

SUMMARY
Animal cloning can be achieved through somatic cell nuclear transfer (SCNT), although the live birth rate is relatively low. Recent studies have identified H3K9me3 in donor cells and abnormal Xist activation as epigenetic barriers that impede SCNT. Here we overcome these barriers using a combination of Xist knockout donor cells and overexpression of Kdm4 to achieve more than 20% efficiency of mouse SCNT. However, post-implantation defects and abnormal placentas were still observed, indicating that additional epigenetic barriers impede SCNT cloning. Comparative DNA methylome analysis of IVF and SCNT blastocysts identified abnormally methylated regions in SCNT embryos despite successful global reprogramming of the methylome. Strikingly, allelic transcriptomic and ChIP-seq analyses of pre-implantation SCNT embryos revealed complete loss of H3K27me3 imprinting, which may account for the postnatal developmental defects observed in SCNT embryos. Together, these results provide an efficient method for mouse cloning while paving the way for further improving SCNT efficiency.
Graphical Abstract

In Brief
Matoba et al. show that mouse cloning efficiency can be significantly improved by simultaneously overcoming two reprogramming barriers, Xist and H3K9me3, although efficiency is still low in comparison with IVF. Comprehensive analyses showed that SCNT embryos can have complete loss of H3K27me3 imprinting, likely explaining this low efficiency.
INTRODUCTION
Mammalian oocytes are capable of reprogramming somatic cells into a totipotent state through somatic cell nuclear transfer (SCNT) (Ogura et al., 2013; Wakayama et al., 1998; Wilmut et al., 1997). Because SCNT enables cloning of animals, this technique has great potential in agro-biotechnology and conservation of endangered species (Hochedlinger and Jaenisch, 2003; Yang et al., 2007). However, the extremely low cloning rate makes the actual use of this technique difficult. For example, in the case of the mouse, only about 30% of SCNT embryos develop to blastocysts, and only 1 %–2% of embryos transferred to surrogate mothers can reach term (Loi et al., 2016; Ogura et al., 2013). Furthermore, the surviving embryos frequently exhibit abnormalities in extraembryonic tissues such as the placenta and umbilical cord (Ao et al., 2017; Loi et al., 2006; Smith et al., 2012; Tanaka et al., 2001). These observations strongly suggest that SCNT reprogramming has some inevitable failure that impedes embryo development. Although previous studies have identified various epigenetic abnormalities, such as DNA methylation (Dean et al., 2001; Yamagata et al., 2007), histone modifications (Wang et al., 2007; Zhang et al., 2009), and genomic imprinting (Mann et al., 2003; Okae et al., 2014), in SCNT embryos, pinpointing a specific epigenetic abnormality to a specific defect of the SCNT embryos remains a daunting task because of the lack of understanding of the reprogramming process.
We have previously identified histone H3 lysine 9 trimethylation (H3K9me3) in donor somatic cells as a major epigenetic barrier that impedes SCNT reprogramming (Matoba et al., 2014). We and others showed that H3K9me3 in donor cells prevents transcriptional activation of the associated regions at zygotic genome activation (ZGA), leading to developmental arrest of SCNT embryos at preimplantation stages in both mouse and human (Chung et al., 2015; Liu et al., 2016; Matoba et al., 2014). Importantly, removal of the H3K9me3 barrier by overexpressing a H3K9me3-specific demethylase, Kdm4d, allows SCNT embryos to develop to the blastocyst stage at a rate similar to that of in vitro fertilization (IVF) (Matoba et al., 2014). Consequently, the overall cloning efficiency, term rate, is increased by 8–9 times. Although the use of Kdm4d in SCNT results in an implantation rate comparable with that of IVF, only less than 15% of the implanted SCNT embryos develop to term (Matoba et al., 2014). Moreover, abnormally large placentas are still observed in Kdm4d-injected SCNT embryos (Matoba et al., 2014), indicating that the H3K9me3 reprogramming barrier mainly impedes preimplantation development and that other barriers affect postimplantation development.
We have previously identified Xist as one such barrier important for postimplantation development of mouse SCNT embryos (Inoue et al., 2010; Matoba et al., 2011). Xist is an X-linked non-coding RNA expressed exclusively from the paternal X chromosome in preimplantation IVF embryos but is activated from both paternal and maternal X chromosomes after SCNT (Inoue et al., 2010). Abnormal expression of Xist from the maternal X chromosome leads to ectopic X chromosome inactivation (XCI) and global transcriptional alteration in preimplantation embryos, resulting in postimplantation developmental failure of SCNT embryos (Inoue et al., 2010). Importantly, this developmental failure caused by ectopic Xist expression can be overcome by using Xist knockout (KO) somatic cells as donor cells or by injecting small interfering RNA against Xist into 1-cell male SCNT embryos, leading to a 8–10 times increase in term rate (Inoue et al., 2010; Matoba et al., 2011).
In this study, we tested whether a combined approach, using Xist KO donor cells coupled with Kdm4d mRNA injection, can improve the cloning efficiency. We show that the combined approach resulted in very high cloning efficiencies in different donor cell types, reaching over 20%. However, many of the SCNT embryos still exhibit postimplantation developmental arrest, and the surviving embryos have an abnormally large placenta, suggesting that some reprogramming defects still persist. Comparative methylome, RNA sequencing (RNA-seq), and chromatin immunoprecipitation sequencing (ChIP-seq) analyses revealed abnormal DNA methylation and loss of histone H3 lysine 27 trimethylation (H3K27me3)-dependent imprinting in SCNT blastocyst embryos, which likely are the cause of the observed developmental defects.
RESULTS
Kdm4d Injection Does Not Alleviate SCNT-Associated Abnormal Xist Activation
Previous studies have demonstrated that the inactive X chromosome is marked by its punctate staining with an anti-H3K27me3 antibody (Plath et al., 2003). Consistently, such punctate staining is detectable only in female (XX) cells, but not in male (XY) cells, in IVF embryos (Inoue et al., 2010). In contrast, we observed such punctate staining in SCNT embryos when male Sertoli cells were used as donor cells (Figure 1A), suggesting abnormal Xist activation in SCNT embryos, which is consistent with a previous report (Inoue et al., 2010).
Figure 1. Combined Use of Xist KO Donor Cells and Kdm4d mRNA Injection Does Not Completely Restore the Developmental Potential of SCNT Embryos.

(A) Representative images of IVF and SCNT blastocysts stained with anti-H3K27me3, anti-Cdx2, and anti-Oct4 antibodies and DAPI. Arrows indicate punctate H3K27me3 signals representing ectopically inactivated X chromosomes. Three to five embryos were examined for each condition. XX and XY represent female and male, respectively. Note that the ectopic XCIs can be observed regardless of Kdm4d mRNA injection. Scale bar, 50 μm.
(B) Bar graphs showing the ratio of cells with or without punctate H3K27me3 signals (representing inactivated X chromosomes) in IVF and SCNT blastocysts. Each column represents a single blastocyst. XX and XY represent female and male, respectively.
(C) Bar graphs showing the pup rate of SCNT embryos examined by cesarian section on E19.5. Note that a combination of using Xist KO donor cells with Kdm4d mRNA injection additively improves the term rate of SCNT embryos with cumulus cells, Sertoli cells, and MEF cells as donors, n represents the number of embryos transferred to pseudopregnant females.
(D) An image of an adult male mouse derived by SCNT using Xist KO Sertoli cells combined with Kdm4d mRNA injection, and its pups, generated through natural mating with a wild-type female.
(E) Boxplots showing the weight of placentae examined by cesarian section on E19.5. The whiskers represent the maximum and minimum, n represents the number of placentae examined. The p value was calculated by t test. ***p < 0.001. ns, not significant.
(F) Representative images of histological sections of a term placenta stained with periodic acid-Schiff (PAS). Microscopic images for each sample were combined into a single panel by adjusting the scale. Note that the PAS-positive spongiotrophoblast layer has invaded the labyrinthine layer in the SCNT placenta regardless of the genotype of the Xist allele in donor cells. Scale bar, 1mm.
Importantly, Kdm4d injection does not alter the punctate staining pattern or its frequency compared with no-injection control SCNT embryos (Figures 1A and 1B). These results demonstrate that H3K9me3 in donor cells and ectopic activation of Xist in SCNT embryos are two independent barriers in SCNT reprogramming.
Combinational Use of Xist Mutant Donor Cell and Kdm4d mRNA Injection Greatly Improves Cloning Efficiency
The fact that the two reprogramming barriers are independent of each other prompted us to ask whether a combined approach by using Xist KO donor cells and injecting Kdm4d will have a synergistic or additive effect to achieve increased cloning efficiency. After confirming that the ectopic XCI in the SCNT blastocysts was fixed by using Xist KO donor cells (Figures S1A and S1B), we performed embryo transfer using cumulus cells as nuclear donors. In the wild-type control with the B6D2F1 (BDF1) background, only 1.2% of embryos transferred to the surrogate mother reached term (Figure 1C; Table S1), which is consistent with previous reports (Inoue et al., 2003; Kishigami et al., 2006; Matoba et al., 2014). Kdm4d mRNA injection increased the pup rate to 8.4%, consistent with our previous observation (Matoba et al., 2014). When cumulus cells derived from Xist heterozygous female mice were used as donor cells and combined with Kdm4d mRNA injection, the pup rate increased to 18.7% (Figure 1C; Table S1). Similarly, the pup rate of Sertoli cell-derived SCNT embryos (male) was improved from 1.8% to 9.1% by Kdm4d mRNA injection and further increased to 23.5% by combining Xist KO with Kdm4d mRNA injection (Figure 1C; Table S1). Importantly, this additive effect was also observed using male mouse embryonic fibroblast (MEF) cells of a different Xist mutant line (Marahrens et al., 1997) in a hybrid (129S1/Svj × CAST/EiJ) genetic background (Figure 1C; Table S1). The pups generated using the combined approach grew up to adulthood and showed normal fertility (Figure 1D; Table S1).
Although we achieved a high cloning efficiency by using a combined approach, even the highest cloning efficiency of 23.5% is still less than half of the IVF pup rate, which is more than 50% of transferred embryos. Indeed, 65% (37 of 57) of the implanted embryos arrested during postimplantation development in the combined Sertoli cell SCNT group (Table S1). A careful morphological examination at different embryonic stages revealed that the embryo arrest starts just after implantation and gradually increases as development proceeds (Figure S1C–S1E). Moreover, morphological and histological analyses revealed that the large placental phenotype (Figure 1E), which is associated with invasion of the periodic acid-Schiff (PAS)-positive spongiotrophoblast cells into the labyrinth layer, was not rescued by Kdm4d mRNA injection or the combined approach (Figures 1E and 1F; Table S1). Thus, despite the combined positive effect of Xist KO and Kdm4d mRNA injection in improving SCNT embryo development, other reprogramming barriers may contribute to the developmental failure of these SCNT embryos.
Extensive DNA Methylation Reprogramming Is Achieved in Combinational Reprogrammed SCNT Blastocysts
Because the combinational reprogrammed SCNT embryos begin to exhibit developmental defects right after implantation (Figure S1C), we postulated that reprogramming-related epigenetic defects would have already existed in the SCNT blastocysts, although they appear morphologically normal. To identify such epigenetic defects, we first generated whole-genome bisulfite sequencing (WGBS) data of SCNT blastocysts derived from Xist KO MEF cells (129S1/Svj × CAST/EiJ background) combined with Kdm4d mRNA injection and compared them with those of genetically matched IVF blastocysts (Figure 2A),
Figure 2. Extensive Reprogramming of DNA Methylation in SCNT Blastocysts.
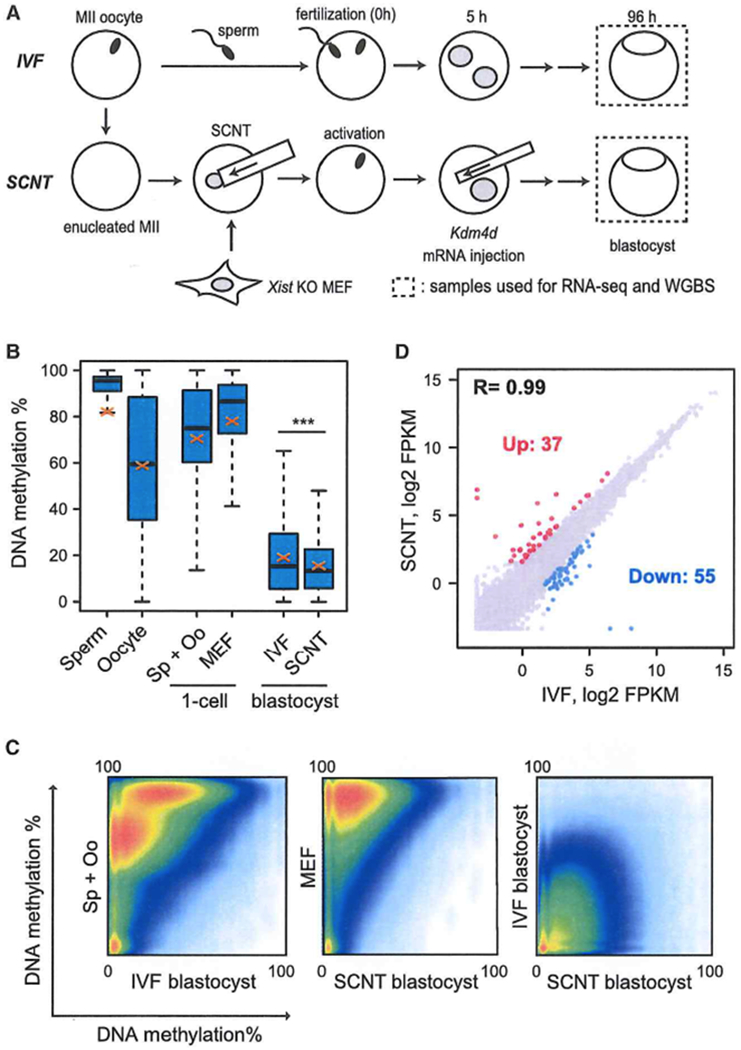
(A) Schematic illustration of the experimental approach. Blastocysts generated by IVF or SCNT (combination of Xist KO donor and Kdm4d injection) were used for whole-genome bisulfite sequencing (WGBS) and RNA-seq.
(B) Boxplots comparing the DNA methylation levels of all covered CpGs across the genome of SCNT and IVF blastocysts as well as MEFs, zygotes, sperm, and oocytes. In this and subsequent box plots, thick lines in boxes indicate the medians, red crosses stand for the mean, and the whiskers represent the 2.5th and 97.5th percentiles. ***p < 2.2e–16 by t test. Sp+Oo represents the average value of sperm and oocytes. WGBS datasets of MEF, sperm, and oocytes were obtained from GEO: GSE56151 and GEO: GSE56697.
(C) Scatterplot comparing the DNA methylation levels between each sample. Note that the heavily methylated donor MEF cell genome is globally reprogrammed by SCNT, resulting in a similar DNA methylation profile as that of IVF blastocysts.
(D) Scatterplot comparing the gene expression profiles of IVF and SCNT blastocysts. Genes upregulated (Up, FC > 3.0) and downregulated (Down, FC > 3.0) in SCNT embryos are colored red and blue, respectively.
We obtained DNA methylation information of 20.6 and 20.9 million CpG sites from IVF and SCNT blastocysts, respectively. For comparison, we also obtained WGBS datasets for MEFs (Yu et al., 2014), sperm, and oocytes (Wang et al., 2014) from a public database. First, we calculated the average DNA methylation level of all covered CpGs and found that the average methylation levels in sperm and oocytes are 82.2% and 58.8%, respectively. The average methylation level of sperm and oocytes, which served as the starting methylation level of IVF zygotes, was 70.5% (Figure 2B). However, the highly methylated gametes were globally reprogrammed to a low methylation level by the blastocyst stage (19.1 %), likely because of the active and passive demethylation processes taking place during preimplantation development (Guo et al., 2014; Shen et al., 2014).
Next we calculated the DNA methylation levels of MEF cells and SCNT blastocysts of the commonly covered CpGs and found that the donor MEF cells are highly methylated (78.0%), but the SCNT blastocysts were lowly methylated, with a methylation level (15.6%) similar to that of IVF blastocysts (19.1%), indicating successful global reprogramming of DNA methylation state (Figure 2B). Indeed, pairwise comparisons of DNA methylation revealed that both IVF and SCNT blastocysts possess extremely low DNA methylation compared with that in gametes or MEF cells (Figure 2C). Not only the global methylation level but also the distribution of methylated CpG is similar (Figure S2A). Consistent with a similar global DNA methylation pattern, RNA-seq revealed a highly similar transcriptome between IVF and SCNT blastocysts (correlation coefficient [R] = 0.99) (Figure 2D; Figure S2B). Indeed, among the 8,921 genes detected (fragments per kilobase of transcript per million mapped reads [FPKM] > 3 in at least one sample), only 92 genes were differentially expressed (fold change [FC] > 3) in SCNT blastocysts (Figure 2D; Table S2), These results indicate that DNA methylation and the transcriptome are largely reprogrammed by SCNT at the blastocyst stage.
Identifying and Characterizing Differentially Methylated Regions in SCNT Blastocysts
Despite successful global reprogramming of the DNA methylome, the mean methylation level of SCNT blastocysts (15.6%) was slightly but significantly lower (p < 2.2e–16, t test) than that of the IVF blastocysts (19.1%) (Figure 2B). We thus performed genome-wide scanning analysis (10 CpGs as a minimum window size) to identify differentially methylated regions (DMRs) between SCNT and IVF blastocysts, which uncovered 56,240 DMRs with an absolute methylation difference greater than 10% (Figure 3A). The majority of these DMRs (48,315 or 85.9%) showed lower DNA methylation in SCNT compared with that of IVF and were termed hypoDMRs (Table S3). We also found 7,925 regions with higher DNA methylation in SCNT compared with that of IVF and were termed hyperDMRs (Table S3). Interestingly, the average length of hyperDMRs (741 bp) is much shorter than that of hypoDMRs (5,743 bp) (Figure 3B). Indeed, a representative hyperDMR is present in the promoter or enhancer region as a sharp peak, whereas a representative hypoDMR covers an entire gene-coding region (Figure S3A and S3B). Consistently, hyperDMRs and hypoDMRs exhibit distinct genomic distributions with hyperDMR enriched in intergenic regions, whereas hypoDMR was enriched in the gene body (Figure 3C).
Figure 3. Identification and Characterization of DMRs in SCNT Blastocysts.

(A) Boxplots showing the DNA methylation levels of SCNT and IVF blastocysts at hyper- and hypoDMRs. Thick lines in boxes indicate the medians, and red crosses stand for the mean. The number of DMRs is also indicated.
(B) Boxplots comparing the lengths of hyper- and hypoDMRs.
(C) Pie chart distribution of hyper- and hypoDMRs in the genome.
(D) Average DNA methylation levels of the indicated samples at hypoDMRs compared with their flanking regions.
(E) Paternal (Pat) and maternal (Mat) allele-specific DNA methylation levels of IVF and SCNT blastocysts at hypoDMRs compared with their flanking regions
(F) Pat and Mat allele-specific DNA methylation levels of IVF and SCNT embryos at the indicated developmental stages at hypoDMRs compared with their flanking regions.
(G) Average DNA methylation levels of the indicated samples at hyperDMRs compared with their flanking regions.
(H) Average DNA methylation levels of the indicated samples at hyperDMRs compared with their flanking regions. Datasets used were from GEO: GSE11034.
To understand how these DMRs are formed and whether they could contribute to the postimplantation developmental failure of SCNT embryos, we first focused our analysis on the hypoDMRs. In sperm, the methylation level at hypoDMRs was significantly higher than at the flanking regions (~90% versus ~80%) (Figure 3D). In oocytes, the methylation difference between hypoDMRs and the flanking regions is even greater (~75% versus ~60%) (Figure 3D). In contrast, the methylation difference between hypoDMRs and the flanking regions is much smaller in MEFs (Figure 3D). Thus, the hypoDMRs of SCNT blastocyst correlate well with their relatively higher DNA methylation levels in gametes, which remain at a higher level in IVF blastocysts should both SCNT and IVF embryos go through the same number of replication-dependent dilutions. Consistent with this notion, a visual inspection of representative hypoDMRs in genome browser view revealed that the methylation peaks in oocytes clearly overlap with those in IVF blastocysts (Figure S3B). Allelic DNA methylation analysis also supports this notion because the methylation pattern in IVF blastocysts are strongly biased to the maternal allele (Figure 3E). Indeed, analysis of published WGBS datasets of preimplantation embryos (Wang et al., 2014) revealed that the maternal allele maintains its high DNA methylation level at hypoDMRs until the 4-cell stage, whereas the paternal allele quickly loses its methylation at these regions (Figure 3F). However, this maternal allele-biased methylation is quickly lost after implantation, even in IVF embryos (Figure S3C), indicating that these methylation differences exhibited in SCNT preimplantation embryos are unlikely to make a major contribution to the postimplantation development failure.
Next we analyzed the hyperDMRs. The methylation levels at hyperDMRs and flanking regions are similar (~50%) in oocytes, whereas they are significantly lower than at the flanking regions (~55% versus ~80%) in sperm (Figure 3G). Despite the difference in methylation levels, both hyperDMRs and flanking regions are demethylated to a very low level (~20%) in IVF blastocysts (Figure 3G). In contrast, hyperDMRs are heavily methylated (~80%), with even higher methylation level than that of the flanking regions in MEFs (Figure 3G). The fact that hyperDMRs are heavily methylated in MEFs but not in gametes suggests that low methylation at these regions might be a unique feature of the germline. Indeed, analyses of public DNA methylome datasets of different cell types revealed that hyperDMRs are indeed heavily methylated in all somatic cell types analyzed but less methylated only in spermatocytes, spermatids, and oocytes (Figure 3H). Consistently, gene ontology (GO) analysis of the genes associated with hyperDMRs revealed significant enrichment of germline-related functions such as spermatogenesis and gametogenesis (Figure S3D). HyperDMRs appear to be demethylated during primordial germ cell (PGC) development by Tet1 (Yamaguchi et al., 2013) because hydroxymethylcytosines (5hmC) was significantly enriched in the hyperDMRs in PGCs (Figure S3E). These results suggest that hyperDMRs are mostly related to germline development but not embryonic development.
DNA Methylation-Dependent Imprinting Genes Are Largely Normal in SCNT Blastocysts
Defective placental development is a central feature in SCNT embryos. Previous studies have established that genomic imprinting plays a critical role in placental development (Coan et al., 2005). Therefore, besides the identified DMRs that can potentially contribute to the post-implantation defects of the SCNT embryos, it is essential to evaluate the effect of our combinatorial approach on genomic imprinting. To this end, we first focused our analysis on the DNA methylation level of the 23 known imprinting control regions (ICRs) because allelic expression of imprinted genes is largely controlled by allelic ICR methylation (Kobayashi et al., 2012). Comparative DNA methylation analysis revealed that, although DNA methylation levels are slightly reduced at most ICRs, 21 of the 23 ICRs maintained at least half that of the IVF blastocyst level (Figure 4A; Table S4), indicating that DNA methylation-mediated genomic imprinting is largely maintained. Indeed, most of the 20 ICRs with sufficient allele-specific methylation information (more than 5 detected CpG in both alleles of both IVF and SCNT blastocysts) showed consistent allele-specific DNA methylation trend between IVF and SCNT blastocysts (Figure 4B). Visual inspection of representative ICRs (Dlk1-Meg3, Impact, Slc38a4, Snrpn-Snurf, Peg10-Sgce, and H19) confirmed that they largely maintain an allelic DNA methylation pattern in SCNT blastocysts, with the exception of Impact, whose ICR is abnormally hypermethylated (Figure 4C; Figure S4).
Figure 4. Imprinting Status of the Canonical Imprinting Genes and Their ICRs.

(A) Bar graphs showing relative DNA methylation levels of the 23 known imprinting control regions (ICRs) in SCNT blastocysts. The methylation level of IVF blastocysts was set as 1. The dashed line indicates 50% of the IVF blastocyst methylation level. Note that 21 of 23 ICRs maintained at least 50% of the IVF methylation levels in SCNT blastocysts, but Snrpn-Snurf and Slc38a4 ICRs (marked as red) showed less than 50% that of the IVF level.
(B) Bar graph showing allelic bias of DNA methylation at 20 ICRs with sufficient allele-specific methylation information (more than 5 detected CpGs in both alleles of both IVF and SCNT blastocysts). Note that most ICRs maintained allelic biased DNA methylation in SCNT blastocysts, except the Impact ICR.
(C) Genome browser view showing DNA methylation levels flanking the Dlk1-Meg3 ICR on mouse chromosome 12, The red box represents the Dlk1-Meg3 ICR (Tomizawa et al., 2011). Note that the paternally biased DNA methylation pattern at the Dlk1-Meg3 ICR is maintained in SCNT embryos. M, maternal allele; P, paternal allele.
(D) Bar graphs showing relative gene expression levels of known imprinted genes in SCNT blastocysts. Shown are 45 imprinted genes reliably detectable in IVF blastocysts (FPKM >1). The expression level of IVF blastocysts was set as 1. Genes were classified as up, down, and unchanged based on their expression levels in SCNT embryos compared with IVF embryos (FC > 1.5).
(E) Bar graphs showing the ratio of allelic expression (Mat divided by Pat) of known imprinted genes in IVF and SCNT blastocysts. Shown are 6 maternally expressed genes (MEGs; Mat/Pat > 2.0) that are expressed at a reliably detectable level with sufficient SNP-tracked reads (FPKM > 1, mean SNP reads > 10 in either sample) in IVF blastocysts. Asterisks represent 100% bias to the Mat allele. Note that all 6 MEGs maintained Mat allelic bias in SCNT blastocysts.
(F) Bar graphs showing the ratio of allelic expression (Pat divided by Mat) of known imprinted genes in IVF and SCNT blastocysts. Shown are 13 paternally expressed genes (PEGs; Pat divided by Mat > 2.0) that are expressed at a reliably detectable level with sufficient SNP-tracked reads (FPKM > 1, mean SNP reads > 10 in either sample) in IVF blastocysts. Asterisks represent 100% bias to the Pat allele. Arrows indicate genes that lost Pat biased expression in SCNT blastocysts. Red marks indicate H3K27me3-dependent imprinted genes.
To further evaluate the potential role of DNA methylation-mediated genomic imprinting in the SCNT defects, we analyzed our RNA-seq datasets focusing on the 126 known imprinted genes. Of the 45 imprinted genes reliably detectable in IVF blastocysts (FPKM > 1), only 6 were significantly upregulated in SCNT blastocysts compared with that in IVF blastocysts (FC > 1.5) (Figure 4D). Allelic expression analysis (FPKM > 1, mean SNP reads > 10 in either sample) revealed that, among the 36 imprinted genes with a sufficient number of SNP reads, 6 showed maternal allele biased (maternal divided by paternal > 2.0) and 13 showed paternal-allele biased (paternal divided by maternal > 2.0) expression in IVF blastocysts (Figures 4E and 4F, blue bars). All 6 maternally expressed genes (MEGs) maintained their maternally biased expression in SCNT blastocysts (Figure 4E). Among the 13 paternally expressed genes (PEGs), 7 lost allelic bias and became biallelically expressed in SCNT blastocysts (arrows in Figure 4F). Interestingly, the 7 PEGs that lost imprinted expression in SCNT blastocysts included Slc38a4, Sfmbt2, Jade1, and Gab1 (red in Figure 4F), whose imprinted expression is known to be independent of DNA methylation but dependent on maternally deposited H3K27me3 (Inoue et al., 2017a).
Loss of H3K27me3-Dependent Imprinting in SCNT Blastocysts
The above observation prompted us to focus our analysis on the H3K27me3-dependent imprinted genes (Inoue et al., 2017a). As a first step, we analyzed allelic expression of H3K27me3-dependent imprinted genes using our blastocyst RNA-seq datasets. Among the 76 genes that exhibit H3K27me3-dependent imprinted expression in the morula embryos (Inoue et al., 2017a), 26 are expressed at a reliably detectable level (FPKM > 1) in our IVF blastocysts. Interestingly, the majority of them (15 of 26) are significantly upregulated in SCNT blastocysts (FC > 1.5) (Figure 5A). Allelic expression analysis revealed that, of the 23 genes with sufficient SNP reads (FPKM > 1, mean SNP reads > 10 in either sample), 17 showed paternally biased expression (paternal divided by maternal > 2.0) in IVF blastocysts under the genetic background analyzed (129S1/Svj × CAST/EiJ). Strikingly, all 17 PEGs lost paternal allele-biased expression and showed biallelic expression in SCNT blastocysts (Figure 5B). These results suggest that the H3K27me3 imprint marks that control the imprinted expression of these genes are likely lost in SCNT embryos.
Figure 5. Loss of H3K27me3-Dependent Imprinting in SCNT Blastocysts.
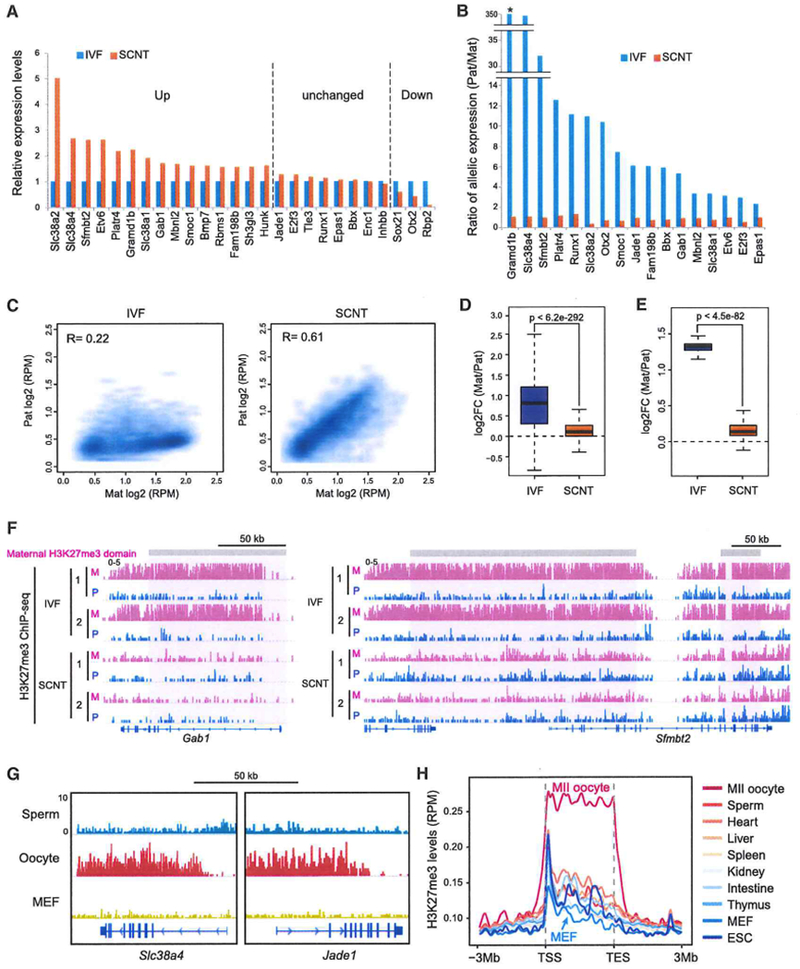
(A) Bar graphs showing relative gene expression levels of H3K27me3-imprinted genes in SCNT blastocysts. Shown are the 26 genes expressed in IVF blastocyst at a reliably detectable level (FPKM > 1). The expression level of IVF blastocysts was set as 1. Genes were classified as up, down, and unchanged by expression changes in SCNT compared with that in IVF blastocysts (FC > 1.5).
(B) Bar graphs showing the ratio (Pat divided by Mat) of allelic expression of the H3K27me3-imprinted genes in IVF and SCNT blastocysts. Among the 26 expressed genes (FPKM > 1), 17 genes with more than 10 SNP reads in either sample are shown. The asterisk represents 100% bias to the Pat allele. Note that all 17 genes lost their Pat allelic bias in SCNT blastocysts.
(C) Scatterplots comparing Mat and Pat H3K27me3 values at 4,135 Mat PI3K27me3-domains identified in IVF embryos. Note that the Mat bias of H3K27me3 at these domains is lost in SCNT embryos.
(D) Boxplots comparing the ratio (Mat/Pat) of allelic H3K27me3 enrichment at the 4,135 Mat H3K27me3 domains in IVF and SCNT morulae. The p value was calculated by t test.
(E) Boxplots comparing the ratio (Mat/Pat) of allelic H3K27me3 enrichment at the 76 H3K27me3-dependent imprinted genes in IVF and SCNT morulae. The p value was calculated by t test.
(F) Genome browser view of H3K27me3 ChIP-seq signals at two H3K27me3-dependent imprinted genes, Gab1 and Sfmbt2.
(G) Genome browser views of H3K27me3 ChIP-seq signals at representative H3K27me3-dependent imprinted genes in sperm, oocytes, and MEF cells.
(H) The average H3K27me3 ChIP-seq intensity of various cell types (oocytes, sperm, MEFs, and embryonic stem cells [ESCs]) and tissues at the 76 H3K27me3-imprinted genes compared with 3-Mb flanking regions. Datasets used were from GEO: GSE49847 and GEO: GSE76687.
To provide direct evidence that loss of imprint (LOI) gene expression is indeed due to loss of H3K27me3 imprinting, we performed ChIP-seq on H3K27me3 using IVF and SCNT morula embryos. To distinguish parental alleles, we used BDF1 × PWK hybrid cells as donors. Using 50–60 morula embryos, we obtained over 40,000 highly reproducible H3K27me3 peaks (Figure S5A). H3K27me3 exhibited very similar global distribution patterns in IVF and SCNT embryos, with extremely low enrichment at CpG island (CGI) or promoters (Figure S5B), consistent with a previous report (Zheng et al., 2016). Interestingly, this distribution pattern is in direct contrast to those of somatic cells, including those of the liver and heart, and MEFs, which have higher enrichment at CGI and CGI-associated promoters (Figure S5B). Additionally, somatic cell promoter-associated H3K27me3 is globally depleted in SCNT embryos to a level similar to that of IVF embryos (Figure S5C). These results suggest that, at a global level, somatic H3K27me3 distribution pattern is reprogrammed by SCNT into a state similar to that of IVF embryos at the morula stage.
We then examined the allele-specific enrichment of H3K27me3 in SCNT embryos. Consistent with previous reports (Inoue et al., 2017b; Zheng et al., 2016), we identified 4,135 large maternally biased H3K27me3 domains in IVF embryos (Table S5; Figure 5C). Remarkably, H3K27me3 distribution in these domains was largely equalized in SCNT embryos (Figures 5C and 5D). Moreover, consistent with loss of allelic gene expression, the maternal H3K27me3 bias is completely lost at the 76 H3K27me3-dependent imprinted genes in SCNT embryos (Figure 5E). Visual inspection confirmed that the maternally biased large H3K27me3 domains observed in the IVF embryos are lost in the SCNT embryos (Figure 5F; Figure S5D). These results clearly demonstrate that H3K27me3-dependent imprinted genes completely lose their imprints in the SCNT embryos.
We next asked why these H3K27me3-dependent imprinted genes lose imprinting in SCNT blastocysts. Because maternal allele-specific H3K27me3 domains are deposited during oogenesis, we postulated that the H3K27me3 pattern in donor MEFs may differ from that in oocytes. Analysis of available H3K27me3 ChIP-seq datasets of fully grown oocytes and MEF cells revealed that the H3K27me3 domains at these imprinted genes in oocytes are completely absent in MEF cells (Figure 5G; Figure S5E). We further expanded our analysis to other somatic cell types and found that the H3K27me3 domains in these imprinted genes are generally absent from somatic cell types analyzed and, therefore, are unique to the oocyte genome (Figure 5H). These results strongly suggest that lack of H3K27me3 methylation at the maternal allele of these imprinted genes in donor somatic cells is likely the cause of LOI after SCNT.
DISCUSSION
Despite successful cloning of more than 20 mammalian species by SCNT (Loi et al., 2016; Ogura et al., 2013), the cloning efficiency is uniformly low, and developmental abnormalities, including placenta overgrowth, are observed in essentially all cloned mammalian species (Ao et al., 2017; Loi et al., 2006; Ogura et al., 2013; Smith et al., 2012; Tanaka et al., 2001). It has been speculated that epigenetic abnormalities are responsible for the developmental failure of the cloned animals. In this study, we combined two approaches to overcome two previously identified reprogramming barriers that impede development of mouse SCNT embryos (Inoue et al., 2010; Matoba et al., 2014). The combinatorial use of Xist KO donor somatic cells and Kdm4d mRNA injection indeed increased the overall cloning efficiency (term rate) by 13- to 16-fold (e.g., 23% using Sertoli cells). This efficiency is remarkable because it is close to that of intra-cytoplasmic sperm or round spermatid injection (ICSI or ROSI), where similar nuclear injection is involved (Ogonuki et al., 2010; Ogura et al., 2005). This achievement clearly demonstrates that H3K9me3 in donor cells and abnormal activation of Xist represent two major barriers of SCNT cloning and, therefore, establishes a foundation for understanding the molecular mechanisms of SCNT-mediated reprogramming.
Despite the remarkable improvement, many of the SCNT embryos generated using the combinational approach failed to develop after implantation. Moreover, placental overgrowth was still observed, regardless of the donor cell types, indicating that additional barriers exist for high-efficiency animal cloning. To identify these additional reprogramming barriers, we narrowed down the time when the developmental failure begins and generated a WGBS dataset from SCNT blastocysts right before the developmental phenotype appears. Comparative DNA methylome analysis revealed successful global DNA methylome reprogramming by SCNT, indicating that the DNA demethylation machineries in ooplasm and cleavage embryos are similarly functional in SCNT embryos compared with IVF embryos. Nevertheless, detailed analysis of DNA methylomes revealed many DMRs between IVF and SCNT blastocysts. Interestingly, hyperDMRs are enriched in genomic regions demethylated in the germline, which is consistent with the fact that germ cell-specific genes are demethylated by TET1 during germ cell development, particularly at the PGC stage (Vincent et al., 2013; Yamaguchi et al., 2012). However, SCNT bypasses this demethylation process. Our list of hyperDMR-associated genes does not include the germline genes reported to be quickly demethylated at 1-cell SCNT embryos (Chan et al., 2012), indicating that some germline genes are demethylated after SCNT. On the other hand, hypoDMRs mainly overlap with regions that are methylated in oocytes. Maternal DNA methylation at these regions appears to escape the demethylation processes, particularly before the 8-cell stage, in IVF embryos. The underlying mechanism for the maternal allele-specific maintenance of DNA methylation before the 8-cell stage is of interest for future study. Collectively, it appears that the SCNT-specific DMRs, either high or low, are formed because of the unique feature of gametogenesis, which is inherited by the blastocysts through normal fertilization. Although maternally biased methylation appears to be mostly equalized after implantation, some maternal DNA methylation passed down from oocytes through fertilization has been shown to play important roles in trophoblast development (Branco et al., 2016). Therefore, loss of the oocyte-like DNA methylation pattern in SCNT blastocysts may partially contribute to developmental defects of SCNT embryos.
Our analysis of DNA methylation-imprinted genes revealed that most ICRs largely maintain their imprinting status as well as their allelic expression pattern in SCNT blastocysts. One exception is Impact ICR, which gained DNA methylation at the paternal allele to become biallelically methylated after SCNT. Although we had previously observed frequent LOI at Dlk1-Meg3 ICR in embryonic day 13.5 (E13.5) SCNT embryos and its high correlation with embryonic lethality (Okae et al., 2014), this ICR maintained normal allelic DNA methylation at the blastocyst stage, suggesting that occasional disruption at this ICR may take place after implantation. In contrast to the largely maintained status of DNA methylation imprinting, the recently discovered H3K27me3 imprinted genes are completely dysregulated and exhibit biallelic expression in SCNT blastocysts. Consistent with their biallelic expression, the maternal allelic H3K27me3 domains observed in IVF embryos are globally lost in SCNT embryos. The list of dysregulated non-canonical imprinted genes in SCNT blastocysts included Slc38a4, Sfmbt2, and Gab1, consistent with a previous report of LOI of these three genes in the placentas of E13.5 SCNT embryos (Okae et al., 2014). Given that all of the three genes and a microRNA cluster located at the Sfmbt2 locus have been shown to play important roles in placental growth (Inoue et al., 2017c; Itoh et al., 2000; Miri et al., 2013; S.M., unpublished data), LOI of these genes likely contributes to the placenta overgrowth phenotype of SCNT embryos. Moreover, Runx1, Otx2, and Etv6 have been shown to play critical roles in mouse early embryonic development (Acampora et al., 1995; Okuda et al., 1996; Wang et al., 1997); thus, LOI of these genes at the blastocyst stage likely contributes to the embryonic lethality phenotype of SCNT embryos. The causes of LOI of the H3K27me3 imprinted genes in SCNT are likely due to the absence of H3K27me3 at these loci in the donor somatic cells. Further detailed analysis of the regulatory mechanisms of the H3K27me3-imprinted genes will provide clues for improving SCNT embryo development.
In summary, in addition to establishing the most efficient mouse cloning method by combining Kdm4d mRNA injection with the use of Xist KO donor cells, we also uncovered H3K27me3 imprinting as a potential barrier preventing efficient animal cloning. Based on the clear association of LOI at the H3K27me3 imprinted genes in mouse SCNT blastocysts and their critical functions in embryonic development, LOI at the H3K27me3 imprinted genes most likely accounts for the postimplantation phenotypes of SCNT embryos, although we cannot exclude the possibility of potential contribution of abnormal DNA methylation we identified in this study. Given that defective postimplantation development and abnormal placental phenotypes in SCNT embryos are commonly observed in mammalian species (Ao et al., 2017; Loi et al., 2006; Smith et al., 2012), analysis of H3K27me3-dependent imprinting status in cloned embryos of other species is of potential interest (Matoba and Zhang, 2018).
STAR★METHODS
CONTACT FOR REAGENTS AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Yi Zhang (yzhang@genetics.med.harvard.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
B6D2F1/J (BDF1) adult female mice at 9-15 week-old were used for the collection of recipient oocytes for SCNT. For mouse embryonic fibroblast (MEF) cell preparation, Xist KO adult female mice maintained in 129S1/Svlmj background (Marahrens et al., 1997) were mated with CAST/EiJ adult males to generate Xist heterozygous KO embryos in 129/CAST F1 background. Male MEF cells were used for SCNT. For cumulus (female) and Sertoli cell (male) preparation, Xist KO adult female mice in C57BL76N background (Sado et al., 2005) were mated with DBA/2 adult males to generate Xist heterozygous KO mice in BDF1 background. Since the sex of SCNT samples depends on that of the donor cells, and different donor cell types were used for each sex (male: MEF and Sertoli, female: cumulus), we did not perform analysis on the influence of sex in SCNT. For H3K27me3 ultra low input native ChIP-seq (ULI-NChlP-seq), Xist heterozygous KO female mice in BDF1 background and PWK males were used to produce Xist heterozygous KO male pups in hybrid genetic background as SCNT nuclear donors. All animal experiments were approved by the Institutional Animal Care and Use Committees of Harvard Medical School and RIKEN Tsukuba Institute. The mice were housed under controlled conditions; daily light period 07:00 to 21:00, temperature at 23 ± 1°C and humidity at 55% with free access to water and food.
Donor cell preparation
Primary MEF cells were derived from Xist KO male mouse embryos at 13.5 dpc. After removal of head and all organs, minced tissue from remaining corpus was dissociated in 500 μL of 0.25% Trypsin with 1 mM EDTA (Thermo Fisher Scientific # 25200056) for 10 min at 37°C. Cell suspension was diluted with equal amount of DMEM (Thermo Fisher Scientific # 11995-073) containing 10% FBS and Penicillin/Streptomycin (Thermo Fisher Scientific # 15140-022) and pipetted up and down 20 times. The cell suspension was diluted with fresh medium and plated onto 100 mm dishes and cultured at 37°C. Two days later, MEF cells were harvested and frozen. Frozen stocks of MEF cells were thawed and used for experiments after one passage.
Cumulus cells were collected from wild-type (WT) and Xist heterozygous KO adult females at 8-20 week-old (RIKEN Bioresource Research Center, RBRC01260) through superovulation by injecting 7.5 IU of pregnant mare serum gonadotropin (PMSG; Millipore # 367222) and 7.5 IU of human chorionic gonadotropin (hCG; Millipore # 230734). Fifteen to seventeen hours after the hCG injection, cumulus-oocyte complexes (COCs) were collected from the oviducts and treated briefly with HEPES-buffered potassium simplex-optimized medium (KSOM) containing 300 U/ml bovine testicular hyaluronidase (Calbiochem # 385931) to obtain dissociated cumulus cells.
Sertoli cells were collected from testes of 3-5 day-old WT or Xist KO male mice as described (Matoba et al., 2011). Testicular masses were incubated in PBS containing 0.1 mg/ml collagenase (Thermo Fisher Scientific # 17104-019) for 30 min at 37°C followed by 5 min treatment with 0.25% Trypsin with 1 mM EDTA at room temperature. After washing for four times with PBS containing 3 mg/ml bovine serum albumin, the dissociated cells were suspended in HEPES-KSOM medium.
METHODS DETAILS
Kdm4d mRNA synthesis
Kdm4d mRNA was synthesized by in vitro transcription (IVT) as described previously (Matoba et al., 2014). Briefly, a pcDNA3.1 plasmid containing full length mouse Kdm4d followed by poly(A)83 (Addgene # 61553) was linearized by Xbal. After purification, the linearized plasmid DNA was used as a template for IVT using mMESSAGE mMACHINE T7 Ultra Kit (Thermo Fisher Scientific # AM1345). The synthesized mRNA was dissolved in nuclease-free water and quantified by NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies). After the mRNA is diluted to 1500 ng/μl, aliquots were stored at −80°C.
SCNT
Mouse somatic cell nuclear transfer was carried out as described previously (Matoba et al., 2014; Ogura et al., 2000). Briefly, recipient MII oocytes were collected from adult BDF1 female mice through superovulation by injecting 7.5 IU of PMSG and 7.5 IU of hCG. Fifteen to seventeen hours after the hCG injection, cumulus-oocyte complexes (COCs) were collected from the oviducts and treated briefly with HEPES-KSOM containing 300 U/ml bovine testicular hyaluronidase to obtain MII oocytes. Isolated MII oocytes were enucleated in HEPES-buffered KSOM medium containing 7.5 μg/ml of cytochalasin B (Calbiochem # 250233) by using Piezo-driven micromanipulator (Primetech # PMM-150FU). The nuclei of cumulus or Sertoli cells were injected into the enucleated oocytes by using Piezo-driven micromanipulator. MEF cells were fused with enucleated oocytes by inactivated Sendai virus envelope (GenomOne CF; Ishihara Sangyo #CF001). After 1h incubation in KSOM, reconstructed SCNT oocytes were activated by incubating in Ca-free KSOM containing 3 mM strontium chloride (SrCl2) and 5 μg/ml cytochalasin B for 1 h, and further cultured in KSOM with 5 μg/ml cytochalasin B for 4 h. Activated SCNT embryos were washed 5 h after the onset of SrCl2 treatment (hours post activation, hpa) and cultured in KSOM in a humidified atmosphere with 5% CO2 at 37.8°C. Some SCNT embryos were injected with ~10 pl of 1500 ng/μl mouse Kdm4d mRNA at 5-6 hpa by using a Piezo-driven micromanipulator. SCNT experiment was performed at least twice for each condition.
IVF
COCs containing MII oocytes were collected from 129S1/Svlmj or BDF1 female mice by superovulation, and were co-incubated with CAST/EiJ or PWK sperm, respectively, in HTF medium. Four to five hours after initiating co-incubation, zygotes were transferred to KSOM for further cultivation.
Embryo transfer
Two-cell stage SCNT embryos were transferred to the oviducts of pseudopregnant (E0.5) ICR females. The pups were recovered by caesarian section on the day of delivery (E19.5) and nursed by lactating ICR females. Some females were sacrificed at E4.5 and E10.5 for examining embryonic development. The number of pseudopregnant females used as recipients (replicate) is indicated in Table S1.
Histological analysis of placenta
Placentae at E19.5 were fixed in 4% paraformaldehyde (PFA) 4°C overnight and routinely embedded in paraffin. Serial sections (4 μm in thickness) were subjected to periodic acid Schiff (PAS) staining. Each stained section was scanned with BZ-9000 microscopy (Keyence Japan) and processed with BZ-II Analyzer software (Keyence Japan). One to twenty placentae for each condition was examined (Figure 1E). Representative placental images for each sample were combined into a single panel by adjusting the scale (Figure 1F).
Immunostaining for H3K27me3 in blastocysts
Blastocysts were fixed with 4% paraformaldehyde (PFA) for 20 min at room temperature. After washing with PBS containing 10 mg/ml bovine serum albumin (BSA; Merck # 12657) (PBS/BSA), the fixed embryos were permeabilized by 15 min incubation with 0.5% Triton X-100. After blocking in PBS/BSA for 1 h at room temperature, they were incubated in a mixture of primary antibodies including rabbit anti-H3K27me3 antibody (1/500, Millipore # 07-449), goat anti-Oct4 antibody (1/500, SantaCruz # sc-8628) and mouse anti-Cdx2 antibody (1/100, BioGenex # AM392-5M) at 4°C overnight. Following three washes with PBS/BSA, the embryos were incubated with a mixture of secondary antibodies including fluorescein isothiocyanate-conjugated anti-mouse IgG (1/400, Jackson Immuno-Research), Alexa Flour 546 donkey anti-rabbit IgG (1/400, Thermo Fisher Scientific) and Alexa Flour 647 donkey anti-goat IgG (1/400, Thermo Fisher Scientific) for 1 h at room temperature. Finally, they were mounted with Vectashield with 4’,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories # H-1200). The fluorescent signals were observed using a laser-scanning confocal microscope (Zeiss LSM510) and an EM-CCD camera (Hamamatsu ImagEM). Three to five embryos were examined for each condition.
WGBS
IVF and SCNT embryos of the blastocyst stage (96 hours after fertilization or activation) were directly subjected to bisulfite conversion using the EZ DNA Methylation-Direct Kit (Zymo Research # D5020). Thirty-nine and 36 embryos were used for preparing the IVF and SCNT samples, respectively. A small amount (0.01 ng) of unmethylated Lambda DNA (Promega # D152A) was added to each sample before bisulfite conversion to serve as spike-in controls for evaluating bisulfite conversion efficiency. Sequencing libraries were prepared using the EpiGnome Methyl-Seq kit (Epicenter # EGMK81312) following the manufacturer’s instructions. Libraries were only amplified for 12 cycles, and were then purified using Agencourt AM Pure XP beads (Beckman Coulter # A63880). Final libraries were subjected to single-end 100 bp sequencing on a HiSeq 2500 sequencer (Illumina) with PhiX spike-in control. We obtained about 300 million mapped reads from each sample with 99.2% of bisulfite conversion efficiency.
RNA-seq
Six IVF or SCNT embryos of the blastocyst stage (96 hours after fertilization or activation) were directly lysed and used for cDNA synthesis using the SMARTer Ultra Low Input RNA cDNA preparation kit (Clontech, 634936). After amplification, the cDNA samples were fragmented using a Covaris M220 sonicator (Covaris). Sequencing libraries were made with the fragmented cDNA using NEBNext Ultra DNA Library Prep Kit for Illumina according to manufacturer’s instructions (New England Biolabs, E7370). Single-end 50 bp sequencing was performed on a HiSeq 2500 sequencer (Illumina). Two biological replicates were analyzed for each condition.
H3K27me3 ULI-NChIP-seq
Ultra-low input native ChIP-seq (ULI-NChIP-seq) using H3K27me3 antibody was performed as described previously (Brind’Amour et al., 2015; Inoue et al., 2017b). In brief, SCNT embryos were generated from Sertoli cells of Xist heterozygous knockout pups in BDF1 × PWK hybrid genetic background through Kdm4d mRNA injection. IVF embryos were generated using BDF1 oocytes and PWK sperm. Fifty to sixty IVF or SCNT embryos at the morula stage were collected at 72 hpa and pooled as a single sample with two biological replicates. After digestion of zona pellucida by acid Tyrode’s solution, polar bodies were removed by brief treatment with 0.25% trypsin 1 mM EDTA followed by several times of quick pipetting. The embryos were then washed three times with PBS containing 0.2% BSA (Merck # 12657) and processed for chromatin isolation and fragmentation by Micrococcal Nuclease (MNase: New England Biolabs # M0247S). 10% of MNase-treated chromatin solution was used as input. Rabbit anti-H3K27me3 antibody (Diagenode # C15410069) was used for immunoprecipitation. Sequencing libraries were constructed using NEBNext Ultra II DNA Library Prep Kit for Illumina (New England Biolabs # E7645S) following manufacturer’s instructions. Single-end 150 bp sequencing was performed on a HiSeq 2500 sequencer (Illumina).
QUANTIFICATION AND STATISTICAL ANALYSIS
Biological replicates
The number of biological replicates and sex of samples for each analysis are indicated in the relevant figure legends and Results. Briefly, three to five blastocysts were used for immunostaining against H3K27me3 for XCI status, 29 to 179 2cell-embryos were transferred to pseudopregnant female mice to examine postimplantation development (the number of pseudopregnant females is shown in Table S1), 1 to 20 placentae were examined for their weight and histology, 2 pooled samples (6 blastocysts per sample) were used for RNA-seq, 2 pooled samples (50 to 60 morulae per sample) were used for ULI-NChIP-seq. A single pooled sample (39 IVF- and 36 SCNT-blastocysts) was used for WGBS analysis due to the technical difficulty in preparing enough number of SCNT embryos using MEF cells.
Statistical analysis of placental weight
Statistical analysis was performed by Student’s t test (two-tailed, equal variance) using Microsoft Excel. P values less than 0.05 were treated as significant.
WGBS and RRBS data analysis
WGBS and reduced representation bisulfite sequencing (RRBS) reads were first trimmed using trim_galore to remove low-quality sequences and adaptor sequences. Bismark (version 0.15.0) was used to align reads to a bisulfite converted reference genome (mm9). The coverage depth and methylation level of each cytosine were extracted from the aligned reads with bismark_methylation_extractor. When calculating methylation level for CpG sites, information from both strands was combined, and a coverage of at least five reads was required. DMRs were identified using methpipe (version 3.4.3) and were further filtered requiring at least 10 CpG sites and at least 10% methylation difference. Functional annotation of DMR associated genes (i.e., genes with a DMR located in the TSS ± 3kb region) was performed with clusterProfiler (version 2.4.3) in R. For allele specific methylation analysis of known ICRs, all detected CpGs within one ICR were pooled together, and a coverage of at least 5 detected CpGs in both alleles was required for further methylation comparison.
RNA-seq data analysis
RNA-seq data were mapped to the mouse genome (mm9) with TopHat (version 2.0.14) with parameters “–no-coverage-search–no-novel-juncs–library-type=fr-unstranded.” Uniquely mapped reads were subsequently assembled into transcripts guided by the reference annotation (UCSC gene models) with Cufflinks (version 2.2.1). Expression levels of each gene was quantified with normalized FPKM (fragments per kilobase of exon per million mapped fragments). The Pearson correlation coefficient of gene expression level was calculated to indicate the correlation between duplicates.
ChIP-seq and DIP-seq data analysis
Downloaded ChIP-seq and DIP-seq reads, as well as H3K27-3 ULI-NChIP-seq reads generated in this study, were mapped to the mouse genome (mm9) using Bowtie (version 2.1.0) with parameters “-D 20 -R 3 -N 1 -L 20 -i S,1,0.50” to obtain only those reads that are mapped uniquely with at most 3 mismatches. Peak calling of H3K27me3 ChIP-seq data was performed using macs2 (version 2.1.1) with parameters “–broad–nomodel–nolambda–down-sample–broad-cutoff 0.01.” To visualize the signals in the genome browser, we generated wig track files for each dataset with MACS2 (version 2.1.1) by extending the uniquely mapped reads (keeping at most two read at the same genomic position) to 200 bp toward the 3′ end and binning the read count to 50 bp intervals. Tag counts were further normalized in each bin to the total number of uniquely mapped reads (reads per million reads, RPM). The ‘computeMatrix’ program from deepTools was used to compute the ChIP-seq and DIP-seq signals over the DMRs or ICRs and their flanking regions.
Allele specific analysis
After mapping the hybrid WGBS and RNA-seq data (129S1/Svj × CAST/EiJ), as well as H3K27me3 ULI-NChIP-seq data (BDF1 × PWK) to the mouse genome (mm9), custom Perl scripts were used to split mapped reads to their parental origin on the basis of SNP information downloaded from the Mouse Genomes Project (ftp://ftp-mouse.sanger.ac.uk/REL-1211-SNPs_Indels/). For BDF1, only high quality homozygous SNPs between ‘C57BL/6NJ’ and ‘DBA/2J’ are considered. The allelic reads were then processed individually.
Identification of maternal H3K27me3 domains
“bedtools makewindows” was used to generate 1-kb bins for the entire mm9 genome, and the RPKM value for each bin was calculated by “bedtools multiBigwigSummary” using SNP tracked H3K27me3 ChiP-seq reads. All bins were classified to three categories of “no signal,” “biallelic,” and “maternal-biased” using a signal cutoff of 1 and a fold change cutoff of 4. A sliding window approach was used to identify domains that were enriched for “maternal-biased” H3K27me3 bins. The criteria used were as follows: Within a window of 20 kb, the minimum number of “maternal-biased” bins was three, and the percentage of “maternal-biased” bins was larger than 40%. Overlapped windows were merged by “bedtools merge.” A total of 4,135 domains were identified in the genome.
Genomics distribution of H3K27me3 peaks
The H3K27me3 peaks were assigned into functional group based on directly overlapping and by the order of promoter (transcription start sites ± 1k), CGI, exons, introns, intergenic repeats and other regions using an annotation of Refseq. To calculate the expected chance of the peaks overlap with promoters, we generate a random set of control by shuffle the peaks within each chromosome.
Statistical analysis of sequencing datasets
Statistical analyses and plots were implemented with R (version 3.4.1, http://www.r-project.org). Pearson correlation coefficient was calculated using the ‘cor’ function with default parameters. Student’s t test (two-tailed, equal variance) was performed using the ‘t.test’ function with default parameters.
Sequencing data visualization
ChIP-seq signals and DNA methylation levels were visualized as custom tracks in the Integrative Genomics Viewer genome browser.
DATA AND SOFTWARE AVAILABILITY
The accession number for the WGBS, RNA-seq and ULI-NChIP-seq datasets reported in this study is GEO: GSE112546.
Published datasets used in this study
WGBS data of gametes, early embryos, and MEFs was obtained from GEO: GSE56697 (Wang et al., 2014), GEO: GSE56151 (Yu et al., 2014), and GEO: GSE76505 (Zhang et al., 2018). RRBS data of different cells and somatic tissues were obtained from GEO: GSE11034 and GEO: GSE43719 (Soumillon et al., 2013). H3K27me3 ChiP-seq data were obtained from GEO: GSE49847 (Yue et al., 2014), GEO: GSE76687 (Zheng et al., 2016) and GEO: GSE66526 (Keniry et al., 2016). DIP-seq data of 5mC and 5hmC during PGC development were downloaded from SRA: SRP016940 (Hackett et al., 2013).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-H3K27me3 (for immunostaining) | Millipore | Cat#07-449; RRID:AB_310624 |
| Rabbit polyclonal anti-H3K27me3 (for ULI-NChIP-seq) | Diagenode | Cat#C15410069 |
| Goat polyclonal anti-Oct4 | SantaCruz | Cat#sc-8628; RRID:AB_653551 |
| Mouse monoclonal anti-Cdx2 | BioGenex | Cat#AM392-5M; RRID:AB_2650531 |
| Fluorescein isothiocyanate(FITC)-conjugated anti-mouse IgG | Jackson ImmunoResearch | Cat#715-096-151; RRID:AB_2340796 |
| Alexa Fluor 546 donkey anti-rabbit IgG | Thermo Fisher Scientific | Cat#A10040; RRID:AB_2534016 |
| Alexa Fluor 647 donkey anti-goat IgG | Thermo Fisher Scientific | Cat# A21447; RRID:AB_2535864 |
| Bacterial and Virus Strains | ||
| pcDNA3-flag-mKdm4d-polyA | Matoba et al., 2014 | Addgene #61553 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Xbal | New England Biolabs | Cat#R0145S |
| 0.25% Trypsin with 1 mM EDTA | Thermo Fisher Scientific | Cat#25200056 |
| DMEM | Thermo Fisher Scientific | Cat#11995-073 |
| Penicillin/Streptomycin | Thermo Fisher Scientific | Cat#15140-022 |
| pregnant mare serum gonadotropin (PMSG) | Sigma-Aldrich | Cat#367222; CAS 9002-70-4 |
| human chorionic gonadotropin (hCG) | Sigma-Aldrich | Cat#230734; CAS 9002-61-3 |
| Hyaluronidase, Bovine Testes | Sigma-Aldrich | Cat#385931; CAS 37326-33-3 |
| Collagenase, Type IV | Thermo Fisher Scientific | Cat#17104-019 |
| Cytochalasin B | Calbiochem | Cat#250233; CAS 14930-96-2 |
| GenomOne CF | Ishihara Sangyo | Cat#CF001 |
| Cold Schiff’s Reagent | Wako Pure Chemical Industries | Cat#039-14645 |
| Vectashield with 4’,6-diamidino-2-phenylindole (DAPI) | Vector Laboratories | Cat#H-1200 |
| unmethylated Lambda DNA | Promega | Cat#D152A |
| Agencourt AMPure XP beads | Beckman Coulter | Cat#A63880 |
| Micrococcal Nuclease (MNase) | New England Biolabs | Cat#M0247S |
| Bovine serum albumin (BSA) | Merck | Cat#12657 |
| Critical Commercial Assays | ||
| EpiGnome Methyl-Seq kit | Epicenter | Cat#EGMK81312 |
| EZ DNA Methylation-Direct Kit | Zymo Research | Cat#D5020 |
| SMARTer Ultra Low Input RNA cDNA preparation kit | Clontech | Cat#634936 |
| NEBNext Ultra DNA Library Prep Kit for Illumina | New England Biolabs | Cat#E7370 |
| mMESSAGE mMACHINE T7 Ultra Kit | Thermo Fisher Scientific | Cat#AM1345 |
| NEBNext Ultra II DNA Library Prep Kit for Illumina | New England Biolabs | Cat# E7645S |
| Deposited Data | ||
| WGBS, RNA-seq and ULI-NChIP-seq data | This paper | GEO: GSE112546 |
| Experimental Models: Organisms/Strains | ||
| Mouse: B6D2F1(BDF1) females, 9-15 weeks of age | Japan SLC, Inc | N/A |
| Mouse: B6;129-Xisttm5Sado adult females | RIKEN BRC Sado et al., 2005 | RBRC02655 |
| Mouse: 129S1/Svlmj; Xisttm1Jae adult females | Marahrens et al., 1997 | N/A |
| Mouse: DBA/2 adult males | Japan SLC, Inc | N/A |
| Mouse: Cast/EiJ adult males | The Jackson Laboratory | Cat#000928 |
| Mouse: PWK adult males | RIKEN BRC | RBRC00213 |
| Software and Algorithms | ||
| Bismark (version 0.15.0) | Krueger and Andrews, 2011 | https://www.bioinformatics.babraham.ac.uk/projects/bismark/ |
| MethPipe (version 3.4.3) | Song et al., 2013 | http://smithlabresearch.org/software/methpipe/ |
| TopHat (version 2.0.14) | Trapnell et al., 2009 | https://ccb.jhu.edu/software/tophat/index.shtml |
| Cufflinks (version 2.2.1) | Trapnell et al., 2010 | https://github.com/cole-trapnell-lab/cufflinks |
| Bowtie2 (version 2.1.0) | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
Highlights.
Use of Kdm4d overexpression and Xist KO donor cells improves SCNT efficiency
Post-implantation defects are still observed in non-viable SCNT embryos
DNA methylome analyses reveal specific DMRs in SCNT embryos
Allelic analyses reveal loss of H3K27me3 imprinting in preimplantation SCNT embryos
ACKNOWLEDGMENTS
We thank Azusa Inoue and Aritra Bhattacherjee for critical reading of the manuscript. This project was supported by the NIH (R01HD092465) and HHMI (to Y.Z.), JSPS KAKENHI grant JP16H06146 (to S.M.), and Zhejiang Provincial Natural Science Foundation of China grant LR18C060001 (to L.S.). L.S. is a scholar of the Thousand Young Talents Program of China. L.J. is supported by a Charles A. King Trust postdoctoral research fellowship. Y.Z. is an investigator of the Howard Hughes Medical Institute.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes five figures and five tables and can be found with this article online at https://doi.Org/10.1016/j.stem.2018.06.008.
DECLARATION OF INTERESTS
Y.Z. is a scientific founder of NewStem Biotechnology. Y.Z. and S.M. are inventors of a patent regarding the role of Kdm4 in improving cloning efficiency.
REFERENCES
- Acampora D, Mazan S, Lallemand Y, Avantaggiato V, Maury M, Simeone A, and Brûlet P (1995). Forebrain and midbrain regions are deleted in Otx2−/− mutants due to a defective anterior neuroectoderm specification during gastrulation. Development 121, 3279–3290. [DOI] [PubMed] [Google Scholar]
- Ao Z, Liu D, Zhao C, Yue Z, Shi J, Zhou R, Cai G, Zheng E, Li Z, and Wu Z (2017). Birth weight, umbilical and placental traits in relation to neonatal loss in cloned pigs. Placenta 57, 94–101. [DOI] [PubMed] [Google Scholar]
- Branco MR, King M, Perez-Garcia V, Bogutz AB, Caley M, Fineberg E, Lefebvre L, Cook SJ, Dean W, Hemberger M, and Reik W (2016). Maternal DNA Methylation Regulates Early Trophoblast Development. Dev. Cell 36, 152–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brind’Amour J, Liu S, Hudson M, Chen C, Karimi MM, and Lorincz MC (2015). An ultra-low-input native ChIP-seq protocol for genome-wide profiling of rare cell populations. Nat. Commun 6, 6033. [DOI] [PubMed] [Google Scholar]
- Chan MM. Smith ZD, Egli D, Regev A, and Meissner A (2012). Mouse ooplasm confers context-specific reprogramming capacity. Nat. Genet 44, 978–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung YG, Matoba S, Liu Y, Eum JH, Lu F, Jiang W, Lee JE, Sepilian V, Cha KY, Lee DR, and Zhang Y (2015). Histone Demethylase Expression Enhances Human Somatic Cell Nuclear Transfer Efficiency and Promotes Derivation of Pluripotent Stem Cells. Cell Stem Ceil 17, 758–766. [DOI] [PubMed] [Google Scholar]
- Coan PM, Burton GJ, and Ferguson-Smith AC (2005). Imprinted genes in the placenta–a review. Placenta 26 (Suppl A), S10–S20. [DOI] [PubMed] [Google Scholar]
- Dean W, Santos F, Stojkovic, Zakhartchenko V, Walter J, Wolf E, and Reik W (2001). Conservation of methylation reprogramming in mammalian development: aberrant reprogramming in cloned embryos. Proc. Natl. Acad. Sci. USA 98, 13734–13738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F, Li X, Liang D, Li T, Zhu P, Guo H, Wu X, Wen L, Gu T-P, Hu B, et al. (2014). Active and passive demethylation of male and female pronuclear DNA in the mammalian zygote. Cell Stem Cell 15, 447–459. [DOI] [PubMed] [Google Scholar]
- Hackett JA, Sengupta R, Zylicz JJ, Murakami K, Lee C, Down TA, and Surani MA (2013). Germline DNA demethylation dynamics and imprint erasure through 5-hydroxymethylcytosine. Science 339 448–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochedlinger K, and Jaenisch R (2003). Nuclear transplantation, embryonic stem cells, and the potential for cell therapy. N. Engl. J. Med 349, 275–286. [DOI] [PubMed] [Google Scholar]
- Inoue K, Ogonuki N, Mochida K, Yamamoto Y, Takano K, Kohda T, Ishino F, and Ogura A (2003). Effects of donor cell type and genotype on the efficiency of mouse somatic cell cloning. Biol. Reprod 69, 1394–1400. [DOI] [PubMed] [Google Scholar]
- Inoue K, Kohda T, Sugimoto M, Sado T, Ogonuki N, Matoba S, Shiura H, Ikeda R, Mochida K, Fujii T, et al. (2010). Impeding Xist expression from the active X chromosome improves mouse somatic cell nuclear transfer. Science 330, 496–499. [DOI] [PubMed] [Google Scholar]
- Inoue A, Jiang L, Lu F, Suzuki T, and Zhang Y (2017a). Maternal H3K27me3 controls DNA methylation-independent imprinting. Nature 547, 419–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue A, Jiang L, Lu F, and Zhang Y (2017b). Genomic imprinting of Xist by maternal H3K27me3. Genes Dev 31, 1927–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K, Hirose M, inoue H, Hatanaka Y, Honda A, Hasegawa A, Mochida K, and Ogura A (2017c). The Rodent-Specific MicroRNA Cluster within the Sfmbt2 Gene Is Imprinted and Essential for Placental Development. Cell Rep 19, 949–956. [DOI] [PubMed] [Google Scholar]
- Itoh M, Yoshida Y t Nishida K, Narimatsu M, Hibi M, and Hirano T (2000). Role of Gab1 in heart, placenta, and skin development and growth factor- and cytokine-induced extracellular signal-regulated kinase mitogen-activated protein kinase activation. Mol. Cell. Biol 20, 3695–3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keniry A, Gearing LJ, Jansz N, Liu J, Holik AZ, Hickey PF, Kinkel SA, Moore DL, Breslin K, Chen K, et al. (2016). Setdb1-mediated H3K9 methylation is enriched on the inactive X and plays a role in its epigenetic silencing. Epigenetics Chromatin 9, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishigami S, Mizutani E, Ohta H, Hikichi T, Thuan NV, Wakayama S, Bui H-T, and Wakayama T (2006). Significant improvement of mouse cloning technique by treatment with trichostatin A after somatic nuclear transfer. Biochem. Biophys. Res. Commun 340, 183–189. [DOI] [PubMed] [Google Scholar]
- Kobayashi H, Sakurai T, Imai M, Takahashi N, Fukuda A, Yayoi O, Sato S, Nakabayashi K, Hata K, Sotomaru Y, et al. (2012). Contribution of intragenic DNA methylation in mouse gametic DNA methylomes to establish oocyte-specific heritable marks. PLoS Genet 8, e1002440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger F, and Andrews SR (2011). Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27, 1571–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Liu X, Wang C, Gao Y, Gao R, Kou X, Zhao Y, Li J, Wu Y, Xiu W, et al. (2016). Identification of key factors conquering developmental arrest of somatic cell cloned embryos by combining embryo biopsy and single-cell sequencing. Cell Discov 2, 16010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loi P, Clinton M, Vackova I, Fulka J Jr., Feil R, Palmieri C, Della Salda L, and Ptak G (2006). Placental abnormalities associated with post-natal mortality in sheep somatic cell clones. Theriogenology 65,1110–1121. [DOI] [PubMed] [Google Scholar]
- Loi P, Iuso D, Czernik M, and Ogura A (2016). A New, Dynamic Era for Somatic Cell Nuclear Transfer? Trends Biotechnol 34, 791–797. [DOI] [PubMed] [Google Scholar]
- Mann MRW, Chung YG, Nolen LD, Verona RI, Latham KE, and Bartolomei MS (2003). Disruption of imprinted gene methylation and expression in cloned preimplantation stage mouse embryos. Biol. Reprod 69, 902–914. [DOI] [PubMed] [Google Scholar]
- Marahrens Y, Panning B, Dausman J, Strauss W, and Jaenisch R (1997). Xist-deficient mice are defective in dosage compensation but not spermatogenesis. Genes Dev 11, 156–166. [DOI] [PubMed] [Google Scholar]
- Matoba S, Inoue K, Kohda T, Sugimoto M, Mizutani E, Ogonuki N, Nakamura T, Abe K, Nakano T, Ishino F, and Ogura A (2011). RNAi-mediated knockdown of Xist can rescue the impaired postimplantation development of cloned mouse embryos. Proc. Natl. Acad. Sci. USA 108, 20621–20626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matoba S, Liu Y, Lu F, Iwabuchi KA, Shen L, Inoue A, and Zhang Y (2014). Embryonic development following somatic cell nuclear transfer impeded by persisting histone methylation. Cell 159, 884–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matoba S, and Zhang Y (2018). Somatic Cell Nuclear Transfer Reprogramming: Mechanisms and Applications. Cell Stem Cell 23 Published online July 19, 2018. 10.1016/j.stem.2018.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miri K, Latham K, Panning B, Zhong Z, Andersen A, and Varmuza S (2013). The imprinted polycomb group gene Sfmbt2 is required for trophoblast maintenance and placenta development. Development 140, 4480–4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogonuki N, Mori M, Shinmen A, Inoue K, Mochida K, Ohta A, Ogura A, and Baltz JM (2010). The effect on intracytoplasmic sperm injection outcome of genotype, male germ cell stage and freeze-thawing in mice. PLoS ONE 5, e11062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura A, Inoue K, Ogonuki N, Noguchi A, Takano K, Nagano R, Suzuki O, Lee J, Ishino F, and Matsuda J (2000). Production of male cloned mice from fresh, cultured, and cryopreserved immature Sertoli cells. Biol. Reprod 62, 1579–1584. [DOI] [PubMed] [Google Scholar]
- Ogura A, Ogonuki N, Miki H, and Inoue K (2005). Microinsemination and Nuclear Transfer Using Male Germ Cells. Int. Rev. Cytol 246,189–229. [DOI] [PubMed] [Google Scholar]
- Ogura A, Inoue K, and Wakayama T (2013). Recent advancements in cloning by somatic cell nuclear transfer. Philos. Trans. R. Soc. Lond. B Biol. Sci 368, 20110329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okae H, Matoba S, Nagashima T, Mizutani E, Inoue K, Ogonuki N, Chiba H, Funayama R, Tanaka S, Yaegashi N, et al. (2014). RNA sequencing-based identification of aberrant imprinting in cloned mice. Hum. Mol. Genet 23, 992–1001. [DOI] [PubMed] [Google Scholar]
- Okuda T, van Deursen J, Hiebert SW, Grosveld G, and Downing JR (1996). AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell 84, 321–330. [DOI] [PubMed] [Google Scholar]
- Plath K, Fang J, Mlynarczyk-Evans SK, Cao R, Worringer KA, Wang H, de la Cruz CC, Otte AP, Panning B, and Zhang Y (2003). Role of histone H3 lysine 27 methylation in X inactivation. Science 300, 131–135. [DOI] [PubMed] [Google Scholar]
- Sado T, Hoki Y, and Sasaki H (2005). Tsix silences Xist through modification of chromatin structure. Dev. Cell 9, 159–165. [DOI] [PubMed] [Google Scholar]
- Shen L, Inoue A, He J, Liu Y, Lu F, and Zhang Y (2014). Tet3 and DNA replication mediate demethylation of both the maternal and paternal genomes in mouse zygotes. Cell Stem Cell 75, 459–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith LC, Suzuki J Jr., Goff AK, Filion F, Therrien J, Murphy BD, Kohan-Ghadr HR, Lefebvre R, Brisville AC, Buczinski S, et al. (2012). Developmental and epigenetic anomalies in cloned cattle. Reprod. Domest. Anim 47 (Suppl 4), 107–114. [DOI] [PubMed] [Google Scholar]
- Song Q, Decato B, Hong EE, Zhou M, Fang F, Qu J, Garvin T, Kessler M, Zhou J, and Smith AD (2013). A reference methylome database and analysis pipeline to facilitate integrative and comparative epigenomics. PLoS ONE 8, e81148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soumillon M, Necsulea A, Weier M, Brawand D, Zhang X. Gu H, Barthès P, Kokkinaki M, Nef S, Gnirke A, et al. (2013). Cellular source and mechanisms of high transcriptome complexity in the mammalian testis. Cell Rep 3, 2179–2190. [DOI] [PubMed] [Google Scholar]
- Tanaka S, Oda M, Toyoshima Y, Wakayama T, Tanaka M. Yoshida N, Hattori N, Ohgane J, Yanagimachi R, and Shiota K (2001). Placentomegaly in cloned mouse concepti caused by expansion of the spongiotrophoblast layer. Biol. Reprod 65, 1813–1821. [DOI] [PubMed] [Google Scholar]
- Tomizawa S, Kobayashi H, Watanabe T, Andrews S, Hata K, Kelsey G, and Sasaki H (2011). Dynamic stage-specific changes in imprinted differentially methylated regions during early mammalian development and prevalence of non-CpG methylation in oocytes. Development 138, 811–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, and Salzberg SL (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25,1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, and Pachter L (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol 28, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent JJ, Huang Y, Chen P-Y, Feng S, Calvopiña JH, Nee K, Lee SA, Le T, Yoon AJ, Faull K, et al. (2013). Stage-specific roles for tet1 and tet2 in DNA demethylation in primordial germ cells. Cell Stem Cell 12, 470–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakayama T, Perry AC, Zuccotti M, Johnson KR, and Yanagimachi R (1998). Full-term development of mice from enucleated oocytes injected with cumulus cell nuclei. Nature 394, 369–374. [DOI] [PubMed] [Google Scholar]
- Wang LC, Kuo F, Fujiwara Y, Gilliland DG, Golub TR, and Orkin SH (1997). Yolk sac angiogenic defect and intra-embryonic apoptosis in mice lacking the Ets-related factor TEL. EMBO J 16, 4374–4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Kou Z, Zhang Y, and Gao S (2007). Dynamic reprogramming of histone acetylation and methylation in the first cell cycle of cloned mouse embryos. Biol. Reprod 77, 1007–1016. [DOI] [PubMed] [Google Scholar]
- Wang L, Zhang J, Duan J, Gao X, Zhu W, Lu X, Yang L, Zhang J, Li G, Ci W, et al. (2014). Programming and inheritance of parental DNA methylomes in mammals. Cell 157, 979–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilmut I, Schnieke AE, McWhir J, Kind AJ, and Campbell KH (1997). Viable offspring derived from fetal and adult mammalian cells. Nature 385, 810–813. [DOI] [PubMed] [Google Scholar]
- Yamagata K, Yamazaki T. Miki H, Ogonuki N, Inoue K, Ogura A, and Baba T (2007). Centromeric DNA hypomethylation as an epigenetic signature discriminates between germ and somatic cell lineages. Dev. Biol 312, 419–426. [DOI] [PubMed] [Google Scholar]
- Yamaguchi S, Hong K, Liu R, Shen L, Inoue A, Diep D, Zhang K, and Zhang Y (2012). Tet1 controls meiosis by regulating meiotic gene expression. Nature 492, 443–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi S, Shen L, Liu Y, Sendler D, and Zhang Y (2013). Role of Tet1 in erasure of genomic imprinting. Nature 504, 460–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Smith SL, Tian XC, Lewin HA, Renard JP, and Wakayama T (2007). Nuclear reprogramming of cloned embryos and its implications for therapeutic cloning. Nat. Genet 39, 295–302. [DOI] [PubMed] [Google Scholar]
- Yu W, Briones V, Lister R, McIntosh C, Han Y, Lee EY, Ren J, Terashima M, Leighty RM, Ecker JR, and Muegge K (2014). CG hypomethylation in Lsh−/− mouse embryonic fibroblasts is associated with de novo H3K4me1 formation and altered cellular plasticity. Proc. Natl. Acad. Sci. USA 111, 5890–5895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue F, Cheng Y, Breschi A, Vierstra J, Wu W, Ryba T, Sandstrom R, Ma Z, Davis C, Pope BD, et al. ; Mouse ENCODE Consortium (2014). A comparative encyclopedia of DNA elements in the mouse genome. Nature 515, 355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Wang F, Kou Z, Zhang Y, and Gao S (2009). Defective chromatin structure in somatic cell cloned mouse embryos. J. Biol. Chem 284, 24981–24987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Xiang Y, Yin Q, Du Z, Peng X, Wang Q, Fidalgo M, Xia W, Li Y, Zhao ZA, et al. (2018). Dynamic epigenomic landscapes during early lineage specification in mouse embryos. Nat. Genet 50, 96–105. [DOI] [PubMed] [Google Scholar]
- Zheng H, Huang B, Zhang B, Xiang Y, Du Z, Xu Q, Li Y, Wang Q, Ma J, Peng X, et al. (2016). Resetting Epigenetic Memory by Reprogramming of Histone Modifications in Mammals. Mol. Cell 63, 1066–1079. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.