

Abstract
Background.
Dysregulation of arousal is symptomatic of numerous psychiatric disorders. Previous research has shown that the activity of dopamine (DA) neurons in the ventral periaqueductal gray (vPAG) tracks with arousal state, and lesions of vPAGDA cells increase sleep. However, the circuitry controlling these wake-promoting DA neurons is unknown.
Methods.
The present study combines Designer Receptors Exclusively Activated by Designer Drugs (DREADDs), behavioral pharmacology, electrophysiology, and immunoelectron microscopy in male and female mice to elucidate mechanisms in the vPAG that promote arousal.
Results.
DREADD-induced activation of locus coeruleus (LC) projections to the vPAG or vPAGDA neurons promoted arousal. Similarly, agonist stimulation of vPAG α1-adrenergic receptors (α1ARs) increased latency to fall asleep, while α1AR blockade had the opposite effect. α1AR stimulation drove vPAGDA activity in a glutamate-dependent, action potential-independent manner. Compared to other dopaminergic brain regions, α1ARs were enriched on astrocytes in the vPAG, and mimicking α1AR transmissin specifically in vPAG astrocytes via Gq-DREADDs was sufficient to increase arousal. In general, the wake-promoting effects observed were not accompanied by hyperactivity.
Conclusions.
These experiments reveal that vPAG α1ARs increase arousal, promote glutamatergic input onto vPAGDA neurons, and are abundantly expressed on astrocytes. Activation of LC inputs, vPAG astrocytes, or vPAGDA neurons increase sleep latency but do not produce hyperactivity. Together, these results support an arousal circuit whereby noradrenergic transmission at astrocytic α1ARs activates wake-promoting vPAGDA neurons via glutamate transmission.
Keywords: dopamine, norepinephrine, arousal, sleep, wakefulness, astrocytes, DREADDs
Introduction
Arousal is a fundamental component of adaptive behavior. Arousal increases when animals must stay alert to forage, mate, or avoid predation. In addition to simple goal-directed behaviors, arousal is an integral part of higher cognition including learning and decision making (1, 2). By contrast, suppression of arousal is required for rest, sleep, and energy conservation. Dysregulation of arousal is symptomatic of multiple neurological and neuropsychiatric disorders such as schizophrenia, depression, and substance abuse (3–7) and can have serious consequences. For example, 5% of adults surveyed reported falling asleep while driving during the past 30 days (8), and sleep disorders, sleepiness, and fatigue cause approximately 20% of car accidents resulting in serious injury or death (9). Therefore, an urgent need exists to understand how the brain mediates arousal and to develop better treatments for impairments in arousal.
In addition to playing critical roles in motivated and motor behaviors, dopamine (DA) is involved in arousal (10–12). Excessive sleepiness is a common non-motor symptom of Parkinson’s disease (13). Recently, optogenetic stimulation of ventral tegmental area (VTA) or dorsal raphe (DRN) DA neurons was shown to promote wakefulness, whereas chemogenetic inhibition of these neurons increased sleep (14, 15). Over a decade ago, neighboring ventral (also known as ventrolateral) periaqueductal gray (vPAG) DA neurons (vPAGDA) were shown to be active during wakefulness and inactive during sleep, and lesioning these neurons resulted in significant increases in time spent asleep (16). While the vPAG is anatomically interconnected with numerous brain regions involved in arousal (16), the functional neural circuitry that modulates vPAGDA activity and its wake-promoting effects remain largely unknown.
Decades of research indicate that the noradrenergic locus coeruleus (LC) modulates arousal (17–19). The LC is active during wakefulness and relatively inactive during sleep (20), optogenetic stimulation of the LC promotes wakefulness (21), and noradrenergic manipulations can alter arousal (22). The LC is an ideal candidate to control wake-promoting vPAGDA neurons because 1) the LC projects to the vPAG (16, 23, 24), 2) α1-adrenergic receptors (α1ARs) are present in the vPAG (25, 26), and 3) norepinephrine (NE) transmission via the Gq-coupled α1AR can modulate the activity of other DA neuron populations including the rostral linear nucleus (RLi) (27), substantia nigra (28, 29), and VTA (30).
The present study tested the hypothesis that LC-vPAG circuitry modulates arousal by examining 1) whether chemogentic activation of vPAGDA and LC projections to the vPAG promote arousal, 2) whether vPAG α1ARs modulate arousal and vPAGDA activity, 3) the precise cellular identity and location of α1AR expression in the vPAG, and 4) the functional contribution of vPAG astrocytes, which contain α1ARs, in modulating arousal.
Materials and Methods
Subjects and behavioral testing
All procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of Emory University. To investigate the role of LC-vPAG circuitry on arousal, we examined latency to fall asleep following chemogenetic or pharmacological manipulations in adult male and female mice. We included measures of horizontal locomotion to serve as a control for potential hyperactivity following wake-promoting manipulations. C57BL/6J mice were used for EEG and terazosin experiments. The Dbh −/− mice used for phenylephrine experiments were generated as previously described (31–33) and maintained on a mixed 129/SvEv and C57BL/6J background. TH-Cre mice (originally obtained from Jackson Laboratories, Bar Harbor, ME) bred on a C57BL/6J background were used for Designer Receptors Exclusively Activated by Designer Drugs (DREADD) experiments to limit AAV- hSyn-DIO-hM3D(Gq)-mCherry expression to TH-containing cells (34). GFAP-hM3Dq mice (breeding pairs generously provided by Ken McCarthy) were bred on a C57BL/6J background and used to test the functional role of vPAG astrocytes in mediating arousal. All genotypes were confirmed by PCR.
Guide cannula were implanted over the vPAG, and AAV-hSyn-DIO-hM3D(Gq)- mCherry was virally expressed in the LC or vPAG (see Supplemental Information). Sleep latency testing was conducted in the home cage of individually-housed mice. Animals were kept on a 12:12 light/dark cycle, and testing began 2–3 h into the light cycle when pressure to sleep is high. A video camera recorded mouse behavior for at least 3 h. When mice fall asleep, they exhibit a distinct posture and breathing pattern that can readily be identified by a trained observer (12). Consistent with previous research (12, 35), sleep latency was quantified as the duration of time post intraperitoneal or intracranial injection that mice were awake until their first sleep bout, defined as exhibiting the sleep posture continuously for 2 min and 75% of the 10-min time period beginning at sleep onset.
To physiologically validate the behaviorally scored sleep posture, sleep latency was quantified independently through EEG/EMG and through behavior by a trained observer (Figure 1A-C; see Supplemental Information). Behavioral and EEG measures of sleep latency were nearly identical (Figure 1C; r = 0.9058, p = 0. 0019), confirming that the behavioral scoring of sleep latency by a trained observer is a valid method of determining sleep onset (12, 35). The one “outlier” was due to the duration of the initial sleep bout; EEG confirmed that the mouse was asleep during the time behaviorally scored as asleep but fell just short of the 2-min criteria.
Figure 1. EEG validation of behavioral scoring of sleep-onset latency.
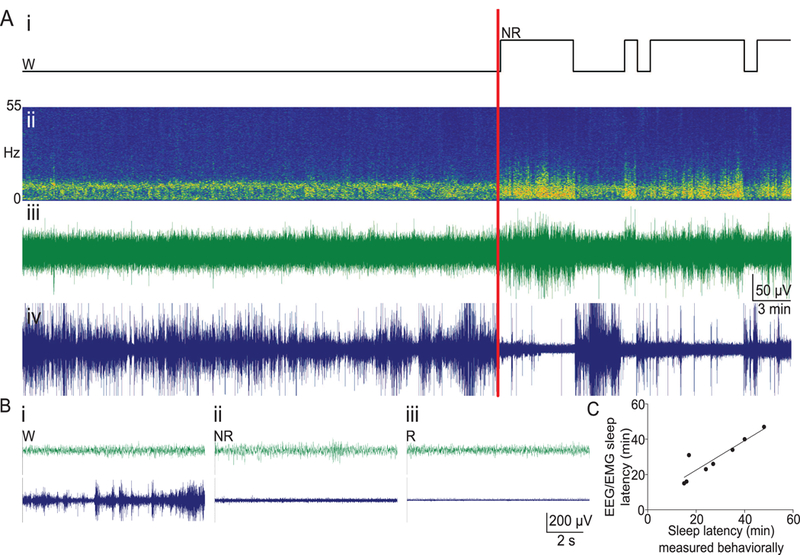
A) Representative EEG/EMG data from one mouse. (i) Hypnogram of a 60 min segment of a mouse with video and EEG/EMG scoring. Wake (W) and non-REM sleep (NR) are shown, with a red vertical line indicating the time of the independently-scored behavioral onset of sleep from the video. (ii) Fast-Fourier-based spectrogram shows predominantly theta activity during wakefulness and an increased in low-frequency EEG power at the onset of sleep, correlating with changes in (iii) EEG and (iv) EMG. B) Example traces with a 30 sec time-base showing EEG and EMG activity during (i) wakefulness, (ii) non-REM sleep, and (iii) REM sleep. C) There is a significant correlation between EEG/EMG and behavioral/video scoring of sleep onset (n = 8, r = 0.9058, p = 0.0019). One outlier resulted from using 20 sec epochs for EEG/EMG scoring and a 2-min requirement for sleep onset for video scoring. Voltage/time scales in (A) and (B) apply to both EEG and EMG traces.
Locomotor activity was measured via photobeam breaks in automated chambers (San Diego Instruments Inc., La Jolla, CA) as described (33, 36). Ambulations, defined as consecutive photobeam breaks, were quantified for 2 h. Testing began immediately following intracranial infusions or 20 min post intraperitoneal injection (to ensure drug reached the brain by the start of the test). For within-subject testing of drug and vehicle, locomotor tests were separated by a week to prevent habituation to the chambers. Drug order was counterbalanced across subjects. Additional methods including surgical details are described in Supplemental Information.
Electrophysiology
Procedures were approved by the IACUC of Vanderbilt University. Electrophysiology experiments were conducted on brain slices from TH-eGFP mice [Tg(Th-EGFP)DJ76Gsat/Mmnc] on a C57BL/6J background. Breeding pairs were obtained from the Mutant Mouse Regional Resource Center (MMRRC; Chapel Hill, NC). Brain slices were prepared and electrophysiological recordings were conducted as previously described (27) (details in Supplemental Information).
Immunoelectron microscopy
Procedures were approved by the IACUC of Christopher Newport University. vPAG sections containing DA cells were prepared from C57BL/6J mice. Immunoelectron microscopy was conducted as previously described (37, 38) and detailed in Supplemental Information.
Results
Chemogenetic activation of vPAG DA neurons promotes wakefulness
To test whether excitation of vPAGDA neurons increases arousal, Cre-dependent hM3Dq DREADDs were virally expressed in the vPAG (Figure 2A-D and S1A). DREADDs were expressed in approximately 75% of TH+ cells in the vPAG of TH-Cre+ mice, but were not detectable in wild-type littermates (Figure 2B-C), demonstrating that the virus is specific to Cre- containing cells. By targeting the rostal vPAG, we minimized overlap with neighboring dopaminergic nuclei such as the DRN and RLi (Figure 2D, S1); DREADD expression was contained to the vPAG in 67% of transfected mice. In 33% of DREADD-expressing subjects, transfection extended to the DRN and RLi; however, their behavioral data did not differ from subjects with DREADD expression confined to the vPAG (Figure S1B), indicating that activation of vPAGDA neurons alone is sufficient for the behavioral results described below. mCherry was not detected in the VTA or substantia nigra of any subject (data not shown).
Figure 2. Activation of vPAG DA neurons promotes wakefulness without causing hyperactivity.
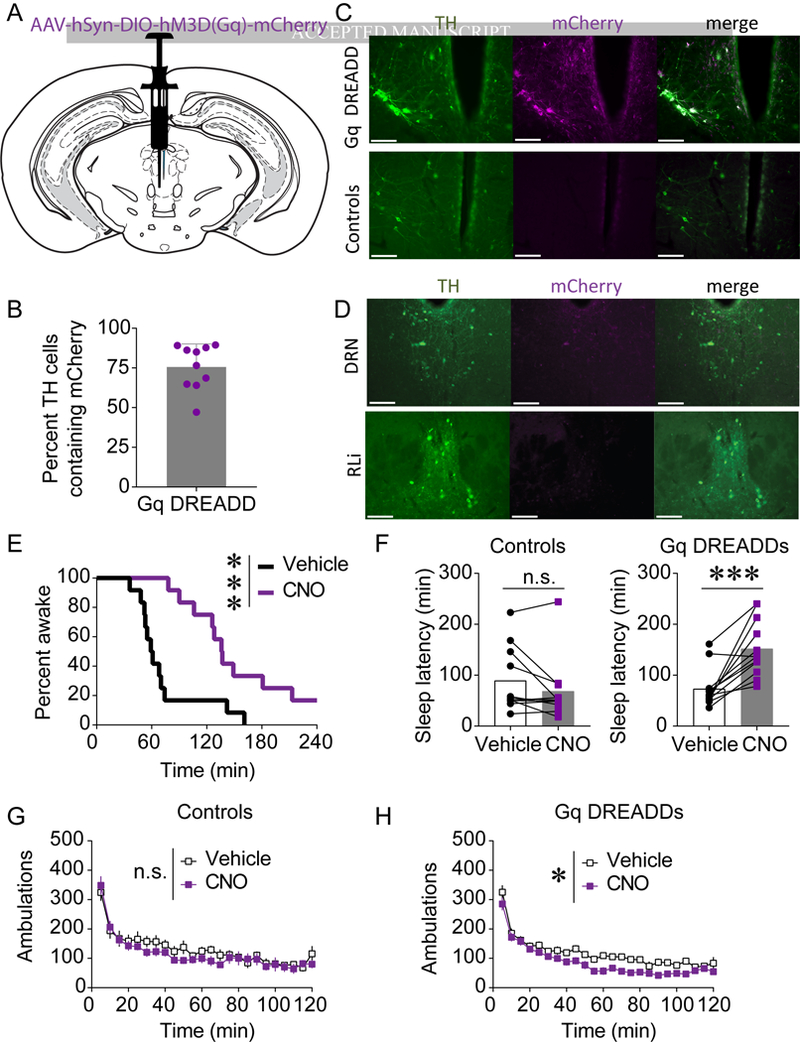
A) AAV-hSyn-DIO-hM3D(Gq)-mCherry was injected into the vPAG of TH-Cre+ mice and wild-type littermates. B) mCherry was expressed in a large proportion of TH-containing vPAGDA cells. C) mCherry (magenta) was expressed in TH-Cre+ mice, but not TH-Cre-mice, and overlapped with TH (green). D) DREADD expression in TH-Cre+ mice did not extend into the DNR or RLi in 67% of subjects. All scale bars = 100 μm. E-F) CNO (1 mg/kg, i.p.) promoted wakefulness in DREADD-expressing mice (E-F) but not in non-DREADD-expressing controls (F; n=11–12; n.s. = not statistically different; ***p < 0.001). G-H) CNO modestly decreased locomotor activity in DREADD-expressing mice (H; n = 12, main effect of drug *p < 0.05) but not in non-DREADD-expressing controls (G; n = 11, n.s. = not statistically significant).
CNO-induced activation of DREADDs robustly promoted wakefulness. Two out of 12 CNO-treated mice expressing DREADDs did not meet sleep criteria within 4 h, whereas all subjects fell asleep during the test when treated with vehicle (Figure 2E). CNO-induced activation of vPAGDA neurons significantly increased sleep latency in DREADD-expressing mice, but had no effect on mice not expressing DREADDs (Figure 2F; controls, t(10) = 1.822, p = 0.098; Gq DREADD-expressing mice, t(11) = 4.987, p = 0.0004), demonstrating that the effects of CNO were due to DREADD activation and not any potential off-target effects of the ligand (39, 40). Activation of vPAGDA neurons did not increase locomotor activity, indicating that vPAGDA neurons can promote wakefulness without causing hyperactivity. CNO did not affect locomotion in controls (Figure 2G; main effect of drug F(1,10) = 1.093, p = 0.320), and chemogenetic stimulation of vPAGDA neurons slightly reduced locomotor activity (Figure 2H; main effect of drug, F(1,11) = 16.959, p = 0.0017).
Chemogentic activation of the LC or LC terminals in the vPAG promotes wakefulness
hM3Dq DREADDs were expressed in the LC of TH-Cre mice (Figure 3A). Whereas all mice treated with vehicle fell asleep, 3 out of 8 CNO (1 mg/kg, i.p.)-treated subjects failed to meet sleep criteria within 3 h (Figure 3B). Overall, CNO-induced DREADD activation of the LC significantly increased sleep latency (Figure 3C; t(7) = 2.444, p = 0.0445).
Figure 3. Chemogenetic stimulation of the LC or LC projections to the vPAG promotes wakefulness.
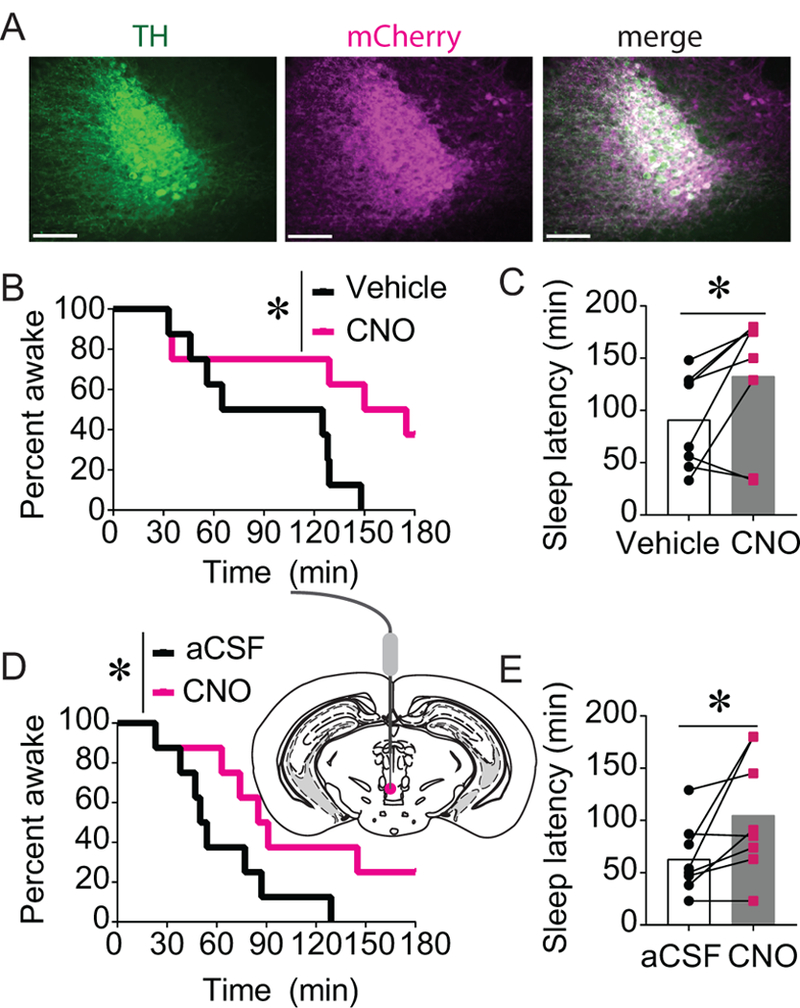
A) AAV-hSyn-DIO-hM3D(Gq)-mCherry was injected into the LC of TH-Cre+ mice. Scale bars = 100 μm. B-C) CNO (1 mg/kg)-induced activation of Gq DREADDs in the LC significantly increased sleep latency (n = 8; *p < 0.05). D-E) Site-specific infusion of CNO (1 μg/0.3 μL) into the vPAG to stimulate LC projections to the vPAG similarly increased sleep latency (n=8; *p < 0.05).
To test whether activation of LC projections to the vPAG is sufficient to promote wakefulness, CNO (1 μg/0.3 μL) or aCSF was infused into the vPAG of mice expressing hM3Dq DREADDs in LC neurons. Activation LC terminals in the vPAG significantly increased latency to sleep (Figure 3E; t(7) = 2.464, p = 0.0432), and 2 out of 8 mice did not meet sleep criteria within 3 h (Figure 3D).
α1ARs modulate vPAG DA neuron activity and arousal
Electrophysiological recordings of vPAGDA neurons in brain slices from TH-GFP mice revealed that the α1AR agonist phenylephrine (3 μM) significantly increased the frequency of spontaneous excitatory postsynaptic currents (sEPSCs) onto vPAGDA neurons (Figure 4A-B; baseline compared to peak phenylephrine, t(6) = 2.739, p = 0.034), suggesting that noradrenergic excitation of vPAGDA neurons is mediated by changes in glutamate transmission.
Figure 4. Stimulation of α1ARs increases glutamatergic drive onto vPAGDA neurons in an AMPA receptor-dependent, action potential-independent manner.
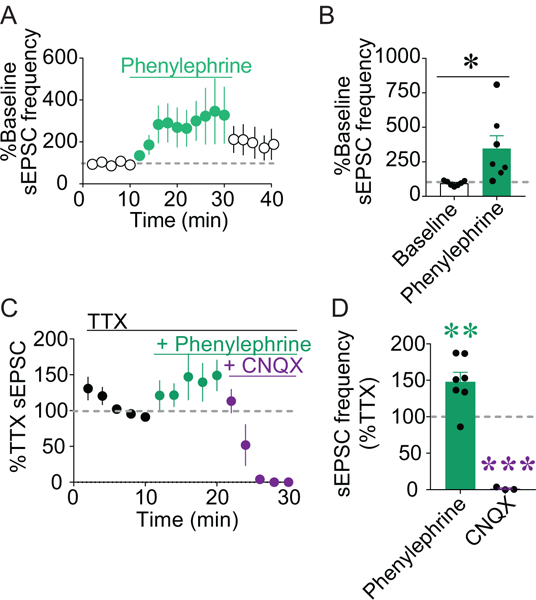
A-B) Bath application of phenylephrine (3 μM) significantly increased sEPSC frequency in vPAGDA neurons (n = 7; *p < 0.05). C) TTX (1 μM), phenylephrine (3 μM), and CNQX (10 μM) were cumulatively applied to slices and sEPSCs were recorded from vPAGDA neurons. D) Phenylephrine in the presence of TTX increased sEPSC frequency compared to sEPSCs during TTX alone (n = 7; **p < 0.01), and CNQX abolished sEPSCs (n = 3; ***p < 0.001).
To determine whether α1AR-mediated excitation of vPAGDA neurons requires α-amino- 3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor activation or neuronal action potentials, we assessed the effects of phenylephrine (3 μM) on sEPSCs in the presence of the AMPA antagonist CNQX (10 μM) and the voltage-gated sodium channel blocker tetrodotoxin (TTX; 1 μM). The facilitation of sEPSCs in vPAGDA neurons by phenylephrine persisted following bath application of TTX but was inhibited by CNQX (Figure 4C-D; F(2,14) = 43.66, p = 0.000001; Dunnett’s post hoc test compared to TTX: phenylephrine p = 0.0028, CNQX p = 0.0001).
Because activation of vPAG α1ARs was sufficient to increase vPAGDA neuron activity, we next tested whether in vivo manipulations of NE/α1AR transmission alters arousal. To confirm that endogenous NE promotes arousal, we tested sleep latency and locomotor activity in dopamine β-hydroxylase knockout mice (Dbh −/−) that completely lack NE. Consistent with previous research (12, 35, 41), Dbh −/− mice exhibited decreased sleep latencies (Figure 5A; t(15) = 3.481, p = 0.0034) and blunted novelty-induced locomotion (Figure 5B; main effect of genotype, F(1,15) = 14.496, p = 0.0017) compared to Dbh +/− littermates with normal levels of NE. To determine whether activation of vPAG α1ARs was sufficient to promote arousal, aCSF or phenylephrine (4 μg/0.3 μL) was infused into the vPAG via a cannula immediately before the start of behavioral tests (Figures 5C and S2A). Stimulation of vPAG α1ARs with phenylephrine significantly increased sleep latency in Dbh +/− control mice as well as in Dbh −/− mice (Figure 5D; Dbh +/−, t(7) = 2.562, p = 0.037; Dbh −/−, t(8) = 3.937, p = 0.004) without causing hyperactivity (Figure 5E; Controls, main effect of drug, F(1,7) = 0.149, p = 0.711; Dbh −/−, main effect of drug, F(1,8) = 0.033, p = 0.858).
Figure 5. vPAG α1ARs bidirectionally modulate wakefulness.
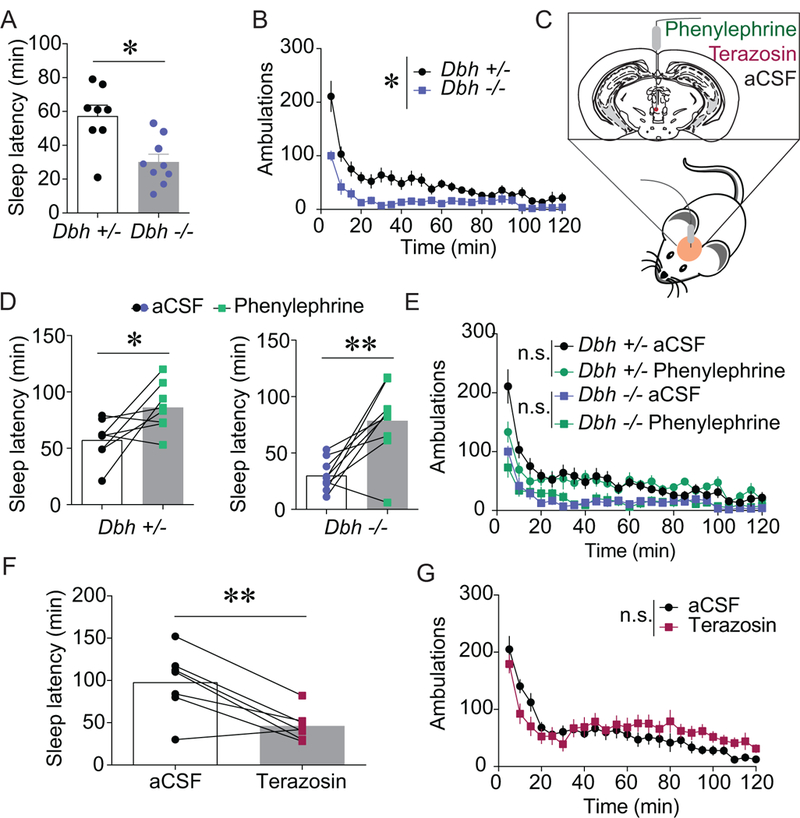
A-B) Mice lacking NE (Dbh −/−; n = 9) fell asleep faster than NE-competent littermates (Dbh +/−; n = 8) following a control injection and were less active in a novel environment (*p < 0.05). C) An α1AR agonist, antagonist, or aCSF was site-specifically infused into the vPAG (D-G). D) Phenylephrine (4 μg/0.3 μL) significantly increased sleep latency in control (n = 8; *p < 0.05) and Dbh −/− (n = 9; **p < 0.01) mice compared to aCSF (0.3 μL) infusion. E) Phenylephrine (4 μg/0.3 μL) site-specifically administered into the vPAG had negligible effects on novelty-induced locomotion (controls, n = 8; Dbh −/−, n = 9; main effects of drug, n.s. = not statistically different). F-G) Intra- vPAG terazosin (3 μg/0.3 μL) significantly decreased sleep latency (F; n = 7; **p < 0.01) but there was no main effect on novelty-induced locomotion (G; n=11; n.s. = not statistically different) in C57BL/6J mice.
To test whether endogenous α1AR tone is important for arousal, aCSF or the α1AR antagonist terazosin (3 μg/0.3 μL) was site-specifically infused into the vPAG (Figure S2B). Blockade of vPAG α1ARs significantly decreased latency to fall asleep (Figure 5F; t(6) = 4.235, p = 0.0055). Terazosin did not significantly alter novelty-induced locomotion (Figure 5G; main effect of time, F(1,10) = 0.377; p = 0.552). Because the vPAG abuts the aqueduct, the arousal-altering effects of intra-vPAG infusions theoretically could be attributed to terazosin actions in other brain regions from the drug circulating through the ventricles following missed injections or diffusion. Therefore, we purposely infused terazosin into the ventricles but saw no alteration in sleep latency (Figure S3A; t(7) = 0.038, p = 0.971) or locomotor activity (Figure S3B; main effect of drug, F(1,7) = 0.001, p = 0.992). Additionally, to test whether antagonism of α1ARs reduces arousal in other wake-promoting dopaminergic nuclei, we also infused terazosin into the VTA, but this manipulation did not alter sleep latency (Figure S3C; t(7) = 1.139, p = 0.292) or locomotor activity (Figure S3D; main effect of drug, F(1,7) = 0.611, p = 0.460). Together, these results reveal that noradrenergic transmission at α1ARs specifically in the vPAG bidirectionally modulates arousal.
vPAG αlARs are enriched on astrocytes
Although α1ARs are expressed in the vPAG (25, 26), their precise anatomical distribution is unknown. Therefore, we conducted immunoelectron microscopy on the same portion of the vPAG in which α1AR agonists and antagonists elicited changes in arousal (Figure 6A). In the vPAG, α1AR immunoreactivity was located primarily presynaptically in unmyelinated axons and in putative glial elements suspected to be astrocytes based upon morphology (Figure 6B-C).
Figure 6. Anatomical location of vPAG α1ARs determined with immunoelectron microscopy.
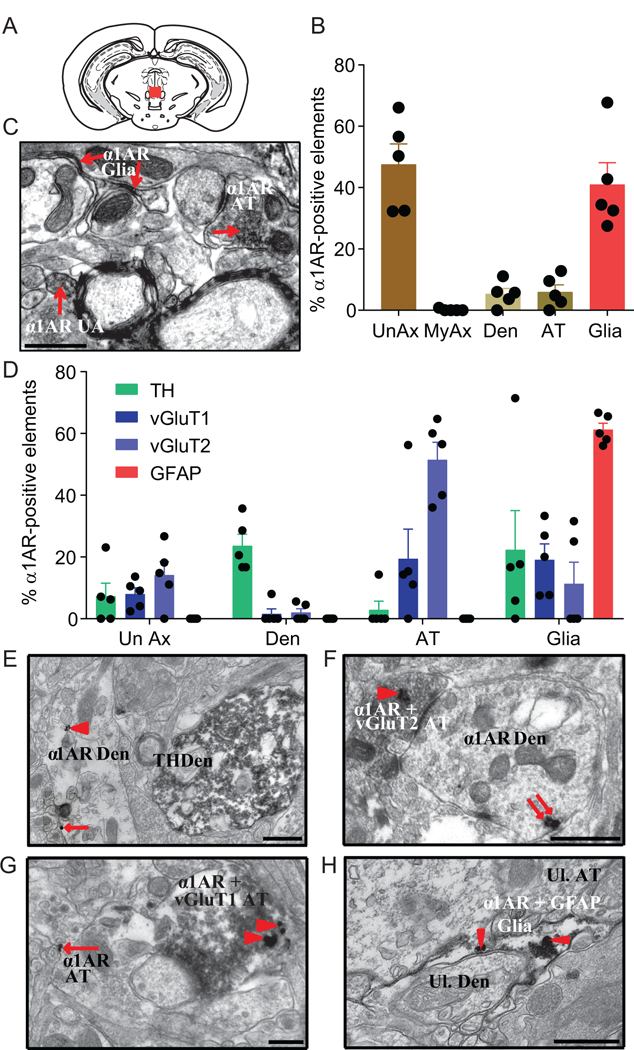
A) Red box signifies the portion of the vPAG used for EM experiments. B) Histogram depicting the mean percentage (±SEM) of unmyelinated axons (UnAx), myelinated axons (MyAx), dendrites (Den), axon terminals (AT), and glia found within the vPAG that contained immunoperoxidase labeling for the α1AR. C) Representative electron micrograph with immunoperoxidase labeling (arrows) for the α1AR in an unmyelinated axon (UnAx), axon terminal (AT), glial element. D) Percentage of α1AR-labeled elements co-labeled with TH, vGluT1, vGluT2, or GFAP. E) Representative micrograph of an α1AR-labeled dendrite (Den) with immunogold (arrowheads point to intracellular gold particles; arrows point to plasma membrane bound gold particles) and a TH-positive dendrite filled with immunoperoxidase labeling. F) Representative micrograph of a double-labeled AT with intracellular gold particles for the α1AR and immunoperoxidase for vGluT2 synapsing on a α1AR labeled dendrite. G) Representative micrograph of a double-labeled axon terminal (AT) for the α1AR in immunogold and vGluT1 with immunoperoxidase synapsing on an unlabeled dendritic process. In addition, pictured is an α1AR-labeled AT with plasma membrane bound gold particles. H) Representative micrograph of a double-labeled astrocyte, containing GFAP with immunoperoxidase (patchy dark gray/black area within astrocyte) and intracellular α1ARs in immunogold (arrowheads point to α1ARs), wrapping around an unlabeled dendrite (Ul. Den). All scale bars = 0.5 μm.
Next, to determine the neurochemical identity of α1AR-containing elements, vPAG slices were double labeled for α1ARs using immunogold and either TH (DA cells), vGluT1 (glutamatergic terminals originating from neurons in the cortex and hippocampus), vGluT2 (glutamatergic terminals originating from neurons in the thalamus and other hindbrain and midbrain structures), or GFAP (astrocytes) using immunoperoxidase (37, 42–44). The most abundant α1AR-containing elements were vGluT2-positive axon terminals and GFAP-positive glia, whereas only modest levels of other combinations of co-labeling were evident (Figure 6D-H). Co-localization of α1ARs and TH was rare.
Gq signaling in vPAG astrocytes is sufficient to promote arousal
Because the overwhelming majority of α1ARs in other dopaminergic brain regions are located on presynaptic neuronal elements (37, 38), α1AR stimulation can activate astrocytes, and astrocytes in other brain regions modulate neuronal activity and sleep (45–48), we were particularly struck by the large proportion of vPAG α1ARs found on astrocytes (Figure 6). To test whether vPAG α1ARs modulate arousal, we mimicked local astrocytic α1AR-Gq transmission using intra-vPAG administration of CNO in GFAP-hM3Dq transgenic mice that express Gq-coupled DREADDs under the astrocytic GFAP promoter (49). Previous research established that expression of DREADDs in the brains of GFAP-hM3Dq mice is limited to astrocytes, and application of CNO increases astrocytic intracellular Ca2+ in these mice but not in wild-type littermates (49). aCSF or CNO (1 μg/0.3 μL) was infused through a guide cannula targeting the vPAG. CNO significantly increased sleep latency in DREADD-expressing mice but not littermate controls (Figure 7A-B; controls, t(8) = 1.066, p = 0.318; GFAP-hM3Dq, t(6) = 2.156, p = 0.0385). This increase in wakefulness was not due to hyperactivity (Figure 8C-D; controls, main effect of drug F(1,8) = 0.808, p = 0.395; GFAP-hM3Dq, main effect of drug F(1,7) = 7.302, p = 0.0305). Combined, these data show that stimulating Gq signaling in vPAG astrocytes is sufficient to promote wakefulness.
Figure 7. Activation of Gq signaling vPAG astrocytes promotes arousal.
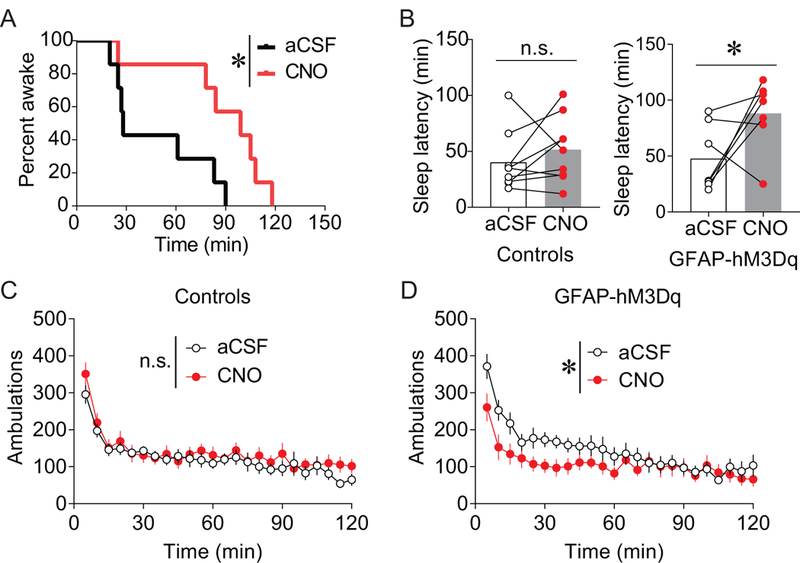
A-B) aCSF or the DREADD ligand CNO (1 μg/0.3 μL) was site-specifically infused into the vPAG of mice expressing Gq-coupled DREADDs in GFAP positive cells (astrocytes). CNO/DREADD-induced activation of vPAG astrocytes increased latency to fall asleep in DREADD-expressing mice (A-B) but not in controls (B; controls, n = 9; n.s. = not statistically different; GFAP-hM3Dq, n = 7; *p < 0.05). C-D) CNO-induced activation of DREADDs in astrocytes reduced novelty-induced locomotion (D; n = 8; *p < 0.05), and CNO had no effect in non-DREADD expressing littermates (C; n= 9; n.s. = not statistically different).
Figure 8. Hypothesized LC-vPAG arousal circuit.
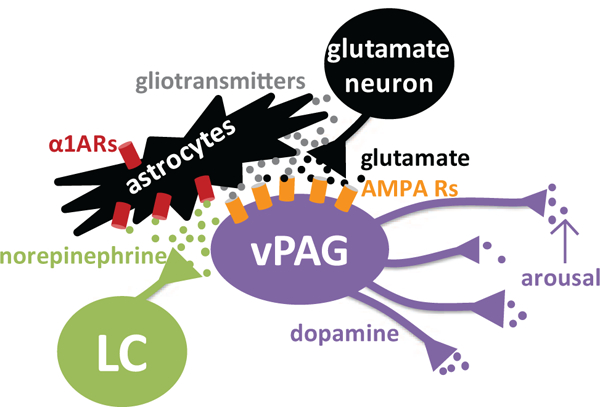
The LC releases NE in the vPAG, which activates astrocytic α1ARs. Astrocytes could release gliotransmitters or otherwise act upon neuronal glutamate terminals to elicit glutamate release in an action potential-independent mechanism, or astrocytes could release glutamate to directly activate DA neurons. Then, glutamate via AMPA receptors drives the activity of wake-promoting vPAGDA neurons to promote arousal. These vPAGDA neurons project to and modulate other brain regions involved in arousal.
Discussion
Using a combination of electrophysiology, immunoelectron microscopy, pharmacological manipulations, and chemogenetics, the present study elucidates mechanisms underlying noradrenergic control of wake-promoting vPAGDA neurons. Transmission at α1ARs increases the excitability of vPAGDA neurons in a glutamate-dependent, action potential-independent manner, and facilitates arousal. vPAG α1ARs are enriched on astrocytes, and Gq activation of vPAG astrocytes is sufficient to promote wakefulness. Together, these experiments support a novel circuit whereby NE increases arousal via astrocytic α1AR-mediated excitation of vPAGDA neurons (Figure 8).
vPAGDA neurons promote wakefulness
Using c-Fos as a measure of activation, previous research found that vPAGDA neurons were active when rodents were awake and inactive when asleep, and lesioning vPAGDA neurons increased time asleep during both day and night (16). The present study demonstrates that chemogenetic activation of vPAGDA neurons promotes arousal by increasing latency to fall sleep without causing hyperactivity. A recent study reported that inhibition of DRN/caudal vPAGDA neurons promotes sleep, while activation of these neurons promotes wakefulness (15). Whereas those authors optogenetically targeted the DRN, we used a chemogenetic approach and targeted rostral vPAGDA neurons. Together, these studies demonstrate that vPAGDA neurons can bidirectionally modulate arousal.
vPAGDA neurons project to multiple brain regions involved in arousal (16). One plausible functional target is the wake-promoting orexin neurons in the lateral hypothalamus (50, 51). Alternatively, orexin’s effects may be upstream of vPAGDA neurons and function via a relay from the LC, because the LC mediates some aspects of orexin-induced arousal (52, 53). The ventrolateral preoptic nucleus facilitates sleep (54, 55) and reciprocally connects with vPAGDA (16), making it another potential functional neuroanatomical partner of vPAGDA neurons. Finally, because vPAGDA neurons can co-release glutamate (34), their wake-promoting properties could involve dual neurotransmitter signal integration by downstream cells.
The finding that DREADD-induced stimulation of vPAGDA neurons promotes wakefulness and causes a small, but statistically significant, decrease in novelty-induced locomotion highlights that wakefulness and locomotor activity are dissociable behaviors. DRN/caudal vPAGDA neurons are activated by a variety of salient stimuli (15). If rostral vPAGDA cells similarly signal salient stimuli, DREADD-induced activation of these neurons may increase other behaviorals that compete with horizontal ambulations.
Noradrenergic transmission via α1ARs modulates vPAGDA neuron activity and arousal
vPAGDA neurons receive input from multiple brain regions that control sleep-wakefulness patterns, such as the LC, lateral hypothalamus, ventrolateral preoptic nucleus, prefrontal cortex, substantia innominata, laterodorsal tegmental nucleus, and DRN (16, 24, 56); however, the functionally significant circuits had not been identified. The present study tested whether vPAGDA neuron-mediated arousal is modulated by noradrenergic transmission from the LC.
Consistent with previous work demonstrating that optogenetic stimulation of the LC promotes wakefulness (21), DREADD-induced activation of the LC also increases wakefulness (Figure 3B-C). CNO infused into projection areas of DREADD-expressing neurons can activate fibers/terminals (57), and we found that intra-vPAG administration of CNO significantly increased latency to sleep (Figure 3D-E) with a magnitude similar to activating all DREADD-expressing LC cells and fibers via systemic CNO administration (Figure 3B-C), demonstrating that specifically activating LC projections to the vPAG is sufficient to promote wakefulness.
Although NE signals through three different AR subtypes (α1, α2, and β), we focused on the α1AR for two reasons. First, this subtype mediates noradrenergic excitatory drive onto DA neurons in the VTA (30), substantia nigra (28, 29), and RLi (27). Second, α1AR activity in other brain regions is implicated in arousal (58–60). Stimulation of vPAG α1ARs increased vPAGDA neuron activity via glutamate and promotes wakefulness in NE-deficient mice (Figures 4–5). Furthermore, local antagonism of α1ARs decreased arousal, consistent with previous research (61). Infusion of terazosin into the vPAG, but not the ventricles or VTA, attenuates arousal (Figures 5F, S3), demonstrating neuroanatomical specificity. Additionally, these experiments indicate that α1ARs do not regulate arousal in all dopaminergic populations; the wake-promoting effects of VTA DA neurons (14) are unlikely under the control of endogenous α1AR tone. Combined, these results illustrate that the vPAG is a locus where NE modulates DA neuron activity and promotes behavioral arousal through α1ARs.
Previous research has shown noradrenergic receptors in other regions modulate arousal. Agonism of α1ARs or βARs in the medial septal area or medial preoptic area promotes wakefulness (60) as does agonism of α1ARs, and to a lesser extent βARs, in the lateral hypothalamus (17); however, infusions in the substantia innominata do not (62). The present study reveals that endogenous noradrenergic tone at α1ARs modulates wakefulness, and intra-vPAG phenylephrine elevates sleep latencies of NE-competent mice as well as mice completely lacking NE. The magnitude of intra-vPAG-induced wakefulness in Dbh −/− mice is noteworthy given that adrenergic receptors in other brain regions contribute functionally to wakefulness. Because arousal is important for adaptive behavior and survival, the brain may have evolved redundant mechanisms involving ARs. Alternatively, each brain region might contribute uniquely to arousal.
α1ARs on vPAG astrocytes modulate wakefulness
Most α1ARs in the VTA, substantia nigra, nucleus accumbens, and prefrontal cortex localize to neuronal elements, with negligible glial expression (37, 38). By contrast, an equally large proportion of α1ARs are found on glia and axons in the vPAG (Figure 6B). Most α1AR-positive glia co-labeled with GFAP, suggesting that astrocytes are a major target of noradrenergic transmission in the vPAG (Figure 6D,H). Importantly, these astrocytes are functionally relevant in modulating arousal. Mimicking α1AR-Gq signaling specifically in vPAG astrocytes using Gq DREADDs promoted wakefulness without causing hyperactivity (Figure 7). Activation of vPAG astrocytes in fact slightly decreased locomotion, similarly to previously reported global activation of astrocytes (49). Recent studies reported that astrocytes in the suprachiasmatic nucleus (63), posterior hypothalamus (64), and hippocampus (47) modulate arousal. The present study reveals that the vPAG is also a critical node in the astrocytic network that controls arousal, adding to research endorsing the ability of astrocytes and gliotransmission to modulate behavior (65–67).
The LC projects broadly, and the LC/NE system is known to regulate astrocyte functions. NE can depolarize astrocytes and this effect is blocked by an α1AR antagonist (68). NE or phenylephrine increases calcium in hippocampal astrocytes (69), and electrical or behaviorally-induced stimulation of the LC provokes calcium transients in cortical astrocytes (70), whereas administering an α1AR antagonist or the LC-specific neurotoxin DSP-4 abolishes astrocytic calcium transients (71, 72). The present study supports the hypothesis that astrocytes respond to arousing events via noradrenergic transmission at α1ARs (73) and implicates vPAG astrocytes as behaviorally relevant targets of the LC.
Astrocytes can release gliotransmitters including glutamate, GABA, D-serine, and ATP to influence local neurons (46, 74). Phenylephrine increased glutamatergic sEPSCs onto vPAGDA neurons, this effect was preserved in the presence of TTX, and CNQX abolished phenylephrine- induced increases in sEPSCs on vPAGDA neurons (Figure 4). The magnitude of phenylephrine-induced sEPSCs was smaller in the presence of TTX. However, TTX reduces not only neuronal activity-dependent glutamate release but also all other neuronally-released neurotransmitters, likely altering the basal tone of numerous neurotransmitter systems that could modulate phenylephrine’s effects. The fact that phenylephrine’s excitatory effects persist in the presence of TTX are consistent with nonconical regulation of glutamate release, such as NE providing excitatory drive onto wake-promoting vPAGDA neuron activity through α1AR-induced activation of local astrocytes (Figure 8). Astrocytes could act on glutamatergic terminals to drive vPAGDA activity through gliotransmission (75) or other mechanisms (76). Because Gq signaling in astrocytes can cause glutamate release (77), a possible alternative is that astrocytes release glutamate directly onto vPAGDA neurons. NE at α1ARs increases the amplitude of mEPSCs in hypothalamic neurons by inducing the release of ATP from astrocytes (78), and astrocytes can promote neuronal plasticity by releasing ATP which acts on purinergic receptors (79). Future experiments could investigate the involvement of ATP and other gliotransmitters in this circuit. Additionally, the potential contribution of axonally-expressed α1ARs remains to be determined.
Technological considerations
Several of our conclusions rely on results of chemogenetic experiments, and we are cognizant of recent controversies surrounding CNO and the critical need for proper controls (39, 40, 80). We validated the specificity of the DREADD-CNO system in our study by including vehicle administration in DREADD-expressing mice and CNO administration in non-DREADD expressing mice. Only CNO administration in DREADD-expressing subjects produced behavioral effects.
Conclusion
The present study demonstrates that wake-promoting vPAGDA neurons are physiologically and functionally under noradrenergic control. vPAG α1ARs drive glutamatergic excitation of vPAGDA neurons, are located on astrocytes, and promote wakefulness. This circuit could be a target for developing therapeutics to treat arousal disturbances and explain a mechanism for some currently available treatments. For example, modafinil (Provigil®) promotes wakefulness without causing hyperactivity (81) via contributions from both NE and DA (12). Future studies should examine the source and functional contributions of glutamatergic neuronal inputs to the vPAG, and which vPAGDA projections are critical for increasing arousal. Additionally, studies targeting DRN and vPAGDA neurons demonstrate that these cells are important in promoting wakefulness by salient stimuli (15), sociability (82), and antinociception (34). Mounting evidence suggests that vPAGDA neurons are not merely an extension of the VTA; further studies should continue to examine the functions of these understudied populations of DA neurons.
Supplementary Material
Acknowledgements
Funding for this work was provided by the National Institute of Neurological Disorders and Stroke (R01NS102306 to DW, DGW, and DAM; F32NS098615 to KAPS; and a P50NS071669 pilot grant to KAPS and DW awarded by the Emory Morris K. Udall Center of Excellence for Parkinson’s Disease Research). The electron microscopy studies were supported by Faculty Development Funds provided by Christopher Newport University (to DAM) and were conducted at Virginia Commonwealth University (Department of Anatomy & Neurobiology Microscopy Facility, supported, in part, by funding from NIH-NINDS Center Core Grant 5 P30 NS047463 and, in part, by funding form NIH-NCI Cancer Center Support Grant P30 CA016059), Emory University (Yerkes National Primate Research Center supported in part, by NIH-Animal Resources Program Yerkes Base Grant P510D011132), and the University of Richmond. Thanks to Christine Lacy, Ricardo Quintanilla, James W. Bogenpohl, and Jean-Francois Pare for technical assistance. We thank C. Strauss for helpful editing of the manuscript.
Footnotes
Disclosures
Sinda Fekir is now affiliated with the Neuroscience Graduate Program, Brown University, Providence, RI 02912. Heather A. Mitchell is now affiliated with the Rodent Models Core, University of Wisconsin-Madison, Madison, WI 53706
All authors report no biomedical financial interests or potential conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Aston-Jones G, Cohen JD (2005): Adaptive gain and the role of the locus coeruleus- norepinephrine system in optimal performance. J Comp Neurol. 493:99–110. [DOI] [PubMed] [Google Scholar]
- 2.de Gee JW, Colizoli O, Kloosterman NA, Knapen T, Nieuwenhuis S, Donner TH (2017): Dynamic modulation of decision biases by brainstem arousal systems. eLife. 6:e23232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rothman SM, Mattson MP (2012): Sleep disturbances in Alzheimer’s and Parkinson’s diseases. Neuromolecular Medicine. 14:194–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Horn WT, Akerman SC, Sateia MJ (2013): Sleep in Schizophrenia and Substance Use Disorders: A Review of the Literature. J Dual Diagn. 9:228–238. [Google Scholar]
- 5.vanBemmel AL (1997): The link between sleep and depression: The effects of antidepressants on EEG sleep. J Psychosomat Res. 42:555–564. [DOI] [PubMed] [Google Scholar]
- 6.Roth T (2007): Insomnia: definition, prevalence, etiology, and consequences. J Clin Sleep Med. 3:S7. [PMC free article] [PubMed] [Google Scholar]
- 7.Bonnet MH, Arand DL (2010): Hyperarousal and insomnia: State of the science. Sleep Medicine Reviews. 14:9–15. [DOI] [PubMed] [Google Scholar]
- 8.CDC (2011): Morbidity and Mortality Weekly Report. Weekly. 60:233–242. [PubMed] [Google Scholar]
- 9.Committee on Sleep Medicine and Research (2006): Sleep Disorders and Sleep Deprivation: An Unmet Public Health Problem. National Academies Press. [PubMed] [Google Scholar]
- 10.Mendoza J, Challet E (2014): Circadian insights into dopamine mechanisms. Neuroscience. 282:230–242. [DOI] [PubMed] [Google Scholar]
- 11.Oishi Y, Lazarus M (2017): The control of sleep and wakefulness by mesolimbic dopamine systems. Neurosci Res. 118:66–73. [DOI] [PubMed] [Google Scholar]
- 12.Mitchell HA, Bogenpohl JW, Liles LC, Epstein MP, Bozyczko-Coyne D, Williams M, et al. (2008): Behavioral responses of dopamine β-hydroxylase knockout mice to modafinil suggest a dual noradrenergic-dopaminergic mechanism of action. Pharmacol Biochem Behav. 91:217–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arnulf I, Leu-Semenescu S (2009): Sleepiness in Parkinson’s disease. Parkinsonism & Related Disorders. 15:S101–S104. [DOI] [PubMed] [Google Scholar]
- 14.Eban-Rothschild A, Rothschild G, Giardino WJ, Jones JR, de Lecea L (2016): VTA dopaminergic neurons regulate ethologically relevant sleep-wake behaviors. Nature Neuroscience. 19:1356–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cho JR, Treweek JB, Robinson JE, Xiao C, Bremner LR, Greenbaum A, et al. (2017): Dorsal Raphe Dopamine Neurons Modulate Arousal and Promote Wakefulness by Salient Stimuli. Neuron.1205–1219. [DOI] [PubMed] [Google Scholar]
- 16.Lu J, Jhou TC, Saper CB (2006): Identification of wake-active dopaminergic neurons in the ventral periaqueductal gray matter. J Neurosci. 26:193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berridge CW, Schmeichel BE, Espana RA (2012): Noradrenergic modulation of wakefulness/arousal. Sleep Medicine Reviews. 16:187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Berridge CW, Waterhouse BD (2003): The locus coeruleus-noradrenergic system: modulation of behavioral state and state-dependent cognitive processes. Brain Res Rev. 42:33–84. [DOI] [PubMed] [Google Scholar]
- 19.Mitchell HA, Weinshenker D (2010): Good night and good luck: Norepinephrine in sleep pharmacology. Biochem Pharmacol. 79:801–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aston-Jones G, Bloom F (1981): Activity of norepinephrine-containing locus coeruleus neurons in behaving rats anticipates fluctuations in the sleep-waking cycle. J Neurosci. 1:876–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carter ME, Yizhar O, Chikahisa S, Nguyen H, Adamantidis A, Nishino S, et al. (2010): Tuning arousal with optogenetic modulation of locus coeruleus neurons. Nature Neuroscience. 13:1526–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.España RA, Schmeichel BE, Berridge CW (2016): Norepinephrine at the nexus of arousal, motivation and relapse. Brain Res. 1641:207–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grzanna R, Molliver M (1980): The locus coeruleus in the rat: an immunohistochemical delineation. Neuroscience. 5:21–40. [DOI] [PubMed] [Google Scholar]
- 24.Jones BE, Moore RY (1977): Ascending projections of locus coeruleus in rat. II. Autoradiographic study. Brain Res. 127:23–53. [PubMed] [Google Scholar]
- 25.Jones LS, Gauger LL, Davis JN (1985): Anatomy of brain alpha1 □ adrenergic receptors: In vitro autoradiography with [125I] □heat. J Comp Neurol. 231:190–208. [DOI] [PubMed] [Google Scholar]
- 26.Pieribone VA, Nicholas AP, Dagerlind A, Hokfelt T (1994): Distribution of alpha 1 adrenoceptors in rat brain revealed by in situ hybridization experiments utilizing subtype-specific probes. J Neurosci. 14:4252–4268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Williams MA, Li C, Kash TL, Matthews RT, Winder DG (2014): Excitatory drive onto dopaminergic neurons in the rostral linear nucleus is enhanced by norepinephrine in an α1 adrenergic receptor-dependent manner. Neuropharmacology. 86:116–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grenhoff J, North RA, Johnson SW (1995): Alpha1 □adrenergic effects on dopamine neurons recorded intracellularly in the rat midbrain slice. Eur J Neurosci. 7:1707–1713. [DOI] [PubMed] [Google Scholar]
- 29.Grenhoff J, Nisell M, Ferre S, Aston-Jones G, Svensson T (1993): Noradrenergic modulation of midbrain dopamine cell firing elicited by stimulation of the locus coeruleus in the rat. J Neural Transm. 93: 11–25. [DOI] [PubMed] [Google Scholar]
- 30.Goertz RB, Wanat MJ, Gomez JA, Brown ZJ, Phillips PE, Paladini CA (2015): Cocaine increases dopaminergic neuron and motor activity via midbrain α1 adrenergic signaling. Neuropsychopharmacology. 40:1151–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thomas SA, Marck BT, Palmiter RD, Matsumoto AM (1998): Restoration of norepinephrine and reversal of phenotypes in mice lacking dopamine β□hydroxylase. J Neurochem. 70:2468–2476. [DOI] [PubMed] [Google Scholar]
- 32.Thomas SA, Matsumoto AM, Palmiter RD (1995): Noradrenaline is essential for mouse fetal development. Nature. 374:643–646. [DOI] [PubMed] [Google Scholar]
- 33.Schank JR, Ventura R, Puglisi-Allegra S, Alcaro A, Cole CD, Liles LC, et al. (2006): Dopamine [beta]-hydroxylase knockout mice have alterations in dopamine signaling and are hypersensitive to cocaine. Neuropsychopharmacology. 31:2221. [DOI] [PubMed] [Google Scholar]
- 34.Li C, Sugam JA, Lowery-Gionta EG, McElligott ZA, McCall NM, Lopez AJ, et al. (2016): Mu opioid receptor modulation of dopamine neurons in the periaqueductal gray/dorsal raphe: A role in regulation of pain. Neuropsychopharmacology. 41: 2122–2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hunsley MS, Palmiter RD (2004): Altered sleep latency and arousal regulation in mice lacking norepinephrine. Pharmacology Biochemistry and Behavior. 78:765–773. [DOI] [PubMed] [Google Scholar]
- 36.Mitchell HA, Ahern TH, Liles LC, Javors MA, Weinshenker D (2006): The effects of norepinephrine transporter inactivation on locomotor activity in mice. Biol Psychiatry. 60:1046–1052. [DOI] [PubMed] [Google Scholar]
- 37.Mitrano DA, Schroeder JP, Smith Y, Cortright JJ, Bubula N, Vezina P, et al. (2012): Alpha-1 adrenergic receptors are localized on presynaptic elements in the nucleus accumbens and regulate mesolimbic dopamine transmission. Neuropsychopharmacology. 37:2161–2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rommelfanger KS, Mitrano DA, Smith Y, Weinshenker D (2009): Light and electron microscopic localization of alpha-1 adrenergic receptor immunoreactivity in the rat striatum and ventral midbrain. Neuroscience. 158:1530–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.MacLaren DAA, Browne RW, Shaw JK, Krishnan Radhakrishnan S, Khare P, España RA, et al. (2016): Clozapine N-Oxide Administration Produces Behavioral Effects in Long-Evans Rats: Implications for Designing DREADD Experiments. eNeuro. 3:0219–0216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gomez JL, Bonaventura J, Lesniak W, Mathews WB, Sysa-Shah P, Rodriguez LA, et al. (2017): Chemogenetics revealed: DREADD occupancy and activation via converted clozapine. Science. 357:503–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hunsley M, Palmiter R (2003): Norepinephrine-deficient mice exhib-it normal sleep-wake states but have shorter sleep latency after mild stress and low doses of amphetamine. Sleep. 26:521–526. [PubMed] [Google Scholar]
- 42.Fremeau RT, Troyer MD, Pahner I, Nygaard GO, Tran CH, Reimer RJ, et al. (2001): The expression of vesicular glutamate transporters defines two classes of excitatory synapse. Neuron. 31:247–260. [DOI] [PubMed] [Google Scholar]
- 43.Raju DV, Smith Y (2005): Differential localization of vesicular glutamate transporters 1 and 2 in the rat striatum. In: Bolam JP, Ingham CA, Magill PJ, editors. Basal Ganglia; VIII, pp 601–610. [Google Scholar]
- 44.Eng LF (1985): Glial fibrillary acidic protein (GFAP) - The major protein of glia intermediate filaments in differentiated astrocytes. J Neuroimmunol. 8:203–214. [DOI] [PubMed] [Google Scholar]
- 45.Frank MG (2013): Astroglial regulation of sleep homeostasis. Curr Opin Neurobiol. 23:812–818. [DOI] [PubMed] [Google Scholar]
- 46.Bazargani N, Attwell D (2017): Amines, Astrocytes, and Arousal. Neuron. 94:228–231. [DOI] [PubMed] [Google Scholar]
- 47.Papouin T, Dunphy JM, Tolman M, Dineley KT, Haydon PG (2017): Septal Cholinergic Neuromodulation Tunes the Astrocyte-Dependent Gating of Hippocampal NMDA Receptors to Wakefulness. Neuron. 94:840–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Halassa MM, Florian C, Fellin T, Munoz JR, Lee SY, Abel T, et al. (2009): Astrocytic modulation of sleep homeostasis and cognitive consequences of sleep loss. Neuron. 61:213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Agulhon C, Boyt KM, Xie AX, Friocourt F, Roth BL, McCarthy KD (2013): Modulation of the autonomic nervous system and behaviour by acute glial cell Gq protein □ coupled receptor activation in vivo. J Physiol. 591:5599–5609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tsujino N, Sakurai T (2013): Role of orexin in modulating arousal, feeding, and motivation. Front Behav Neurosci. 7:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sutcliffe JG, de Lecea L (2002): The hypocretins: Setting the arousal threshold. Nature Reviews Neuroscience. 3:339–349. [DOI] [PubMed] [Google Scholar]
- 52.Carter ME, Brill J, Bonnavion P, Huguenard JR, Huerta R, de Lecea L (2012): Mechanism for Hypocretin-mediated sleep-to-wake transitions. Proc Natl Acad Sci. 109:E2635–E2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schwartz MD, Nguyen AT, Warrier DR, Palmerston JB, Thomas AM, Morairty SR, et al. (2016): Locus coeruleus and tuberomammillary nuclei ablations attenuate hypocretin/orexin antagonist-mediated REM sleep. eNeuro. 3:0018–0016.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Saper CB, Fuller PM, Pedersen NP, Lu J, Scammell TE (2010): Sleep state switching. Neuron. 68:1023–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lu J, Greco MA, Shiromani P, Saper CB (2000): Effect of lesions of the ventrolateral preoptic nucleus on NREM and REM sleep. J Neurosci. 20:3830–3842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marchand JE, Hagino N (1983): Afferents to the periaqueductal gray in the rat - A horseradish-peroxidase study. Neuroscience. 9:95–106. [DOI] [PubMed] [Google Scholar]
- 57.Mahler SV, Vazey EM, Beckley JT, Keistler CR, McGlinchey EM, Kaufling J, et al. (2014): Designer receptors show role for ventral pallidum input to ventral tegmental area in cocaine seeking. Nature Neuroscience. 17:577–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Berridge C, Espana R (2000): Synergistic sedative effects of noradrenergic α 1-and β-receptor blockade on forebrain electroencephalographic and behavioral indices. Neuroscience. 99:495–505. [DOI] [PubMed] [Google Scholar]
- 59.Buzsaki G, Kennedy B, Solt V, Ziegler M (1991): Noradrenergic control of thalamic oscillation: the role of α□2 receptors. Eur J Neurosci. 3:222–229. [DOI] [PubMed] [Google Scholar]
- 60.Berridge CW, Isaac SO, España RA (2003): Additive wake-promoting actions of medial basal forebrain noradrenergic α1- and β-receptor stimulation. Behav Neurosci. 117:350–359. [DOI] [PubMed] [Google Scholar]
- 61.Stone EA, Lin Y, Ahsan R, Quartermain D (2004): Gross mapping of α 1-adrenoceptors that regulate behavioral activation in the mouse brain. Behav Brain Res. 152:167–175. [DOI] [PubMed] [Google Scholar]
- 62.Berridge CW, Bolen SJ, Manley MS, Foote SL (1996): Modulation of Forebrain Electroencephalographic Activity in Halothane-Anesthetized Rat via Actions of Noradrenergic β-Receptors within the Medial Septal Region. J Neurosci. 16:7010–7020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brancaccio M, Patton AP, Chesham JE, Maywood ES, Hastings MH (2017): Astrocytes control circadian timekeeping in the suprachiasmatic nucleus via glutamatergic signaling. Neuron. 93:1420–1435.e1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pelluru D, Konadhode RR, Bhat NR, Shiromani PJ (2015): Optogenetic stimulation of astrocytes in the posterior hypothalamus increases sleep at night in C57BL/6J mice. Eur J Neurosci. 43:1298–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Oliveira JF, Sardinha VM, Guerra-Gomes S, Araque A, Sousa N (2015): Do stars govern our actions? Astrocyte involvement in rodent behavior. Trends in Neurosciences. 38:535–549. [DOI] [PubMed] [Google Scholar]
- 66.Reissner KJ, Brown RM, Spencer S, Tran PK, Thomas CA, Kalivas PW (2014): Chronic Administration of the Methylxanthine Propentofylline Impairs Reinstatement to Cocaine by a GLT-1-Dependent Mechanism. Neuropsychopharmacology. 39:499–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen NY, Sugihara H, Kim J, Fu ZY, Barak B, Sur M, et al. (2016): Direct modulation of GFAP-expressing glia in the arcuate nucleus bi-directionally regulates feeding. eLife. 5:e18716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bowman CL, Kimelberg HK (1987): Pharmalogical properties of the norepinephrine-induced depolarization of astrocytes in primary culture - evidence for the involvement of an alpha-adrenergic receptor. Brain Res. 423:403–407. [DOI] [PubMed] [Google Scholar]
- 69.Duffy S, MacVicar B (1995): Adrenergic calcium signaling in astrocyte networks within the hippocampal slice. J Neurosci. 15:5535–5550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bekar LK, He W, Nedergaard M (2008): Locus Coeruleus alpha-Adrenergic-Mediated Activation of Cortical Astrocytes In Vivo. Cereb Cortex. 18:2789–2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ding FF, O’Donnell J, Thrane AS, Zeppenfeld D, Kang HY, Xie LL, et al. (2013): alpha(1)-Adrenergic receptors mediate coordinated Ca2+ signaling of cortical astrocytes in awake, behaving mice. Cell Calcium. 54:387–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Paukert M, Agarwal A, Cha J, Doze VA, Kang JU, Bergles DE (2014): Norepinephrine controls astroglial responsiveness to local circuit activity. Neuron. 82:1263–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kjaerby C, Rasmussen R, Andersen M, Nedergaard M (2017): Does global astrocytic calcium signaling participate in awake brain state transitions and neuronal circuit function? Neurochem Res. 42:1810–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Parpura V, Basarsky TA, Liu F, Jeftinija K, Jeftinija S, Haydon PG (1994): Glutamate-mediated astrocyte-neuron signalling. Nature. 369:744–747. [DOI] [PubMed] [Google Scholar]
- 75.Savtchouk I, Volterra A (2018): Gliotransmission: Beyond Black-and-White. J Neurosci. 38:14–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fiacco TA, McCarthy KD (2018): Multiple lines of evidence indicate that gliotransmission does not occur under physiological conditions. J Neurosci. 38:3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Scofield MD, Boger HA, Smith RJ, Li H, Haydon PG, Kalivas PW (2015): Gq-DREADD selectively initiates glial glutamate release and inhibits cue-induced cocaine seeking. Biol Psychiatry. 78:441–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gordon GRJ, Baimoukhametova DV, Hewitt SA, Rajapaksha W, Fisher TE, Bains JS (2005): Norepinephrine triggers release of glial ATP to increase postsynaptic efficacy. Nature Neuroscience. 8:1078–1086. [DOI] [PubMed] [Google Scholar]
- 79.Gordon GR, Iremonger KJ, Kantevari S, Ellis-Davies GC, MacVicar BA, Bains JS (2009): Astrocyte-mediated distributed plasticity at hypothalamic glutamate synapses. Neuron. 64:391–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Manvich DF, Webster KA, Foster SL, Farrell MS, Ritchie JC, Porter JH, et al. (2018): The DREADD agonist clozapine N-oxide (CNO) is reverse-metabolized to clozapine and produces clozapine-like interoceptive stimulus effects in rats and mice. Scientific Reports. 8:3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cope ZA, Minassian A, Kreitner D, MacQueen DA, Milienne-Petiot M, Geyer MA, et al. (2017): Modafinil improves attentional performance in healthy, non-sleep deprived humans at doses not inducing hyperarousal across species. Neuropharmacology. 125:254–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Matthews GA, Nieh EH, Vander Weele CM, Halbert SA, Pradhan RV, Yosafat AS, et al. (2016): Dorsal raphe dopamine neurons represent the experience of social isolation. Cell. 164:617–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.