

Abstract
The renin-angiotensin system tightly controls aldosterone synthesis. Dysregulation is evident in hypertension (primary aldosteronism, low-renin and resistant hypertension), but also can exist in normotension. Whether chronic, mild aldosterone autonomy can elicit hypertension remains untested. Previously, we reported that global genetic deletion of two pore-domain potassium channels (K2P), TASK-1 and TASK-3, from mice produces striking aldosterone excess, low renin and hypertension. Here, we deleted TASK-1 and TASK-3 channels selectively from zG cells and generate a model of mild aldosterone autonomy with attendant hypertension that is aldosterone-driven and Angiotensin II-independent. This study shows that a zG-specific channel defect can produce mild autonomous hyperaldosteronism sufficient to cause chronic blood pressure elevation.
Keywords: aldosterone, hypertension, renin, tandem pore-domain potassium channels, zona glomerulosa
Summary
zG-specific deletion of TASK-1 and TASK-3 channels can produce a mild chronic hyperaldosteronism that is renin-independent and pathogenic.
Aldosterone overproduction is considered autonomous when it is uncoupled from the renin-angiotensin system (RAS), and is a more prevalent clinical problem than previously recognized. Primary aldosteronism is the most severe form of aldosterone autonomy1,2. However, autonomous aldosterone overproduction is evident in low-renin essential hypertension3 and resistant hypertension4, which together account for ~20% of the hypertensive population5. Recently, Vaidya and colleagues exposed a component of renin-independent aldosteronism in normotensive individuals that is mild, subclinical and compensated6. In these subjects, measurement of 24hr. urinary excretion of aldosterone, Na+ and K+ revealed mild overproduction of aldosterone that was not evident from standard measurements of serum aldosterone or aldosterone-to-renin ratios. Moreover, several studies7,8, including the large Framingham Offspring study of 1,688 non-hypertensive persons, showed that serum aldosterone levels within the highest quartile of the normal physiological range predict hypertensive risk. However, it remains unknown whether these high-normal serum aldosterone levels are a precursor or a symptom of subclinical disease, and whether mild aldosterone autonomy without further exacerbation can drive a transition to hypertension6.
Autonomous aldosterone production in human tumorigenic primary aldosteronism (i.e., aldosterone-producing adenoma, APA) contributes to the large increase in the aldosterone-to-renin ratio characteristic of APA2,9,10. A major cause of hyperaldosteronism in APA is genetic variation in genes encoding ion channels and pumps11,12. In APA, single nucleotide polymorphisms identified in Ca2+-regulating genes are predicted to raise intracellular Ca2+, the critical second messenger that drives aldosterone production11,13–18. A subset of somatic mutations in these Ca2+-regulating genes are also predicted to depolarize aldosterone-producing zona glomerulosa (zG) cells, thereby causing an increase in extracellular calcium entry through voltage-dependent Ca2+ channels11,13,14. Therefore, the regulation of membrane voltage by the stabilizing/hyperpolarizing activity of K+ channels exerts an important restraint on aldosterone production. Although significant differences exist in the complement of K+ channels expressed on zG cells across species, the two pore-domain potassium channel family (K2P) is well represented and expressed in human, rodent and bovine ZG cells, albeit with differing predominant subtypes (human19–21: TASK-1 and TASK-2; rat and mouse22–25: TASK-1 and TASK-3; bovine26: TREK-1). As background, or “leak” channels, K2P channels display little voltage dependence, remaining active at negative membrane voltages, and thus are ideally suited to regulate the negative resting membrane voltage of cells, including zG cells (negative to −75 mV)27.
We previously reported that combined global deletion of TASK-1 and TASK-3 channels in mice produces exaggerated hyperaldosteronism, with a large component of autonomous aldosterone production that was not suppressed by high dietary sodium intake, nor corrected with candesartan, an Ang II receptor 1 antagonist24. However, because TASK channels are widely distributed throughout the central nervous system and the periphery27, it remains to be determined whether zG-specific deletion of TASK channels, or any ion channel, can generate RAS-independent hyperaldosteronism in vivo and mediate chronic blood pressure elevation28. Herein, we report that zG-selective deletion of TASK-1 and TASK-3 channels in mice produces a surprisingly mild aldosterone autonomy that is sufficient to raise blood pressure chronically. Thus, we find that subtle intrinsic zG cell dysfunction that leads to mild RAS-independent hyperaldosteronism can be pathogenic, underscoring the clinical importance of early detection of even mild autonomous aldosterone production in normotension.
Methods
The authors declare that all supporting materials are available within the article and in the online-only Data Supplement. Additional information on techniques is available from the corresponding authors upon request.
Mice
Mice were housed in a temperature and humidity controlled vivarium on a 12:12 light:dark cycle. All experiments were carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and approved by the University of Virginia Animal Care and Use Committee.
Generation of mouse lines.
AS (aldosterone synthase)+/Cre ::TASK-1fl/fl ::TASK-3fl/fl mice. AS+/Cre mice29 were bred to TASK-1fl/fl ::TASK-3fl/fl24 (Jackson: 025928 and 026191) generating AS+/Cre ::TASK-1+/fl ::TASK-+/fl mice and then intercrossed to generate AS+/Cre ::TASK-1fl/fl ::TASK-3fl/fl mice (zG-TASK-KO). Heterozygosity at the AS allele was maintained in all experimental animals including control mice (AS+/Cre ::TASK-1+/+ ::TASK-3+/+) AS+/Cre :: R26RmTmG/ mTmG mice. AS+/Cre mice were crossed with Gt(ROSA)26Sortm4(ACTB-tdTomato-EGFP)Luo/ J reporter line (Jackson: 007676) to generate AS+/Cre :: R26R+/ mTmG. These mice express membrane-targeted red fluorescence before and membrane-targeted green fluorescence after Cre-mediated recombination (Fig 2).
Figure 2.
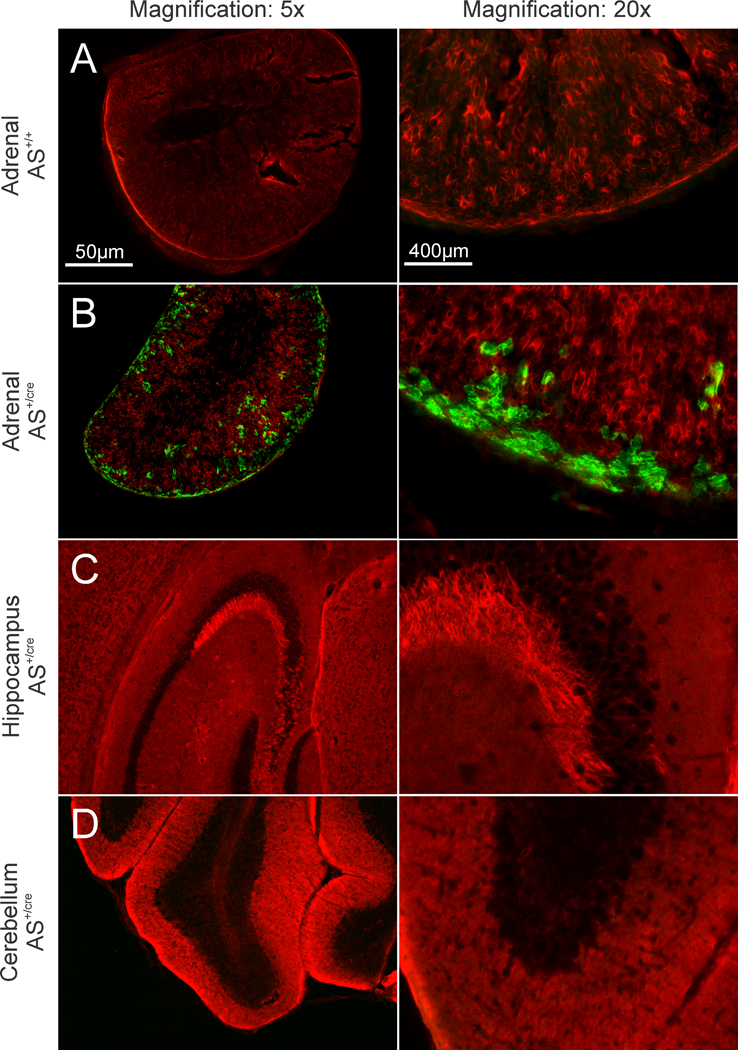
Visualization of GFP (green) and tdTomato (red) in R26R+/ mTmG mice, amplified by immunostaining (anti-GFP and anti-DsRed). Wide-field images of 40µm tissue slices from control mice (AS+/+, A) and mice expressing Cre-recombinase in aldosterone synthase expressing cells (AS+/Cre, B-D). Green fluorescence demonstrates Cre expression in the zG layer (B) but not in the hippocampus (C) nor cerebellum (D) of AS+/Cre mice. Images were captured at 5x (left column; scale bar = 50µm) or 20x magnification (right column; scale bar = 400µm).
Genotyping
Genomic DNA was isolated by alkaline lysis (2.5 M NaOH) at 95°C for 45 min, followed by neutralization in 40 mM Tris HCl (final concentration). Sequences of primers and PCR conditions used for genotyping are listed in Table S1. Briefly, aldosterone synthase (AS) alleles were genotyped using three primer PCR reactions29 yielding either WT allele PCR product (525 bp) or mutant-Cre allele PCR product (625 bp). TASK-1 allele detection resulted in 77 bp WT product or 101 bp floxed product, and TASK-3 allele detection resulted in 93 bp WT product or 127 bp floxed product. For detection of deleted TASK-1 and TASK-3 alleles at the genomic level24, DNA from AS+/Cre ::TASK-1fl/fl ::TASK-3fl/f adrenal gland was isolated and amplified using the same primers as for genotyping. The recombined TASK-1 allele, missing exon 2, resulted in amplification of specific 218 bp PCR product (Table S1) and recombined TASK-3 allele results in a 189 bp product. The R26RmTmG allele was detected by PCR amplification of the GFP coding region and results in a 284 bp product (Table S1).
Tissue Preparation and Microscopy
Gene-expression Analysis.
Adrenal glands were dissected from terminally anesthetized mice (ketamine, 15mg, i.p.), flash-frozen, sectioned on a cryostat at 8 μm, and mounted onto specialized microscope slides containing a polyethylene napthalate window. A laser capture system (AS/LMD, Leica Microsystems, Inc.) was used to visualize and carefully sample zG tissue for subsequent qRT-PCR analysis. RNA was isolated with the PicoPure RNA isolation kit (Arcturus) and cDNA was generated with iScript reverse transcription kit (Biorad) and qRT-PCR performed in quadruplicate using an iCycler, with iQ SYBR Green SuperMix reagents (BioRad). In preliminary experiments, a dilution series of cDNA was used with each primer set to establish conditions (primer concentration, annealing temperature) to yield >90% efficiency; in addition, the PCR product was run on an agarose gel and sequenced to confirm its identity. Melt curve analysis and no-template controls were included with each run.
Immunohistochemistry.
AS+/Cre R26R+/ mTmG and AS+/+ R26R+/ mTmG mice 22–40 days old were intracardially perfused with 0.1 M phosphate buffered saline followed by 4% paraformaldehyde (PFA), and tissue harvested for immunohistochemistry. Adrenal glands, hippocampus, and cerebellums were sectioned (40µm thick) on a vibratome (Leica), mounted on slides, and stored at 4°C until processed. To amplify GFP and tdTomato signals, we incubated tissue with a polyclonal GFP antibody raised in chicken (1:750, Aves, product gfp-1020) and polyclonal DsRed antibody raised in rabbit (Takara Bio, product 632496) followed by fluorescent secondary antibodies Alexa Fluor 488 goat anti-chicken (1:250, Jackson ImmunoResearch, product 103–545-155) and Cy3 donkey anti-rabbit (1:250, Jackson ImmunoResearch, product 711–165-152). Images were captured on a Zeiss Imager.Z1 microscope using Neurolucida software (MBF Bioscience).
Urine and Plasma Analysis
Metabolic Cage Experiments.
Adult male mice age 80–150 days were used for all experiments. To control for differences in genetic background, experiments were performed using littermates. Metabolic cage experiments and blood analysis were conducted as described previously30. Briefly, mice were housed individually in metabolic cages designed for urine collection. After a habituation period of 4 days in metabolic cages on normal chow diet, mice had access to NS (0.3% Na) or HS (4% Na) chow diet for 1 week. Urine was sampled every twenty-four hours for the last 4 days of diet for aldosterone assay, and drinking water and urine volume were recorded. In some experiments, candesartan was delivered via drinking water (10mg/kg/day). Candesartan stock solution was prepared 1 mg/ml in a mixture containing 10% PEG 400, 5% ethanol, 2% cremaphor EL, 83% water (wt/wt). On the last day of collection, tail vein blood was sampled for renin measurement and retro-orbital sinus sampled for blood chemistry analysis.
Measurements of Electrolyte and Hormone Levels.
Methods for urine and plasma analysis were conducted as previously described30. Urinary aldosterone was measured using an aldosterone I125 radioimmunoassay (Tecan, Morrisville, NC), and standardized to urinary creatinine (Jaffe’ colorimetric assay, Cayman Chemicals, Ann Arbor, MI) in order to account for differences in GFR and urine volume. Plasma renin concentration was determined by RIA (Diasorin, Stillwater, MN). Blood chemistries were determined from retro-orbital sinus sampling using heparinized capillary tubes for analysis with an iStat hand held analyzer (EC8+ cartridge, Heska, Fort Collins, CO). Urinary Na+ and K+ were measured by flame photometry (IL943 Automatic Flame Photometer, Instrumentation Laboratory, Inc.).
Blood pressure
Telemetry.
Blood pressure was recorded from conscious, freely behaving mice, as previously described30. Pressure transmitters (Data Sciences International telemetry system; DSI, St Paul, MN) were threaded through the left carotid artery and implanted in the aortic arch. After 7 days of surgical recovery, blood pressure and heart rate were recorded for 5 min every hour on NS diet for 4 days before challenge with HS diet for 7 days. Instead of HS, some mice were given either candesartan for 4 days (10 mg/kg/day) or spironolactone for 2 weeks (20 mg/kg/day) delivered in the drinking water. Spironolactone was dissolved in 100% ETOH to make a 20 mg/ml stock; final drinking solution contained less than 0.3% ETOH.
Statistical Analysis
All data in graphs are presented as the mean ± SEM. Where multiple comparisons were made, a one-way ANOVA was used to test for significance among groups with Bonferroni multiple comparison post hoc test. A Student’s t test was used for analysis of a single dependent variable compared between two groups. Significance was determined at P<0.05.
Results
Aldosterone synthase (AS) catalyzes the three-step terminal conversion of deoxycorticosterone to aldosterone, and its expression in the adrenal gland is restricted to cortical cells within the zG layer31,32. We used AS-Cre mice that specifically express Cre-recombinase from the AS genetic locus29 to generate zG-TASK-KO mice in which the K2P channels (TASK-1 and TASK-3) are deleted from zG cells (zG-TASK-KO). qRT-PCR analysis of RNA prepared from zG regions isolated by laser capture microdissection confirmed genetic deletion of TASK channels from the zG layer (Figure 1A). Relative to control littermates (CON), Kcnk3 (TASK-1) and Kcnk9 (TASK-3) mRNA expression in zG-TASK-KO mice is 0.05-fold ± 0.05 and 0.12-fold ± 0.04, respectively. By contrast, in cerebellum, where TASK channel transcripts are abundant in the rodent (rat, mouse)33, TASK-1 and TASK-3 mRNA are preserved in zG-TASK-KO mice (CON vs. zG-TASK-KO, N.S.; Figure 1B).
Figure 1.
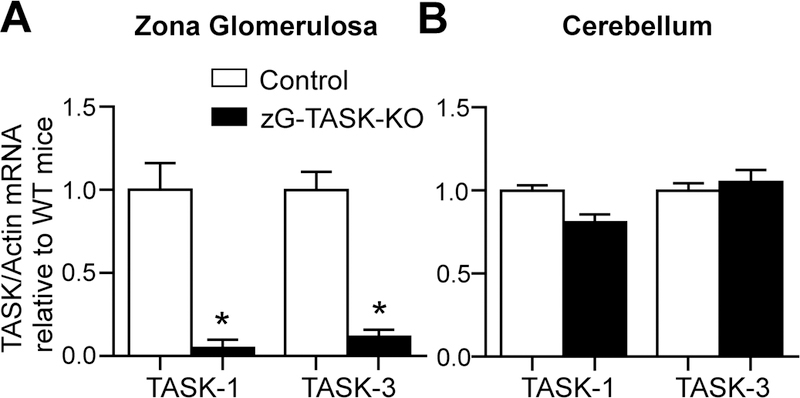
TASK-1 and TASK-3 deletion in zG-TASK-KO mice is restricted to the zG layer. Expression of TASK-1 and TASK-3 mRNA in zG layer (A, n=5; p<0.001) and cerebellum (B, n=5; N.S.) of zG-TASK-KO (black bars) and CON (white bars) mice relative to CON mice. TASK mRNA is standardized to Actin mRNA. Data presented as mean±s.e.; asterisks indicate p<0.05.
We also used a Cre-dependent reporter mouse to assess Cre-recombinase activity in select brain regions, where extra-adrenal mRNA for AS has been detected and where TASK channels have been located 33,34. For this, we bred AS+/Cre mice to the R26R mTmG /mTmG reporter line to generate AS+/Cre :: R26R+/ mTmG mice. In these mice, cells express one of two membrane-targeted fluorescent reporter proteins depending on Cre-expression, either tdTomato before, or GFP after, Cre-mediated recombination. We used indirect immunofluorescence with anti-DsRed and anti-GFP antibodies to improve detection sensitivity. As expected, green fluorescence is evident throughout the zG layer of AS+/Cre :: R26R+/ mTmG mice (Figure 2B, left and right panels), but not in AS+/+ wild-type littermates (Figure 2A, left and right panels, WT). From serial brain sections of AS+/Cre R26R+/ mTmG mice, we assayed the dentate gyrus and CA3 cells of the hippocampus and the Purkinje cells of the cerebellum 33,34. These regions of expression previously identified in rat are divergent from the human where Cyp11b2 transcripts localize to the corpus callosum, the spinal cord, thalamus and caudate nucleus35. Immunostaining of the mouse hippocampus showed no GFP expression in the dentate gyrus or CA3 layer pyramidal cells (Figures 2C, left and right panels). Similarly, GFP was absent from the Purkinje layer of the cerebellum (Figures 2D, left and right panels). The absence of AS-driven Cre-recombinase activity is consistent with previous studies reporting AS (Cyp11b2) expression by PCR in the brain of the rat, but not the mouse36,37. Thus, although TASK-1 and TASK-3 channels are widely distributed throughout the rodent CNS33, we find no evidence for AS expression in mouse brain regions where TASK channels are expressed, further supporting selective deletion of TASK channels in the zG of the zG-TASK-KO mouse line.
Analysis of blood chemistries from retro-orbital samples (a mixture of venous and arterial blood), revealed no significant differences between zG-TASK-KO and control mice (Table 1). Electrolyte balance was equivalent between genotypes, as was blood gas analysis for pH, bicarbonate, and PCO2. In addition, zG-TASK-KO mice maintained a normal hematocrit and hemoglobin concentration compared with control littermates. Thus, our analyses indicate that zG-specific deletion of TASK channels results in no obvious abnormalities in pulmonary or renal function.
Table 1.
| Electrolytes/ Chemistries/ Gasses |
Control (N=10) |
KO (N=9) |
||
|---|---|---|---|---|
| Mean | SE | Mean | SE | |
| K+ (mmol/L) | 4.42 | 0.11 | 4.54 | 0.37 |
| Na+ (mmol/L) | 149.10 | 0.41 | 148.44 | 0.77 |
| iCa2+(mmol/L) | 1.25 | 0.02 | 1.25 | 0.03 |
| Cl- (mmol/L) | 110.25 | 1.31 | 109.50 | 0.65 |
| pH | 7.19 | 0.02 | 7.23 | 0.02 |
|
HCO3 (mmol/L) |
23.99 | 1.15 | 26.03 | 0.41 |
|
Anion Gap (mmol/L) |
20.25 | 1.70 | 17.00 | 0.41 |
| BUN (mg/dl) | 25 | 1.08 | 25 | 2.74 |
|
PCO2 (mmHg) |
62.03 | 2.46 | 66.58 | 4.69 |
| PO2 (mmHg) | 39.00 | 1.29 | 46.20 | 5.09 |
|
TCO2 (mmol/L) |
25.90 | 1.17 | 27.89 | 0.48 |
| sO2 (%) | 59.00 | 3.04 | 68.00 | 7.24 |
| HCT (%pcv) | 40.50 | 0.31 | 42.78 | 0.85 |
| Hb (g/dl) | 13.76 | 0.10 | 14.53 | 0.29 |
| Weight (g) | 32.2 | 1.19 | 32.6 | 0.75 |
|
UNa/Creat (mmol/mg) |
0.10 | 0.02 | 0.11 | 0.02 |
|
UK/Creat (mmol/mg) |
0.75 | 0.06 | 0.75 | 0.05 |
To determine if zG-specific deletion of TASK-1 and TASK-3 channels alters aldosterone production in vivo, we evaluated the 24hr. urinary excretion of aldosterone (normalized to creatinine) in zG-TASK-KO mice and age-matched littermate controls. This measure provides a representative assessment of daily aldosterone secretory activity that minimizes effects of stress and diurnal variability of aldosterone production that is inherent in “spot” sampling38. Hence, it is well-suited to uncover subtle changes in aldosterone output6. On a normal salt diet (NS, 0.3% Na+), zG-TASK-KO mice produced significantly more aldosterone than control mice. However, surprisingly, the magnitude of this overproduction was only 28% of that observed with global TASK-KO mice on the same diet (global-TASK-KO: 42.6± 6.9 ng/mg-aldo/creat30; zG-TASK-KO 11.8±0.8 ng/mg-aldo/creat; Figure 3A). This comparative difference was not the result of haplo-insufficiency as mice with one copy of the intact AS allele produce aldosterone equivalently to WT mice on multiple salt diets16. Thus, the large difference in aldosterone production between global- and zG-TASK-KO mice could indicate that extra-adrenal removal of TASK channels prevents intrinsic compensatory mechanisms within zG cells and/or, more likely, that global deletion of TASK channels magnifies the intrinsic dysfunction caused by the deletion of TASK channels from the zG.
Figure 3.

Hyperaldosteronism in zG-TASK-KO is RAS-independent. A, Aldosterone production in zG-TASK-KO mice is greater than littermate controls (n=10 CON, 8 KO; p=0.003), but modest relative to global-TASK KO mice (n=38 CON,12 KO; p<0.001). Plasma renin (B, n=6; NS-diet.), and aldo-to-renin ratios (C, n=6, NS-diet.) remain unchanged. D, RAS suppression with ATR1 blockade (Candesartan=10mg/kg/day in drinking water; n=6; p=0.0015) or high sodium diet (HS-diet, 4% Na+; n=10 CON, 8 KO; p<0.001) does not normalize aldosterone output between genotypes. Dotted lines indicate aldosterone production of each genotype on NS-diet. Data presented as mean±s.e. of 24-hr urinary aldosterone standardized to urinary creatinine; asterisks indicate p<0.05.
As expected, the mild hyperaldosteronism observed in zG-TASK-KO mice did not reduce plasma renin (CON vs. zG-TASK-KO, N.S.; Figure 3B) nor significantly increase the aldosterone-to-renin ratio (CON vs. zG-TASK-KO, N.S.; Figure 3C), in contrast to the hyperaldosteronism produced by global deletion of TASK channels24,30. Nevertheless, zG-specific deletion of TASK channels produced significant aldosterone autonomy. Suppression of RAS with either AT1 receptor antagonism (candesartan [C] administration) or high dietary salt (HS) intake, did not normalize production between genotypes (Figure 3D; dotted lines indicate production in the absence of candesartan ). Rather, each treatment amplified the relative difference in aldosterone output between genotypes (compare fold-change of zG-TASK-KO/CON on NS (1.3±0.1), NS + C (2.0±0.2) or HS (2.5±0.1), P<0.5; Figure 3A & 3D) revealing a large Ang II-independent component of aldosterone production in zG-TASK-KO mice that was greater than the Ang II-dependent component. Thus, our data indicate that intrinsic zG dysfunction can produce mild aldosterone autonomy in vivo.
To determine if mild aldosterone autonomy is sufficient to raise blood pressure chronically, we implanted pressure transmitters in the aortic arch and measured blood pressure by telemetry in conscious freely behaving mice. On a normal salt diet, zG-TASK-KO mice had significantly higher systolic and diastolic blood pressures than control mice at night (Figure 4A). During rest/inactivity (day) systolic and diastolic blood pressures decreased normally in both genotypes but higher pressures in zG-TASK-KO mice persisted (Figure 4B). Notably, zG-TASK-KO mice were not significantly more active than control mice (not shown: 24hr-activity (counts/min); CON: 5.3 ± 0.7; KO: 6.2 ± 0.6, N.S.), had similar heart rates (Figure 4C) and had similar levels of urinary corticosterone (Figure S1). Thus, the blood pressure elevation in zG-TASK-KO mice was not attributable to differences in activity patterns, or endocrine rhythms that might prevent normal circadian or activity-dependent effects on blood pressure 39–41. Rather, we found that blood pressure elevation in zG-TASK-KO mice was aldosterone-dependent. Administration of spironolactone, a mineralocorticoid receptor antagonist, normalized blood pressure between genotypes (Figure 4D) producing a greater reduction of systolic pressure in zG-TASK-KO mice than control mice (△CON: −1.0 ± 2.7 mmHg; △zG-TASK-KO: −14.3 ± 6.3 mmHg; P =.037). By contrast, candesartan administration did not normalize blood pressure between genotypes (Figure 4D). In both mouse strains, candesartan lowered mean arterial pressures equivalently (△MAP CON: −22.3 ± 1.25 mmHg; △MAP zG-TASK-K: −18.7 ± 2.2 mmHg; N.S.), concomitant with reduced aldosterone output (Figure 3D, △CON: −5.2 ± 1.3 ng/mg; △zG-TASK-K: −5.1 ± 0.7 ng/mg; N.S.). These data indicate that even mild autonomous aldosterone production caused by intrinsic zG cell dysfunction can be pathogenic in vivo.
Figure 4.

Blood pressure elevation in zG-TASK-KO mice is Ang II-independent. Diurnal variation in BP is normal in zG-TASK-KO and CON mice (NS-diet; n=8–9); BP in the active/wake phase (A, night) decreases during sleep (B, day). During both night (A) and day (B) systolic (night: p=0.013; day: p=0.001) and diastolic (night: p=0.005; day: p=0.002) BPs are elevated in zG-TASK-KO mice. C, HR remains unchanged. D, Candesartan (C=10mg/kg/day) reduced BP of both genotypes equivalently, failing to normalize BP (n=5–6; p<0.001). MR antagonism with spironolactone (20mg/kg/day) confirmed aldosterone-dependence of BP elevation (n=5–6; N.S.). Data presented as mean±s.e.; asterisks indicate p<0.05.
Discussion
In this study, we provide a new in vivo model of mild autonomous hyperaldosteronism resulting from the zG-specific deletion of cell-intrinsic background K+ channels. Notably, the subtle hyperaldosteronism in this model was not sufficient to suppress plasma renin, produce hypokalemia or metabolic alkalosis, nor increase the aldosterone to renin ratio. However, it was sufficient to chronically elevate blood pressure. Together, these data provide proof-of-principle that mild, Ang II-independent hyperaldosteronism resulting from a zG-specific defect is sufficient to chronically elevate blood pressure and can be detected by measuring 24-hr. urinary aldosterone excretion.
Our animal model supports recent revelations that human primary aldosteronism (PA) is a disease continuum of autonomous aldosterone production, ranging from mild hyperaldosteronism evident in subclinical PA to extreme hyperaldosteronism manifest in aldosterone-producing adenoma3,6,42,43. This expanded understanding suggests that the prevalence of non-tumorigenic, primary aldosteronism is much higher than previously recognized1,10,38. Notably, PA carries cardiovascular and cerebrovascular risks that are disproportionate to the degree of hypertension7,8,44–46. Thus, understanding mild forms of PA has taken on new clinical importance.
The deletion of K2P channels, TASK-1 and TASK-3, from zG cells produced a hyperaldosteronism that was exclusively independent of RAS. Specifically, we found that blockade of the ATII receptor with candesartan reduced aldosterone production and blood pressure in zG-TASK-KO and control mice, but failed to restore them to control levels in zG-TASK-KO mice. This failure to normalize following treatment with candesartan indicates that elevated aldosterone levels in zG-specific TASK-KO are not explained by Ang II regulation of zG cells. This is in stark contrast to our previous reports revealing a marked adrenal hypersensitivity to Ang II in global TASK-1/3 KO mice24 and global-TASK-3 KO mice30. In the global-TASK-1/3 KO mouse, an increase in the Ang II-dependent component of aldosterone production, combined with elevated autonomous (Ang II-independent) production, drives a striking 4-fold increase in aldosterone output. Our studies do not address the putative extra-adrenal mechanism(s) underlying adrenal Ang II-hypersensitivity in the global-TASK-1/3 KO mice, but it is not explained by genetic background as marked production was observed in global-TASK-1/3 KO mice on a mixed (129Sv/C57BL/6J)24 or congenic (C57BL/6J)30 background. Our data from adult zG-TASK-KO mice, together with our previously published data in adult mice, indicate that there is likely a distinct (i.e., non-zG) site that requires the disruption of both TASK-147 and TASK-330 channels to sustain aldosterone overproduction at very high levels. Notably, global-TASK-1 KO and global-TASK-3 KO adult mice produce aldosterone at levels that are similar to that we report here for zG-TASK-KO mice30, albeit aldosterone levels in zG-TASK-KO neonates may be higher, consistent with developmental differences in the adrenal gene expression profile48,49.
Mild, purely renin-independent, aldosterone overproduction evoked BP changes (△MAP: +9.5 mmHg) in zG-TASK-KO mice that appeared disproportionately large for the level of urinary aldosterone excretion; i.e., a 4-fold increase in aldosterone production generated by global TASK-KO mice evoked changes in blood pressure that were only 2-fold greater (△MAP: +19 mmHg). Moreover, these evoked-changes in blood pressure were fully correctable by mineralocorticoid receptor antagonism indicating that aldosterone is likely the sole causative factor producing Ang II-independent hypertension in zG-TASK-KO mice.
The aldosterone-induced increase in blood pressure could derive from an increase in vascular volume, sympathetic drive or MR-dependent vascular reactivity. Although identifying the mechanism by which aldosterone autonomy chronically elevates blood pressure in zG-TASK-KO mice was beyond the goals of our study, we provide some limited speculation based on our data. A) Aldosterone-induced increase in volume: We found that zG-TASK-KO mice were not overtly volume overloaded. Mice weight remained unchanged, and neither hematocrit, BUN nor hemoglobin levels were lower in zG-TASK-KO mice (Table 1).In addition, urinary Na+ excretion was not reduced and urinary K+ excretion was not increased, as one could expect from aldosterone-induced renal actions (Table 1). Nevertheless, a modest increase in vascular volume of zG-TASK KO mice would not be detected by these measures and cannot be excluded. B) Aldosterone-induced increase in sympathetic outflow: We found that HR of hypertensive zG-TASK KO mice was not different from that of control mice and that MR antagonism, but not AT1R antagonism, normalized blood pressure between genotypes. In many studies in humans and in rodents, MR activation of the sympathetic nervous system is a component of aldosterone-induced hypertension. Sympathetic overactivity is evident in patients harboring aldosterone-producing adenomas50, in those with mild hypertension following diuretic therapy51 and in rats after the hypertension-producing intracerebroventricular infusion of aldosterone52. In our mouse model, we do not know if central or peripheral MRs play a role in modulating sympathetic activity, but it remains a likely possibility. C) Aldosterone-induced increase in vascular reactivity: Our data does not address this possibility but it is worth noting that MRs are expressed in smooth muscle cells and MR activation directly increases vascular contractility 53–56, in contrast to MRs in the endothelium that contribute to dysfunction only in the setting of established cardiovascular disease57. These actions could provide additional extra-renal and extra-neural mechanisms for BP regulation by which mild aldosterone autonomy could produce normokalemic Ang II-independent hypertension. A determination of the relative contribution of these actions of aldosterone to blood pressure elevation in zG TASK-KO mice awaits further experimentation.
Recently, guidelines for the categorization of human hypertension have become more stringent with the recognition of graded associations between higher blood pressures and increased risks for cardiovascular disease 58. Our zG-TASK-KO mouse strain indicates that even mild dysregulated (autonomous) aldosterone production can cause a rise in BP that remains uncompensated by renal actions and thus can be an important contributor to cardiovascular pathogenesis.
Perspectives
Here, we report the first mouse model with a zG-specific defect28 that causes renin-independent mild hyperaldosteronism. This mouse line may be a useful tool to explore how chronic, mild aldosterone autonomy causes cardiovascular damage, and to find new cellular targets for its control. Accordingly, this mouse model may provide additional insights into the pathology and management of the early stages of the PA-disease continuum.
Supplementary Material
Novelty and Significance
What is New?
zG cell-specific dysfunction alone can produce hyperaldosteronism in vivo.
Extra-adrenal K2P-channel deletion amplifies zG-specific dysfunction.
Chronic mild aldosterone autonomy is sufficient to drive Ang II-independent hypertension.
What is Relevant?
PA is a disease continuum that begins with aldosterone dysregulation in normotension. zG cell-specific K2P-channel deletion provides a mouse model of chronic mild aldosterone autonomy that shows its potential for causing hypertension and highlights the need for early PA detection.
Acknowledgements
Sources of Funding
This study was supported by National Institute of Health R01 grants: HL089717 to P.Q. Barrett and D.A. Bayliss, DK100653A to D.T. Breault and DK113632 to T.H.Le.
Footnotes
Disclosures
None.
Contributor Information
Nick A. Guagliardo, Departments of Pharmacology, University of Virginia School of Medicine, Charlottesville, VA
Junlan Yao, Departments of Pharmacology, University of Virginia School of Medicine, Charlottesville, VA.
Eric J. Stipes, Departments of Pharmacology, University of Virginia School of Medicine, Charlottesville, VA
Sylvia Cechova, Medicine/Division of Nephrology, University of Virginia School of Medicine, Charlottesville, VA.
Thu H. Le, Medicine/Division of Nephrology, University of Virginia School of Medicine, Charlottesville, VA
Douglas A. Bayliss, Departments of Pharmacology, University of Virginia School of Medicine, Charlottesville, VA
David T. Breault, Pediatrics/Division of Endocrinology, Boston Children’s Hospital, Harvard Medical School, Boston, MA
Paula Q. Barrett, Departments of Pharmacology, University of Virginia School of Medicine, Charlottesville, VA
References
- 1.Funder JW, Carey RM, Mantero F, Murad MH, Reincke M, Shibata H, Stowasser M, Young WF Jr. The management of primary aldosteronism: Case detection, diagnosis, and treatment: An endocrine society clinical practice guideline. J Clin Endocrinol Metab 2016;101:1889–1916. [DOI] [PubMed] [Google Scholar]
- 2.Stowasser M Update in primary aldosteronism. J Clin Endocrinol Metab 2015;100:1–10. [DOI] [PubMed] [Google Scholar]
- 3.Kater CE, Biglieri EG. The syndromes of low-renin hypertension: “Separating the wheat from the chaff”. Arq Bras Endocrinol Metabol 2004;48:674–681. [DOI] [PubMed] [Google Scholar]
- 4.Nishizaka MK, Zaman MA, Calhoun DA. Efficacy of low-dose spironolactone in subjects with resistant hypertension. Am J Hypertens 2003;16:925–930. [DOI] [PubMed] [Google Scholar]
- 5.Piaditis G, Markou A, Papanastasiou L, Androulakis, II, Kaltsas G. Progress in aldosteronism: A review of the prevalence of primary aldosteronism in pre-hypertension and hypertension. Eur J Endocrinol 2015;172:R191–203. [DOI] [PubMed] [Google Scholar]
- 6.Baudrand R, Guarda FJ, Fardella C, Hundemer G, Brown J, Williams G, Vaidya A. Continuum of renin-independent aldosteronism in normotension. Hypertension 2017;69:950–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meneton P, Galan P, Bertrais S, Heudes D, Hercberg S, Menard J. High plasma aldosterone and low renin predict blood pressure increase and hypertension in middle-aged caucasian populations. J Hum Hypertens 2008;22:550–558. [DOI] [PubMed] [Google Scholar]
- 8.Vasan RS, Evans JC, Larson MG, Wilson PW, Meigs JB, Rifai N, Benjamin EJ, Levy D. Serum aldosterone and the incidence of hypertension in nonhypertensive persons. N Engl J Med 2004;351:33–41. [DOI] [PubMed] [Google Scholar]
- 9.Wisgerhof M, Brown RD, Hogan MJ, Carpenter PC, Edis AJ. The plasma aldosterone response to angiotensin ii infusion in aldosterone-producing adenoma and idiopathic hyperaldosteronism. J Clin Endocrinol Metab 1981;52:195–198. [DOI] [PubMed] [Google Scholar]
- 10.Funder JW. Primary aldosteronism and salt. Pflugers Arch 2015;467:587–594. [DOI] [PubMed] [Google Scholar]
- 11.Choi M, Scholl UI, Yue P, et al. K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science 2011;331:768–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zennaro MC, Boulkroun S, Fernandes-Rosa F. An update on novel mechanisms of primary aldosteronism. J Endocrinol 2015;224:R63–77. [DOI] [PubMed] [Google Scholar]
- 13.Azizan EA, Poulsen H, Tuluc P, et al. Somatic mutations in atp1a1 and cacna1d underlie a common subtype of adrenal hypertension. Nat Genet 2013;45:1055–1060. [DOI] [PubMed] [Google Scholar]
- 14.Beuschlein F, Boulkroun S, Osswald A, et al. Somatic mutations in atp1a1 and atp2b3 lead to aldosterone-producing adenomas and secondary hypertension. Nat Genet 2013;45:440–444, 444e441–442. [DOI] [PubMed] [Google Scholar]
- 15.Boulkroun S, Beuschlein F, Rossi GP, et al. Prevalence, clinical, and molecular correlates of kcnj5 mutations in primary aldosteronism. Hypertension 2012;59:592–598. [DOI] [PubMed] [Google Scholar]
- 16.Fernandes-Rosa FL, Williams TA, Riester A, et al. Genetic spectrum and clinical correlates of somatic mutations in aldosterone-producing adenoma. Hypertension 2014;64:354–361. [DOI] [PubMed] [Google Scholar]
- 17.Mulatero P, Tauber P, Zennaro MC, et al. Kcnj5 mutations in european families with nonglucocorticoid remediable familial hyperaldosteronism. Hypertension 2012;59:235–240. [DOI] [PubMed] [Google Scholar]
- 18.Scholl UI, Goh G, Stolting G, et al. Somatic and germline cacna1d calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nat Genet 2013;45:1050–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lenzini L, Prisco S, Gallina M, Kuppusamy M, Rossi GP. Mutations of the twik- related acid-sensitive k+ channel 2 (task-2) promoter in human primary aldosteronism. Endocrinology 2018;159:1352–1359. [DOI] [PubMed] [Google Scholar]
- 20.Lenzini L, Rossi GP. The molecular basis of primary aldosteronism: From chimeric gene to channelopathy. Curr Opin Pharmacol 2015;21:35–42. [DOI] [PubMed] [Google Scholar]
- 21.Nogueira EF, Gerry D, Mantero F, Mariniello B, Rainey WE. The role of task1 in aldosterone production and its expression in normal adrenal and aldosterone-producing adenomas. Clin Endocrinol (Oxf) 2010;73:22–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Czirjak G, Enyedi P. Formation of functional heterodimers between the task-1 and task-3 two-pore domain potassium channel subunits. J Biol Chem 2002;277:5426–5432. [DOI] [PubMed] [Google Scholar]
- 23.Czirjak G, Enyedi P. Task-3 dominates the background potassium conductance in rat adrenal glomerulosa cells. Mol Endocrinol 2002;16:621–629. [DOI] [PubMed] [Google Scholar]
- 24.Davies LA, Hu C, Guagliardo NA, Sen N, Chen X, Talley EM, Carey RM, Bayliss DA, Barrett PQ. Task channel deletion in mice causes primary hyperaldosteronism. Proc Natl Acad Sci U S A 2008;105:2203–2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lotshaw DP. Biophysical and pharmacological characteristics of native two-pore domain task channels in rat adrenal glomerulosa cells. J Membr Biol 2006;210:51–70. [DOI] [PubMed] [Google Scholar]
- 26.Enyeart JA, Danthi SJ, Enyeart JJ. Trek-1 k+ channels couple angiotensin ii receptors to membrane depolarization and aldosterone secretion in bovine adrenal glomerulosa cells. Am J Physiol Endocrinol Metab 2004;287:E1154–1165. [DOI] [PubMed] [Google Scholar]
- 27.Bayliss DA, Barrett PQ. Emerging roles for two-pore-domain potassium channels and their potential therapeutic impact. Trends Pharmacol Sci 2008;29:566–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aragao-Santiago L, Gomez-Sanchez CE, Mulatero P, Spyroglou A, Reincke M, Williams TA. Mouse models of primary aldosteronism: From physiology to pathophysiology. Endocrinology 2017;158:4129–4138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Freedman BD, Kempna PB, Carlone DL, Shah M, Guagliardo NA, Barrett PQ, Gomez-Sanchez CE, Majzoub JA, Breault DT. Adrenocortical zonation results from lineage conversion of differentiated zona glomerulosa cells. Dev Cell 2013;26:666–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guagliardo NA, Yao J, Hu C, Schertz EM, Tyson DA, Carey RM, Bayliss DA, Barrett PQ. Task-3 channel deletion in mice recapitulates low-renin essential hypertension. Hypertension 2012;59:999–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Malee MP, Mellon SH. Zone-specific regulation of two messenger rnas for p450c11 in the adrenals of pregnant and nonpregnant rats. Proc Natl Acad Sci U S A 1991;88:4731–4735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oertle M, Muller J. Two types of cytochrome p-450(11 beta) in rat adrenals: Separate regulation of gene expression. Mol Cell Endocrinol 1993;91:201–209. [DOI] [PubMed] [Google Scholar]
- 33.Talley EM, Solorzano G, Lei Q, Kim D, Bayliss DA. Cns distribution of members of the two-pore-domain (kcnk) potassium channel family. J Neurosci 2001;21:7491–7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.MacKenzie SM, Clark CJ, Fraser R, Gomez-Sanchez CE, Connell JM, Davies E. Expression of 11beta-hydroxylase and aldosterone synthase genes in the rat brain. J Mol Endocrinol 2000;24:321–328. [DOI] [PubMed] [Google Scholar]
- 35.Yu L, Romero DG, Gomez-Sanchez CE, Gomez-Sanchez EP. Steroidogenic enzyme gene expression in the human brain. Mol Cell Endocrinol 2002;190:9–17. [DOI] [PubMed] [Google Scholar]
- 36.Stromstedt M, Waterman MR. Messenger rnas encoding steroidogenic enzymes are expressed in rodent brain. Brain Res Mol Brain Res 1995;34:75–88. [DOI] [PubMed] [Google Scholar]
- 37.Taves MD, Gomez-Sanchez CE, Soma KK. Extra-adrenal glucocorticoids and mineralocorticoids: Evidence for local synthesis, regulation, and function. Am J Physiol Endocrinol Metab 2011;301:E11–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Funder JW. Primary aldosteronism: The next five years. Horm Metab Res 2017;49:977–983. [DOI] [PubMed] [Google Scholar]
- 39.James GD, Pickering TG. The influence of behavioral factors on the daily variation of blood pressure. Am J Hypertens 1993;6:170S–173S. [DOI] [PubMed] [Google Scholar]
- 40.Smolensky MH, Haus E. Circadian rhythms and clinical medicine with applications to hypertension. Am J Hypertens 2001;14:280S–290S. [DOI] [PubMed] [Google Scholar]
- 41.Smolensky MH, Hermida RC, Portaluppi F. Circadian mechanisms of 24-hour blood pressure regulation and patterning. Sleep Med Rev 2017;33:4–16. [DOI] [PubMed] [Google Scholar]
- 42.Gordon RD, Laragh JH, Funder JW. Low renin hypertensive states: Perspectives, unsolved problems, future research. Trends Endocrinol Metab 2005;16:108–113. [DOI] [PubMed] [Google Scholar]
- 43.Stowasser M Aldosterone excess and resistant hypertension: Investigation and treatment. Curr Hypertens Rep 2014;16:439. [DOI] [PubMed] [Google Scholar]
- 44.Cohn JN, Colucci W. Cardiovascular effects of aldosterone and post-acute myocardial infarction pathophysiology. Am J Cardiol 2006;97:4F–12F. [DOI] [PubMed] [Google Scholar]
- 45.Schiffrin EL. Effects of aldosterone on the vasculature. Hypertension 2006;47:312–318. [DOI] [PubMed] [Google Scholar]
- 46.Ohno Y, Sone M, Inagaki N, et al. Prevalence of cardiovascular disease and its risk factors in primary aldosteronism: A multicenter study in japan. Hypertension 2018;71:530–537. [DOI] [PubMed] [Google Scholar]
- 47.Manichaikul A, Rich SS, Allison MA, Guagliardo NA, Bayliss DA, Carey RM, Barrett PQ. Kcnk3 variants are associated with hyperaldosteronism and hypertension. Hypertension 2016;68:356–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bandulik S, Tauber P, Penton D, Schweda F, Tegtmeier I, Sterner C, Lalli E, Lesage F, Hartmann M, Barhanin J, Warth R. Severe hyperaldosteronism in neonatal task3 potassium channel knockout mice is associated with activation of the intraadrenal renin-angiotensin system. Endocrinology 2013;154:2712–2722. [DOI] [PubMed] [Google Scholar]
- 49.Heitzmann D, Derand R, Jungbauer S, et al. Invalidation of task1 potassium channels disrupts adrenal gland zonation and mineralocorticoid homeostasis. EMBO J 2008;27:179–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kontak AC, Wang Z, Arbique D, Adams-Huet B, Auchus RJ, Nesbitt SD, Victor RG, Vongpatanasin W. Reversible sympathetic overactivity in hypertensive patients with primary aldosteronism. J Clin Endocrinol Metab 2010;95:4756–4761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Raheja P, Price A, Wang Z, Arbique D, Adams-Huet B, Auchus RJ, Vongpatanasin W. Spironolactone prevents chlorthalidone-induced sympathetic activation and insulin resistance in hypertensive patients. Hypertension 2012;60:319–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xue B, Beltz TG, Yu Y, Guo F, Gomez-Sanchez CE, Hay M, Johnson AK. Central interactions of aldosterone and angiotensin ii in aldosterone- and angiotensin ii-induced hypertension. Am J Physiol Heart Circ Physiol 2011;300:H555–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.DuPont JJ, Jaffe IZ. 30 years of the mineralocorticoid receptor: The role of the mineralocorticoid receptor in the vasculature. J Endocrinol 2017;234:T67–T82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.DuPont JJ, McCurley A, Davel AP, McCarthy J, Bender SB, Hong K, Yang Y, Yoo JK, Aronovitz M, Baur WE, Christou DD, Hill MA, Jaffe IZ. Vascular mineralocorticoid receptor regulates microrna-155 to promote vasoconstriction and rising blood pressure with aging. JCI Insight 2016;1:e88942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McCurley A, Jaffe IZ. Mineralocorticoid receptors in vascular function and disease. Mol Cell Endocrinol 2012;350:256–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Galmiche G, Pizard A, Gueret A, El Moghrabi S, Ouvrard-Pascaud A, Berger S, Challande P, Jaffe IZ, Labat C, Lacolley P, Jaisser F. Smooth muscle cell mineralocorticoid receptors are mandatory for aldosterone-salt to induce vascular stiffness. Hypertension 2014;63:520–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mueller KB, Bender SB, Hong K, Yang Y, Aronovitz M, Jaisser F, Hill MA, Jaffe IZ. Endothelial mineralocorticoid receptors differentially contribute to coronary and mesenteric vascular function without modulating blood pressure. Hypertension 2015;66:988–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Whelton PK, Carey RM, Aronow WS, et al. 2017. acc/aha/aapa/abc/acpm/ags/apha/ash/aspc/nma/pcna guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: Executive summary: A report of the american college of cardiology/american heart association task force on clinical practice guidelines. Hypertension 2018;71:1269–1324. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.