

Abstract
BACKGROUND:
The slow and rapid delayed rectifier K+ currents (IKs and IKr, respectively) are responsible for repolarizing the ventricular action potential (AP) and preventing abnormally long APs that may lead to arrhythmias. Although differences in biophysical properties of the two currents have been carefully documented, the respective physiological roles of IKr and IKs are less established. In this study, we sought to understand the individual roles of these currents and quantify how effectively each stabilizes the AP and protects cells against arrhythmias across multiple species.
METHODS:
We compared 10 mathematical models describing ventricular myocytes from human, rabbit, canine, and guinea pig. We examined variability within heterogeneous cell populations, tested the susceptibility of cells to proarrhythmic behavior, and studied how IKs and IKr responded to changes in the AP.
RESULTS:
We found that: (1) models with higher baseline IKs exhibited less cell-to-cell variability in action potential duration (APD); (2) models with higher baseline IKs were less susceptible to early afterdepolarizations (EADs) induced by depolarizing perturbations; (3) as APD is lengthened, IKs increases more profoundly than IKr, thereby providing negative feedback that resists excessive AP prolongation; and (4) the increase in IKs that occurs during β-adrenergic stimulation is critical for protecting cardiac myocytes from EADs under these conditions.
CONCLUSIONS:
Slow delayed rectifier current is uniformly protective across a variety of cell types. These results suggest that IKs enhancement could potentially be an effective antiarrhythmic strategy.
Keywords: electrophysiology, K-channel, potassium channels, action potential, arrhythmia, mathematical modeling, early afterdepolarizations, Arrhythmias, Electrophysiology, Basic Science Research, Computational Biology, Ion Channels/Membrane Transport
Graphical Abstract
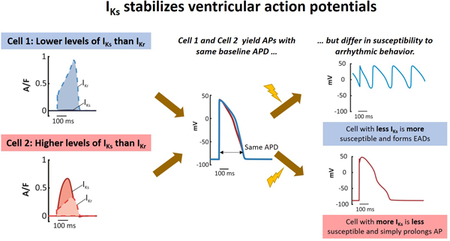
INTRODUCTION
In ventricular myocytes, action potentials (APs) are terminated by outward K+ currents that repolarize the cell membrane, i.e. bring membrane potential back to its resting level. It is well-established that the rapid and slow delayed rectifier currents, IKr and IKs, respectively, are the K+ currents most responsible for membrane repolarization. Early cardiac electrophysiology literature had described only a single time and voltage-dependent K+ current, called the delayed rectifier, before Sanguinetti and Jurkiewicz convincingly showed, in 1990, that the rapid and slow components were two distinct currents.1 Subsequent studies determined the proteins responsible for each current (Kv7.1 for IKs and Kv11.1 for IKr) and thoroughly characterized differences in gating between the two currents. Despite this extensive prior research, the individual physiological roles of the currents remain unclear. Studies have generally focused on either: (1) promiscuous binding of drugs to the channel responsible for IKr, which can cause drug-induced arrhythmias;2, 3 or, (2) the dramatic increase in IKs that is seen with sympathetic stimulation and activation of β-adrenergic receptors.4 Although the importance of these special conditions is well-appreciated, the distinct roles of IKr and IKs under baseline physiological conditions remain incompletely understood.
Both IKr and IKs are voltage-gated outward currents that are activated by membrane depolarization. In other words, the ion channels are closed at the resting membrane potential, then open during APs, passing current that drives the membrane towards its resting potential. This means that IKr and IKs can provide negative feedback that protects cells against perturbations. For instance, by itself an increase in inward current would prolong the AP. However, if this depolarizing perturbation also increases the activation of IKr and/or IKs, the augmented outward K+ current can counteract this effect. Conversely, if K+ currents cannot increase in response to a depolarizing perturbation, abnormally long APs and arrhythmogenic early afterdepolarizations (EADs) may result.
We recently proposed that IKs is superior to IKr at stabilizing the ventricular AP.5 This hypothesis was based on an experimental and computational study that examined how guinea pig ventricular myocytes respond to augmentation and inhibition of specific ionic currents. Important results in this study were that cells with high IKs and low IKr exhibit (1) smaller responses to perturbations and (2) resistance to arrhythmogenic EADs, compared with cells that have low IKs and high IKr. Two important questions, however, remained undetermined. First, although the hypothesis was consistent with results obtained in guinea pig myocytes, we could not conclude whether this might be generalizable other species. Second, the mechanism by which IKs stabilized APs was unclear.
Here we have addressed these unresolved questions through extensive simulations of ventricular APs. By investigating a variety of models describing myocytes from different species, we found that IKs is universally stabilizing compared to IKr. In addition, we characterize how this occurs due to stronger negative feedback exerted by IKs. These results improve our understanding of how cells respond to potentially arrhythmogenic perturbations and have important implications for therapies that aim to protect hearts from arrhythmias.
METHODS
The authors declare that all computer simulation code are available on the at the first author’s github site: https://github.com/meeravarshneya1234/IKs_stabilizes_APs.
Models & Software
We analyzed 10 ventricular myocyte mathematical models representing4 species – canine, guinea pig, rabbit, and human. The models employed are those published by: (1) Hund & Rudy;6 (2)Fox, McHarg& Gilmour;7(3) Heijman, Volders, Westra, & Rudy;8(4) Livshitz& Rudy;9 (5)Devenyi, Ortega, Groenendaal, Krogh-Madsen, Christini&Sobie;5 (6) Shannon, Wang, Puglisi, Weber &Bers;10(7) ten Tusscher, Noble, Noble &Panfilov;11(8) ten Tusscher&Panfilov;12 (9)Grandi, Pasqualini&Bers;13 (10) O’Hara, Virag, Varro & Rudy.14For shorthand, each model is referred to by the name of the paper’s first author, with TT04 and TT06 used to distinguish the two models published by ten Tusscher and coworkers. Several models include variants describing cells from various layers of the ventricular wall --for each of these models we employed the endocardial variant. All models were simulated in MATLAB v2016b with a 64-bit Intel processor. The model equations were solved using the ode15sMATLAB function with relative tolerance of 10−3 and absolute tolerance of 10−6.
Stimulation Protocol
All models were paced at 1 Hz with a 2 ms current injection stimulus with amplitude equal to 2 times that model’s threshold (Table S1). To ensure that steady-state was reached, each model was paced for 2000 consecutive beats, and the first beat where the action potential duration (APD) varied by less than +/−1% for 500 consecutive beats was considered the minimum number of beats to reach steady state (Table S1). The state variables from that beat were used as the initial conditions.
High IKs and Low IKs model variants
In the O’Hara model14, we created a High IKs variant and a Low IKs variant to compare with the published endocardial cell baseline model. The High IKs and Low IKs variants, which produced the same APD as the baseline model at 1 Hz pacing, were developed by increasing or decreasing the IKs conductance parameter (GKs) 10-fold, then adjusting the IKr conductance parameter (GKr) until the identical APD was observed. This process required GKr to be scaled by factors of 0.39 and 1.08 to produce the High IKs and Low IKs models, respectively. A similar approach was followed to create a High IKs version of the Grandi model and a Low IKs version of the TT04 model, with IKs levels altered by a factor of 20 and IKr adjusted to maintain constant APD.
Parameter randomization to generate heterogeneous populations
To determine how levels of IKs influence the variability in AP morphology observed across a heterogeneous population, we imposed random variation in parameters controlling ionic current levels. As previously described,5, 15–17 model parameters were log-normally distributed such that log-transformed parameter scaling factors had a mean of zero and standard deviation of 0.2. The parameters that were varied in each model controlled ionic current magnitudes or rates of intracellular ion transport. For instance, GKr is the conductance that determines the maximal amplitude of IKr. The complete lists of parameters varied in each model are presented in Tables S2-S11. With each model, a population of 300 model variants was generated, and each cell in the population was stimulated at 1 Hz. Cells that failed to repolarize were removed from the population and replaced with alternative model variants to ensure a total of 300 cells in each population. See Table S12 for details on how many APs failed during each test. APD was calculated for each model variant as the interval between the stimulus and the return of the membrane potential below −75 mV. For each heterogeneous population, our primary goal was to determine the degree of APD variability across the population. Because the shape of the APD distribution could differ between models, we quantified this with a non-parametric statistic that we termed the APD Spread:
Perturbations to prolong action potentials with depolarizing currents
To determine cellular arrhythmia susceptibility, we induced two depolarizing, potentially proarrhythmic perturbations in each model. The first was to increase the magnitude of the L-type Ca2+ current (ICaL) by scaling GCaL, the parameter that controls this current in each model. This scaling factor was progressively increased until proarrhythmic dynamics were observed in any of the last 10 beats during 1 Hz pacing, and the smallest ICaL scaling factor that produced these dynamics was defined as the model’s proarrhythmia threshold (Figure S1). EADs were defined as any change in the voltage derivative from negative to positive occurring more than 100 ms after AP initiation. Proarrhythmic dynamics generally appeared as EADs, except in the TT06 model, where repolarization failure occurred. To compare proarrhythmia thresholds across models, we computed the integrated ICaL at the level immediately preceding the threshold. Models in which smaller levels of integrated ICaL produced arrhythmic dynamics were considered more susceptible.
The second perturbation was to test each model’s response to current injection. We performed this by injecting a constant inward current into each model while the membrane voltage was greater than −60 mV. We increased the level of this injection in increments of 0.1 µA/µF until proarrhythmic behaviors (mainly repolarization failures) were apparent. Similar to the calcium perturbation, sensitivity to a small injection of current meant that the model was highly susceptible
AP Clamp Simulation
To examine how IKs and IKr respond when APs are perturbed, we performed AP clamp simulations. The goal of these simulations was to keep AP shape and cycle length constant while varying APD. To accomplish this, we recorded the steady-state AP during 1 Hz pacing. This waveform was then split into depolarized and resting epochs, corresponding to periods where the membrane potential was above or below −75 mV, respectively. The two epochs were either stretched or compressed using the interp1 function in MATLAB to make the AP longer or shorter while keeping the total duration constant at 1000 ms (Figure S2). These waveforms with stretched or compressed APs were then used as command waveforms in a voltage clamp version of each of the models. IKs and IKr were recorded after applying 100 AP clamp command waveforms to the myocyte.
β-Adrenergic Stimulation
To determine how regulation of IKs by β-adrenergic stimulation affects AP variability and stability, we performed simulations with the model of Heijman et al,8which describes how signaling pathways downstream of β-adrenergic receptors influence electrophysiology and calcium in canine ventricular myocytes. We considered a population of 300 model variants that did not have EADs at baseline, before and after application of 1 µM isoproterenol (ISO), a concentration that produces a saturating β-adrenergic signaling response. The initial conditions for each population were obtained by running the Heijman model to steady state (Table S1) at 1 Hz, with and without 1 µM ISO. We also compared these groups to population simulations In addition to simulating the population of 300 cells with no ISO and with saturating ISO, we simulated 8 separate conditions in which ISO was present, but one protein kinase A phosphorylation of one of that kinase’s targets was prevented. The no ISO population contained thirteen APs that formed EADs, which were removed from each population. We also applied increases in L-type Ca2+ current, as described above, to assess arrhythmia susceptibility under different conditions.
RESULTS
Within a single human ventricular myocyte model, increasing the contribution of IKs is protective
To determine whether our hypothesis that IKs is protective applied to human cells, we investigated the functional consequences of the balance between IKr and IKs in the O’Hara model,14the most recent representation of human ventricular myocytes. We created multiple variants of this model by increasing or decreasing the IKs conductance 10-fold while adjusting IKr levels to maintain a constant APD (Figure 1A). To quantify the difference, we calculated the “IKs Fraction” as integrated IKs (during the AP) divided by the sum of integrated IKs and integrated IKr (Figure 1B). The IKs Fractions of the High (red), Baseline (black), and Low IKs(blue) models are 0.635, 0.085, and 0.0088 respectively. Based on previous results,5we hypothesized that the High IKs model would be less sensitive to perturbations that alter APD.
Figure 1. Altering the contribution of IKs within the same model influences population variability.
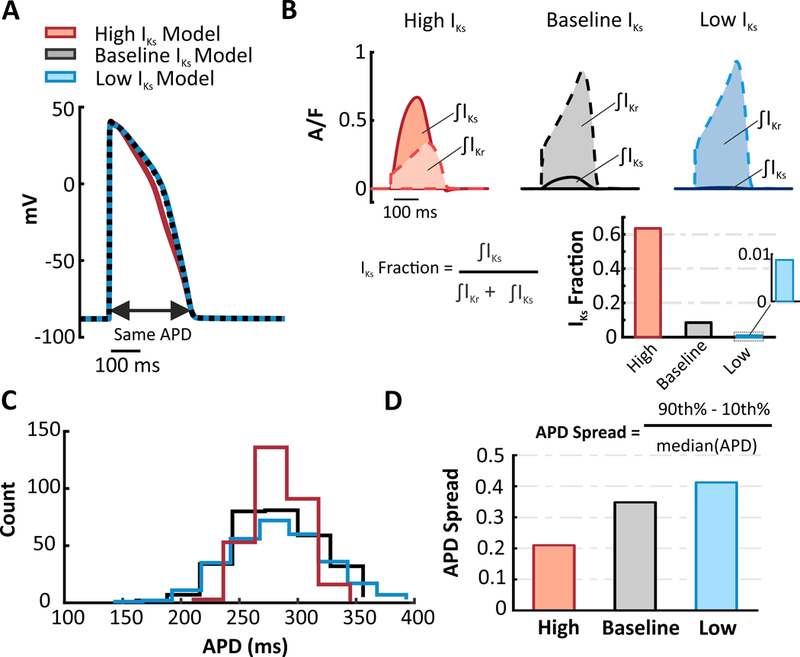
(A) In the O’Hara human ventricular myocyte model, High IKs and Low IKs versions were developed by increasing or decreasing (10 fold) IKs and adjusting IKr to produce an identical APD. (B) IKs and IKr waveforms of the High, Baseline, and Low IKs models. The shaded regions represent the integrated currents (area under the curve, or AUC) during the APs. The current AUCs were used to calculate the fraction of repolarizing current contributed by IKs(IKs Fraction)in each model. (C) Using each of the three model variants as the baseline model, model populations (300 cells each) were generated by randomly varying parameters. Distributions of APD show that greater IKs promotes less variability within a population. (D) APD Spread was calculated for each of the populations to quantify the variability within the APD distributions.
We first simulated a heterogeneous population of myocytes in each model variant. The populations were created by randomly varying the model parameters that control ion transfer rates (see prior studies15–18 and Methods). Imposing an equal degree of parameter variability on all three models led to much greater variability in AP shape in the Low IKs model compared to the High IKs model. To quantify the differences, we calculated the spread of the APD distribution (APD Spread), as described in Methods. APD histograms (Figure 1C) and the APD Spread (Figure 1D) confirm that increasing the contribution of IKs leads to reduced variability across a heterogeneous population, even when IKr is adjusted to maintain the same baseline APD.
To test each model variant’s susceptibility to proarrhythmic EADs, we progressively increased ICaL to prolong the AP until, at a threshold perturbation level, EADs occurred (Figure 2). We found that the Low IKs model was the most susceptible to EADs, and the High IKs model was the most resistant (Figure 2A-C).Over a range of ICaL Factors, the higher IKs model requires a greater “hit” to induce EADs (Figure 2D). This further supports the hypothesis that increasing the relative contribution of IKs in the O’Hara model stabilizes the AP in the face of depolarizing perturbations.
Figure 2. Altering the contribution of IKs within the same model influences arrhythmia susceptibility.
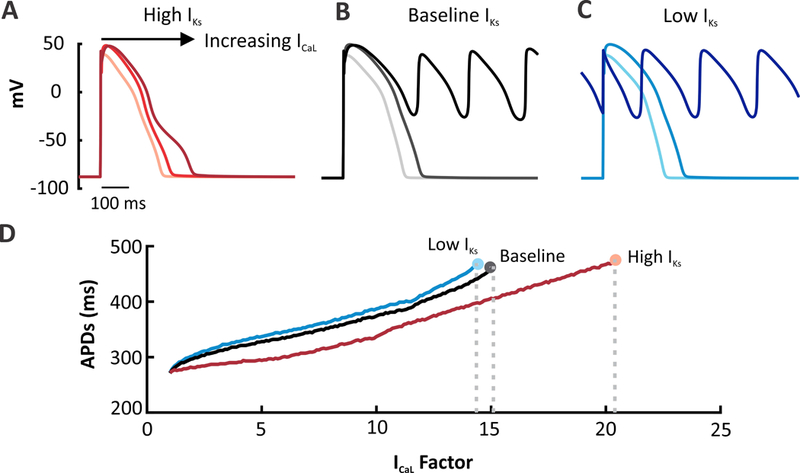
ICaL was progressively increased in each model to induce EADs and arrhythmic behavior. ICaL factor refers to the increase in channel permeability coefficient (1.0 equals control level).(A) APs simulated in the High IKs version of the O’Hara model at baseline and with ICaL augmentation (7.5 and 15.5 times). Plot shows the 92nd beat simulated at a 1 Hz pacing rate. (B) APs simulated in the baseline O’Hara model with the same ICaL perturbations. Plot shows the 91st beat. (C) Low IKs version simulated under the same conditions. Greater APD prolongation and increased number of EADs are produced in the low IKs version whereas the high model cells are protected from arrhythmogenic dynamics. Plot shows the 91st beat. (D) All three versions of the model were run under a wide range of ICaL factors and plotted against APD. The end of each line represents the last factor before an EAD forms The High IKs model requires a greater increase in the total ICaL current to induce an EAD.
Between myocyte models, high IKs leads to less APD variability across a heterogeneous population
To further determine how levels of IKs affect cellular behavior, we analyzed two additional human ventricular myocyte models,TT04 and Grandi. Although the two models describe cells from the same ventricular layer (endocardium), we observed striking differences in how IKr and IKs contribute to repolarization in the two models. Ionic current waveforms shown in Figure3A illustrate that IKr is much greater than IKs in the Grandi model (purple) whereas the reverse is true in TT04 (orange), with a much larger IKs Fraction (0.62) in TT04 than in Grandi (0.04) (Figure 3B).The corresponding current waveforms from the O’Hara model are replotted for comparison. To investigate the functional importance of the balance between IKr and IKs, we simulated heterogeneous populations of myocytes in the TT04, O’Hara, and Grandi models (Figure3C). Imposing an equal degree of parameter variability in the threemodels led to much greater variability in AP shape in the Grandi compared with the TT04 model. APD Spread was considerably greater in the Grandi model (0.601) than in the TT04 model (0.175), with O’Hara intermediate between the two (0.348),consistent with the results shown above.
Figure 3. Dramatic differences seen between two human myocyte models in K+ currents and population variability.
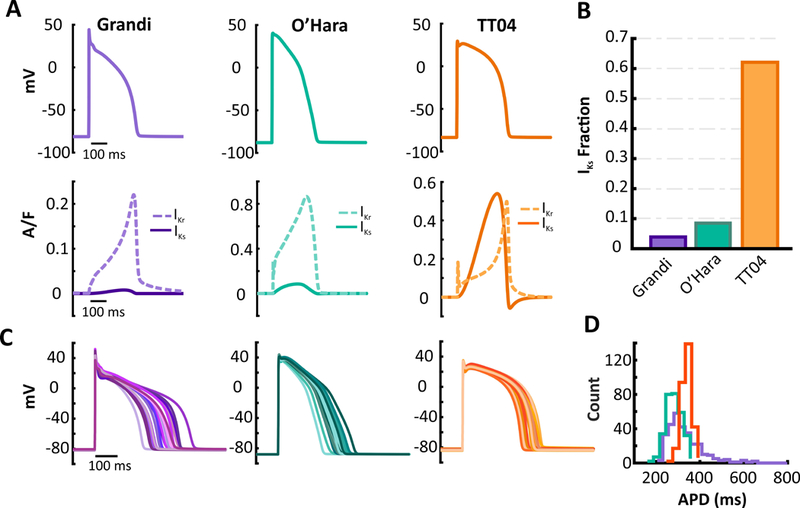
(A)Graphs of AP, IKs, and IKr waveforms in the Grandi (purple), O’Hara (turquoise – replotted from Figure 1 for comparison), and the TT04 (orange) models under baseline conditions. Plots show the 100th beat simulated at a 1 Hz pacing rate. (B) TT04 model has higher levels of IKs or IKs Fraction than the O’Hara and Grandi models. (C) Representative APs from a population of 300 model variants generated by imposing random variability in parameters in the Grandi, O’Hara (replotted from Figure 1 for comparison)and TT04 models. (D) Comparison of the distribution of APD from the 300 model variants. Greater variability is seen in the Grandi model.
Less arrhythmic behavior is observed in models with higher IKs levels
We next investigated how the TT04 model and the Grandi model differed in their susceptibility to arrhythmogenic behavior such as EADs. We perturbed each model by applying progressively increasing levels of ICaL to prolong the AP until EADs were seen. While the Grandi model (baseline low IKs) developed EADs after a relatively small increase in ICaL (1.6 times baseline), the TT04 model (baseline high IKs) required a much larger increase (27.8 times baseline).However, because the baseline ICaL magnitude differed between the 2 models, this multiplicative factor does not provide a fair basis of comparison. To compare susceptibility to EADs, we integrated the ICaL waveform at the level immediately under the EAD threshold (labeled “below EAD threshold” in Figure 4B). These integrated currents, shown in Figure 4C, indicate that approximately 11 times more ICaL is required to induce EADs in the TT04 model compared with the Grandi model. Results from the O’Hara model, replotted from Figure 2 for comparison, show that it sits between the Grandi and TT04 models in arrhythmia susceptibility. These results further support the hypothesis that high IKs levels, as in TT04, make these cells less sensitive to perturbations.
Figure 4. Susceptibility to Early Afterdepolarizations (EADs) in Grandi and TT04 models.
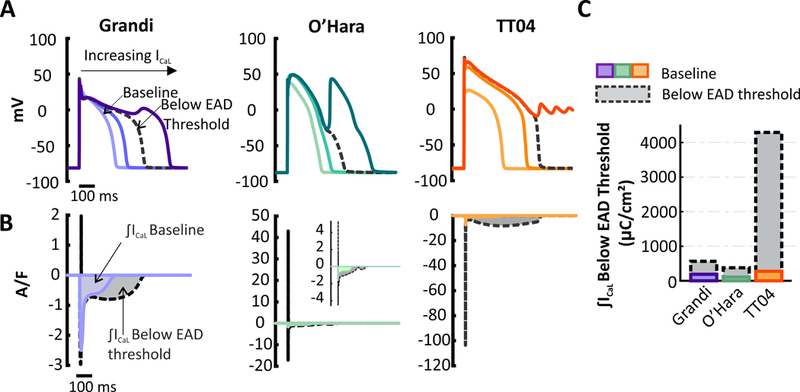
(A) APs simulated in Grandi (purple), O’Hara (turquoise), andTT04 (orange) models with different degrees of ICaL current augmentation. Plots represent the 91st beat at 1-Hz pacing rate. (B)ICaL waveforms in the two models. The colored curves show the baseline ICaL whereas the black dashed curves plot ICaL immediately before the first EAD occurred. The shaded regions illustrate the area under the curve (AUC). (C) Bar graph of the current AUCs from (B) show that small increases in ICaL induce EADs in the O’Hara and Grandi models(lower IKs models) whereas large increases are required to induce EADs in TT04 (high IKs model).
For comparison with the O’Hara model variants shown in Figures 1 and 2, we created a High IKs version of the Grandi model and a Low IKs version of the TT04 model. Results, shown in Figures S3 and S4, are consistent with those presented above. The Low IKs version of TT04 exhibited high variability within a population and greater arrhythmia susceptibility than the original TT04 model, whereas opposite effects were seen with the High IKs version of the Grandi model.
IKs levels influence sensitivity to perturbations across models and species
To test whether the protective effect of high IKs levels can be considered a general phenomenon, we examined population variability and the susceptibility to EADs in 7 additional models that describe canine, rabbit, guinea pig, and human ventricular myocytes (see Methods Section). In each model, we calculated the IKs Fraction from IKr and IKs waveforms during steady-state pacing at 1 Hz (Figures 3 & S5). Based on Figure 1 and previous results,5 we predicted that a larger IKs Fraction would be associated with smaller population variability and a higher EAD threshold across these models. Figure 5 shows that these trends are indeed observed: models with low baseline IKs (e.g. Grandi, Devenyi, Fox) exhibited high population heterogeneity when parameters are varied (Figures 5A & S6) and susceptibility to EADs when stressed with a depolarizing stimulus(Figure 5B & S7),whereas models with higher baseline IKs (e.g. TT04, TT06, Livshitz) showed lower population variability and resistance to EADs. Table S13 presents summary statistics on the population distributions produced by all models. Moreover, because ICaL formulations as well as magnitudes vary between models, we tested an additional cellular perturbation. We injected a constant inward current into each model and recorded the value that caused a proarrhythmic event. Similar to the calcium perturbation (Figure 5B), we find that there is a positive correlation between the current required to induce arrhythmic behavior and the IKs Fraction in each model (Figure 5C & S8). The IKs Fraction also exhibited a negative correlation with the percent change in APD caused by a constant inward current of 0.1 A/F --models with lower baseline IKs levels had a greater prolongation in APD compared to models with higher baseline IKs (Figure S9, bottom).Thus, across a range of mathematical models, high levels of IKs protect ventricular myocytes from multiple types of perturbations that affect APs.
Figure 5. Comparison of population variability & susceptibility to EADs across models from multiple species.
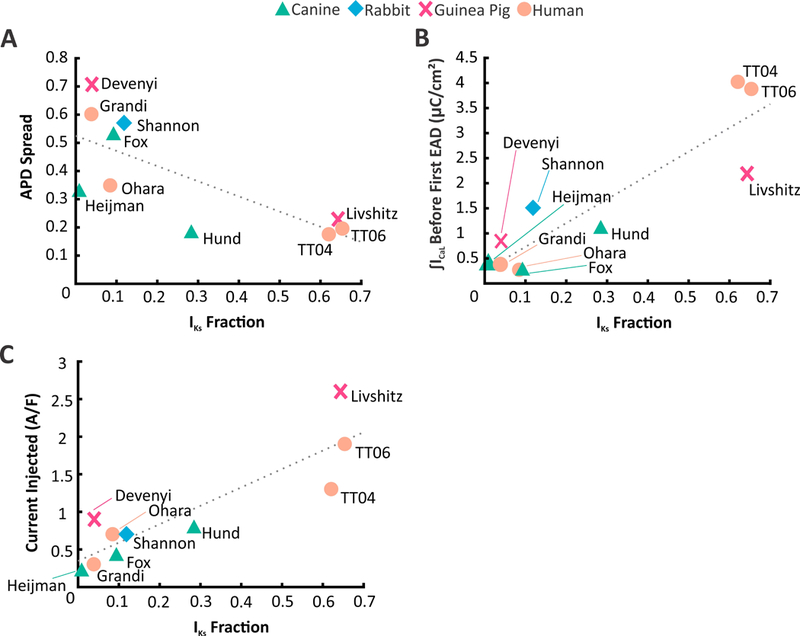
(A) APD Spread plotted against IKs Fraction of ten ventricular myocyte models from four different species. APD Spread assesses the APD distribution of the populations developed in Figures 3&S2. It is defined as the difference of the 90th Percentile and 10th Percentile divided by the median. Models with low IKs have greater population variability than models with high IKs. (B) Integrated ICaL levels just below the EAD threshold (calculated in Figures 5& S4) plotted against IKs Fraction. Low IKs models were more susceptible to EADs than high IKs models.(C) Injecting constant inward current into each model, when the Vm is above –60 mV, induced arrhythmic behavior to different extents. Similar to (B) models with low IKs were more sensitive to a slight injection of inward current as compared to models with high IKs.
IKs provides more effective negative feedback than IKr in response to prolonged APs
Although the results presented thus far are internally consistent, they do not address the mechanism by which high levels of IKs provide resistance to perturbations. We hypothesized that a depolarizing, AP-prolonging perturbation would lead to a greater increase in IKs than IKr, due to differences in gating kinetics between the two currents. To test this idea, we performed AP clamp simulations in all 10 models. The results for the O’Hara model are displayed in Figure 6, while summary data for remaining models can be found in Figure S10. The command voltage waveform was either a stretched or compressed version of the AP obtained during steady state pacing at 1 Hz. This allowed us to vary the APD while keeping the cycle length constant and minimally perturbing the AP shape (Figure 6A; see Methods and Figure S2 for simulation details). The resultant current waveforms and gating variables are displayed in Figure 6B. These show that AP prolongation causes a large increase in both the IKs maximal amplitude and the area under the curve (AUC; top). Gating variable time courses to the right illustrate that the peak levels reached by both activation variables increase when the command AP is two times longer. In contrast, the IKr waveform becomes stretched or compressed in parallel with the AP waveform, with no change in the peak level. This occurs because the rapid activation and inactivation variables simply remain at the same extreme values for a longer period of time. As a result of these kinetic differences, increases or decreases in APD have a much larger effect on IKs than on IKr, illustrated by the integrated currents, shown on a logarithmic scale in Figure 6C. We found similar trends as seen in Figure 6C in the remaining 9 models (Figure S10). Thus, in the face of an AP-prolonging perturbation, IKs provides more counteracting negative feedback than IKr.
Figure 6. AP clamp simulations illustrate changes in K+ currents with alterations in AP shape.
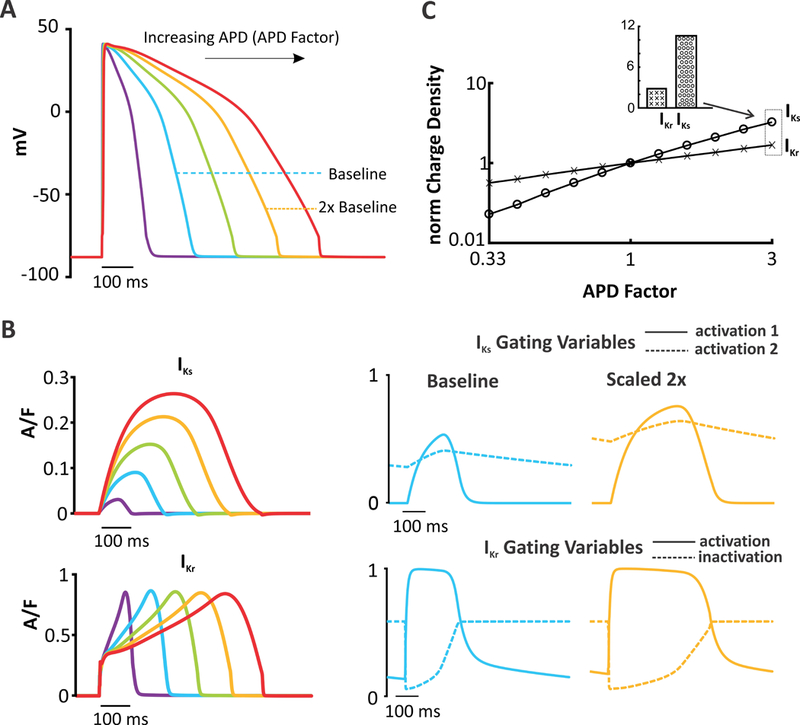
(A) AP clamp was performed in the O’Hara model by compressing or stretching the baseline AP waveform (see Methods for details). Example waveforms are shown with AP durations of 0.5,1.5, 2 & 2.5 times the baseline value. (B) IKr& IKs current graphs and corresponding gating graphs produced after imposing the AP waveforms shown in (A). APs are applied at 1 Hz, and the response to the last AP in a train of 100 APs is shown. (C) Integrated currents (area under the curve, AUC) of each K+ currents, normalized to the integrated current of the baseline AP, plotted as a function of APD. As APD increases, there is a much greater relative increase in IKs compared with IKr.
IKs stabilizes the AP during β-adrenergic stimulation
In large animals such as dogs and humans, IKs is thought to be relatively insignificant at baseline but to increase in magnitude substantially during β-adrenergic stimulation.19–21However, because β-adrenergic stimulation results in changes to numerous ion transport pathways,22 it is not clear if augmentation of IKs will be sufficient to protect cells from arrhythmias. To test this, we utilized the canine ventricular myocyte model of Heijman et al,8 which combines electrophysiology and Ca2+ cycling with β-adrenergic signaling pathways. We ran 3 identical heterogeneous population of 300 myocytes under 3 different conditions:(1) in the absence of β-adrenergic stimulation(no ISO);(2) saturating β-adrenergic stimulation with 1 µM isoproterenol (ISO); and (3)β-adrenergic stimulation + IKs phosphorylation blocked (ISO + IKs-ØP).The addition of ISO greatly reduced the variability in AP shape seen across the population (Figure 7A and 7B). When ISO was applied and phosphorylation of IKs was prevented (Figure 7C), variability increased, indicating the importance of IKs phosphorylation for stabilizing AP shape. To quantify the variability, we calculated the APD Spread for each population. For the no ISO, ISO, and ISO + IKs-ØP populations, the APD Spread was 0.301, 0.118, and 0.209 respectively (Figure 7D). Together these results show that increased IKs during β-adrenergic stimulation acts to stabilize APs. We also individually blocked phosphorylation of the remaining seven PKA targets to determine how each target separately contributes to the stability of the overall population (Figure S11). No other PKA target had as large an effect as IKs, indicating that PKA phosphorylation of this channel contributes the most to the reduced APD Spread observed with ISO.
Figure 7. IKs stabilizes the AP during β-adrenergic stimulation.
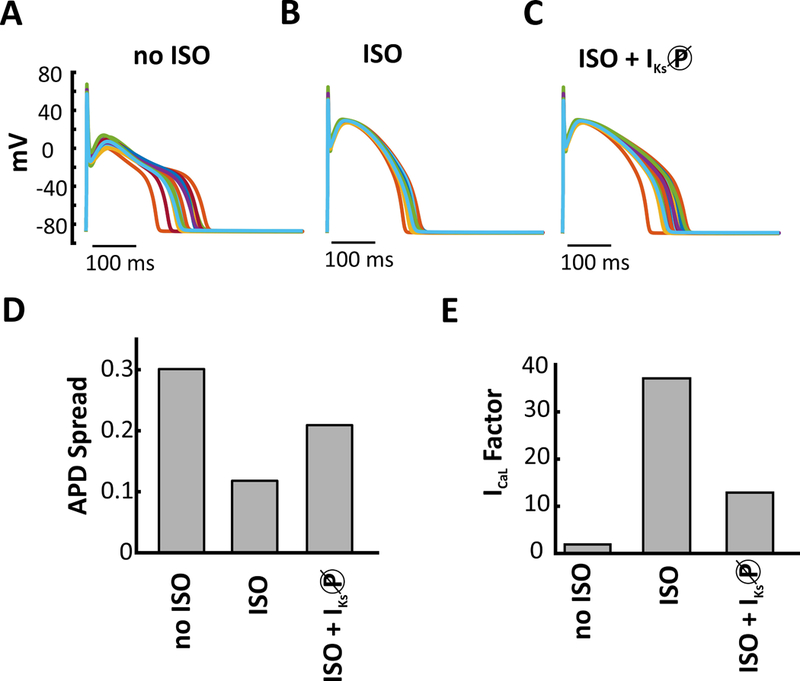
Representative APs from three populations (300 model variants) to demonstrate how the addition of 1 μM isoproterenol (ISO), and phosphorylation of IKs, influences AP variability. (A) baseline conditions no added ISO (B) added ISO (C) added ISO and blocked PKA phosphorylation of IKs. (D) ISO decreases AP variability compared with control. Blocking phosphorylation of IKs under ISO conditions increases APD variability. (E) Calcium perturbation applied on no ISO, ISO, and ISO + IKs-P block APs shows that inhibiting IKs phosphorylation makes the cell more susceptible to arrhythmic activity.
To extend this analysis, we applied the L-type Ca2+ current perturbation under the three conditions (no ISO, ISO, and ISO + IKs-P block). We found that progressively increasing the levels of ICaL causes the no ISO AP to fail immediately. Adding ISO causes an increase in both levels of IKs and ICaL. Though levels of ICaL were already quite high further increase in ICaL did not induce EADs in the ISO AP until an ICaL factor of 37.1 (Figure 7E).However block of PKA phosphorylation of IKs increases the susceptibility of the cell and it forms EADs at a factor of 12.9 (Figure 7E).. Thus, augmentation of IKs during beta-adrenergic stimulation is necessary to prevent arrhythmic activity.
DISCUSSION
A previous study from our group proposed that AP stability and arrhythmia susceptibility depend on the balance between the two delayed rectifier currents IKr and IKs, based on simulations and experiments in guinea pig ventricular myocytes.5 Here we sought to uncover mechanistic explanations and determine whether these results could be generalized across species. To achieve these aims, we carefully studied ten ventricular myocyte models that simulate the electrophysiology of canine, rabbit, guinea pig, and human cells.5–14 Within a given model, we found that increasing the relative contribution of IKs reduced the variability observed in a heterogeneous population and made myocytes less susceptible to EADs. The same trend held when comparing across models such that those with higher baseline IKs exhibited reduced population variability and EAD susceptibility. We also found that an increase in APD leads to a more profound augmentation of IKs than IKr; this effect provides negative feedback that can protect cells from excessive AP prolongation. Finally, we demonstrated that the increase in IKs seen with β-adrenergic stimulation protects ventricular myocytes from arrhythmogenic dynamics under these conditions. Overall, this study confirms and reinforces the strongly protective effect of cardiac IKs, a finding with important clinical and therapeutic implications.
Insights gained through a comparison between models
It is well-appreciated that because ventricular myocytes from different species contain different complements of ionic currents, the cells will show different AP morphologies and behaviors.23 An initially surprising result, however, was the fact that that two mathematical models of the same cell type could nonetheless display considerable differences in response to simulated perturbations such as channel-blocking drugs.24 However, while several studies have catalogued important differences between models,24–27 it has been more challenging to determine the reasons for these discrepancies. In this study, similar to previous work,17, 28 we attempted to uncover general principles by simulating a wide variety of models describing ventricular myocytes from different species. We found that the relative importance of IKs in a given model, quantified as the “IKs Fraction,” correlates with both the population variability and the arrhythmia susceptibility displayed by that model (Figure 5). This shows the insight that can be gained by examining several models that collectively exhibit phenotypic variability.
Although the model comparisons provided important support for our hypothesis about the protective role of IKs, the question still remains as to why IKs levels can be so different across models, especially those describing cells from the same species. A review of the literature suggests two potential explanations for why voltage clamp studies examining IKs might obtain divergent results. One stems from the fact that β-adrenergic stimulation, and subsequent channel phosphorylation by PKA, greatly increases the current flowing through IKs.4, 29 Thus, myocytes isolated in two different labs may show widely divergent IKs amplitudes due to differences in baseline channel phosphorylation. A second possible explanation is suggested by the recent study of Bartos et al.,30 who observed a strong dependence of IKs on intracellular [Ca2+]. Since IKs is frequently measured under conditions of buffered [Ca2+], and this can vary between studies, this factor may also contribute to the wide range of IKs amplitudes observed in the models that we investigated.
Consequences of IKs for understanding heterogeneity between individuals
One metric we investigated was APD Spread, the variability in APD across a heterogeneous population of myocytes (Figures 1C-D, 3C-D, S4). This quantity is of interest both because it helps us understand phenotypic variability between individuals and because it can allow for inferences about variability observed at the molecular level. In general, investigations into heterogeneity are interested in both functional variability, observed in quantities such as QT intervals, and molecular heterogeneity, such as differences in channel expression between cells. In this study we imposed equal molecular heterogeneity in all models and then measured, as an output, the resulting differences in phenotypic variability. An equally valid strategy is to determine from data an appropriate range for functional heterogeneity, then select “calibrated” populations of models that match the heterogeneity.31,32,33A consequence of our results is that when this approach is taken, the amount of molecular heterogeneity seen in calibrated populations will differ considerably between models and depend to a large extent on the model’s baseline level of IKs. In other words, the level of IKs in a model plays a major role in the mapping between molecular and phenotypic heterogeneity.
Mechanisms underlying the protective role of IKs
To determine why IKs protects cells from arrhythmias, we built upon prior studies that examined the biophysical properties of K+ currents. For example, Varro et al predicted in 2000 that IKs can provide negative feedback during AP prolongation that can counteract the effects of the depolarizing perturbation.34 Similarly, Rocchetti and coworkers observed an increase in IKs with rapid pacing and concluded that this resulted from a greater percentage of time spent at depolarized potentials.35 Studies such as these informed our hypothesis that the gating kinetics of IKs would make it especially well-suited, compared with IKr, to increase in magnitude in response to AP prolongation. The AP clamp simulations shown in Figure 6 demonstrate this; a 3-fold lengthening of the AP, for instance, causes a roughly 3-fold increase in the charge carried by IKr but a more than 10-fold increase in the charge carried by IKs.
These results illustrate a subtle aspect of the commonly-invoked concept of “repolarization reserve,” an idea initially articulated by Roden.36 It is important to note that repolarization reserve, properly understood, does not refer simply to the sum of the repolarizing currents in a myocyte. Instead, because repolarization reserve describes the ability of the myocyte to respond to an AP-prolonging, potentially arrhythmogenic perturbations, the response of ionic currents to such perturbations must also be considered. For instance, a previous study from our group showed that certain ionic current alterations can prolong the AP while simultaneously improving the cell’s ability to respond to block of IKr.37Results such as these imply that a cell with a longer AP does not necessarily have less repolarization reserve than a cell with a shorter AP --the repolarization reserve cannot be assessed unless one knows how the two cells respond to perturbations. The results presented here show that the biophysical properties of IKs allow it to provide repolarization reserve.
Role of IKs during β-adrenergic stimulation
When β-adrenergic receptors in ventricular myocytes are stimulated, a number of ion transport pathways are altered to produce a positive inotropic and lusitropic response. Some of these changes, such as increased ICaL and phosphorylation of ryanodine receptors, can be potentially proarrhythmic. The magnitude of IKs also substantially increases during β-adrenergic stimulation, and this effect is thought to help counteract the AP-prolonging actions of increased ICaL. Our results suggest that this IKs augmentation may have a further role of stabilizing the AP beyond simply reducing APD. Indeed, previous studies have suggested that differences in the time courses of changes in ICaL and IKs after β-adrenergic receptor activation may lead to a transient increase in arrhythmia risk.38Similarly, patients with Long-QT Syndrome Type 1 (LQT1), caused by loss of function mutations in the gene that encodes for the IKs channel α-subunit (KCNQ1), experience arrhythmias during β-adrenergic stimulation.39 Our results indicate that increased IKs during β-adrenergic stimulation acts to stabilize APs (Figure 7), consistent with our other results showing a key role for IKs in stabilizing the AP under baseline conditions. Therefore, while IKs plays only a minor role in the electrophysiology of canine and human ventricular myocytes in the absence of beta-adrenergic stimulation, its activation during β-adrenergic stimulation may be critically important for suppressing arrhythmias under these conditions.
Therapeutic Implications
Our results offer important insight into potential strategies for reducing arrhythmia risk with therapeutics. Block of IKr, by a variety of drugs, is well-known to prolong QT intervals and increase arrhythmia risk, the so-called drug-induced Long QT Syndrome.3 Consistent with these clinical findings, cellular experiments and simulations in a variety of species generally show that IKr block causes more severe AP prolongation than IKs block.40, 41 Based on these results, we anticipate that an IKs-enhancing drug could potentially cause only minimal AP shortening at baseline, yet still effectively increase AP stability and protect cells from stressors that prolong the AP, such as IKr-blocking drugs, increased ICaL, or hypokalemia. Following on results identifying autoantibodies against the IKs α-subunit that augment the current;42 a small molecule IKs-enhancing drug could potentially have antiarrhythmic efficacy.
Study Limitations and Conclusions
The simulations presented here have demonstrated that the population variability and EAD susceptibility exhibited by a particular myocyte model depends to a large extent on its level of IKs (Figure 5). The scatter in the results shown in Figure 5, however, indicates that additional factors, not addressed here, must contribute. For instance, the Livshitz model is more vulnerable to EADs than the TT06 model, even though these have similarly high IKs levels, whereas the Devenyi model shows more variability than the Grandi model, even though these have comparably low IKs levels. These discrepancies likely result from differences between models in additional cardiac ionic currents besides IKs. It is also important to note that the kinetic formulations of both IKr and IKs may differ between two competing models, which can reflect either true species differences or inconsistencies in the original voltage clamp data used to build the models. Although these factors were not explored in this study, the systematic comparison between models that we have presented identifies issues that future studies can address. Similarly, both the AP clamp simulations shown in Figure 6 and the EAD susceptibility simulations presented in Figures 2 and 4represent predictions that can be tested experimentally to support or refute the potentially protective role of IKs.
In summary, we examined the roles of IKr and IKs in AP population variability and arrhythmia vulnerability across a variety of cardiac myocyte models representing several species. The results reinforce the hypothesis that IKs is superior to IKr at stabilizing the AP during a perturbation and preventing excessive APD prolongation that can eventually lead to EADs and ventricular arrhythmias. Overall, the study supports the idea that enhancement of IKs may be an effective antiarrhythmic strategy.
Supplementary Material
WHAT IS KNOWN?
Action potential (AP) repolarization is controlled by the rapid and slow delayed rectifier potassium channels (IKr and IKs).
An inadequate response from IKr and IKs during a depolarizing perturbation can lead to AP prolongation and the formation of early afterdepolarizations (EADs).
WHAT THE STUDY ADDS?
This study uses ten ventricular myocyte models of four different species to elucidate the role played by IKs in protecting cells from early afterdepolarizations (EADs).
An increased ratio of IKs:IKr decreases the heterogeneity displayed within a population of cells and reduces susceptibility to arrhythmic behavior.
During an AP-prolonging perturbation, IKs provides more counteracting negative feedback than IKr and stabilizes the cell to prevent the onset of EAD formation.
Acknowledgments:
The authors thank Dr. David J. Christini, Dr. Trine Krogh-Madsen, and Mr. Francis Ortega for helpful discussions and suggestions.
Sources of Funding: This work was supported by the National Heart Lung and Blood Institute [U01 HL136297 to EAS] and the National Institute of General Medical Sciences[P50 GM071558 to EAS]. MV and RAD were supported, at different times, by a training grant from the National Institute of General Medical Sciences[T32GM062754].
Footnotes
Disclosures: None
References:
- 1.Sanguinetti MC, Jurkiewicz NK. Two components of cardiac delayed rectifier K+ current. Differential sensitivity to block by class III antiarrhythmic agents. J Gen Physiol 1990;96:195–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mitcheson JS, Chen J, Lin M, Culberson C, Sanguinetti MC. A structural basis for drug-induced long QT syndrome. Proc Natl Acad Sci U S A 2000;97:12329–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roden DM. Pharmacogenetics and drug-induced arrhythmias. Cardiovasc Res 2001;50:224–31. [DOI] [PubMed] [Google Scholar]
- 4.Banyasz T, Jian Z, Horvath B, Khabbaz S, Izu LT, Chen-Izu Y. Beta-adrenergic stimulation reverses the I Kr-I Ks dominant pattern during cardiac action potential. Pflugers Arch 2014;466:2067–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Devenyi RA, Ortega FA, Groenendaal W, Krogh-Madsen T, Christini DJ, Sobie EA. Differential roles of two delayed rectifier potassium currents in regulation of ventricular action potential duration and arrhythmia susceptibility. J Physiol 2017;595:2301–2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hund TJ, Rudy Y. Rate dependence and regulation of action potential and calcium transient in a canine cardiac ventricular cell model. Circulation 2004;110:3168–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fox JJ, McHarg JL, Gilmour RF. Ionic mechanism of electrical alternans. Am J Physiol Heart Circ Physiol 2002;282:H516–30. [DOI] [PubMed] [Google Scholar]
- 8.Heijman J, Volders PG, Westra RL, Rudy Y. Local control of β-adrenergic stimulation: Effects on ventricular myocyte electrophysiology and Ca2+-transient. J Mol Cell Cardiol 2011;50:863–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Livshitz L, Rudy Y. Uniqueness and stability of action potential models during rest, pacing, and conduction using problem-solving environment. Biophys J 2009;97:1265–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shannon TR, Wang F, Puglisi J, Weber C, Bers DM. A mathematical treatment of integrated Ca dynamics within the ventricular myocyte. Biophys J 2004;87:3351–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.ten Tusscher KH, Noble D, Noble PJ, Panfilov AV. A model for human ventricular tissue. Am J Physiol Heart Circ Physiol 2004;286:H1573–89. [DOI] [PubMed] [Google Scholar]
- 12.ten Tusscher KH, Panfilov AV. Alternans and spiral breakup in a human ventricular tissue model. Am J Physiol Heart Circ Physiol 2006;291:H1088–100. [DOI] [PubMed] [Google Scholar]
- 13.Grandi E, Pasqualini FS, Bers DM. A novel computational model of the human ventricular action potential and Ca transient. J Mol Cell Cardiol 2010;48:112–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O’Hara T, Virág L, Varró A, Rudy Y. Simulation of the undiseased human cardiac ventricular action potential: model formulation and experimental validation. PLoS Comput Biol 2011;7:e1002061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sobie EA. Parameter sensitivity analysis in electrophysiological models using multivariable regression. Biophys J 2009;96:1264–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Devenyi RA, Sobie EA. There and back again: Iterating between population-based modeling and experiments reveals surprising regulation of calcium transients in rat cardiac myocytes. J Mol Cell Cardiol 2016;96:38–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cummins MA, Dalal PJ, Bugana M, Severi S, Sobie EA. Comprehensive analyses of ventricular myocyte models identify targets exhibiting favorable rate dependence. PLoS Comput Biol 2014;10:e1003543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sarkar AX, Sobie EA. Regression analysis for constraining free parameters in electrophysiological models of cardiac cells. PLoS Comput Biol 2010;6:e1000914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jost N, Virág L, Comtois P, Ordög B, Szuts V, Seprényi G, Bitay M, Kohajda Z, Koncz I, Nagy N, Szél T, Magyar J, Kovács M, Puskás LG, Lengyel C, Wettwer E, Ravens U, Nánási PP, Papp JG, Varró A, Nattel S. Ionic mechanisms limiting cardiac repolarization reserve in humans compared to dogs. J Physiol 2013;591:4189–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Volders PG, Stengl M, van Opstal JM, Gerlach U, Spätjens RL, Beekman JD, Sipido KR, Vos MA. Probing the contribution of IKs to canine ventricular repolarization: key role for beta-adrenergic receptor stimulation. Circulation 2003;107:2753–60. [DOI] [PubMed] [Google Scholar]
- 21.Kang C, Badiceanu A, Brennan JA, Gloschat C, Qiao Y, Trayanova NA, Efimov IR. β-adrenergic stimulation augments transmural dispersion of repolarization via modulation of delayed rectifier currents in the human ventricle. Sci Rep 2017;7:15922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saucerman JJ, McCulloch AD. Cardiac beta-adrenergic signaling: from subcellular microdomains to heart failure. Ann N Y Acad Sci 2006;1080:348–61. [DOI] [PubMed] [Google Scholar]
- 23.O’Hara T, Rudy Y. Quantitative comparison of cardiac ventricular myocyte electrophysiology and response to drugs in human and nonhuman species. Am J Physiol Heart Circ Physiol 2012;302:H1023–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cherry EM, Fenton FH. A tale of two dogs: analyzing two models of canine ventricular electrophysiology. Am J Physiol Heart Circ Physiol 2007;292:H43–55. [DOI] [PubMed] [Google Scholar]
- 25.Elshrif MM, Cherry EM. A quantitative comparison of the behavior of human ventricular cardiac electrophysiology models in tissue. PLoS One 2014;9:e84401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Niederer SA, Fink M, Noble D, Smith NP. A meta-analysis of cardiac electrophysiology computational models. Exp Physiol 2009;94:486–95. [DOI] [PubMed] [Google Scholar]
- 27.Romero L, Carbonell B, Trenor B, Rodríguez B, Saiz J, Ferrero JM. Systematic characterization of the ionic basis of rabbit cellular electrophysiology using two ventricular models. Prog Biophys Mol Biol 2011;107:60–73. [DOI] [PubMed] [Google Scholar]
- 28.Soltis AR, Saucerman JJ. Robustness portraits of diverse biological networks conserved despite order-of-magnitude parameter uncertainty. Bioinformatics 2011;27:2888–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saucerman JJ, Healy SN, Belik ME, Puglisi JL, McCulloch AD. Proarrhythmic consequences of a KCNQ1 AKAP-binding domain mutation: computational models of whole cells and heterogeneous tissue. Circ Res 2004;95:1216–24. [DOI] [PubMed] [Google Scholar]
- 30.Bartos DC, Morotti S, Ginsburg KS, Grandi E, Bers DM. Quantitative analysis of the Ca2+ -dependent regulation of delayed rectifier K+ current IKs in rabbit ventricular myocytes. J Physiol 2017;595:2253–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Britton OJ, Bueno-Orovio A, Van Ammel K, Lu HR, Towart R, Gallacher DJ, Rodriguez B. Experimentally calibrated population of models predicts and explains intersubject variability in cardiac cellular electrophysiology. Proc Natl Acad Sci U S A 2013;110:E2098–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muszkiewicz A, Britton OJ, Gemmell P, Passini E, Sánchez C, Zhou X, Carusi A, Quinn TA, Burrage K, Bueno-Orovio A, Rodriguez B. Variability in cardiac electrophysiology: Using experimentally-calibrated populations of models to move beyond the single virtual physiological human paradigm. Prog Biophys Mol Biol 2016;120:115–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou X, Bueno-Orovio A, Orini M, Hanson B, Hayward M, Taggart P, Lambiase PD, Burrage K, Rodriguez B. In Vivo and In Silico Investigation Into Mechanisms of Frequency Dependence of Repolarization Alternans in Human Ventricular Cardiomyocytes. Circ Res 2016;118:266–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Varro A, Baláti B, Iost N, Takács J, Virág L, Lathrop DA, Csaba L, Tálosi L, Papp JG. The role of the delayed rectifier component IKs in dog ventricular muscle and Purkinje fibre repolarization. J Physiol 2000;523 Pt 1:67–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rocchetti M, Besana A, Gurrola GB, Possani LD, Zaza A. Rate dependency of delayed rectifier currents during the guinea-pig ventricular action potential. J Physiol 2001;534:721–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roden DM. Taking the “idio” out of “idiosyncratic”: predicting torsades de pointes. Pacing Clin Electrophysiol 1998;21:1029–34. [DOI] [PubMed] [Google Scholar]
- 37.Sarkar AX, Sobie EA. Quantification of repolarization reserve to understand interpatient variability in the response to proarrhythmic drugs: a computational analysis. Heart Rhythm 2011;8:1749–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xie Y, Grandi E, Puglisi JL, Sato D, Bers DM. β-adrenergic stimulation activates early afterdepolarizations transiently via kinetic mismatch of PKA targets. J Mol Cell Cardiol 2013;58:153–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.O-Uchi J, Rice JJ, Ruwald MH, Parks XX, Ronzier E, Moss AJ, Zareba W, Lopes CM. Impaired IKs channel activation by Ca2+-dependent PKC shows correlation with emotion/arousal-triggered events in LQT1. J Mol Cell Cardiol 2015;79:203–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jost N, Virág L, Bitay M, Takács J, Lengyel C, Biliczki P, Nagy Z, Bogáts G, Lathrop DA, Papp JG, Varró A. Restricting excessive cardiac action potential and QT prolongation: a vital role for IKs in human ventricular muscle. Circulation 2005;112:1392–9. [DOI] [PubMed] [Google Scholar]
- 41.Lengyel C, Varró A, Tábori K, Papp JG, Baczkó I. Combined pharmacological block of IKr and IKs increases short-term QT interval variability and provokes torsades de pointes. Br J Pharmacol 2007;151:941–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li J, Seyler C, Wiedmann F, Schmidt C, Schweizer PA, Becker R, Katus HA, Thomas D. Anti-KCNQ1 K⁺ channel autoantibodies increase IKs current and are associated with QT interval shortening in dilated cardiomyopathy. Cardiovasc Res 2013;98:496–503. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.