

Abstract
Glucose-sensitive neurons have long been implicated in glucose homeostasis, but how glucose-sensing information is used by the brain in this process remains uncertain. Here, we propose a model in which 1) information relevant to the circulating glucose level is essential to the proper function of this regulatory system, 2) this input is provided by neurons located outside the blood-brain barrier (BBB) (since neurons situated behind the BBB are exposed to glucose in brain interstitial fluid, rather than that in the circulation), and 3) while the efferent limb of this system is comprised of neurons situated behind the BBB, many of these neurons are also glucose-sensitive. Precedent for such an organizational scheme is found in the thermoregulatory system, which we draw upon in this framework for understanding the role played by brain glucose sensing in glucose homeostasis.
eTOC Blurb
Bentsen and colleagues present a discussion of the mechanism by which the brain responds to and controls glycemia. While neurons in the brain are glucose-sensitive, glucose concentrations behind the blood-brain barrier (BBB) are only a fraction of plasma glucose concentrations. Hence, input to the brain regarding circulating glucose is likely detected by structures lying outside the BBB.
I. Introduction
The hypothesis that the brain, working in cooperation with pancreatic islets and liver, plays a key role in day-to-day control of glucose homeostasis makes teleological sense in light of the brain’s reliance on circulating glucose as its primary fuel source. Consistent with this concept is compelling evidence of the brain’s ability to influence determinants of glucose homeostasis (e.g., the secretion of insulin and glucagon and other glucoregulatory hormones, tissue insulin sensitivity and insulin-independent glucose disposal) in ways that can powerfully impact the circulating glucose level (Deem et al., 2017). To carry out this role, the brain (like pancreatic beta cells) must have the capacity to sense ambient glucose levels and transduce this information into adaptive, homeostatic responses. Consistent with this possibility is the discovery of “glucose-sensitive” neurons in the 1960s (Anand et al., 1964; Oomura et al., 1964), which have since been well-characterized (reviewed in (Fioramonti et al., 2017)).
Glucose-sensitive neurons can be subdivided into “glucose-excited” and “glucose-inhibited” neuronal subsets, and they are concentrated in hypothalamic areas implicated in whole-body glucose homeostasis (for reviews, see (Fioramonti et al., 2017; Pozo and Claret, 2018; Zhou et al., 2018)). Moreover, many of these neurons use the same glucose-sensing molecular machinery found in pancreatic beta cells. What is less clear, however, is the extent to which the brain senses changes in circulating glucose by sampling from peripheral glucose sensors, as opposed to sensing the glucose to which neurons are exposed in brain interstitial fluid (ISF). The goal of this Perspectives article is to synthesize information relevant to this and related questions into a model for understanding the roles played by glucose-responsive neurons - and brain glucose sensing more broadly - in glucose homeostasis.
II. What specific function does brain glucose sensing serve?
As a first step to answering this question, one must consider where a glucose-responsive neuron sits with respect to the blood-brain barrier (BBB). This is because the concentration of glucose in brain ISF (behind the BBB) is believed to be only a fraction (perhaps as low as 20 to 30%) of the circulating level (outside the BBB) (Dunn-Meynell et al., 2009; Fioramonti et a l 2017). Furthermore, a change in the level of circulating glucose is reflected in the brain ISF compartment in a manner that is both dampened and delayed (Hwang et al., 2017; Langlet et al., 2013). As one example, a recent study in humans showed that although brain glucose levels (measured by magnetic resonance spectroscopy) increased as expected following a fixed (or “clamped”) increase in the plasma glucose level, the rise was much slower than in plasma, such that 60–90 minutes passed before a new steady-state was reached (Hwang et al., 2017). While acknowledging the need for additional data on this topic, available evidence strongly suggests that glucose levels in brain ISF do not faithfully reflect the temporal dynamics of changes in the circulating level. Yet the vast majority of glucose-sensitive neurons in the brain, including those in both the hindbrain and in hypothalamic areas such as the ventromedial nucleus (VMN) that are implicated in glucose homeostasis (Kang et al., 2006; Zhou et al., 2018), are exposed only to brain ISF and not to plasma.
From these considerations, we infer that if the brain does in fact play a key role in whole-body glucose homeostasis, the neurons involved in the afferent glucose-sensing limb of this regulatory system must either be situated outside of the BBB or have axons that project across the BBB. But if this is true, how then does the capacity to detect and respond to changing glucose levels in surrounding ISF - levels that are related to, but not a direct reflection of the circulating glucose level - enable glucose-sensitive neurons situated behind the BBB to participate in glucose homeostasis? How can changes in the activity of glucose-sensitive neurons in the VMN, for example, contribute to glucose homeostasis if they are unable to sense perturbations in circulating glucose owing to their anatomical location? In an attempt to answer these questions, we turn to the organization of the thermoregulatory system.
III. Lessons learned from the thermoregulatory system.
As a general rule, homeostatic control systems for key biological variables require inputs that both accurately represent the current and also predict the future states of those variables (Ramsay and Woods, 2016). One consequence of this type of arrangement is that the variable in question does not actually have to change for corrective responses to be engaged that keep it stable. For this to occur, the variable to be regulated (or biological proxy variables) must be sensed in a rapid and faithful manner, so that adaptive responses can be mounted in response to even the slightest change in the level of that parameter (in some cases, before the defended parameter has even begun to change). The model proposed herein views the glucose level in brain ISF as the variable to be regulated, with control over the circulating glucose level being crucial to this regulation.
A good example of this type homeostatic control is provided by the thermoregulatory system, in that information regarding changing cutaneous temperature is continuously transmitted from thermosensory afferents innervating the skin (reviewed in (Morrison, 2016)). This information is in turn communicated from spinal cord and brainstem via an ascending pathway to neurons in the hypothalamic preoptic area (POA). Neurons in the POA process this cutaneous thermosensory input and, via projections to the hypothalamic dorsomedial nucleus, transduce it into adaptive responses (e.g., activation of brown adipose tissue, shivering, cutaneous vasoconstriction) that preserve constancy of the internal (i.e., brain) temperature (Tan and Knight, 2018).
Owing to the high fidelity of this system, normal mice placed into a cold environment are able to profoundly increase heat production and reduce heat dissipation, despite virtually no detectable change in average core temperature (Kaiyala et al., 2015). This impressive feat would not be possible if temperature sensing were performed primarily by neurons in the brain, since the brain temperature would have to change before a homeostatic response could be engaged. Such a system would ensure that brain temperature changes continuously - the opposite of the desired outcome, and opposite to what is observed (Bratincsak and Palkovits, 2005).
The analogy between homeostatic control of body temperature and blood glucose levels is made even more compelling by the fact that subsets of POA neurons that comprise the integrative/effector limb of the thermoregulatory system are themselves capable of sensing and responding to changes of ambient temperature (Tan and Knight, 2018). Extending the analogy further, both “cold-sensitive” and “warm-sensitive” neurons exist in the POA (Morrison, 2016; Tan and Knight, 2018), analogous to glucose-excited and glucose-inhibited neurons in the VMN and other hypothalamic areas.
For clarity, we note that “thermosensitive” and “thermoresponsive” POA neurons can be distinguished from one another, in that the former applies to neurons whose membrane potential and firing rate is directly sensitive to a change in ambient temperature, whereas the latter applies to neurons in a circuit that is responsive to a change of external temperature (whether or not the neurons are intrinsically thermosensitive). Thus, housing mice in a warm environment activates neurons in the POA, but only a subset of these are truly “warm-sensitive”, because others that are not intrinsically thermosensitive can also be activated as part of circuit that responds to the change in external temperature. By extension, although the terms “glucose-sensitive” and “glucose-responsive” are sometimes used interchangeably in the literature, we define the former as neurons whose membrane potential and firing rate is directly sensitive to a change in ambient glucose, whereas that latter applies more broadly to neurons in a glucose-responsive circuit.
There is little question that activation (or inhibition) of intrinsically thermosensitive POA neurons can have a powerful impact on the thermoregulatory system. For example, if the POA is cooled directly (via implantation of device known as a “thermode”), potent thermogenic responses are elicited that mimic those observed when the animal is placed in a cold environment (Mohammed et al., 2018). Yet under physiological conditions, these POA neurons do not participate as temperature sensors; instead, they function as thermoregulatory actuators whose activity is governed by afferent information conveyed by thermosensory neurons situated outside of the brain (Morrison, 2016; Tan and Knight, 2018). This arrangement allows adaptive responses to be engaged before brain temperature changes, a prerequisite if the goal is to preserve constancy of brain temperature.
Why then should these POA neurons possess molecular machinery that enables them to sense and respond to changes of ambient (brain) temperature? While the answer is unknown, a likely possibility is that the intrinsic thermosensing capacity of POA neurons serves as a back-up or “fail-safe” system that can be engaged in the event that brain temperature changes - which happens only if the physiological system has either failed or is overwhelmed by environmental extremes. An alternative hypothesis is that information related to brain temperature offers a baseline against which afferent information from outside the brain can be compared, thereby enabling “anticipatory” responses to be engaged before the brain temperature has changed. These two possibilities are not mutually exclusive, and each may apply.
The overarching point is that even though subpopulations of POA neurons exist that are themselves thermosensitive, they do not generate the primary afferent temperature sensing information used under physiological conditions (Morrison, 2016; Tan and Knight, 2018). Instead, their thermosensitive properties identify them as components of the integrative/effector limb that transduces relevant sensory input into efferent responses crucial to effective thermoregulation, with the overarching goal of ensuring that brain temperature does not change (Bratincsak and Palkovits, 2005).
IV. A parallel to glucose sensing by the brain?
Just as brain temperature is biologically defended by a highly effective thermoregulatory system, brain glucose levels appear to remain relatively constant over time, even in the face of fluctuating plasma levels (Dunn-Meynell et al., 2009; Fioramonti et al., 2017; Langlet et al., 2013). Based on the above considerations, it is difficult to envision how sensing of glucose of within brain ISF alone could constitute an afferent signal that accounts for this degree of homeostatic control. Stated differently, reliance on afferent input based primarily on local glucose levels in brain ISF ensures that brain glucose levels must change before effector responses are engaged. Beyond this bedrock principle of physiology, the delayed and blunted change of brain ISF glucose levels following a corresponding change in the plasma level would further limit the effectiveness of a system designed to maintain stability in the supply of glucose to the brain. Therefore, just as effective brain control of core body temperature relies on cutaneous (as opposed to brain) temperature sensing, we submit that effective brain control of whole-body glucose homeostasis is contingent upon the capacity to sense the circulating glucose level, rather than relying primarily on glucose sensing in brain ISF.
Of course, it is possible that under physiological conditions, the endocrine pancreas controls glucose homeostasis without help from the brain, and that brain glucose sensing impacts whole-body glucose homeostasis only under pathological conditions sufficient to impact glucose levels in brain ISF (i.e., hypoglycemia). Indeed, this possibility constitutes a major obstacle to broad acceptance of the notion that the control of glucose homeostasis is achieved via cooperative interaction between brain and islet, as we and others have proposed (Schwartz et al., 2013).
But glucose-stimulated insulin secretion is clearly regulated by autonomic input under physiological conditions (Deem et al., 2017), and glucose-sensitive neurons are in fact found in sites outside the brain that are exposed to the circulation. As one example, neurons innervating the hepatic portal vein have well-described glucose-sensing properties (Donovan, 2002). As such, these neurons are uniquely positioned to function in meal-related glucose sensing, since calories absorbed from the GI tract are absorbed directly into the hepatic portal vein before entering systemic circulation.
Indeed, the resulting gradient between portal and systemic glucose levels has been identified as an afferent “portal” signal that triggers the marked increase of liver glucose uptake that occurs during a meal (Moore et al., 2012). Moreover, physiological control of hepatic glucose uptake during a meal appears to be governed by a brain mechanism activated in response to the portal signal created by the meal-induced influx of glucose into the hepatic portal vein from the GI tract. The underlying mechanism appears to involve reduced sympathetic outflow to the liver, which in turn activates liver glucokinase, an enzyme that catalyzes glucose phosphorylation and is rate-limiting for hepatic glucose uptake (Coate et al., 2013; Dicostanzo et al., 2006)
As the GI tract is also endowed with glucose-sensing neurons that can transmit afferent information relevant to nutrient ingestion (Williams et al., 2016), this input together with that that from the hepatic portal vein has been described as portal-mesenteric vein (PMV) glucose sensing. Activation of this PMV glucose sensing circuit (e.g., in response to hypoglycemia in the portal-splanchnic circulation) activates neurons in the NTS via spinal afferent nerves (predominantly sympathetic), rather than the vagus nerve (Chan and Sherwin, 2014; Jokiaho et al., 2014; Watts and Donovan, 2010). From the hindbrain, ascending catecholaminergic projections relay this afferent information to the hypothalamus, where it is specifically implicated in the response to hypoglycemia that is slow in onset (Chan and Sherwin, 2014; Jokiaho et al., 2014; Watts and Donovan, 2010). Neurons in the NTS also project extensively to the parabrachial nucleus, which contains neurons that project in turn to the VMN and are involved the response to glucopenia (Flak et al., 2014; Garfield et al., 2014).
However, since PMV glucose sensing is not particularly useful as a measure of the systemic glucose level in the non-fasted state, a search for other glucose-sensitive neurons that are exposed to the systemic circulation is warranted. Consistent with this notion, neurons with glucose-sensing properties have been identified in brain areas located within or in close proximity to circumventricular organs (CVOs), which sit outside the BBB and hence are exposed directly to the circulating glucose level. Among these brain areas are the ventromedial portion of the arcuate nucleus and adjacent median eminence (ARC-ME), as well as the nucleus of the solitary tract (NTS) and area postrema in the hindbrain (Boychuk et al., 2015; Murphy et al., 2009; Wang et al., 2004). The extent to which such neurons constitute a relevant source of afferent input to the brain’s glucoregulatory system warrants careful study.
Returning to the question posed earlier, if the afferent limb of the brain system controlling glucose homeostasis involves neurons that sense circulating glucose levels directly, what role in glucose homeostasis might be played by glucose-sensitive neurons situated behind the BBB? One possibility is that the primary role of such neurons is unrelated to glucose-sensing per se, at least under physiological conditions, and that this glucose-sensing capacity is a marker of hypothalamic effector neurons situated downstream of neurons that constitute the afferent limb of the system. Another possibility mentioned earlier is that glucose sensing by neurons situated behind the BBB constitutes a “baseline” signal to which incoming input generated in response to changing plasma glucose levels is compared as part of an integrative process that controls the efferent limb of the system. These models are again analogous to the key role played by POA neurons that, despite being thermosensitive themselves, rely upon afferent input from the skin to mount effector responses that enable effective thermoregulation.
Accordingly, the afferent and efferent limbs of the brain’s glucoregulatory system should be distinguishable from one another, with the afferent limb being comprised of neurons situated outside of, or with access across, the BBB and the efferent limb situated in areas behind the BBB, such as the VMN (by analogy to the organization of neurocircuits involved in thermoregulation). After all, changes in the activity of glucose-sensitive neurons in the VMN and elsewhere can potently influence both the plasma glucose level and the secretion of insulin and glucagon (via changes in autonomic output to liver and pancreas) (Fioramonti et al., 2017; Kang et al., 2006; Meek et al., 2016; Pozo and Claret, 2018; Zhou et al., 2018). Moreover, the activity of glucoregulatory neurons in the VMN comprising the efferent limb of the system is known to be influenced by ascending input from projections from the parabrachial nucleus (Flak et al., 2014; Garfield et al., 2014) (a brain area richly supplied with projections from the NTS).
With this background, it is not difficult to envision afferent input regarding glucose levels in the GI tract and hepatic portal vein being transmitted via spinal afferents to the hindbrain (Chan and Sherwin, 2014; Donovan, 2002; Jokiaho et al., 2014; Watts and Donovan, 2010), and from there to the VMN (Flak et al., 2014; Garfield et al., 2014). At the same time, information relevant to systemic (rather than PMV) glucose levels may be transmitted to the VMN by way of glucose-sensing neurons located outside the BBB, potentially including the ventromedial arcuate nucleus, median eminence, NTS, area postrema, or some combination thereof. On theoretical grounds, neurons in these areas are ideally positioned to provide acute, direct signals regarding glucose levels in the circulation that the brain uses to mount efferent responses.
This organizational scheme, illustrated in Figure 1, warrants consideration in part because if proven correct, it will have a major impact on the interpretation of previous experimental outcomes. As one example, a key role for VMN glucose-sensitive neurons in the counterregulatory response (CRR) to hypoglycemia has been inferred from classic studies demonstrating that 1) the CRR to hypoglycemia is sharply blunted by infusion of glucose directly into the VMN (Borg et al., 1997), and 2) induction of glucopenia locally in the VMN is sufficient to elicit CRRs even in the absence of systemic hypoglycemia (Borg et al., 1995). These findings appear to show that VMN glucose-sensing is both necessary and sufficient to explain the adaptive response to hypoglycemia.
Figure 1. Schematic illustration of negative feedback control of glucose homeostasis by the brain.
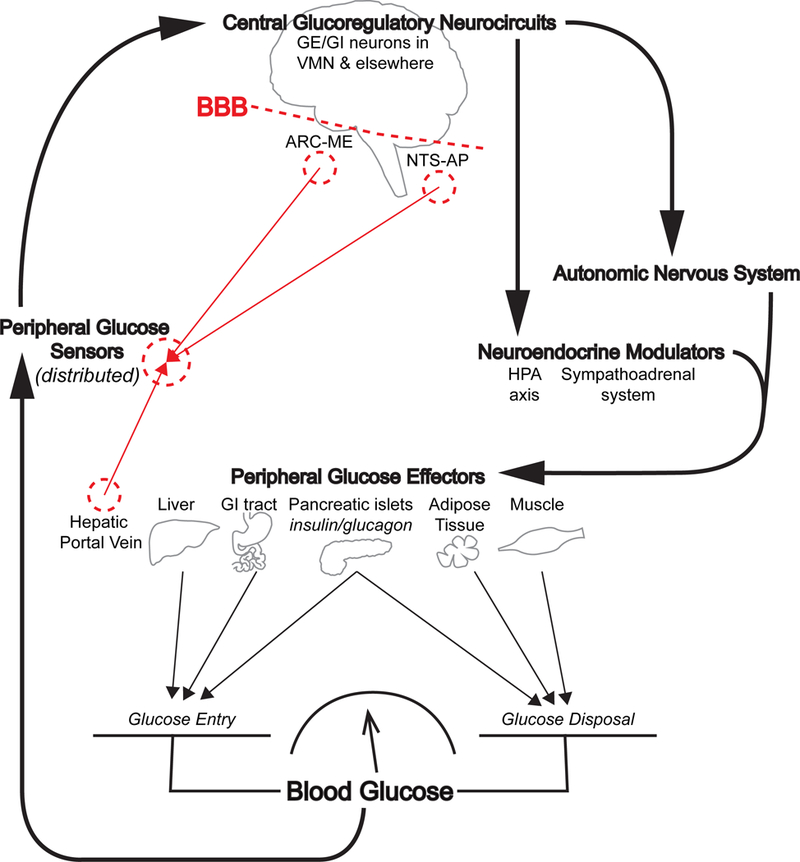
Neurons that sense the circulating glucose level (Peripheral Glucose Sensors, which may include nerves innervating the hepatic portal vein as well as neurons in circumventricular areas such as the arcuate nucleus-median eminence (ARC-ME) and nucleus of the solitary tract-area postrema (NTS-AP)) are proposed to constitute the afferent “sensory” limb of this regulatory system. This afferent information is transmitted to Central Glucoregulatory Circuits in the hypothalamic ventromedial nucleus (VMN) and other areas on the “brain side” of the blood brain barrier (BBB). These neurons are proposed to comprise the integrative/efferent limb of the brain’s glucoregulatory system. While some of these neurons are glucose-excited (GE) or glucose-inhibited (GI), they are not anatomically-positioned to sense glucose in the circulation, and hence do not play a primary role in brain glucose sensing. Instead, these neurons are responsive both to changes in the concentration of glucose in local brain interstitial fluid (which is not closely related to the circulating level) and to input from afferent glucose-sensing neurons, and they project onto and regulate the output from neuroendocrine (including the hypothalamic-pituitary-adrenal (HPA) axis and sympathoadrenal system) and autonomic control systems. Autonomic and neuroendocrine outputs in turn affect liver, GI tract, pancreatic islets, skeletal muscle and adipose tissue in a highly-coordinated manner that ultimately determines the balance between glucose entry into and disposal from the bloodstream, which in turn determines the circulating glucose level.
However, a different interpretation can be drawn from these studies if glucose-sensitive VMN neurons are viewed as components of the efferent (or integrative), rather than the afferent, limb of the glucoregulatory system. Specifically, local glucose infusion into the VMN is predicted by either model to block circuit activation during systemic hypoglycemia, because neurons comprising the efferent limb are themselves glucose-sensitive. The effect would be analogous to using optogenetic silencing of VMN neurons that drive the CRR to prevent effective counterregulation, as has been reported (Meek et al., 2016). Thus, even if the source of afferent input is derived from neurons situated elsewhere, activation of the efferent limb can be blocked by the experimentally-induced increase of glucose concentrations locally in the VMN.
By the same token, induction of glucopenia locally in the VMN is predicted to activate the circuit (and hence generate CRRs) independently of physiologically-relevant afferent input, because VMN neurons comprising the effector limb are themselves inherently glucosesensitive and can be potently activated by local glucopenia - even if this is not how glucopenia is detected under physiological conditions. Indeed, selective activation of VMN neurons is sufficient to robustly engage CRRs and thereby cause hyperglycemia in otherwise normal animals (Meek et al., 2016) (Faber et al., 2018), so it is not surprising that the same response is elicited by local glucopenia. But such an outcome does not establish a role for VMN glucose-responsive neurons as primary sensors of hypoglycemia - only that the activation of these neurons potently drives the CRR.
In other words, we interpret these studies to show that direct manipulation of the effector limb (by manipulating local glucose levels in local brain ISF) can activate (or block) effector responses opposite to what would otherwise occur if the system were functioning properly and, as before, a parallel to the organization of the thermoregulatory system can be drawn. For example, to investigate whether thermal sensing by POA neurons is required for the adaptive response to a change of ambient temperature, one may wish to determine if thermos-induced cooling of the POA blocks the adaptive response to heat exposure. A positive outcome from this study, however, would not necessarily imply that hypothalamic warming is required for an intact response to a warm environment. Instead, it merely confirms that the effector limb of the thermoregulatory neurocircuit is itself comprised of thermosensitive neurons, and that experimentally manipulating the activity of these neurons can engage the system in ways that are sufficiently powerful to overwhelm its normal function (Mohammed et al., 2018).
Insight into the validity of this model can potentially be gained by determining if homeostatic responses to a change of circulating glucose levels are mounted prior to a change of glucose levels in brain ISF. Of particular interest is the adrenomedullary system’s response time (as evidenced by epinephrine release): during insulin-induced hypoglycemia, this response occurs within seconds of the fall of blood glucose levels. Although the time course over which hypothalamic glucose levels fall in this setting is unknown, it seems possible (or even likely) that the epinephrine response precedes the corresponding decline of VMN glucose levels. Should we find that glucose levels in brain ISF begin to fall only after plasma epinephrine levels have increased, one may safely conclude that the adrenomedullary response was initiated in response to glucose-sensing by neurons situated outside of the BBB. A complementary approach might be to modify a non-metabolizeable glucose analogue (e.g., 2-deoxyglucose or gold thioglucose) so as to render it unable to cross the BBB. If systemic administration of such a compound elicits CRRs similar to those observed in response to brain-permeable glucose analogues, one infers that peripheral glucose-sensing mechanisms are capable of triggering CRRs in the absence of central glucopenia. Consistent with this notion, CRRs can be induced by experimental glucopenia localized to the hepatic portal circulation (Donovan, 2002).
In sum, we suggest that a brain system for effective whole-body glucose homeostasis (Figure 1) cannot rely upon afferent input derived primarily from local concentrations of glucose in brain ISF any more than effective thermoregulation can be achieved by a system based on afferent input regarding the temperature of the brain itself. From this perspective, efforts to distinguish between the afferent and efferent limbs of this system are a key priority going forward.
V. Why does it matter which neurons sense the plasma glucose level?
To more fully understand and investigate the role played by the brain in whole-body glucose homeostasis, a crucial next step is to distinguish neuronal systems that provide afferent glucose-sensing information from neurons comprising the effector limb that transduces this afferent input into homeostatic responses. That this distinction has yet to be made shines a light on the gulf that exists between our current understanding of the brain’s glucoregulatory system and the comparably far more advanced understanding of the thermoregulatory system.
A second consideration pertains to the possibility that defective brain glucose sensing contributes to the defense of an elevated level of glycemia in the setting of type 2 diabetes. Although links between defective brain glucose-sensing, obesity and type 2 diabetes have been identified (Tarussio et al., 2014), formal testing of this hypothesis cannot proceed without knowing where to look for the defect. Finally, a more complete understanding of the organization and function of this brain control system may open the door to effective new drug development strategies that target the brain to treat diabetes and related metabolic disorders.
VI. Looking to the future.
Significant challenges will need to be overcome if we are to distinguish neuronal subsets comprising the afferent limb from those that comprise the efferent limb of a distributed glucoregulatory network, especially if many of the latter neurons are themselves glucosesensitive. An important first step is to establish in greater detail the temporal relationship between a change of glucose levels in plasma and the corresponding change in brain ISF, and to determine whether such changes precede or follow adaptive glucoregulatory responses.
An important unanswered question pertains to how best to distinguish those neurons that sense the circulating glucose level from those that sense glucose present in local ISF. While electrophysiology has been a technological mainstay of the brain glucose sensing field for decades, this approach cannot readily distinguish these two neuronal populations from one another. Dynamic monitoring of neuronal activity in vivo (e.g., via calcium imaging or fiber photometry) can in theory identify neuronal subsets whose activity is responsive to a change of circulating glucose levels, but again cannot distinguish glucose-sensitive from glucose-responsive neurons that are situated a few synapses downstream that are activated subsequently, but rapidly, as part of a glucoregulatory neural network.
One promising option is to determine if network responsiveness to changing blood glucose levels is prevented by interventions that selectively disrupt the glucose-sensing function of neurons that comprise the afferent limb of the system. To this end, efforts to identify, characterize and functionally disrupt glucose-sensitive neurons located in or near CVOs (such as the arcuate nucleus, median eminence or NTS) may offer an important next step towards a more complete understanding of brain control of glucose homeostasis.
A final possibility worth exploring is the role played by astrocytes and perhaps other glial cells in brain glucose sensing. Unlike most neurons, astrocytes have access to the circulation via end-foot processes that enwrap capillaries and pericytes that form the BBB. Via the glucose transporter GLUT1, these astrocyte foot-processes efficiently transport glucose across the BBB, and this step is implicated in physiological brain glucose transport (Magistretti and Allaman, 2018). Upon entry into astrocytes, glucose can be stored as glycogen or oxidized via glycolysis, either for ATP generation or to produce lactate that can in turn be made available to neurons for use as a fuel. Indeed, a large (and somewhat contentious) literature suggests that astrocyte-derived lactate, rather than glucose, constitutes a primary fuel source for many neurons, particularly when they are activated (Magistretti and Allaman, 2018). Combined with evidence pointing to a role for astrocytes in the ARC and other brain areas as nutrient sensors (Rogers et al., 2018; Young and McKenzie, 2004) and that astrocytes also play an obligatory role in the response of hindbrain neurons to glucoprivation (Young and McKenzie, 2004), a potential role for astrocytes in brain glucose sensing and its role in glucose homeostasis can be considered. Indeed, work from Rossetti and colleagues (Lam et al., 2005) supports a model in which the glucose-lowering effect of intrahypothalamic glucose administration is dependent on its conversion to lactate by astrocytes. In addition, astrocyte-specific deletion of insulin receptors has been shown to impair whole-body glucose homeostasis (Garcia-Caceres et al., 2016). Future investigation into both the extent to which and mechanisms whereby astrocytes contribute to brain glucose sensing in the control of glucose homeostasis may prove fruitful.
Acknowledgements
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) grants DK101997 (M.W.S), DK083042 (M.W.S.), DK035816 (M.W.S.) and Novo Nordisk, Inc. M.A.B. is supported by The Novo Nordisk Foundation (NNF17OC0024328) and Novo Nordisk Foundation Center for Basic Metabolic Research (NNF10CC1016515) by an unconditional grant from the Novo Nordisk Foundation to the University of Copenhagen, and ZM is supported by NIDDK grant DK108596, National Institute of Neurological Disorders and Stroke grant NRCDP K12, and the Barrow Neurological Foundation. The authors gratefully acknowledge thoughtful input provided by Drs. Karl J. Kaiyala, Steve Woods, Rudy Leibel, Shaun Morrison and Daniel Porte, Jr.
Footnotes
Declaration of Interest
The authors declare no competing interests. Research support provided by Novo Nordisk, Inc.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anand BK, Chhina GS, Sharma KN, Dua S, and Singh B (1964). Activity of Single Neurons in the Hypothalamic Feeding Centers: Effect of Glucose. The American journal of physiology 207, 1146–1154. [DOI] [PubMed] [Google Scholar]
- Borg MA, Sherwin RS, Borg WP, Tamborlane WV, and Shulman GI (1997). Local ventromedial hypothalamus glucose perfusion blocks counterregulation during systemic hypoglycemia in awake rats. The Journal of clinical investigation 99, 361–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borg WP, Sherwin RS, During MJ, Borg MA, and Shulman GI (1995). Local ventromedial hypothalamus glucopenia triggers counterregulatory hormone release. Diabetes 44, 180–184. [DOI] [PubMed] [Google Scholar]
- Boychuk CR, Gyarmati P, Xu H, and Smith BN (2015). Glucose sensing by GABAergic neurons in the mouse nucleus tractus solitarii. Journal of neurophysiology 114, 999–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bratincsak A, and Palkovits M (2005). Evidence that peripheral rather than intracranial thermal signals induce thermoregulation. Neuroscience 135, 525–532. [DOI] [PubMed] [Google Scholar]
- Chan O, and Sherwin RS (2014). Is there cross talk between portal and hypothalamic glucose-sensing circuits? Diabetes 63, 2617–2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coate KC, Kraft G, Irimia JM, Smith MS, Farmer B, Neal DW, Roach PJ, Shiota M, and Cherrington AD (2013). Portal vein glucose entry triggers a coordinated cellular response that potentiates hepatic glucose uptake and storage in normal but not high-fat/high-fructose-fed dogs. Diabetes 62, 392–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deem JD, Muta K, Scarlett JM, Morton GJ, and Schwartz MW (2017). How Should We Think About the Role of the Brain in Glucose Homeostasis and Diabetes? Diabetes 66, 1758–1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dicostanzo CA, Dardevet DP, Neal DW, Lautz M, Allen E, Snead W, and Cherrington AD(2006). Role of the hepatic sympathetic nerves in the regulation of net hepatic glucose uptake and the mediation of the portal glucose signal. Am J Physiol Endocrinol Metab 290, E9–E16. [DOI] [PubMed] [Google Scholar]
- Donovan CM (2002). Portal vein glucose sensing. Diabetes, nutrition & metabolism 15, 308–312; discussion 313–304. [PubMed] [Google Scholar]
- Dunn-Meynell AA, Sanders NM, Compton D, Becker TC, Eiki J, Zhang BB, and Levin BE (2009). Relationship among brain and blood glucose levels and spontaneous and glucoprivic feeding. The Journal of neuroscience : the official journal of the Society for Neuroscience 29, 7015–7022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber CL, Matsen ME, Velasco KR, Damian V, Phan BA, Adam D, Therattil A, Schwartz MW, and Morton GJ (2018). Distinct Neuronal Projections from the Hypothalamic Ventromedial Nucleus Mediate Glycemic and Behavioral Effects. Diabetes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fioramonti X, Chretien C, Leloup C, and Penicaud L (2017). Recent Advances in the Cellular and Molecular Mechanisms of Hypothalamic Neuronal Glucose Detection. Frontiers in physiology 8, 875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flak JN, Patterson CM, Garfield AS, D’Agostino G, Goforth PB, Sutton AK, Malec PA, Wong JT, Germani M, Jones JC, et al. (2014). Leptin-inhibited PBN neurons enhance responses to hypoglycemia in negative energy balance. Nature neuroscience 17, 1744–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Caceres C, Quarta C, Varela L, Gao Y, Gruber T, Legutko B, Jastroch M, Johansson P, Ninkovic J, Yi CX, et al. (2016). Astrocytic Insulin Signaling Couples Brain Glucose Uptake with Nutrient Availability. Cell 166, 867–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garfield AS, Shah BP, Madara JC, Burke LK, Patterson CM, Flak J, Neve RL, Evans ML,Lowell BB, Myers MG Jr., et al. (2014). A parabrachial-hypothalamic cholecystokinin neurocircuit controls counterregulatory responses to hypoglycemia. Cell metabolism 20, 1030–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang JJ, Jiang L, Hamza M, Sanchez Rangel E, Dai F, Belfort-DeAguiar R, Parikh L, Koo BB, Rothman DL, Mason G, et al. (2017). Blunted rise in brain glucose levels during hyperglycemia in adults with obesity and T2DM. JCI insight, e95913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jokiaho AJ, Donovan CM, and Watts AG (2014). The rate of fall of blood glucose determines the necessity of forebrain-projecting catecholaminergic neurons for male rat sympathoadrenal responses. Diabetes 63, 2854–2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiyala KJ, Ogimoto K, Nelson JT, Schwartz MW, and Morton GJ (2015). Leptin signaling is required for adaptive changes in food intake, but not energy expenditure, in response to different thermal conditions. PloS one 10, e0119391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang L, Dunn-Meynell AA, Routh VH, Gaspers LD, Nagata Y, Nishimura T, Eiki J, Zhang BB, and Levin BE (2006). Glucokinase is a critical regulator of ventromedial hypothalamic neuronal glucosensing. Diabetes 55, 412–420. [DOI] [PubMed] [Google Scholar]
- Lam TK, Gutierrez-Juarez R, Pocai A, and Rossetti L (2005). Regulation of blood glucose by hypothalamic pyruvate metabolism. Science 309, 943–947. [DOI] [PubMed] [Google Scholar]
- Langlet F, Levin BE, Luquet S, Mazzone M, Messina A, Dunn-Meynell AA, Balland E, Lacombe A, Mazur D, Carmeliet P, et al. (2013). Tanycytic VEGF-A boosts blood-hypothalamus barrier plasticity and access of metabolic signals to the arcuate nucleus in response to fasting. Cell metabolism 17, 607–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magistretti PJ, and Allaman I (2018). Lactate in the brain: from metabolic end-product to signalling molecule. Nat Rev Neurosci 19, 235–249. [DOI] [PubMed] [Google Scholar]
- Meek TH, Nelson JT, Matsen ME, Dorfman MD, Guyenet SJ, Damian V, Allison MB, Scarlett JM, Nguyen HT, Thaler JP, et al. (2016). Functional identification of a neurocircuit regulating blood glucose. Proceedings of the National Academy of Sciences of the United States of America 113, E2073–2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammed M, Madden CJ, Burchiel KJ, and Morrison SF (2018). Preoptic area cooling increases the sympathetic outflow to brown adipose tissue (BAT) and BAT thermogenesis. American journal of physiology. Regulatory, integrative and comparative physiology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MC, Coate KC, Winnick JJ, An Z, and Cherrington AD (2012). Regulation of hepatic glucose uptake and storage in vivo. Advances in nutrition 3, 286–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison SF (2016). Central control of body temperature. F1000Research 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy BA, Fioramonti X, Jochnowitz N, Fakira K, Gagen K, Contie S, Lorsignol A, Penicaud L, Martin WJ, and Routh VH (2009). Fasting enhances the response of arcuate neuropeptide Y -glucose- inhibited neurons to decreased extracellular glucose. American journal of physiology. Cell physiology 296, C746–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oomura Y, Kimura K, Ooyama H, Maeno T, Iki M, and Kuniyoshi M (1964). Reciprocal Activities of the Ventromedial and Lateral Hypothalamic Areas of Cats. Science 143, 484–485. [DOI] [PubMed] [Google Scholar]
- Pozo M, and Claret M (2018). Hypothalamic Control of Systemic Glucose Homeostasis: The Pancreas Connection. Trends in endocrinology and metabolism: TEM 29, 581–594. [DOI] [PubMed] [Google Scholar]
- Ramsay DS, and Woods SC (2016). Physiological Regulation: How It Really Works. Cell metabolism 24, 361–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers RC, McDougal DH, Ritter S, Qualls-Creekmore E, and Hermann GE (2018). Response of catecholaminergic neurons in the mouse hindbrain to glucoprivic stimuli is astrocyte dependent. American journal of physiology. Regulatory, integrative and comparative physiology 315, R153–R164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz MW, Seeley RJ, Tschop MH, Woods SC, Morton GJ, Myers MG, and D’Alessio D (2013). Cooperation between brain and islet in glucose homeostasis and diabetes. Nature 503, 59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan CL, and Knight ZA (2018). Regulation of Body Temperature by the Nervous System. Neuron 98, 31–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarussio D, Metref S, Seyer P, Mounien L, Vallois D, Magnan C, Foretz M, and Thorens B (2014). Nervous glucose sensing regulates postnatal beta cell proliferation and glucose homeostasis. The Journal of clinical investigation 124, 413–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Liu X, Hentges ST, Dunn-Meynell AA, Levin BE, Wang W, and Routh VH (2004). The regulation of glucose-excited neurons in the hypothalamic arcuate nucleus by glucose and feeding-relevant peptides. Diabetes 53, 1959–1965. [DOI] [PubMed] [Google Scholar]
- Watts AG, and Donovan CM (2010). Sweet talk in the brain: glucosensing, neural networks, and hypoglycemic counterregulation. Frontiers in neuroendocrinology 31, 32–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams EK, Chang RB, Strochlic DE, Umans BD, Lowell BB, and Liberles SD (2016). Sensory Neurons that Detect Stretch and Nutrients in the Digestive System. Cell 166, 209–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JK, and McKenzie JC (2004). GLUT2 immunoreactivity in Gomori-positive astrocytes of the hypothalamus. J Histochem Cytochem 52, 1519–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C, Teegala SB, Khan BA, Gonzalez C, and Routh VH (2018). Hypoglycemia: Role of Hypothalamic Glucose-Inhibited (GI) Neurons in Detection and Correction. Frontiers in physiology 9, 192. [DOI] [PMC free article] [PubMed] [Google Scholar]