-1. (H) ELISA analysis of IL-1β and LHD release in cultured supernatants. PEMs were transfected with dsDNA after 12 hours. (I) Cell extracts were immunoblotted for caspase-1. (J) ELISA analysis of IL-1β and LDH release in cultured supernatants. Error bars indicate SEM. NS, not significant. *P < 0.05 and **P < 0.01 (Student’s t test). Nig, nigericin; Casp-1, caspase-1. Data represent one out of three biological replicates, with three technical replicates each at least.
The complement protein C3b is the natural ligand of VSIG4 (7). Enzyme-linked immunosorbent assays (ELISA) showed that LPS promotes C3 protein expression in mouse PEMs and RAW264.7 cells (fig. S1A), suggesting that C3b, a normal cleavage product of C3 under these conditions, might interact with VSIG4 in macrophages. To test this hypothesis, we stimulated the human monocyte cell line THP-1 with phorbol 12-myristate 13-acetate (PMA) to induce VSIG4 expression, and Western blotting confirmed that human C3b stimulation causes decrease in LPS-induced NLRP3 and proIL-1β levels (fig. S1B). For further validation, we developed a panel of monoclonal antibodies (mAbs) against mouse VSIG4 and identified one specific clone (#VG11) capable of recognizing the VSIG4 protein (fig. S2A). This cloned mAb can also repress LPS-induced proIL-1β and NLRP3 expression in WT PEMs (Fig. 1D), whereas proIL-1β and NLRP3 proteins from Vsig4−/− PEMs were unresponsive to the mAb treatment (fig. S2B). These combined data suggest that VSIG4 initiates inhibitory signals to specifically attenuate Nlrp3 and Il-1β expression in macrophages.
The protein levels of NLRP3 inflammasome components are considered as a rate-limiting element for controlling inflammasome activation. We next examined whether VSIG4 affects caspase-1 activity during NLRP3 inflammasome activation. Notably, substantial increases in caspase-1 activity (Casp-1 p20) and IL-1β accumulation in the cultured medium were observed in LPS-primed Vsig4−/− PEMs treated with ATP, nigericin, or crystalline (silica) as compared with WT controls (Fig. 1, E and F). However, none of these stimulations alone cause measurable changes of caspase-1 activation and IL-1β secretion in the Vsig4−/− PEMs (fig. S3). Besides proIL-1β and proIL-18 cleavage, the dysregulation of caspase-1 activity can also cause pyroptosis, an inflammatory form of lytic cell death (3). As anticipated, NLRP3 inflammasome stimuli significantly enhanced induction of caspase-1–dependent pyroptosis in Vsig4−/− PEMs in comparison with WT controls (Fig. 1F). Conversely, administration of VG11 mAbs suppresses caspase-1 activity (Casp-1 p20) in WT PEMs (Fig. 1G), resulting in decreases of IL-1β secretion and lactate dehydrogenase (LDH) release in response to NLRP3 inflammasome stimuli (Fig. 1H). These data indicate that VSIG4 negatively regulates NLRP3 inflammasome activation.
We also stimulated PEMs with cytosolic double-stranded DNA (dsDNA) for AIM2 inflammasome activation (1), with results showing that bioprocessing caspase-1 (Fig. 1I) and activation of pyroptosis were comparably enhanced between WT and Vsig4−/− PEMs (Fig. 1J), although the secretion of IL-1β in response to dsDNA treatments appeared to be consistently higher in Vsig4−/− PEMs as compared with their WT counterparts (Fig. 1J), indicating plausible increase in IL-1β mRNA expression in Vsig4−/− PEMs (Fig. 1A). These combined data suggest enhanced caspase-1 activation, pyroptosis, and IL-1β secretion from Vsig4−/− macrophages treated by NLRP3 inflammasome stimuli.
VSIG4 inhibits Nlrp3 and Il-1β transcription via A20-mediated NF-κB inactivation
The observation of VSIG4 prominently inhibiting Nlrp3 and IL-1β transcription in macrophages led us to investigate the underlying signaling pathways. A20, a protein capable of down-modulating LPS-induced Nlrp3 transcription through inhibiting NF-κB activation (13), came up as a candidate for transcriptional control of inflammatory genes. Western blot showed lower basal A20 expression in Vsig4−/− PEMs and in liver tissues from Vsig4−/− mice (Fig. 2A). Conversely, forcing Vsig4 overexpression in RAW264.7 cells appears to enhance A20 protein in vitro (Fig. 2B). Silencing A20 resulted in augmentation of LPS-induced NF-κB p65 phosphorylation (p-p65) and therefore up-regulation of NLRP3 and IL-1β protein expression (Fig. 2C). Furthermore, inhibition of NF-κB activity by BAY 11-7082 (5 μM) also decreased LPS-primed NLRP3 and proIL-1β expression in PEMs (Fig. 2D). These data suggest that VSIG4 transfers a feedback signal to suppress NF-κB activation through augmenting A20 activity, thus down-modulating Nlrp3 and Il-1β expression.
Fig. 2. VSIG4 inhibits the transcription of Nlrp3 and Il-1β via activating the AKT-STAT3-A20 axis.
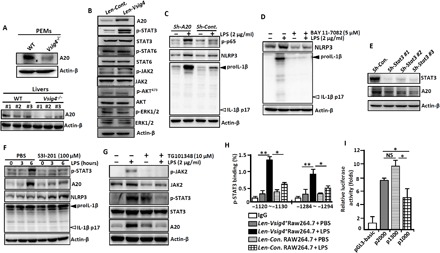
PEMs and liver tissues were isolated from Vsig4−/− mice and their C57BL/6 WT littermates. (A) Western blot analysis of A20 (each number represents an individual mouse). RAW264.7 cells were transfected with Len-Cont. or Len-Vsig4. (B) Cell extracts were immunoblotted for the indicated protein. (C) A20 in RAW264.7 cells was silenced by shRNA, cells were further treated with LPS (2 μg/ml) for an additional 3 hours, and cell extracts were immunoblotted for the indicated protein. (D) PEMs were treated with LPS (2 μg/ml) in the presence of NF-κB inhibitor BAY 11-7082 (5 μM), and cell extracts were immunoblotted for the indicated molecules after 6 hours. (E) The transcription of Stat3 in RAW264.7 cells was silenced by shRNA, and cell extracts were immunoblotted for A20 and STAT3. LPS-primed RAW264.7 cells were treated with (F) the STAT3 inhibitor S3I-201 (100 mM) and (G) the JAK2 inhibitor TG101348 (10 μM), and cell extracts were immunoblotted for the indicated molecules after 12 hours. (H) ChIP-qPCR analysis of the enrichment of p-STAT3 binding to the potential binding sites in the A20 promoter region in Vsig4+RAW264.7 cells after 3 hours of LPS (2 μg/ml) administration. (I) Analysis of luciferase activity of A20 gene promoter constructs by luciferase reporter assay. Error bars indicate SEM. NS, not significant. *P < 0.05 and **P < 0.01 (Student’s t test). Data represent one out of three biological replicates, with three technical replicates each at least.
Searching transcription factor binding sites in the promoter region of the A20 gene using PROMO (http://alggen.lsi.upc.es) reveals multiple motifs for the signal transducers and activators of transcription (STATs) transcription factors. Western blot showed that overexpression of Vsig4 in RAW264.7 cells elicits phosphorylation of STAT3 (p-STAT3) and their upstream Janus kinase 2 (p-JAK2) without affecting the expression of p-STAT6/STAT6, p-AKT/AKT, p-ERK1/2 (phospho–extracellular signal–regulated kinase 1/2), or ERK1/2 (Fig. 2B). Silencing Stat3 in Vsig4+RAW264.7 cells also causes reduction in A20 expression (Fig. 2E). In accordance, the STAT3 inhibitor S3I-201 is capable of down-regulating LPS-induced A20 in Vsig4+RAW264.7 cells and, thereby, enhancing the expression of NLRP3 and proIL-1β (Fig. 2F). Furthermore, the JAK2 inhibitor TG101348 (10 μM) is also capable of down-regulating LPS-induced STAT3 phosphorylation, thus inhibiting A20 expression (Fig. 2G), suggesting that VSIG4 triggers the activated JAK2-STAT3 signaling to augment A20 expression.
Chromatin immunoprecipitation (ChIP)–qPCR assays further demonstrated that VSIG4 mediates direct binding of p-STAT3 to the identified sites in the promoter region of A20 upon LPS stimulation (Fig. 2H). To validate that these putative binding sites of p-STAT3 are critical for controlling A20 expression, a 2000–base pair (bp) (−2000~+1) fragment of the A20 promoter (p2000) and its truncated fragments were cloned into a luciferase reporter vector pGL3-Basic for measurement of promotor activity. By transfecting RAW264.7 cells, we observed that LPS-induced luciferase reporter activity was significantly reduced in p1000 (−1000~+1), the fragmented promoter region lacking the predicted p-STAT3 binding sites, as compared with the full-length p2000 or p1300, the two segments that still contain the p-STAT3 binding sites (Fig. 2I). These data demonstrate that VSIG4 activates the JAK2-STAT3-A20 signaling pathway to inhibit Nlrp3 and IL-1β transcription in macrophages.
VSIG4 interacts with MS4A6D to activate JAK2-STAT3-A20 signaling
To further explore the immediate signaling events of VSIG4 that lead to JAK2-STAT3-A20 activation, we applied a yeast split-ubiquitin screening system (14) using mouse Vsig4 C-terminal ubiquitin fusion (pBT3-SUC-Vsig4) as the bait against a complementary DNA (cDNA) library constructed from mouse PEMs (fig. S4A). Five candidate genes were identified to interact with Vsig4 in a bait-dependent fashion (fig. S4B). Transient transfection experiments confirmed that VSIG4 cross-links with MS4A6D (fig. S4C), a four-pass membrane protein of the Ms4a superfamily that might interact with other surface molecules in the formation of signaling complexes (15).
Immunofluorescence double staining demonstrated that MS4A6D colocalizes with VSIG4 on macrophage membrane (Fig. 3A). Western blot analysis showed that endogenous VSIG4 and MS4A6D also interact in PEMs, and binding VSIG4 by VG11 mAbs appears to further enhance VSIG4/MS4A6D interactions (Fig. 3B). Transient overexpression experiments in RAW264.7 cells confirmed that VSIG4 coimmunoprecipitates with MS4A6D, and further protein truncation assessments showed that amino acid residues between 1 and 46 at the N terminus of MS4A6D are crucial for mediating the interactions (Fig. 3C). Together, our data demonstrate that VSIG4 interacts with MS4A6D.
Fig. 3. VSIG4 cross-links with MS4A6D to activate the JAK2-STAT3-A20 signaling.
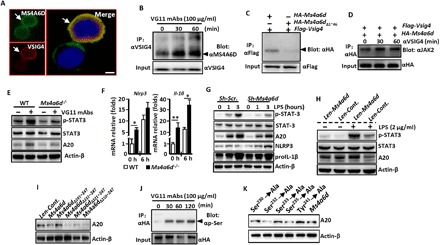
PEMs from C57BL/6 WT mice were isolated. (A) Immunofluoresence double-staining analysis of the coexpression of VSIG4 and MS4A6D. Arrows indicate positive cells, and blue indicates nuclear 4′,6-diamidino-2-phenylindole (DAPI) staining. Scale bar, 20 μm. (B) PEMs were treated with the VG11 mAbs (100 μg/ml) at indicated times, and cell lysates were used for IP with anti-VSIG4 and blotting with anti-MS4A6D. (C) Ms4a6d–HA (hemagglutinin) and Vsig4-Flag were coexpressed in RAW264.7 cells, cell lysates were collected, and VSIG4/MS4A6D interactions were analyzed by Co-IP experiments. (D) Ms4a6d-HA and Vsig4-Flag were cotransfected in RAW264.7 cells, cells were further triggered by VG11 mAbs (100 μg/ml), and MS4A6D and JAK2 interactions were analyzed by Co-IP experiments. (E) PEMs were isolated and further treated with VG11 mAbs (100 μg/ml) for 12 hours, and cell extracts were immunoblotted for p-STAT3/STAT3 and A20. (F) PEMs were isolated from Ms4a6d−/− and WT mice, cells were further treated with LPS (2 μg/ml) for 0 or 3 hours, the transcription of Nlrp3 and Il-1β was analyzed by qRT-PCR. (G) Ms4a6d in Vsig4+RAW264.7 cells was silenced by shRNA, and cell extracts were immunoblotted for the indicated molecules. (H) Overexpression of Ms4a6d in Vsig4+RAW264.7 cells, cells were further treated with LPS (2 μg/ml) for 6 h, and cell extracts were immunoblotted for p-STAT3/STAT3 and A20. (I) Cell extracts were immunoblotted for A20 from Ms4a6d−/− PEMs that were transfected with different Ms4a6d deletion constructs. (J) Vsig4+RAW264.7 cells were treated with VG11 mAbs (100 μg/ml), MS4A6D protein was pulled down by IP experiments, and Ser phosphorylation (p-Ser) was analyzed by Western blot. (K) Ms4a6d−/− PEMs were transfected with different Ms4a6d mutations, and cell extracts were immunoblotted for A20. Error bars indicate SEM. *P < 0.05 and **P < 0.01 (Student’s t test). Data represent one out of three biological replicates, with three technical replicates each at least.
Coimmunoprecipitation (Co-IP) shows that MS4A6D directly binds JAK2, and treatment with VG11 mAbs further enhances MS4A6D/JAK2 interaction in Vsig4+RAW264.7 cells (Fig. 3D), suggesting that MS4A6D directly activates JAK2. Furthermore, the VG11 mAbs can promote STAT3 phosphorylation and augment A20 expression in PEMs from WT mice, whereas PEMs from Ms4a6d−/− animals have opposite effects in response to VG11 mAbs (Fig. 3E). Ms4a6d−/− PEMs manifested significantly higher levels of basal and LPS-induced Nlrp3 and Il-1β transcription than their WT counterparts (Fig. 3F). Similarly, silencing Ms4a6d expression in Vsig4+RAW264.7 cells by short hairpin RNA (shRNA) resulted in suppression of LPS-induced p-STAT3 activation and A20 up-regulation, which, in turn, appear to cause up-regulation of NLRP3 and proIL-1β (Fig. 3G). Conversely, lentiviral overexpressing Ms4a6d in PEMs from WT mice promotes LPS-induced p-STAT3 activation and A20 up-regulation (Fig. 3H). To identify the functional motifs responsible for mediating A20 up-regulation, we created a panel of truncation variants for Ms4a6d and transiently forced their expression in PEMs that were isolated from Ms4a6d−/− mice. Western blot showed that the MS4A6D C-terminal 231 to 241 amino acid residues are critical for mediating A20 expression (Fig. 3I). Co-IP experiments illustrated that VG11 mAbs trigger Ser phosphorylation (p-Ser) of MS4A6D (Fig. 3J). We therefore created a panel of point mutations and observed that Ser232 and Ser235, but not Ser230, Ser233, or Tyr241, substitutions in Ms4a6d-reconstituted Ms4a6d −/− PEMs are essential for VSIG4-mediated A20 induction in macrophages (Fig. 3K). These combined data indicate that MS4A6D is the proximal membrane adaptor protein interacting with VSIG4 to further recruit and mediate signals through the JAK2-STAT3-A20 axis in quenching Nlrp3 and Il-1β transcription.
Vsig4 deficiency deteriorates the severity of EAE in mice
Dysregulation of the NLRP3 inflammasome and overload of IL-1β secretion have been shown to deteriorate the severity of inflammatory neurodegenerative EAE in vivo (16). We therefore tested the effect of VSIG4 deficiency on a mouse EAE model induced through immunization with neuromyelin antigen MOG35~55 peptides and found that Vsig4 deficiency causes acceleration of disease onset and severity as compared with WT controls (Fig. 4A). Histopathological analyses showed that at peak of the disease, the spinal cords from Vsig4−/− mice manifested significantly more infiltrating leukocytes (Fig. 4B). Flow cytometry further confirmed that the numbers of inflammatory leukocytes (including F4/80+ macrophages, Ly6Ghigh neutrophils, and Ly6Chigh monocytes) were significantly increased in the Vsig4−/− spinal cords (Fig. 4, C and D, and fig. S5A). T helper 17 (TH17) and TH1 cells have been described to deteriorate the pathogenesis of EAE through directly promoting demyelination (17). In addition, markedly higher percentages of IL-17A+TH17 cells and interferon-γ (IFN-γ)+TH1 cells were seen in the spinal cords and spleen tissues of Vsig4−/− EAE mice (Fig. 4, E to G, and fig. S5B). These combined data suggest that Vsig4 deficiency accelerates EAE progression.
Fig. 4. Vsig4 deficiency promotes EAE deterioration in mice.
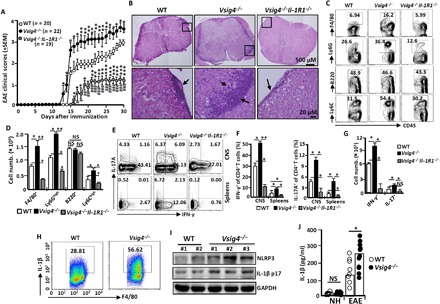
The indicated mice were induced to develop EAE by MOG35~55 peptides and complete Freund's adjuvant (CFA) as described in Materials and Methods. (A) The EAE clinical scores were assessed and compared daily. Data of four independent experiments with similar results were merged. The spinal cords were harvested. (B) Representative morphological sections were stained by HE). Magnifications, ×5 (top) and ×20 (bottom). Arrows indicate the infiltrating leukocytes (n = 10 to 15 per group). The infiltrating cells from spinal cords were isolated and purified. (C) Representative flow cytometry plots for immune cell infiltration with antibodies to the indicated molecules. (D) Absolute numbers of infiltrating cells in the spinal cords were compared (n = 10 to 15 per group). (E) Representative flow cytometry plots for IL-17A– and IFN-γ–secreting CD4+ T cells within the spinal cords (CNS) and spleens. (F) The percentage of IL-17A+CD4+T and IFN-γ+CD4+T cells within the CNS and spleens were compared (n = 10 to 15 per group). (G) Absolute numbers of infiltrating cells IL-17A+CD4+T and IFN-γ+CD4+T cells within spinal cords were compared (n = 5 per group). (H) Representative flow cytometry plots for IL-1β secretion from F4/80+ macrophages within spinal cords. (I) Western blot analysis of NLRP3 and IL-1β levels in spinal cords (each number represents an individual mouse). (J) ELISA analysis of IL-1β in serum (each circle represents an individual mouse). All data shown in (B) to (J) were detected at peak of disease. NH, normal health. Error bars indicate SEM. NS, not significant. *P < 0.05, **P < 0.01, and ***P < 0.001 (Student’s t test). GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
At the peak of EAE disease, macrophages from Vsig4−/− spinal cords appear to express higher IL-1β than the EAE of WT animals (Fig. 4H), concurring with further observations of significantly more NLRP3 and IL-1β protein accumulation in the spinal cords (Fig. 4I) and in the serum (Fig. 4J) of the Vsig4−/− EAE mice. This indicates that exaggerated elevation of IL-1β in Vsig4−/− mice is likely responsible for the deteriorated EAE. In further exploration, we generated the Vsig4−/−Il-1R1−/− double gene knockout (DKO) mice by crossing Vsig4−/− mice with Il-1R1−/− mice. Deletion of Il-1R1 in Vsig4−/− mice dramatically delayed EAE onset with the mice showing ameliorated clinical scores (Fig. 4A). The spinal cords of Vsig4−/−Il-1R1−/− DKO EAE mice manifested only slightly tissue damage (Fig. 4B), along with extremely low level of leukocyte infiltration (Fig. 4, C and D) at the peak of the disease. Additionally, we observed a reduction in IL-17A+TH17 cells and IFN-γ+TH1 cells in the central nervous system (CNS) from these DKO EAE mice as compared with WT and Vsig4−/− littermate controls (Fig. 4, E to G). In addition, treating Vsig4−/− mice under EAE induction with the IL-1Rα, a naturally occurring inhibitory protein that blocks IL-1β biologic activity during inflammation, appeared to markedly ameliorate the severity of EAE (fig. S6A). We also generated the Vsig4−/−Nlrp3−/− DKO mice by crossing Vsig4−/− mice with Nlrp3−/− mice. As expected, these mice also presented with markedly attenuated EAE severity as compared with Vsig4−/− animals (fig. S6B). These combined results demonstrate that hyperactivation of NLRP3 and elevated IL-1β in Vsig4−/− mice are detrimental for EAE pathogenesis.
Vsig4−/− mice are resistant to DSS-induced colitis
Hyperactivation of the NLRP3 inflammasome has also been proposed to exert protective functions in some circumstances. For example, NLRP3 inflammasome activation appears to inhibit DSS-induced histopathological presentations in the gut since mice deficient in NLRP3 inflammasome components have shown more lethality in DSS-induced colitis (18). To examine this question, we exposed Vsig4−/− mice to 3.5% DSS in drinking water ad libitum for six continuous days. Unexpectedly, Vsig4−/− mice appeared to have a significantly higher survival rate after DSS exposure compared to that of WT littermates (Fig. 5A). Furthermore, the DSS-exposed Vsig4−/− mice exhibited considerably less weight loss, significantly lower disease activity index, and dramatically longer colon length presentation compared with the controls (Fig. 5, B and C). Experiments with oral administration of fluorescein isothiocyanate (FITC)–dextran showed that the diffusion of FITC-dextran across the epithelium was significantly lower in DSS-treated Vsig4−/− mice (Fig. 5D). Hematoxylin and eosin (H&E) staining further showed that DSS induced massive inflammatory infiltration, mucosal edema, and hyperplasia in colonic tissues of WT mice, in contrast to the minimal necrosis observed in Vsig4−/− mice. Semiquantitative scoring of these histological parameters revealed that DSS-induced colitis severity in Vsig4−/− mice was significantly less than in that in WT mice (Fig. 5E). Similar results were also observed on Vsig4−/− mice treated with 2.5% DSS (fig. S7), suggesting that Vsig4−/− mice are resistant to DSS-induced inflammatory colitis.
Fig. 5. Vsig4−/− mice are resistant to DSS-induced colitis.
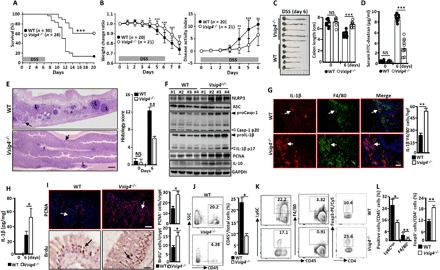
Mice were given 3.5% DSS in their drinking water for 6 days to induce colitis. (A) The survival rate was assessed and compared daily for a total of 20 days. Data of five independent experiments with similar results were merged. (B) Comparative analysis of body weight and disease activity index. Data of four independent experiments with similar results were merged. Mice were euthanized at day 6 of DSS feeding. (C) The colon lengths were calculated and compared. (D) Mice were gavaged with FITC-dextran; these mice were euthanized after 3 hours, and sera were collected to detect the FITC-dextran amounts (each circle represents an individual mouse). (E) H&E staining analysis of histopathological changes (left) and semiquantitative scoring of histopathology (right). The asterisk indicates severe edema/inflammation, the arrows indicate the normal epithelium, and L indicates large lymphoid nodules. Scale bar, 300 μm (n = 15 per group). (F) Colonic homogenates were immunoblotted for the indicated molecules (each number represents an individual mouse). (G) In situ immunofluorescence analysis of IL-1β expression in F4/80+ macrophages. Scale bar, 20 μm. Arrows indicate positive cells, and blue indicates nuclear DAPI staining. (H) ELISA analysis of IL-1β in colonic homogenate. (I) In situ immunofluorescence analysis of PCNA expression and immunohistochemistry analysis of BrdU incorporation. Arrows indicate positive cells, and blue indicates DAPI. Scale bar, 20 μm. (J) Representative flow cytometry plots for CD45+ leukocytes infiltrating colonic tissues (left), and the percentages of CD45+ cells/total cells were compared (n = 10 per group). (K) Representative flow cytometry plots for CD45+F4/80+ macrophages, CD45+Ly6C+ monocytes, and Foxp3+CD4+T cell infiltrating colonic tissues. (L) The percentages of indicated cells within colonic tissues were compared (n = 10 per group). Error bars indicate SEM. *P < 0.05, **P < 0.01, and ***P < 0.001. (A) was analyzed by a log-rank test, and others were compared by Student’s t test. Data presented in (E) to (L) were analyzed at day 6 of DSS treatment.
Colonic homogenates from DSS-treated Vsig4−/− mice revealed increases in NLRP3, Casp-1p10, and IL-1β protein compared with the WT controls (Fig. 5F). In situ immunofluorescence staining also illustrated more proIL-1β production from infiltrated macrophages in the Vsig4−/− colons (Fig. 5G). In addition, ELISA also demonstrated a significant increase in local colonic IL-1β production in Vsig4−/− mice as compared with WT controls at this time point (Fig. 5H). To clarify whether the protective phenotype directly relies on the increase in NLRP3 activation and IL-1β production, we treated Vsig4−/− mice with the soluble NLRP3 inhibitor CY09 (50 μg per mouse) or IL-1Rα (200 ng per mouse) during the course of DSS exposure, finding that interruption of IL-1β signaling or inhibition of NLRP3 activity resulted in significant increases in weight loss and more severe disease scores (fig. S6, C and D). Similarly, DSS-treated Vsig4−/−IL-1R1−/− DKO mice seemed to suffer from more severe colitis than the WT and Vsig4−/− single KO mice (fig. S8). These data suggest that the resistance of Vsig4−/− mice to DSS-mediated colitis is associated with increase in NLRP3 activity and NLRP3-mediated IL-1β production in vivo.
Previous work has suggested that the NLRP3 inflammasome protects DSS-induced colonic inflammatory damage by inducing a compensatory proliferative response of epithelial cells in the gut (18). Both Western blot (Fig. 5F) and in situ immunofluorescence (Fig. 5I) showed that DSS-treated Vsig4−/− mice have significant increases in the proliferation marker proliferating cell nuclear antigen (PCNA) expression in the affected colon tissue and 5-bromo-2′-deoxyuridine (BrdU) incorporation within the colonic crypts. A better preservation of the gut epithelial integrity from DSS-treated Vsig4−/− mice appears to correlate with less infiltration of CD45+ leukocytes (Fig. 5J), especially the CD45+Ly6Chigh monocytes and F4/80+ macrophages in the affected colonic tissues (Fig. 5, K and L). Flow cytometry showed that the colonic tissues from DSS-treated Vsig4−/− mice manifested a higher percentage of CD4+Foxp3+Treg than WT controls (Fig. 5, K and L), and this was associated with higher levels of IL-10 accumulation, as demonstrated by Western blot (Fig. 5F). These data suggest that resistance of Vsig4−/− mice to DSS-induced colitis relies on increased IL-1β, through which it likely induces gut epithelial proliferation/regeneration and colonic Treg infiltration that enhances colonic IL-10 accumulation.
VSIG4 agonist decelerates EAE progression
The aforementioned data indicate that VSIG4 mediates suppressive signals that lead to transcriptional inhibition of Nlrp3 and Il-1β in macrophages, highlighting a plausible target for therapeutic intervention of NLRP3-mediated inflammatory disorders. To test the possibility, C57BL/6 WT mice were intraperitoneally injected with VG11 mAbs (100 μg per mouse per week) or isotype IgG1 mAbs (100 μg per mouse per week) during EAE induction. Antibody stimulation of surface VSIG4 caused significant reduction in disease severity in EAE mice (Fig. 6A). Western blot analysis confirmed that VG11 mAb treatment induces JAK2 and STAT3 phosphorylation, which, in turn, results in enhancement of A20 expression in the spinal cords that leads to inhibition of NLRP3 and IL-1β expression in vivo. This effect is consistent with the reduction in astrocytic cell proliferation as determined by marker glial fibrillary acidic protein expression in the EAE spinal cords (Fig. 6B). The PEMs isolated from VG11 mAbs–treated EAE mice also manifested significant reduction in IL-1β production (Fig. 6C). At the same time, using VG11 mAbs also significantly reduced serum IL-1β accumulation (Fig. 6C), along with decreased local infiltration of proinflammatory cells including CD45+Ly6Ghigh neutrophils and CD45+Ly6Chigh monocytes (Fig. 6D), the IL-17A+TH17 cells, and IFN-γ+TH1 cells (Fig. 6, E and F) in the EAE spinal cords. Together, these data suggest that enhancing VSIG4 signaling might have therapeutic potentials for NLRP3-associated inflammatory disorders.
Fig. 6. Agonist VSIG4 improves the progress of EAE.
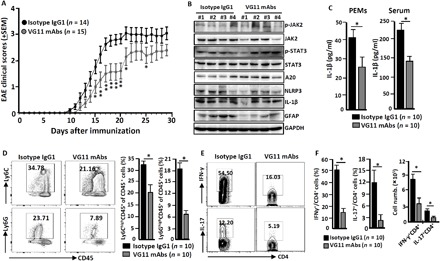
The C57BL/6 WT mice were induced to develop EAE by MOG35~55 peptides and CFA as described in Materials and Methods; these mice were also treated with VG11 mAbs (100 μg per mouse per week) or mouse isotype IgG1 antibodies (100 μg per mouse per week). (A) The EAE clinical scores were assessed and compared daily. Data of three independent experiments with similar results were merged. At peak of disease, mice were euthanized. (B) The homogenate of spinal cords was immunoblotted for the indicated molecules (each number represents an individual mouse). (C) The PEMs and sera were collected from EAE mice. ELISA analysis of IL-1β in culture supernatants of PEMs and sera (n = 10 per group). The infiltrating cells from spinal cords were isolated and purified. (D) Representative flow cytometry plots for immune cell infiltration with antibodies to the indicated proteins (left), and the percentages of infiltrating cells were compared (right). (E) Representative flow cytometry plots for IL-17A– and IFN-γ–secreting CD4+ T cells within spinal cords. (F) The percentage and number of these cells were calculated and compared (right). n = 10 per group. Error bars indicate SEM. *P < 0.05, **P < 0.01 (Student’s t test).
DISCUSSION
The activation of the NLRP3 inflammasome is tightly controlled at the transcriptional and posttranslational levels to prevent unwanted excessive inflammation in the host. Therefore, identification and characterization of unknown players that are involved in the regulation of the NLRP3 inflammasome might provide insights for the development of target therapeutics against NLRP3 inflammasome–associated diseases. We demonstrate here that VSIG4, a B7-associated protein that is specifically expressed in resting macrophages, mediates inhibitory signals to suppress Nlrp3 and Il-1β transcription upon its occupancy with the agonist VG11 mAbs or C3b ligand. We also found that MS4A6D interacts with VSIG4 to form a surface signaling complex, which activates JAK2-STAT3-A20 cascades to inhibit NF-κB activation and repress Nlrp3 and Il-1β transcription. As a common consequence of lacking the negative regulation loop that leads to uncontrolled IL-1β production during inflammatory responses, Vsig4−/− mice become more susceptible to development of EAE yet appear to be protected from DSS-mediated colitis in vivo. Notably, stimulation of VSIG4 by agonistic VG11 mAbs can effectively mitigate the progression of EAE via inhibition of NLRP3 and IL-1β. These combined data suggest that modulating VSIG4 signals has therapeutic potentials for treating NLRP3-associated inflammatory disorders (fig. S9).
The protein levels of NLRP3 inflammasome components are considered to be critical for controlling inflammasome activation. Factors that participate in the posttranslational negative regulation of NLRP3 protein expression and the NLRP3 inflammasome complex assembly have been studied extensively. However, the transcriptional inhibition of NLRP3 inflammasome components remains poorly understood. To determine the mechanisms underlying VSIG4-mediated transcriptional inhibition of Nlrp3 and Il-1β genes in macrophages, we examined the activation status of NF-κB and the ERK/MAPK pathways in PEMs, finding that upon VG11 mAb or C3b occupancy, VSIG4 triggers JAK2-STAT3-A20 axis signaling to inhibit Nlrp3 and Il-1β transcription through NF-κB inhibition (Figs. 2 and 3). By means of the yeast split-ubiquitin screening system and Co-IP experiments, we demonstrated that MS4A6D, as a membrane signaling adapter, can directly interact with VSIG4 and deliver signals for JAK2-STAT3-A20 activation (Fig. 3). MS4A6D is a member of a family that includes 18 highly homologous MS4A genes in mouse chromosome 19 that is syntenic for human chromosome 11. The expression of MS4A6D has been reported in tolerizing dendritic cells (15). Functionally, MS4A protein interacts with other surface molecules for the formation of signaling complexes that sense, create, and propagate biological signals that regulate cell activation, proliferation, and differentiation (15). For example, interaction of MS4A4B with glucocorticoid-induced tumor necrosis factor receptor family-related gene (GITR, also known as TNFRSF18) leads to the augmentation of IL-2 production from T cells in response to stimulations by GITR ligands or anti-GITR mAbs (19). We here also found that silencing Ms4a6d in Vsig4+RAW264.7 cells leads to reduction in A20 and consequential enhancement of NLRP3 and proIL-1β expression in vitro (Fig. 3E), although an unexpected low survival rate of Ms4a6d−/− mouse breeding due to salpingitis appears to hinder the direct evidence for its phenocopying Vsig4−/− animals in EAE and DSS-mediated colitis. On the other hand, a recent study suggests that unsaturated fatty acids, such as oleic acid and arachidonic acid, appear to be the ligands for MS4A6D (20). Whether MS4A6D can directly affect the transcription of Nlrp3 and inflammasome substrates through these ligand interactions remains unclear, and the related research is in progress in our laboratory.
A20, expressed in various types of cells, is a well-known negative regulator of NF-κB activation. It can be induced by proinflammatory cytokines and some microbial products that trigger PRR, and polymorphisms in the A20 gene locus have been identified as risk alleles for multiple human autoimmune diseases (21). A20 has been shown to attenuate NLRP3 inflammasome activity, and microglia A20-deficient mice show exacerbated CNS inflammation due to hyperactivation of the NLRP3 inflammasome that leads to enhanced IL-1β secretion and CNS inflammation (22). Moreover, a recent study showed that macrophages lacking A20 contributes to the pathology of rheumatoid arthritis due to an increase in transcriptional expressions of Nlrp3 and proIL-1β (23), and subsequent studies revealed that proIL-1β and NLRP3 proteins regulated by A20 are dependent on NF-κB– and K63-linked polyubiquitin signaling (6). To explore the molecular mechanism responsible for VSIG4-mediated down-regulation of Nlrp3 and Il-1β transcription, we focused on the expression of A20 in macrophages and showed an inverse correlation between A20 abundance and NLRP3/IL-1β expression. Further studies demonstrated that VSIG4 augments A20 expression via activating the MS4A6D-JAK2-STAT3 signaling pathway, suggesting that this signaling axis could potentially serve as the biomarker and treatment target in controlling the pathogenesis of NLRP3-mediated disorders. It is noteworthy that an obvious decrease in basal expression of A20 in macrophages and liver tissues does not seem to translate into any spontaneous autoinflammatory diseases in Vsig4−/− mice (Fig. 2A). One possibility is that the remaining A20, even at very low levels, is still capable of controlling NF-κB activity in macrophages under laboratory mouse keeping conditions. The second possibility is that higher levels of IL-1β result in augmented IL-10 production from macrophages and other cells that might counteract inflammatory pathogenesis (24, 25), thus preventing the development of spontaneous autoinflammatory disorders in Vsig4−/− mice.
MS is an inflammatory demyelinating disease of the CNS that affects around 2.5 million people worldwide; the dysregulation of NLRP3 and overloading IL-1β have been proposed to play critical roles in the disease pathology (2). EAE is the most commonly used animal model for studying MS, and during the initiation stage, the antigen-reactive IFN-γ+TH1 cells, IL-17A+TH17 cells, and proinflammatory cells within the CNS seem to be encephalitogenic, relying on the presence of IL-1β (26). We demonstrate here that Vsig4−/− mice manifest exaggerated up-regulation of NLRP3 and IL-1β secretion from macrophages in the CNS of EAE, which is in association with disease deterioration attributing to increases of infiltrating proinflammatory F4/80+ macrophages, Ly6Ghigh neutrophils, Ly6Chigh monocytes, IFN-γ+TH1 cells, and IL-17+TH17 cells in the affected CNS and the peripheral lymphoid tissues (Fig. 6). It has been reported that Il-1R1−/− mice exhibit attenuated induction and progression of EAE (27). We therefore generated Vsig4−/−Il-1R1−/− DKO mice to investigate the involvement of VSIG4 in signaling through the IL-1β/IL-1R1 axis for regulation of EAE inflammation. We observed that Vsig4−/−Il-1R1−/− DKO mice manifested reduced disease severity and, in accordance, less CNS infiltration of proinflammatory cells (Fig. 4). Similarly, other animal models including Vsig4−/−Nlrp3−/− DKO mice, Vsig4−/− animals treated with IL-1Rα in vivo, or WT mice treated with the VSIG4 agonistic antibody VG11 all showed attenuation for EAE progression. These combined data suggest that insufficiency of the VSIG4 eliminates a critical negative signaling pathway in controlling NLRP3 inflammasome activation/escalation, thus predisposing the host vulnerable to pathogenic stimulations causing inflammatory damages.
DSS-mediated colitis in animals has been used in the study of inflammatory bowel diseases (IBDs) such as ulcerative colitis and Crohn’s disease in humans. Several reports have established an association between NLRP3 inflammasome activity in the gut and IBDs. A genetic study has identified single-nucleotide polymorphisms associated with reduced NLRP3 expression and IBDs (28). Nevertheless, the roles of the NLRP3 inflammasome in the pathogenesis of murine IBD models are controversial. One study showed that Nlrp3−/− mice were resistant to DSS-induced colitis, presumably due to down-modulation of local inflammation, as evidenced by observation of lower levels of bioactive IL-1β and other proinflammatory cytokines in the homogenates of affected colon tissues (29). In contrast, however, other groups have found that NLRP3 inflammasome activation has a protective effect on DSS-induced colitis (18, 30). The controversy extends to the findings in Caspase-1–, Il-1R1–, and Il-18–deficient mice, for which both caspase-1−/− mice (31) and WT mice treated with the caspase-1 inhibitor pralnacasan (32) show ameliorated colonic inflammation, whereas another study showed that caspase-1−/− mice have aggravated colonic inflammation (33). Moreover, Il-18−/− or Il-18R−/− mice developed more severe colitis than WT controls (34), whereas blocking IL-18 activity by IL-18 binding protein (IL-18bp) attenuates intestinal damage in DSS-induced colitis (35). In addition, Caspase-1−/− mice treated with recombinant IL-18 protein manifested less severity of DSS-mediated colitis when compared with those receiving phosphate-buffered saline (PBS) (18). IL-1β has been proposed to promote epithelial proliferation in the guts following Citrobacter rodentium infection (36), which is supported by evidence showing that Il-1R1−/− mice are more susceptible to DSS-induced colitis owing to reduction in IL-10 production (24). It is not clear whether the subtle differences in these experiments were the cause of the controversial results; however, our current study shows that attaining resistance to DSS-induced colitis is associated with augmentation of NLRP3 inflammasome activation and overburden of IL-1β in Vsig4−/− mice. Accordingly, we also observed that increases in IL-1β/IL-1R1–dependent Treg differentiation, IL-10 production, and epithelial cell proliferation may collectively contribute to protection from DSS-induced colitis (37). These combined data indicate that the resistance of Vsig4−/− mice to DSS-induced colitis relies on the increase in NLRP3 activation and the overproduction IL-1β.
In summary, we have demonstrated that antibody stimulation of VSIG4 induces recruitment of the adaptor protein MS4A6D in the formation of SISC, which activates the cascade JAK2-STAT3-A20 signaling pathway, leading to NF-κB inhibition. Thus, VSIG4 suppresses the expression of NLRP3 and IL-1β in macrophages during the inflammatory response in vitro and in vivo. Together with our previous work showing that forced expression of Vsig4 in WT mice ameliorates MHV-3–induced viral fulminant hepatitis (38), we speculate that targeted intervention of VSIG4 and its associated signaling pathway may generate beneficial therapeutics for various inflammatory disorders.
MATERIALS AND METHODS
Mice
The Vsig4-deficient (Vsig4−/−) mice were provided by M. van Lookeren Campagne (Department of Immunology, Genentech, CA, USA). Nlrp3−/− (#017970), Il-1R1−/− (#003245), and C57BL/6 WT were purchased from the Jackson laboratory (Bar Harbor, Maine, USA). The Ms4a6d−/− mice were generated by Cyagen Biosciences (Guangzhou, China) using the CRISPR/Cas9 technique (fig. S10). Vsig4−/− mice were crossed with Nlrp3−/− and Il-1R1−/− mice to develop Vsig4−/−Nlrp3−/− and Vsig4−/−Il-1R1−/− DKO mice. All mice were backcrossed 10 times onto the B6 background to avoid unpredictable confounders. Mice were maintained in microisolator cages, fed with standard laboratory chow diet and water, and housed in the animal colony at the animal center of the Third Military Medical University (TMMU). All animals received humane care according to the criteria outlined in the Guide for the Care and Use of Laboratory Animals prepared by the National Academy of Sciences and published by the National Institutes of Health (NIH publication 86-23, revised 1985). All of the experiments comply with the animal study protocol approved by the Laboratory Animal Welfare and Ethics Committee of the TMMU (no. SYXK-PLA-20120031).
Antibodies
The rabbit anti-mouse MS4A6D polyclonal antibodies were developed by AbMax Biotechnology Co. Ltd. (Beijing, China). The anti-mouse VSIG4 mAbs (clone VG11) were produced by AbMart Biotechnology Co. Ltd. (Shanghai, China). Other antibodies were commercial and were from indicated companies to be mentioned in succeeding sections.
Cells
The mouse macrophage cell line RAW264.7, human monocyte cell line THP-1, and 293T cells were provided by the Cell Institute of the Chinese Academy of Sciences (Shanghai, China). PEMs were harvested and cultured as described previously (38). To induce NLRP3 activation, PEMs were first stimulated by LPS (2 μg/ml; #L2630, Sigma-Aldrich Co., CA, USA) for 3 hours. Subsequently, cells were further treated for 45 min with 1.5 mM ATP (#987-65-5, Amresco) and 5 μM nigericin (#N7143, Sigma-Aldrich) or for 4 hours with 10 μM silica (#S5130, Sigma-Aldrich), respectively. The AIM2 inflammasomes were activated by transfection with 100 μM dsDNA (2 μg/ml; #P0883, Sigma-Aldrich) plus 0.1% (v/v) Lipofectamine 2000 (Life Technologies) for 18 hours. In the mAb treatment experiments, both RAW264.7 cells and PEMs were treated with VG11 mAbs (50 μg/ml) or mouse IgG1 mAbs (50 μg/ml) for 12 hours. In the inhibitor treatment experiments, macrophages were treated with LPS (2 μg/ml) in the presence of the NF-κB inhibitor BAY 11-7082 (5 μM), the STAT3 inhibitor S3I-201 (50 mM), or the JAK2 inhibitor AG490 (50 μM), respectively. Cells were lysed to detect the expression of caspase-1, NLRP3, and IL-1β by Western blot after 6 hours or otherwise as indicated in figure legends. Supernatants were harvested to analyze the release of IL-1β and LDH by ELISA.
Induction and assessment of MOG35~55 peptides caused EAE
The 13-week-old female mice were immunized subcutaneously with 150 μg of MOG35~55 peptides (Cambridge BioSciences, Cambridge, UK) emulsified in CFA containing heat-killed MTB (0.4 mg per mouse, Chondrex, Redmond, WA, USA). Mice were injected intraperitoneally with 500 ng of pertussis toxin (Kaketsuken, Kumamoto, Japan) on days 0 and 2 (39). Mice were observed daily for clinical signs of disease and euthanized at the peak of disease. Disease severity was recorded as follows: 0, normal; 1, limp tail; 2, wobbly gait; 3, hind limb weakness; and 4, hind limb paralysis. In IL-1Rα treatment experiments, Vsig4−/− mice were subcutaneously injected with IL-1Rα (#200-01RA, 200 ng per mouse per week, PeproTech) or saline solution (vehicle) starting 1 day before MOG35~55 peptide administration. In VG11 mAb therapeutic experiments, C57BL/6 WT female mice were induced to develop EAE by MOG35~55 peptides; mice also received VG11 mAbs (100 μg per mouse per week) or mouse isotype IgG1 antibodies (100 μg per mouse per week) by intraperitoneal injection. At day 30, these mice were euthanized and serum was collected. PEMs were also isolated from these EAE mice and seeded into six-well plates at a density of 2 × 105 cells per well. Cell culture supernatants were collected to detect the secretion of IL-1β after 12 hours. For histopathological analyses, the spinal cords were fixed in 4% formaldehyde, decalcified in EDTA, embedded in paraffin, sectioned, and stained with H&E. In some experiments, mononuclear cells were isolated from the spinal cords and spleen tissues of EAE mice following perfusion with PBS by Percoll density centrifugation, and cell infiltration was detected by flow cytometry.
Induction of DSS-mediated colitis
Acute colitis was induced with 2.5% or 3.5% (w/v) DSS (Mw = 36,000 to 50,000 Da; MP Biomedicals, Santa Ana, CA, USA) dissolved in sterile, distilled water ad libitum for experimental days 1 to 6 followed by normal drinking water until the end of the experiment. The DSS solutions were made fresh every 2 days. Mice were monitored daily for body weight loss and signs of intestinal inflammation. Scoring for stool consistency and occult blood was done as described previously (40). Stool consistency scores were determined as follows: 0, well-formed pellets; 1, semiformed stools that did not adhere to the anus; 2, semiformed stools that adhered to the anus; and 3, liquid stools that adhered to the anus. Bleeding scores were determined as follows: 0, no blood by using hemoccult; 1, positive hemoccult; 2, blood traces in stool visible; and 3, gross rectal bleeding. Stool consistency scores and bleeding scores were added and presented as the clinical score. In some experiments, mice were treated twice daily with CY09 (50 μg per mouse, provided by R. Zhou, The CAS Key Laboratory of Innate Immunity and Chronic Disease, School of Life Sciences, University of Science and Technology of China), IL-1Rα (200 ng per mouse), or saline solution (vehicle) via subcutaneous injection every 3 days (starting 1 day before DSS exposure), respectively. To detect cell proliferation, 3.5% DSS–treated mice received BrdU (100 mg/kg; #B5002, Sigma-Aldrich) via intraperitoneal injection at day 6, and these mice were euthanized and the colonic tissues were collected after 3 hour. To analyze epithelial barrier permeability in vivo, food and water were withdrawn at day 6 of DSS treatment, and these mice were then gavaged with FITC-dextran (60 mg/100 g; Mw = 4000; Sigma-Aldrich). Blood was collected after 3 hours, and FITC-dextran amount in the serum was measured with a fluorescence spectrophotometer setup with emission and excitation wavelengths of 490 and 520 nm, respectively. In some experiments, strips of colon were mechanically crushed, vortexed in 200 ml of tissue protein extraction reagent (Pierce, Rockford, USA) for 1 min, and shock frozen in liquid nitrogen. The homogenate was centrifuged at 12,000g at 4°C for 20 min, and the amount of IL-1β in the colon homogenate was quantified by ELISA. At the same time, the colon tissues were fixed in 4% formaldehyde, decalcified in EDTA, embedded in paraffin, sectioned, and stained with H&E or used for immunohistochemistry and immunofluorescence staining as described later.
Immunohistochemistry and immunofluorescence double staining
Paraffin-embedded tissue blocks were cut into 2.5-μm slices and were mounted on polylysine-charged glass slides. Endogenous peroxidase activity was blocked by exposure to 3.0% H2O2 for 30 min. Antigen retrieval was performed in a citrate buffer (pH 6.0) at 120°C for 10 min. Sections were then incubated at 4°C overnight with anti-Brdu mAb (#B8434, 1:500 per mouse, Sigma-Aldrich). After washing, the sections were incubated with the corresponding secondary antibodies for 2 hours at room temperature. The VECTASTAIN ABC Kit (Vector Laboratories, San Diego, CA, USA) was used to perform the avidin-biotin complex method according to the manufacturer’s instructions. Sections incubated with isotype- and concentration-matched immunoglobulins without primary antibodies were used as isotype controls. Peroxidase activity was visualized with the DAB Elite Kit (K3465, DAKO), and brown coloration of tissues represented positive staining. A similar protocol was used to detect the expression of PCNA in colons.
To detect MS4A6D and VSIG4 colocalization, PEMs were fixed with 4% paraformaldehyde, permeabilized with 0.1% saponin in PBS for 5 min, and blocked with PBS containing 2% bovine serum albumin for 1 hour at 4°C. The cells were then stained with rabbit anti-MS4A6D antibodies and mouse anti-VSIG4 overnight at 4°C and then stained with Alexa Fluor 488–conjugated donkey anti-rabbit IgG (H + L) highly cross-adsorbed secondary antibody (#A-21206, Thermo Fisher Scientific, Billerica, MA, USA) as well as Alexa Fluor 555–conjugated goat anti-mouse IgG (H + L) highly cross-adsorbed secondary antibody (#A-21422, Thermo Fisher Scientific) for 1 hour. A similar protocol was used to detect the colocalization of IL-1β and F4/80 in colons. The results were analyzed using fluorescence microscopy (Zeiss Axioplan 2).
Split-ubiquitin screening with macrophage libraries
The macrophage cDNA libraries in the prey vector pPR3-N used for yeast two-hybrid screening were constructed according to a split-ubiquitin system (Dualsystems Biotech). The Vsig4 encoding sequence were obtained by PCR with specific primers with Sfi I restriction site at respective 5′ terminals and cloned into bait vector pBT3-SUC. The bait constructs were transformed into the yeast reporter strain NMY51 via standard procedures. For screening, the library plasmids were introduced into the yeast cells harboring bait constructs. After 3 days, transformants were grown on selective medium lacking leucine, tryptophan, histidine, and adenine, with addition of 20 mM 3-AT. Library plasmids were isolated from positive clones and retransformed into NMY51 to test bait dependency. Only preys that activated the histidine and adenine reporters in the presence of VSIG4 and not pBT3-SUC were considered true interactors.
Quantitative reverse transcription polymerase chain reaction
Total RNA was extracted from cultured cells or the indicated tissues with TRIzol reagent according to the manufacturer’s instructions (Thermo Fisher Scientific Co., CA, USA). First-strand cDNA was synthesized with the PrimeScript RT-PCR Kit (Takara, Dalian, China). The expression of mRNA encoding for the indicated genes was quantified by qRT-PCR with the SYBR Premix ExTaq Kit (Takara) and was normalized to the expression of β-actin. qRT-PCR was performed with specific primers (table S1). The results were compared by the 2−ΔΔCt method.
shRNA silencing
Lentiviral constructs with shRNAs were directed against murine Ms4a6d, A20, and Stat3 in the pGV112 vectors. The primers are shown in table S1. Lentivirus was prepared by transient transfection of 293T cells with transfer vectors along with third-generation packaging constructs (pHelper 1.0 and pHelper 2.0). The viral titers were determined with serial dilution of virus-containing media on NIH3T3 cells. RAW264.7 cells or PEMs were transfected with unconcentrated virus supernatant overnight in the presence of polybrene (8 mg/ml) and selected in puromycin (0.5 mg/ml).
ChIP and qPCR
Bone marrow–derived macrophages (BMDMs) underwent cross-linking for 10 min with 1% formaldehyde in medium. Chromatin fragments were prepared, followed by immunoprecipitation with anti–p-STAT3 (#ab5073, Abcam, Cambridge, MA, USA) or rabbit IgG isotype mAbs, and coupled to Dynabeads Protein G (#10003D, Thermo Fisher Scientific, CA, USA). DNA was phenol/chloroform extracted, and ethanol was precipitated in the presence of glycogen. DNA was purified with a PCR purification kit (#28104, Qiagen, CA, USA). qPCR was performed with special primers flanking the putative A20-binding sites; the primers are shown in table S1. The input DNA was an aliquot of sheared chromatin before immunoprecipitation and was used for normalization of the samples to the amount of chromatin added to each ChIP.
A20 gene promoter luciferase reporter assay
A 2000-bp (−2000/+1) and shortcut fragment of the A20 gene promoter was cloned into the pGL3-basic expression vector (herein called p2000, p1300, and p1000) using primers described in table S1. RAW264.7 cells were cultured in 12-well plates and were transfected with these plasmids using an Amaxa Nucleofector (4D-Nucleofector, Lonza, Allendale, NJ, USA). At 48 hours after transfection, cells were further treated with LPS (500 ng/ml) for an additional 12 hours. Thereafter, cells were washed with PBS and lysed in a reporter lysis buffer (Invitrogen). Luciferase reporter activities were measured in triplicate using the Dual-Luciferase Reporter Assay System (Promega, Madison, WI, USA) according to the manufacturer’s protocol and quantified using the GloMax 96-well plate luminometer (Promega). The firefly luciferase–to–Renilla luciferase ratios were determined and were defined as the relative luciferase activity. Results are shown as the mean ± SEM of a representative experiment performed in triplicate. To examine the transfection efficiency, RAW264.7 cells were transfected with a pmaxGFP control vector, and transfection efficiency was assessed by in situ green fluorescent protein (GFP) expression according to the manufacturer’s protocol (Lonza).
Lentiviral constructs and transduction
The whole gene expression cDNA for Vsig4 and Ms4a6d, the truncation mutants of Ms4a6d, and Ms4a6d site-directed mutagenesis were amplified with specific PCR primers (table S1). cDNA was cloned into the pCDH-MCS-T2A-copGFP-MSCV (CD523A-1) vector. This vector was mutated using the QuickChange Site-Directed Mutagenesis Kit II (Stratagene, Santa Clara, CA, USA). The lentiviral packaging vectors psPAX2 and pVSVG were purchased from Addgene (Cambridge, MA, USA). The psPAX2 plasmids (2 μg), the expression vectors (2 μg) and the pVSVG plasmids (2 μg) were cotransfected into 293T cells, and the virus supernatants were collected after 48 hours (2000 rpm/min, 3 min). RAW264.7 cells were transduced with unconcentrated virus supernatant overnight in the presence of polybrene (8 mg/ml) and selected in puromycin (0.5 mg/ml).
Flow cytometry
The leukocytes from the spinal cords, spleen, and colonic tissues were collected from the indicated mice. The death cells were excluded first by staining with the LIVE/DEATH Fixable Near-IR Dead Cell Stain Kit (Life Technologies, Eugene, Oregon, USA). For intracellular cytokine staining of IFN-γ, TNF-α, and IL-17 in CD4+T cells and IL-1β from F4/80+ macrophages, cells were stimulated for up to 10 hours with PMA and ionomycin, and brefeldin A was added at the last 4 hours. Cells were fixed and permeabilized using Cytofix/Cytoperm (BD Biosciences) and incubated with fluorescent antibodies (anti–IL-1β, anti–IFN-γ, anti–TNF-α, and anti–IL-17; BD, Becton Drive, NJ, USA) in the dark at 4°C for 30 min. To measure the expression of phenotype markers on the cell surface, suspended cells were incubated for 1 hour at room temperature in the dark using fluorescent antibodies (anti-CD45, anti-Ly6G, anti-Ly6C, anti-F4/80, and anti-B220; eBioscience, San Diego, CA, USA). A total of 10,000 live cells were analyzed by a FACsAria cytometer (BD, Franklin Lakes, NJ, USA). All the flow cytometry data were analyzed using the CellQuest Pro software, and the gate strategies are shown in fig. S5.
ELISA
The IL-1β (#EK0394) ELISA kits were from Boster Ltd. (Wuhan, China). The murine C3 ELISA kit (#ab157711) was purchased from Abcam. The LDH ELISA kit (#A020-2) was from Nanjing Jiancheng Bioengineering Institute (Nanjing, Jiangsu province, China). The concentration of cytokines and LDH was measured by the ELISA kits according to the manufacturers’ instructions.
Western blotting
The expression of GAPDH (#2118, CST, Danvers, MA, USA), actin-β (#3700, CST), Akt (#2920, CST), p-Akt (p-Thy308, #4056; p-Ser473, #4051, CST), STAT3 (#9139, CST), p-STAT3 (p-Tyr705, #9145, CST), Stat6 (#5397, CST), p-Stat6 (p-Tyr641, #56554, CST), p-p65 (#3033, CST), Jak1 (#3344, CST), p-Jak1 (#3331, CST), Jak2 (#3230, CST), p-Jak2 (#3771, CST), ASC (#sc-376916, Santa Cruz Biotechnology), IL-18 (#ab71495, Abcam), IL-1β (#ab9722, Abcam), caspase-1 (AG-20B-0042, Adipogen, San Diego, CA, USA), and NLRP3 (#AG-20B-0014-C100, Adipogen) in macrophages, liver tissues, and spinal cords were measured by Western blot, as described previously (38).
Statistical analysis
Survival data from the in vivo experiments were analyzed by a log-rank test performed on curves generated by GraphPad Prism 7.0 (Software MacKiev). For all other analysis, two-tailed, unpaired Student’s t tests with a 95% confidence interval performed on graphs generated in GraphPad Prism were used. Error bars represent the SEM. A value of P < 0.05 was considered as statistically significant. All results shown are representative of at least three separate experiments.
Supplementary Material
Acknowledgments
We thank R. Zhou (The CAS Key Laboratory of Innate Immunity and Chronic Disease, School of Life Sciences, University of Science and Technology of China) for the NLRP3 inhibitor CY09. Funding: This work was supported by the National Key Research and Development Program of China (2016YFA0502204) and the National Natural Science Foundation of China (NSFC; nos. 91442203, 81361120400, 91442203, 81471862, and 8177169). Author contributions: Y.C. and Y.W. designed the research and analyzed the data; X.H., J.L., and S.G. performed most of the experiments; Y.J. and G.G. participated in the split-ubiquitin screening; L.F., Q.X., and Z.F. conducted the plasmid mutation; C.Y. did the flow cytometry analysis; L.Z. and Y.C. developed the project and prepared the manuscript. Conflict of interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/1/eaau7426/DC1
Fig. S1. Liganding VSIG4 by complement C3b results from decreasing NLRP3 and proIL-1β expression in macrophages.
Fig. S2. The expression of NLRP3 and IL-1β in Vsig4−/− PEMs was not affected by VG11 mAbs.
Fig. S3. Vsig4 deficiency does not cause spontaneous NLRP3 inflammasome activation and IL-1β secretion.
Fig. S4. MS4A6D interacts with VSIG4 by using a yeast split-ubiquitin screen.
Fig. S5. Gating strategies of flow cytometry.
Fig. S6 Inhibition of NLRP3 activity or blocking IL-1β signaling controls the progress of EAE and colitis.
Fig. S7. Vsig4−/− mice are resistant to 2.5% DSS-induced colitis.
Fig. S8. Vsig4−/−Il-1R1−/− mice deteriorated DSS-induced colonic damage.
Fig. S9. Model of VSIG4/MS4A6D signaling complex attenuates NLRP3 inflammasome activation.
Fig. S10. Creating Ms4a6d−/− mice by CRISPR/Cas9-mediated genome engineering.
Table S1. The PCR primers used in this study.
REFERENCES AND NOTES
- 1.Rathinam V. A. K., Fitzgerald K. A., Inflammasome complexes: Emerging mechanisms and effector functions. Cell 165, 792–800 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lamkanfi M., Dixit V. M., Inflammasomes and their roles in health and disease. Annu. Rev. Cell Dev. Biol. 28, 137–161 (2012). [DOI] [PubMed] [Google Scholar]
- 3.Miao E. A., Rajan J. V., Aderem A., Caspase-1-induced pyroptotic cell death. Immunol. Rev. 243, 206–214 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang J., Liu Z., Xiao T. S., Post-translational regulation of inflammasomes. Cell. Mol. Immunol. 14, 65–79 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chuang S. Y., Yang C. H., Chou C. C., Chiang Y. P., Chuang T. H., Hsu L. C., TLR-induced PAI-2 expression suppresses IL-1β processing via increasing autophagy and NLRP3 degradation. Proc. Natl. Acad. Sci. U.S.A. 110, 16079–16084 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Duong B. H., Onizawa M., Oses-Prieto J. A., Advincula R., Burlingame A., Malynn B. A., Ma A., A20 restricts ubiquitination of pro-interleukin-1β protein complexes and suppresses NLRP3 inflammasome activity. Immunity 42, 55–67 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Helmy K. Y., Katschke K. J. Jr., Gorgani N. N., Kljavin N. M., Elliott J. M., Diehl L., Scales S. J., Ghilardi N., van Lookeren Campagne M., CRIg: A macrophage complement receptor required for phagocytosis of circulating pathogens. Cell 124, 915–927 (2006). [DOI] [PubMed] [Google Scholar]
- 8.Vogt L., Schmitz N., Kurrer M. O., Bauer M., Hinton H. I., Behnke S., Gatto D., Sebbel P., Beerli R. R., Sonderegger I., Kopf M., Saudan P., Bachmann M. F., VSIG4, a B7 family-related protein, is a negative regulator of T cell activation. J. Clin. Invest. 116, 2817–2826 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yuan X., Yang B. H., Dong Y., Yamamura A., Fu W., CRIg, a tissue-resident macrophage specific immune checkpoint molecule, promotes immunological tolerance in NOD mice, via a dual role in effector and regulatory T cells. eLife 6, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.He J. Q., Wiesmann C., van Lookeren Campagne M., A role of macrophage complement receptor CRIg in immune clearance and inflammation. Mol. Immunol. 45, 4041–4047 (2008). [DOI] [PubMed] [Google Scholar]
- 11.Jiang H., He H., Chen Y., Huang W., Cheng J., Ye J., Wang A., Tao J., Wang C., Liu Q., Jin T., Jiang W., Deng X., Zhou R., Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J. Exp. Med. 214, 3219–3238 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lane T., Lachmann H. J., The emerging role of interleukin-1β in autoinflammatory diseases. Curr. Allergy Asthma Rep. 11, 361–368 (2011). [DOI] [PubMed] [Google Scholar]
- 13.Ma A., Malynn B. A., A20: Linking a complex regulator of ubiquitylation to immunity and human disease. Nat. Rev. Immunol. 12, 774–785 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johnsson N., Varshavsky A., Split ubiquitin as a sensor of protein interactions in vivo. Proc. Natl. Acad. Sci. U.S.A. 91, 10340–10344 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eon Kuek L., Leffler M., Mackay G. A., Hulett M. D., The MS4A family: Counting past 1, 2 and 3. Immunol Cell Biol 94, 11–23 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Inoue M., Williams K. L., Gunn M. D., Shinohara M. L., NLRP3 inflammasome induces chemotactic immune cell migration to the CNS in experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. U.S.A. 109, 10480–10485 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stromnes I. M., Cerretti L. M., Liggitt D., Harris R. A., Goverman J. M., Differential regulation of central nervous system autoimmunity by TH1 and TH17 cells. Nat. Med. 14, 337–342 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zaki M. H., Boyd K. L., Vogel P., Kastan M. B., Lamkanfi M., Kanneganti T. D., The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity 32, 379–391 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Howie D., Nolan K. F., Daley S., Butterfield E., Adams E., Garcia-Rueda H., Thompson C., Saunders N. J., Cobbold S. P., Tone Y., Tone M., Waldmann H., MS4A4B is a GITR-associated membrane adapter, expressed by regulatory T cells, which modulates T cell activation. J. Immunol. 183, 4197–4204 (2009). [DOI] [PubMed] [Google Scholar]
- 20.Greer P. L., Bear D. M., Lassance J. M., Bloom M. L., Tsukahara T., Pashkovski S. L., Masuda F. K., Nowlan A. C., Kirchner R., Hoekstra H. E., Datta S. R., A family of non-GPCR chemosensors defines an alternative logic for mammalian olfaction. Cell 165, 1734–1748 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boone D. L., Turer E. E., Lee E. G., Ahmad R. C., Wheeler M. T., Tsui C., Hurley P., Chien M., Chai S., Hitotsumatsu O., McNally E., Pickart C., Ma A., The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat. Immunol. 5, 1052–1060 (2004). [DOI] [PubMed] [Google Scholar]
- 22.Voet S., Mc Guire C., Hagemeyer N., Martens A., Schroeder A., Wieghofer P., Daems C., Staszewski O., Vande Walle L., Jordao M. J. C., Sze M., Vikkula H. K., Demeestere D., van Imschoot G., Scott C. L., Hoste E., Gonçalves A., Guilliams M., Lippens S., Libert C., Vandenbroucke R. E., Kim K. W., Jung S., Callaerts-Vegh Z., Callaerts P., de Wit J., Lamkanfi M., Prinz M., van Loo G., A20 critically controls microglia activation and inhibits inflammasome-dependent neuroinflammation. Nat. Commun. 9, 2036 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walle L. V., Van Opdenbosch N., Jacques P., Fossoul A., Verheugen E., Vogel P., Beyaert R., Elewaut D., Kanneganti T.-D., van Loo G., Lamkanfi M., Negative regulation of the NLRP3 inflammasome by A20 protects against arthritis. Nature 512, 69–73 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.González-Navajas J. M., Law J., Nguyen K. P., Bhargava M., Corr M. P., Varki N., Eckmann L., Hoffman H. M., Lee J., Raz E., Interleukin 1 receptor signaling regulates DUBA expression and facilitates Toll-like receptor 9–driven antiinflammatory cytokine production. J. Exp. Med. 207, 2799–2807 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shouval D. S., Biswas A., Kang Y. H., Griffith A. E., Konnikova L., Mascanfroni I. D., Redhu N. S., Frei S. M., Field M., Doty A. L., Goldsmith J. D., Bhan A. K., Loizides A., Weiss B., Yerushalmi B., Yanagi T., Lui X., Quintana F. J., Muise A. M., Klein C., Horwitz B. H., Glover S. C., Bousvaros A., Snapper S. B., Interleukin 1β mediates intestinal inflammation in mice and patients with interleukin 10 receptor deficiency. Gastroenterology 151, 1100–1104 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin C. C., Edelson B. T., New insights into the role of IL-1β in experimental autoimmune encephalomyelitis and multiple sclerosis. J. Immunol. 198, 4553–4560 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schiffenbauer J., Streit W. J., Butfiloski E., LaBow M., Edwards C. III, Moldawer L. L., The induction of EAE is only partially dependent on TNF receptor signaling but requires the IL-1 type I receptor. Clin. Immunol. 95, 117–123 (2000). [DOI] [PubMed] [Google Scholar]
- 28.Villani A. C., Lemire M., Fortin G., Louis E., Silverberg M. S., Collette C., Baba N., Libioulle C., Belaiche J., Bitton A., Gaudet D., Cohen A., Langelier D., Fortin P. R., Wither J. E., Sarfati M., Rutgeerts P., Rioux J. D., Vermeire S., Hudson T. J., Franchimont D., Common variants in the NLRP3 region contribute to Crohn’s disease susceptibility. Nat. Genet. 41, 71–76 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bauer C., Duewell P., Mayer C., Lehr H. A., Fitzgerald K. A., Dauer M., Tschopp J., Endres S., Latz E., Schnurr M., Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome. Gut 59, 1192–1199 (2010). [DOI] [PubMed] [Google Scholar]
- 30.Zaki M. H., Lamkanfi M., Kanneganti T. D., The Nlrp3 inflammasome: Contributions to intestinal homeostasis. Trends Immunol. 32, 171–179 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Siegmund B., Lehr H. A., Fantuzzi G., Dinarello C. A., IL-1β-converting enzyme (caspase-1) in intestinal inflammation. Proc. Natl. Acad. Sci. U.S.A. 98, 13249–13254 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Loher F., Bauer C., Landauer N., Schmall K., Siegmund B., Lehr H. A., Dauer M., Schoenharting M., Endres S., Eigler A., The interleukin-1β-converting enzyme inhibitor pralnacasan reduces dextran sulfate sodium-induced murine colitis and T helper 1 T-cell activation. J. Pharmacol. Exp. Ther. 308, 583–590 (2004). [DOI] [PubMed] [Google Scholar]
- 33.Elinav E., Strowig T., Kau A. L., Henao-Mejia J., Thaiss C. A., Booth C. J., Peaper D. R., Bertin J., Eisenbarth S. C., Gordon J. I., Flavell R. A., NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 145, 745–757 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Takagi H., Kanai T., Okazawa A., Kishi Y., Sato T., Takaishi H., Inoue N., Ogata H., Iwao Y., Hoshino K., Takeda K., Akira S., Watanabe M., Ishii H., Hibi T., Contrasting action of IL-12 and IL-18 in the development of dextran sodium sulphate colitis in mice. Scand. J. Gastroenterol. 38, 837–844 (2003). [DOI] [PubMed] [Google Scholar]
- 35.Sivakumar P. V., Westrich G. M., Kanaly S., Garka K., Born T. L., Derry J. M., Viney J. L., Interleukin 18 is a primary mediator of the inflammation associated with dextran sulphate sodium induced colitis: Blocking interleukin 18 attenuates intestinal damage. Gut 50, 812–820 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lebeis S. L., Powell K. R., Merlin D., Sherman M. A., Kalman D., Interleukin-1 receptor signaling protects mice from lethal intestinal damage caused by the attaching and effacing pathogen Citrobacter rodentium. Infect. Immun. 77, 604–614 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chaudhry A., Samstein R. M., Treuting P., Liang Y., Pils M. C., Heinrich J. M., Jack R. S., Wunderlich F. T., Brüning J. C., Müller W., Rudensky A. Y., Interleukin-10 signaling in regulatory T cells is required for suppression of Th17 cell-mediated inflammation. Immunity 34, 566–578 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li J., Diao B., Guo S., Huang X., Yang C., Feng Z., Yan W., Ning Q., Zheng L., Chen Y., Wu Y., VSIG4 inhibits proinflammatory macrophage activation by reprogramming mitochondrial pyruvate metabolism. Nat. Commun. 8, 1322 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martin B. N., Wang C., Zhang C. J., Kang Z., Gulen M. F., Zepp J. A., Zhao J., Bian G., Do J. S., Min B., Pavicic P. G., el-Sanadi C., Fox P. L., Akitsu A., Iwakura Y., Sarkar A., Wewers M. D., Kaiser W. J., Mocarski E. S., Rothenberg M. E., Hise A. G., Dubyak G. R., Ransohoff R. M., Li X., T cell-intrinsic ASC critically promotes TH17-mediated experimental autoimmune encephalomyelitis. Nat. Immunol. 17, 583–592 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stienstra R., van Diepen J. A., Tack C. J., Zaki M. H., van de Veerdonk F. L., Perera D., Neale G. A., Hooiveld G. J., Hijmans A., Vroegrijk I., van den Berg S., Romijn J., Rensen P. C. N., Joosten L. A. B., Netea M. G., Kanneganti T. D., Inflammasome is a central player in the induction of obesity and insulin resistance. Proc. Natl. Acad. Sci. U.S.A. 108, 15324–15329 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/1/eaau7426/DC1
Fig. S1. Liganding VSIG4 by complement C3b results from decreasing NLRP3 and proIL-1β expression in macrophages.
Fig. S2. The expression of NLRP3 and IL-1β in Vsig4−/− PEMs was not affected by VG11 mAbs.
Fig. S3. Vsig4 deficiency does not cause spontaneous NLRP3 inflammasome activation and IL-1β secretion.
Fig. S4. MS4A6D interacts with VSIG4 by using a yeast split-ubiquitin screen.
Fig. S5. Gating strategies of flow cytometry.
Fig. S6 Inhibition of NLRP3 activity or blocking IL-1β signaling controls the progress of EAE and colitis.
Fig. S7. Vsig4−/− mice are resistant to 2.5% DSS-induced colitis.
Fig. S8. Vsig4−/−Il-1R1−/− mice deteriorated DSS-induced colonic damage.
Fig. S9. Model of VSIG4/MS4A6D signaling complex attenuates NLRP3 inflammasome activation.
Fig. S10. Creating Ms4a6d−/− mice by CRISPR/Cas9-mediated genome engineering.
Table S1. The PCR primers used in this study.