

Abstract
Hepatocellular carcinoma (HCC) is one of the most fatal and fastest growing cancers. Recently, nonalcoholic steatohepatitis (NASH) has been recognized as a major HCC catalyst. However, it is difficult to decipher the molecular mechanisms underlying the pathogenesis of NASH and understand how it progresses to HCC by studying humans. Progress in this field depends on the availability of reliable preclinical models amenable to genetic and functional analyses and exhibit robust NASH to HCC progression. Although numerous mouse models of NASH have been described, many do not faithfully mimic the human disease and few reliably progress to HCC. Here, we review current literature on the molecular etiology of NASH-related HCC and critically evaluate existing mouse models and their suitability for studying this malignancy. We also compare human transcriptomic and histopathological profiles with data from MUP-uPA mice, a reliable model of NASH-driven HCC that has been useful for evaluation of HCC-targeting immunotherapies.
Graphical Abstract
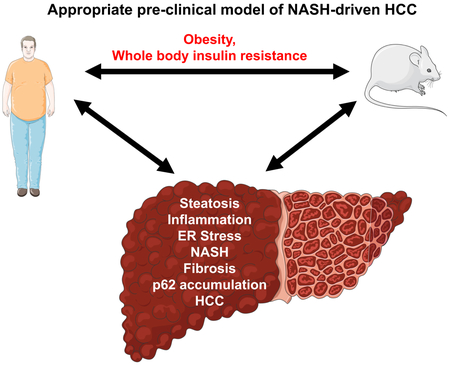
eTOC Blurb
Progress in the understanding of NASH-driven HCC depends on reliable preclinical models that are amenable to genetic and functional analyses and exhibit robust NASH to HCC progression. We review current literature on molecular etiology of NASH-related HCC, and critically evaluate existing mouse models and their suitability for studying this malignancy.
INTRODUCTION
Hepatocellular carcinoma (HCC) is one of the most common and fatal cancers worldwide (Starley et al., 2010). In developed countries, the past three decades have witnessed the incidence of HCC treble, becoming the fastest rising cause of cancer-related deaths (El-Serag and Kanwal, 2014). Initially ascribed to the emergence and spread of hepatitis C virus (HCV), only 50% of the dramatic increase in HCC mortality in developed countries is virus dependent. Up to 50% of newly diagnosed cases of HCC are in ‘virus-absent’ patients (El-Serag, 2011), most of whom present with obesity, non-alcoholic fatty liver disease (NAFLD), and, more commonly, non-alcoholic steatohepatitis (NASH), the most severe manifestation of NAFLD (Cohen et al., 2011). It is estimated that at least 30% of the adult population in developed countries has NAFLD (Lazo et al., 2013), with up to 15% of such patients exhibiting some degree of NASH (Cohen et al., 2011). Alarmingly, approximately 25% of NASH patients progress to cirrhosis, a serious premalignant condition that may require liver transplantation or result in liver failure (McCullough, 2004). Although mortality associated with most types of cancer has steadily declined over the last 40 years, both incidence and deaths due to HCC have substantially increased (Ryerson et al., 2016). Our ability to tackle this emerging epidemic has been severely hampered by the poor understanding of NASH. Even less is known about NASH-driven HCC.
NASH-driven HCC: etiology and mechanisms
Initially, NASH was thought to represent just one end of a broad spectrum of clinical and pathological signs associated with NAFLD. Currently, NAFLD is thought to be initiated by simple hepatosteatosis that arises from an imbalance between triglyceride deposition, mainly due to elevated de novo lipogenesis (DNL), and removal, either by very low density lipoprotein (VLDL)-mediated secretion or via fatty acid (FA) oxidation in mitochondria (Cohen et al., 2011). Such an imbalance is often the outcome of a sedentary lifestyle combined with overeating and excessive consumption of fructose-containing processed foods and beverages, but genetic factors (Dongiovanni et al., 2013) and inflammation of unknown origins (Marengo et al., 2016; Tilg and Moschen, 2006) are also of importance. Nevertheless, lipid droplet accumulation alone does not lead to liver damage and inflammation and it was therefore recognized that simple hepatosteatosis, also called “bland steatosis”, must be accompanied by a necro-inflammatory response, which is characterized by ballooning hepatocytes, liver damage, inflammatory infiltrates and fibrosis. This realization gave rise to the so-called ‘two-hit’ or ‘multiple-hit’ hypothesis, according to which lipid droplet accumulation is only one of several factors that by itself is insufficient for initiation of NASH pathogenesis. Additional factors include oxidative stress, endoplasmic reticulum (ER) stress, or presence of infectious or commensal organisms that trigger liver inflammation (Day, 2006; Day and James, 1998; Font-Burgada et al., 2016; Tilg and Moschen, 2006). Another important factor that distinguishes bland steatosis from NASH is the accumulation of free non-esterified cholesterol in the latter (Farrell and van Rooyen, 2012; Ioannou, 2016). Free cholesterol and its oxidized derivatives are cytotoxic and can synergize with TNF, whose synthesis is markedly elevated in NASH (Nakagawa et al., 2014) to cause mitochondrial dysfunction and liver damage (Verbeek et al., 2015). Exactly how NASH progresses to HCC is not entirely clear and the only reliable mechanistic information comes from studies of mouse models. Like most other drivers of HCC, NASH is associated with chronic liver damage, which triggers so-called compensatory proliferation. The same inflammatory cytokine involved in the transition from bland steatosis to NASH, TNF, was found to drive the proliferation of HCC-initiating cells, mainly through activation of NF-κB signaling (Nakagawa et al., 2014). However, how NASH results in accumulation of oncogenic mutations that lead to HCC initiation is unknown as the mutational profile and signature of NASH-associated HCC (Shalapour et al., 2017) do not seem to differ from those attached to other HCC etiologies (Schulze et al., 2015).
Role of cirrhosis and fibrosis
NASH is a risk factor for both HCC and liver cirrhosis (Adams et al., 2005). Cirrhosis is thought to contribute to the progression from NASH to HCC (Marengo et al., 2016). This is based on a large human clinical study in which less than 1% of patients with NAFLD developed HCC within 8 years after initial diagnosis. However, in NAFLD patients with cirrhosis, the rate of progression was 10% (Yatsuji et al., 2009). It is important to note, however, that patients who develop cirrhosis as a result of NASH have a lower relative risk of developing HCC compared with those with viral- or alcohol-related cirrhosis (Marengo et al., 2016). Although less well-documented, there is emerging evidence that HCC can also present in noncirrhotic NASH patients. From a clinical perspective, it is somewhat dangerous to define patients at risk of developing HCC by the presence or absence of cirrhosis. It should also be emphasized that while cirrhosis is not a universal feature of NASH-driven HCC, liver fibrosis is a cardinal sign of NASH. Therefore, a faithful mouse model of NASH-driven HCC need not necessarily present with cirrhosis but should exhibit extensive “chicken wire”-like pericellular fibrosis (see Table 1). Nonetheless, the mechanisms by which fibrosis, which usually refers to the deposition of collagen fibers, affects HCC development are obscure. Furthermore, the cells that are responsible for collagen deposition in the liver, hepatic stellate cells (HSC), are related to activated myofibroblasts or cancer-associated fibroblasts (CAF), which are known to have broad-spectrum tumor-promoting activities, not all of which are related to collagen deposition (Affo et al., 2017). Activated HSC and CAF produce TGF-β and many other cytokines and chemokines that recruit follicular helper (TFH) cells and naive B cells into the liver and stimulate conversion of the latter to immunosuppressive IgA+ plasma cells, which accumulate in human NASH and promote NASH to HCC progression in mice (Shalapour et al., 2017). Activated HSC also produce IL-6, another cytokine that is elevated during hepatic steatosis and can support HCC progression (Park et al., 2010).
Table 1. Summary of mouse models of NASH and HCC.
Abbreviations: ✓✓: marked expression; ✓: modest expression; X: no expression; ?: not assessed; HFD: high-fat diet; WD: Western Diet; MCD: methionine-and-choline deficient diet; CD: choline-deficient diet; STAM: streptozotocin+HFD-treated mice; DEN: diethylnitrosamine; ob/ob: leptin-deficient mice; db/db: leptin-receptor-deficient mice; SREBP1c Tg: sterol regulatory element-binding protein 1c transgenic mice; Agouti: KK-Ay-a mice; PTEN ko: phosphatase and tensin homologue deleted on chromosome 10 knockout mice; PPARα ko: peroxisome proliferator-activated receptor α knockout mice; AOX ko: acetyl-CoA oxidase deficient mice; MATA1 ko: methionine adenosyl transferase 1A knockout mice; MUP-uPA Tg: mice expressing transiently high amounts of urokinase plasminogen activator in hepatocytes; DIAMOND: isogenic BL6/129 hybrid strain fed an obesogenic WD supplemented by high fructose and sucrose.
| Obesity | Insulin Resistance |
Steatosis | Inflammation/ ER Stress |
NASH | Fibrosis | p62 Accumulation |
HCC | |
|---|---|---|---|---|---|---|---|---|
| Diet Models | ||||||||
| HFD | ✓✓ | ✓✓ | ✓✓ | ✓ | × | × | × | × |
| WD | ✓✓ | ✓✓ | ✓✓ | ✓ | × | × | × | × |
| MCD | × | × | ✓✓ | ✓ | ✓✓ | ✓✓ | ? | × |
| CD | × | × | ✓✓ | ✓ | ✓✓ | ✓✓ | ? | × |
| HFD/fructose | ✓✓ | ✓✓ | ✓✓ | ✓✓ | ✓✓ | ✓✓ | ? | ✓✓ |
| CD HFD | ✓✓ | ✓✓ | ✓✓ | ✓✓ | ✓✓ | ✓ | ? | ✓ |
| Toxins/Diet-Based Models | ||||||||
| STAM | × | ✓✓ | ✓✓ | ✓✓ | ✓✓ | ✓ | ? | ✓✓ |
| HFD/DEN | ✓✓ | ✓✓ | ✓✓ | ✓✓ | × | × | ✓✓ | ✓✓ |
| CD HFD/DEN | ✓✓ | ✓✓ | ✓✓ | ✓✓ | × | × | ? | ✓✓ |
| Genetic/Diet Models | ||||||||
| ob/ob | ✓✓ | ✓✓ | ✓✓ | ✓ | × | × | ? | × |
| db/db | ✓✓ | ✓✓ | ✓✓ | ✓ | × | × | ? | × |
| SREBP1c Tg | × | ✓✓ | ✓✓ | ✓✓ | ✓✓ | ✓✓ | ? | ✓ |
| Agouti | ✓✓ | ✓✓ | ✓✓ | ✓ | × | × | ? | × |
| PTEN ko | × | ✓✓ | ✓✓ | ✓✓ | ✓✓ | ✓✓ | ? | ✓ |
| PPARα ko | × | × | × | ✓✓ | ✓✓ | ✓✓ | ? | ✓ |
| AOX ko | × | × | × | ✓✓ | ✓✓ | × | ? | ✓ |
| MAT1A ko | × | × | × | ✓✓ | ✓✓ | × | ? | ✓ |
| MUP-uPA Tg | ✓✓ | ✓✓ | ✓✓ | ✓✓ | ✓✓ | ✓✓ | ✓✓ | ✓✓ |
| DIAMOND | ✓✓ | ✓✓ | ✓✓ | ✓✓ | ✓✓ | ✓✓ | ? | ✓✓ |
Inflammation and adaptive immunity in HCC
The concept that obesity leads to a state of low-grade chronic inflammation is now well-accepted (Hotamisligil et al., 1993). Work from our group has demonstrated that infiltrating macrophages and resident Kupffer cells produce inflammatory cytokines, such as IL-6 and TNF (Nakagawa et al., 2014; Naugler et al., 2007; Park et al., 2010), which contribute to both NASH and HCC development through c-Jun terminal kinase (JNK) and IκB kinase (IKK) signaling (Font-Burgada et al., 2016). However, the role of adaptive immunity in HCC development has, until recently, been unclear. Using mouse models of NASH that progress to HCC, we found that immunosuppressive IgA+ plasma cells that accumulate in fibrotic livers dismantle CD8+ cytotoxic T cell (CTL)-mediated immunosurveillance, thereby allowing early neoplastic lesions to escape immune elimination and progress to full-blown HCC (Shalapour et al., 2017). The immunosuppressive function of the fibrosis-stimulated IgA+ plasma cells most likely depends on expression of programmed death ligand 1 (PD-L1) and IL-10, which can directly induce CTL exhaustion or dysfunctionality. These findings explain why PD-1 inhibitors, which reverse CD8+ T cell exhaustion, are highly effective for the treatment of HCC (El-Khoueiry et al., 2017), despite the low mutational load associated with this type of cancer (Alexandrov et al., 2013). A different NAFLD-related mechanism was described by Greten and coworkers (Ma et al., 2016), who attributed the decline in liver-resident CD4+ T cells in high-fat diet (HFD)-fed mice that express a MYC transgene (which is not a relevant model of NASH-driven HCC) to the production of linoleic acid by steatotic hepatocytes. Although HFD also leads to a decline in liver-resident CD4+ T cells in MUP-uPA mice, it remains to be determined whether this has any effect on anti-HCC immunity (Shalapour et al., 2017).
Genetic factors that contribute to NASH-driven HCC
A limited number of genetic risk factors have been identified which predispose individuals to the development of NAFLD, the most notable of which is a missense mutation in the patatin-like phospholipase domain-containing 3 (PNPLA3) gene (Romeo et al., 2008). There are also a limited number of rare variants of other genes, such as nonsense mutations in the apolipoprotein B (APOB) gene and the telomerase reverse transcriptase (TERT) gene, that influence disease progression (Dongiovanni et al., 2014). It is important to note that deep sequencing of tumors and adjacent non-tumor tissue from NASH-driven HCC and comparison with HBV/HCV-driven HCC is of importance, but to date, no such studies have been reported. This is of importance considering some of the basic differences in liver metabolism between steatohepatitis and viral hepatitis. For example, hydroxysterols accumulate in NAFLD/NASH-afflicted livers (Li et al., 2017) but not in livers of viral hepatitis patients. Another important player is the liver-specific microRNA 122 (miR-122). Reduced miR-122 expression in HCV-related HCC correlates with increased metastasis and poor prognosis (Filipowicz and Grosshans, 2011), while delivery of miR-122 to a MYC-driven mouse HCC markedly reduces tumorigenesis (Hsu et al., 2012). Paradoxically, inhibition of miR-122 in HFD-fed mice with antisense oligonucleotides markedly improved hepatosteatosis and reduced liver damage, prerequisites for NASH development (Esau et al., 2006). While genetic factors play a role in almost every disease, it is important to note that weight-loss surgery leads to improvement and/or resolution of NASH in approximately 80% of patients (Clark and Diehl, 2003), suggesting that environmental factors may dominate over genetic factors in this disease. However, when it comes to NASH-to-HCC progression, not a single genetic or epidemiological study has led to the identification of factors that specifically affect this transition. Therefore, our knowledge on how NASH progresses to HCC is almost exclusively dependent on preclinical models.
NASH pipeline: hope or hype?
Currently, there are only two options for the treatment of early NASH, one of which is lifestyle modification through diet and exercise. However, adherence to such simple and noninvasive therapy is not overly successful and the benefits of exercise are understudied. The second option, bariatric surgery, is quite effective, but like any major surgery, is associated with potential health risks and is also costly. Nonetheless, NASH is now the second most common cause for liver transplantation in the US and is anticipated to become the leading cause by 2020. The cost and risk of liver transplantation outweighs those associated with all the above-mentioned preventive and early treatment measures. New and effective NASH therapies are therefore an urgent necessity.
Unfortunately, no new drugs have been approved for the prevention or treatment of NASH, although the development of targeted NASH therapies is a highly active field of translational medicine. Given that the NASH drug market is estimated to reach 40 billion USD by 2025, it is not surprising that many companies have embarked on this effort. Accordingly, there are over 35 clinical trials in various phases testing new pharmacological agents in NASH patients, most of which were discussed in detail in a recent review (Lazaridis and Tsochatzis, 2017). Notably, there are four drugs in current Phase III clinical trials: the farnesoid X nuclear receptor (FXR) ligand obeticholicacid or Ocaliva (Intercept Pharmaceuticals), the PPAR α/δ agonist GFT505 or Elafibranor (Genfit), the apoptosis signal-regulating kinase 1 (ASK1) Selonsertib (Gilead Sciences Inc), and the dual chemokine receptor 2/5 (CCR2/CCR5) inhibitor Cenicriviroc (Allergan). It should be noted, however, that in September 2017 the FDA issued a safety communication warning of a risk for serious liver injury and death from incorrect dosing of Ocaliva in patients with primary biliary cholangitis after 19 such deaths have occurred. Elafibranor, Selonsertib and Cenicriviroc are currently still in Phase III with no results posted. Several other NASH-targeting candidate compounds, such as the pan-caspase inhibitor Emricasan (Conatus), were recently found ineffective in proof-of-concept trials, although few other NASH trials are still ongoing. Another clinical candidate that has failed in reducing NASH-associated high blood pressure, although it was found effective in ameliorating hepatic ballooning, is the galectin-3 ligand GR-MD-02 (Galectin Technologies).
Undoubtedly, in order for successful drug development, the biological understanding of NASH needs dramatic improvement and biomarkers that can be used for patient stratification and evaluation of therapeutic outcomes need to be developed. Our opinion is that any significant progress in these fronts depends on the availability of mouse models that closely mimic human NASH. Since mouse models of NASH have been recently reviewed (Haczeyni et al., 2018; Hansen et al., 2017; Takakura et al., 2018), we focus the following discussion on preclinical models for studying NASH-driven HCC, the most dangerous outcome of untreated NASH. In addition to replicating the human pathology and showing robust progression to HCC within a reasonable timeframe (less than 12 months) and without the use of chemical carcinogens, a suitable preclinical model of NASH-driven HCC should also be responsive to the only effective HCC treatment identified thus far, inhibitors of the PD-L1:PD-1 checkpoint (El-Khoueiry et al., 2017). Most likely, future advances in HCC therapy will be based on different ways for increasing the response rates and efficacy of established anti-PD-L1:PD-1 drugs.
Mouse Models of NASH-driven HCC
Dietary intervention models
HFD in which 60% of the caloric value is provided by saturated fats causes simple hepatic steatosis but practically no liver injury or inflammation in most strains of mice (Nakagawa et al., 2014). A considerably higher degree of hepatic injury is elicited by the so-called WD, which contains pork lard, beef tallow, saturated FA, cholesterol, and sucrose. This is particularly apparent in ApoE−/− mice, which are defective in triglyceride export, where NASH-like symptoms and modest fibrosis are observed after WD consumption (Schierwagen et al., 2015). However, no HCC has been reported to date in such mice. More severe NASH-like pathology is produced by feeding mice toxic diets that are deficient in either methionine and choline (MCD) or choline alone (CD) (Machado et al., 2015). Both diets cause rapid weight loss (including loss of both fat mass and lean mass), reduced fasting glycemia, decreased circulating lipidemia, and no insulin resistance or increases in circulating or liver IL-6 (Kammoun et al., 2017). Unfortunately, this phenotype bears little resemblance to the etiology and metabolic features of human NASH. Moreover, neither MCD- nor CD-fed mice develop HCC. The CD model can be improved by combining it with HFD (CD+HFD), resulting in inhibition of weight loss and progression to HCC in about 25% of mice in a manner dependent on NF-κB signaling (Wolf et al., 2014). However, a major drawback of this model is the low penetrance of HCC induction and lack of information regarding factors that differentiate the few mice that progress to HCC from the majority that remains tumor-free.
Recently, it has become apparent that elevated fructose consumption is an important contributor to the NAFLD and NASH epidemic, acting primarily by induction of liver ER stress and DNL (Softic et al., 2016). Accordingly, mice maintained on a diet high in trans-fats and high-fructose corn syrup along with a sedentary lifestyle developed liver inflammation and bridging fibrosis within a year and 60% of them show hepatocellular neoplasms (Dowman et al., 2014). Although this dietary manipulation appears very useful as a model, the metabolic and immune mechanisms that drive neoplastic progression in response to fructose remain to be investigated and the tumors need to be shown to be similar to human HCC.
Toxins plus diet-based models
A new mouse model of NASH-driven HCC was recently developed based on administration of streptozotocin (STZ), a naturally occurring chemical that is toxic to pancreatic insulin-producing β cells that is often used in preclinical settings to induce type 1 diabetes. In this so-called STAM model, low-dose STZ is given to mice immediately after birth, followed by HFD feeding at 4 weeks of age. Simple steatosis appears after 5 weeks and NASH after 7 weeks, followed by fibrosis after 9 weeks, adenomas after 12 weeks and evidence of HCC at 16 weeks (Fujii et al., 2013; Saito et al., 2015). The STAM model has the advantage of being rapid and this may be the main reason for its wide adoption. However, unlike NASH-driven human HCC that almost always develops in the context of obesity, insulin resistance, and type 2 diabetes, STZ-treated mice exhibit type 1 diabetes and are lean. In addition, one cannot rule out an independent carcinogenic effect of STZ, as STAM mice display very mild fibrosis (Shalapour et al., 2017) and STZ is a DNA-damaging alkylating agent (Ridolfi et al., 1991). Thus, this model may not be all that different from the diethylnitrosamine (DEN)+HFD model, which depends on administration of the procarcinogen DEN (Park et al., 2010). As mentioned above, HFD alone does not induce NASH and its combination with DEN does not model NASH-driven HCC. Nonetheless, the DEN+HFD model provided the first demonstration that obesity and simple steatosis increase HCC risk by enhancing local production of IL-6 and TNF. Likewise, the administration of DEN to CD+HFD-fed mice allows for much more efficient HCC induction (Kishida et al., 2016), although it does not model NASH-induced HCC. Another recently developed model has combined Western diet (WD) with weekly dosing of carbon tetrachloride (CCl4) (Tsuchida et al., 2018). This model has the advantage of rapid disease progression, as the mice develop stage 3 fibrosis by 12 weeks and HCC by 24 weeks. Moreover, the model exhibits hepatic transcriptomic alignment with human NASH. It should be noted, however, that CCl4 is a potent hepatotoxin that can induce genotoxicity and oxidative DNA damage in rodents (Alkreathy et al., 2014).
Genetics plus diet-based models
The most popular genetic models of obesity, both leptin (ob/ob)- and leptin-receptor (db/db)-deficient mice do not develop NASH or HCC but are more susceptible to HCC induction on DEN administration (Park et al., 2010). However, several other genetically modified mice that are susceptible to NASH development have been described. SREBP-1c transgenic mice, the agouti (KK-Ay/a) mouse, and mice with global deficiencies in PTEN, PPAR-α, AOX, and MAT1A have all been studied in the context of NASH-driven HCC. As previously reviewed (Takahashi et al., 2012), these models have limitations since they fail to either display obesity (PTEN-, PPAR-α-, AOX-, MAT1A-deficient and SREBP-1c transgenic mice) or NASH (KK-Ay/a mice). Moreover, RNA-Seq analysis had shown that the liver gene expression profile of Pten−/− mice is quite different from other mouse models of NASH (Teufel et al., 2016). Furthermore, Pten−/− mice are insulin hyper-responsive rather than insulin resistant, in marked contrast to patients presenting with NASH.
Recently, we developed a new model for NASH-driven HCC that does not suffer from most of the limitations listed above. This model is based on feeding HFD to MUP-uPA transgenic mice, which express high amounts of urokinase plasminogen activator (uPA) specifically in hepatocytes during the first 6 weeks of life (Nakagawa et al., 2014). Excessive uPA expression induces hepatocyte ER stress and transient liver damage, both of which have been implicated in human NASH development (Lebeaupin et al., 2018). Unlike WT BL6 mice kept on HFD, all HFD-fed MUP-uPA mice develop NASH-like disease with up to 85% of them spontaneously progressing to HCC. This pattern of disease progression allows for biomarker discovery and identification of molecular drivers of HCC progression, since mice can be examined at the onset of NASH and then followed until they separate into those that develop tumors and those that do not. HFD-fed MUP-uPA mice show substantial upregulation of collagen gene expression, HSC activation and upregulation of fibrogenic markers. Classical NASH signs, including ballooning hepatocytes, inflammatory infiltrates, and pericellular and bridging fibrosis indistinguishable from the pattern observed in human NASH, are observed within 4 months of HFD initiation (Nakagawa et al., 2014) (Figure 1A). Both NASH development and HCC progression depend on elevated TNF production, ER stress, and hepatic expression of p62, all of which also participate in the human pathology (Nakagawa et al., 2014; Umemura et al., 2016). Altogether, the HFD-fed MUP-uPA model is almost identical to human NASH in its pathological features (Maurel et al., 2014). As mentioned above, HCC progression in MUP-uPA mice depends on accumulation of immunosuppressive IgA+ plasma cells, a prominent feature of human NASH (Shalapour et al., 2017). The involvement of immunosuppressive IgA+ plasma cells that express PD-L1 and IL-10 makes the HFD-fed MUP-uPA mouse highly suitable for testing various treatments that can further increase the efficacy of PD-L1:PD-1 signaling inhibitors.
Figure 1. Histological and transcriptomic alignment between the MUP-uPA mouse model and human NASH.

(A) Histological comparison of MUP-uPA mice and human NASH; data previously published (Nakagawa et al., 2014). (B) RNA-Seq data from human NASH patients and normal individuals (left) and MUP-uPA mice fed either HFD or normal chow (right). All terms are enriched with a p < 0.05, the color coding represents the p-value for enrichments calculated for the MUP-uPA+HFD mice, the darker red the more significant, and the size of the nodes represents the p-value for enrichments calculated for the human data (all nodes that are only enriched for the human data are light red, all nodes that are only enriched for MUP-uPA are small). (C) Gene alignment between our current data set of NASH patients (n = 3) compared with four other publicly available data sets.
Another recently developed genetically modified mouse that progresses to NASH and HCC is the isogenic B6/129 hybrid strain fed a WD with a high-fructose-sucrose solution (WD-SW diet) (Asgharpour et al., 2016). These mice, nicknamed DIAMOND mice, show zone 3 hepatic steatosis within 8 weeks and steatohepatitis 16-24 weeks after WD-SW initiation, including ballooning hepatocytes and Mallory-Denk bodies (Asgharpour et al., 2016). Of note, 90% of DIAMOND mice develop HCC after 32-52 weeks, which is quite similar to HFD-fed MUP-uPA mice. However, the unique genetic background of DIAMOND mice makes it difficult to cross them with other gene-targeted mice, most of which are on a pure BL6 background, which is required for the genetic identification of additional factors and signaling pathways involved in NASH-to-HCC progression. Nonetheless, DIAMOND mice may be suitable for testing immunotherapies and other interventions, as the mechanisms of tumor development in these mice should be similar to those operating in MUP-uPA mice. This, however, remains to be demonstrated. Without further comparison it seems that both the MUP-uPA+HFD and DIAMOND models are well-suited for studying NASH-driven HCC and are far more physiologically relevant than the STAM model. Nonetheless, HCC development in either MUP-uPA or DIAMOND mice is considerably slower than in STAM mice. Furthermore, the mutational landscape of MUP-uPA (and probably also DIAMOND) mice varies from mouse to mouse (Shalapour et al., 2017). Although this is much more similar to human HCC, which is one of the most heterogenous cancers (Schulze et al., 2015), this heterogeneity may complicate drug development studies.
Role of microbiota
Diet and other environmental factors modulate the composition and metabolic activity of the gut microbiota, which in turn can impact health. In particular, HFD and obesity induce dysbiosis and increase the representation of inflammation-evoking bacteria in the gut (Hildebrandt et al., 2009; Parks et al., 2013; Shalapour et al., 2017). HFD- or fructose-induced dysbiosis may also enhance energy harvest from ingested food and, together with inflammation, promote energy storage and insulin resistance (Conlon and Bird, 2014). Not surprisingly, NASH in both humans and mice was found to be associated with dysbiosis (Brandl and Schnabl, 2017; Shalapour et al., 2017; Yu and Schwabe, 2017). Dysbiosis can contribute to deterioration of the intestinal epithelial barrier, resulting in translocation of inflammation-provoking bacterial products and metabolites (and even intact bacteria) that reach the liver via the portal circulation to further enhance NAFLD and NASH progression. As the gut microbiota is metabolically active, metabolomic studies have identified metabolomic profiles unique to NASH patients (Brandl and Schnabl, 2017). Some of these metabolites, such as the bile acid deoxycholic acid, may also contribute to NASH-to-HCC progression via yet-to-be identified mechanisms (Yoshimoto et al., 2013; Yu and Schwabe, 2017). Gut bacteria and their metabolites also influence adaptive immunity. For example, gut bacteria support the development of IgA+ immunosuppressive plasmocytes (Shalapour et al., 2017), and microbial-derived bile acids control a chemokine-dependent accumulation of hepatic NKT cells and anti-tumor immunity in the liver (Ma et al., 2018), indicating a dual role of bacteria in the context of cancer immunity. Further studies are needed to fully comprehend how the gut microbiota affects hepatic metabolism and immunity and determine whether bacteria play a primary or secondary role in NASH-driven HCC.
Mouse vs human
In addition to histological and metabolic similarities between mouse models and human NASH-driven HCC, transcriptomic alignment between the species is also informative. We therefore analyzed RNA-Seq data from 3 patients with NASH and 3 healthy individuals (previously unpublished data) and compared these with publicly available RNA-Seq data (GSE94097) collected from MUP-uPA mice kept on either normal chow or HFD (Shalapour et al., 2017). Figure 1B shows Gene Ontology pathways that were enriched in both human and mouse NASH, as well as pathways that were exclusive for one species or the other. In the human data, the inflammatory response was more pronounced than in MUP-uPA mice, but it should be acknowledged that none of the analyzed patients were drug naïve, that only a very small data set was analyzed, and that human NASH is extremely heterogeneous. In fact, one should note that the human data set previously described by Teufel et al., 2016 was not unique to NASH and did not make any discrimination between NASH and simple steatosis, as liver samples were obtained from patients undergoing biopsy for suspected NAFLD or NASH. To illustrate the degree of heterogeneity in human data sets, we compared our human NASH RNA-Seq data with four other publicly available NASH data sets and only a low degree of transcriptomic alignment was found (Figure 1C). Notwithstanding the limitations to these analyses, we observed some degree of transcriptomic alignment when comparing the MUP-uPA model with our human data set, notably in regulation of the immune system, innate immune response and the response to cytokine gene sets. These transcriptomic alignments are fully consistent with our recent descriptions of metabolic and signaling similarities between human NASH and the MUP-uPA+HFD model (Shalapour et al., 2017).
Conclusions
NASH-driven HCC is a fast-growing, serious, and complex malignancy that requires better understanding and further study before it reaches epidemic proportions. Preclinical models are a cornerstone of further research, especially given the difficulties in obtaining suitable human specimens representing different stages of the disease. The MUP-uPA model, while not perfect or rapid, exhibits many similarities to the human disease. We, therefore, suggest its adaptation by researchers exploring those aspects of NASH-driven HCC that are best replicated by this model or by the related DIAMOND mouse. Both models are highly suitable for the development of preventive, early treatment, and diagnostic procedures. Regardless of the final model to be adapted by the community, one should be cautious of mechanistic, immunological, or drug development studies that are performed with inappropriate models. Despite their so-called “convenience”, such models should be avoided.
Highlights.
Hepatocellular carcinoma (HCC) is one of the most fatal cancers
The pathogenesis of NASH driven-HCC is difficult to study in humans
Progress in the field depends on reliable preclinical mouse models
To faithfully mimic human NASH-driven HCC several criteria must be met
ACKNOWLEDGEMENTS
This study was funded, in part, by the National Health and Medical Research Council (NHMRC Project Grant APP112227 to MAF and MK), National Institutes of Health (NIH Grants R01AI043477, R01CA118165, R01CA127923, R01CA155120, R01CA198103, R01CA211794, P42ES010337, P30DK063491, U01AA022614 to MK; U01AA027681 to SS and MK), and Southern California Research Center for ALPD & Cirrhosis funded by the National Institute on Alcohol Abuse and Alcoholism (P50AA011999 subaward to SS). MAF is a Senior Principal Research Fellow of the NHMRC (APP1116936). MK is an American Cancer Research Society Professor and holds the Ben and Wanda Hildyard Chair for Mitochondrial and Metabolic Diseases. We thank Jeniffer Lee for manuscript assistance.
Footnotes
DECLARATION OF INTERESTS
MK holds US Patent No. 10,034,462 B2 on the use of MUP-uPA mice for the study of NASH and NASH-driven HCC. All other authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Adams LA, Lymp JF, St Sauver J, Sanderson SO, Lindor KD, Feldstein A, and Angulo P (2005). The natural history of nonalcoholic fatty liver disease: a population-based cohort study. Gastroenterology 129, 113–121. [DOI] [PubMed] [Google Scholar]
- Affo S, Yu LX, and Schwabe RF (2017). The Role of Cancer-Associated Fibroblasts and Fibrosis in Liver Cancer. Annu Rev Pathol 12, 153–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, et al. (2013). Signatures of mutational processes in human cancer. Nature 500, 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkreathy HM, Khan RA, Khan MR, and Sahreen S (2014). CCl4 induced genotoxicity and DNA oxidative damages in rats: hepatoprotective effect of Sonchus arvensis. BMC complementary and alternative medicine 14, 452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asgharpour A, Cazanave SC, Pacana T, Seneshaw M, Vincent R, Banini BA, Kumar DP, Daita K, Min HK, Mirshahi F, et al. (2016). A diet-induced animal model of non-alcoholic fatty liver disease and hepatocellular cancer. J Hepatol 65, 579–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandl K, and Schnabl B (2017). Intestinal microbiota and nonalcoholic steatohepatitis. Curr Opin Gastroenterol 33, 128–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark JM, and Diehl AM (2003). Nonalcoholic fatty liver disease: an underrecognized cause of cryptogenic cirrhosis. Jama 289, 3000–3004. [DOI] [PubMed] [Google Scholar]
- Cohen JC, Horton JD, and Hobbs HH (2011). Human fatty liver disease: old questions and new insights. Science 332, 1519–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conlon MA, and Bird AR (2014). The impact of diet and lifestyle on gut microbiota and human health. Nutrients 7, 17–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day CP (2006). From fat to inflammation. Gastroenterology 130, 207–210. [DOI] [PubMed] [Google Scholar]
- Day CP, and James OF (1998). Steatohepatitis: a tale of two “hits”? Gastroenterology 114, 842–845. [DOI] [PubMed] [Google Scholar]
- Dongiovanni P, Donati B, Fares R, Lombardi R, Mancina RM, Romeo S, and Valenti L (2013). PNPLA3 I148M polymorphism and progressive liver disease. World J Gastroenterol 19, 6969–6978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dongiovanni P, Romeo S, and Valenti L (2014). Hepatocellular carcinoma in nonalcoholic fatty liver: role of environmental and genetic factors. World J Gastroenterol 20, 12945–12955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowman JK, Hopkins LJ, Reynolds GM, Nikolaou N, Armstrong MJ, Shaw JC, Houlihan DD, Lalor PF, Tomlinson JW, Hubscher SG, et al. (2014). Development of hepatocellular carcinoma in a murine model of nonalcoholic steatohepatitis induced by use of a high-fat/fructose diet and sedentary lifestyle. Am J Pathol 184, 1550–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, Kim TY, Choo SP, Trojan J, Welling THR, et al. (2017). Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 389, 2492–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Serag HB (2011). Hepatocellular carcinoma. N Engl J Med 365, 1118–1127. [DOI] [PubMed] [Google Scholar]
- El-Serag HB, and Kanwal F (2014). Epidemiology of hepatocellular carcinoma in the United States: where are we? Where do we go? Hepatology 60, 1767–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esau C, Davis S, Murray SF, Yu XX, Pandey SK, Pear M, Watts L, Booten SL, Graham M, McKay R, et al. (2006). miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab 3, 87–98. [DOI] [PubMed] [Google Scholar]
- Farrell GC, and van Rooyen D (2012). Liver cholesterol: is it playing possum in NASH? American journal of physiology. Gastrointestinal and liver physiology 303, G9–11. [DOI] [PubMed] [Google Scholar]
- Filipowicz W, and Grosshans H (2011). The liver-specific microRNA miR-122: biology and therapeutic potential. Progress in drug research. Fortschritte der Arzneimittelforschung. Progres des recherches pharmaceutiques 67, 221–238. [PubMed] [Google Scholar]
- Font-Burgada J, Sun B, and Karin M (2016). Obesity and Cancer: The Oil that Feeds the Flame. Cell Metab 23, 48–62. [DOI] [PubMed] [Google Scholar]
- Fujii M, Shibazaki Y, Wakamatsu K, Honda Y, Kawauchi Y, Suzuki K, Arumugam S, Watanabe K, Ichida T, Asakura H, et al. (2013). A murine model for non-alcoholic steatohepatitis showing evidence of association between diabetes and hepatocellular carcinoma. Med Mol Morphol 46, 141–152. [DOI] [PubMed] [Google Scholar]
- Haczeyni F, Yeh MM, Ioannou GN, Leclercq IA, Goldin R, Dan YY, Yu J, Teoh NC, and Farrell GC (2018). Mouse models of non-alcoholic steatohepatitis: A reflection on recent literature. J Gastroenterol Hepatol. [DOI] [PubMed] [Google Scholar]
- Hansen HH, Feigh M, Veidal SS, Rigbolt KT, Vrang N, and Fosgerau K (2017). Mouse models of nonalcoholic steatohepatitis in preclinical drug development. Drug Discov Today 22, 1707–1718. [DOI] [PubMed] [Google Scholar]
- Hildebrandt MA, Hoffmann C, Sherrill-Mix SA, Keilbaugh SA, Hamady M, Chen YY, Knight R, Ahima RS, Bushman F, and Wu GD (2009). High-fat diet determines the composition of the murine gut microbiome independently of obesity. Gastroenterology 137, 1716–1724 e1711–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS, Shargill NS, and Spiegelman BM (1993). Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 259, 87–91. [DOI] [PubMed] [Google Scholar]
- Hsu SH, Wang B, Kota J, Yu J, Costinean S, Kutay H, Yu L, Bai S, La Perle K, Chivukula RR, et al. (2012). Essential metabolic, anti-inflammatory, and anti-tumorigenic functions of miR-122 in liver. J Clin Invest 122, 2871–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioannou GN (2016). The Role of Cholesterol in the Pathogenesis of NASH. Trends Endocrinol Metab 27, 84–95. [DOI] [PubMed] [Google Scholar]
- Kammoun HL, Allen TL, Henstridge DC, Kraakman MJ, Peijs L, Rose-John S, and Febbraio MA (2017). Over-expressing the soluble gp130-Fc does not ameliorate methionine and choline deficient diet-induced non alcoholic steatohepatitis in mice. PLoS One 12, e0179099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishida N, Matsuda S, Itano O, Shinoda M, Kitago M, Yagi H, Abe Y, Hibi T, Masugi Y, Aiura K, et al. (2016). Development of a novel mouse model of hepatocellular carcinoma with nonalcoholic steatohepatitis using a high-fat, choline-deficient diet and intraperitoneal injection of diethylnitrosamine. BMC Gastroenterol 16, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazaridis N, and Tsochatzis E (2017). Current and future treatment options in non-alcoholic steatohepatitis (NASH). Expert Rev Gastroenterol Hepatol 11, 357–369. [DOI] [PubMed] [Google Scholar]
- Lazo M, Hernaez R, Eberhardt MS, Bonekamp S, Kamel I, Guallar E, Koteish A, Brancati FL, and Clark JM (2013). Prevalence of nonalcoholic fatty liver disease in the United States: the Third National Health and Nutrition Examination Survey, 1988-1994. Am J Epidemiol 178, 38–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebeaupin C, Vallee D, Hazari Y, Hetz C, Chevet E, and Bailly-Maitre B (2018). Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J Hepatol 69, 927–947. [DOI] [PubMed] [Google Scholar]
- Li C, Deng YQ, Wang S, Ma F, Aliyari R, Huang XY, Zhang NN, Watanabe M, Dong HL, Liu P, et al. (2017). 25-Hydroxycholesterol Protects Host against Zika Virus Infection and Its Associated Microcephaly in a Mouse Model. Immunity 46, 446–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C, Kesarwala AH, Eggert T, Medina-Echeverz J, Kleiner DE, Jin P, Stroncek DF, Terabe M, Kapoor V, ElGindi M, et al. (2016). NAFLD causes selective CD4(+) T lymphocyte loss and promotes hepatocarcinogenesis. Nature 531, 253–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado MV, Michelotti GA, Xie G, Almeida Pereira T, Boursier J, Bohnic B, Guy CD, and Diehl AM (2015). Mouse models of diet-induced nonalcoholic steatohepatitis reproduce the heterogeneity of the human disease. PLoS One 10, e0127991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marengo A, Rosso C, and Bugianesi E (2016). Liver Cancer: Connections with Obesity, Fatty Liver, and Cirrhosis. Annu Rev Med 67, 103–117. [DOI] [PubMed] [Google Scholar]
- Maurel M, Samali A, and Chevet E (2014). Endoplasmic reticulum stress: at the crossroads of inflammation and metabolism in hepatocellular carcinoma development. Cancer Cell 26, 301–303. [DOI] [PubMed] [Google Scholar]
- McCullough AJ (2004). The clinical features, diagnosis and natural history of nonalcoholic fatty liver disease. Clin Liver Dis 8, 521–533, viii. [DOI] [PubMed] [Google Scholar]
- Nakagawa H, Umemura A, Taniguchi K, Font-Burgada J, Dhar D, Ogata H, Zhong Z, Valasek MA, Seki E, Hidalgo J, et al. (2014). ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell 26, 331–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naugler WE, Sakurai T, Kim S, Maeda S, Kim K, Elsharkawy AM, and Karin M (2007). Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science 317, 121–124. [DOI] [PubMed] [Google Scholar]
- Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, Osterreicher CH, Takahashi H, and Karin M (2010). Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 140, 197–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks BW, Nam E, Org E, Kostem E, Norheim F, Hui ST, Pan C, Civelek M, Rau CD, Bennett BJ, et al. (2013). Genetic control of obesity and gut microbiota composition in response to high-fat, high-sucrose diet in mice. Cell Metab 17, 141–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridolfi R, Amaducci L, Derni S, Fabbri L, Innocenti MP, and Vignutelli P (1991). Chemotherapy with 5-fluorouracil and streptozotocin in carcinoid tumors of gastrointestinal origin: experiences with 13 patients. J Chemother 3, 328–331. [DOI] [PubMed] [Google Scholar]
- Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, Boerwinkle E, Cohen JC, and Hobbs HH (2008). Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 40, 1461–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryerson AB, Eheman CR, Altekruse SF, Ward JW, Jemal A, Sherman RL, Henley SJ, Holtzman D, Lake A, Noone AM, et al. (2016). Annual Report to the Nation on the Status of Cancer, 1975–2012, featuring the increasing incidence of liver cancer. Cancer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito K, Uebanso T, Maekawa K, Ishikawa M, Taguchi R, Nammo T, Nishimaki-Mogami T, Udagawa H, Fujii M, Shibazaki Y, et al. (2015). Characterization of hepatic lipid profiles in a mouse model with nonalcoholic steatohepatitis and subsequent fibrosis. Scientific reports 5, 12466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schierwagen R, Maybuchen L, Zimmer S, Hittatiya K, Back C, Klein S, Uschner FE, Reul W, Boor P, Nickenig G, et al. (2015). Seven weeks of Western diet in apolipoprotein-E-deficient mice induce metabolic syndrome and non-alcoholic steatohepatitis with liver fibrosis. Scientific reports 5, 12931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze K, Imbeaud S, Letouze E, Alexandrov LB, Calderaro J, Rebouissou S, Couchy G, Meiller C, Shinde J, Soysouvanh F, et al. (2015). Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat Genet 47, 505–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalapour S, Lin XJ, Bastian IN, Brain J, Burt AD, Aksenov AA, Vrbanac AF, Li W, Perkins A, Matsutani T, et al. (2017). Inflammation-induced IgA+ cells dismantle anti-liver cancer immunity. Nature 551, 340–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Softic S, Cohen DE, and Kahn CR (2016). Role of Dietary Fructose and Hepatic De Novo Lipogenesis in Fatty Liver Disease. Dig Dis Sci 61, 1282–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starley BQ, Calcagno CJ, and Harrison SA (2010). Nonalcoholic fatty liver disease and hepatocellular carcinoma: a weighty connection. Hepatology 51, 1820–1832. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Soejima Y, and Fukusato T (2012). Animal models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J Gastroenterol 18, 2300–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takakura K, Oikawa T, Tomita Y, Mizuno Y, Nakano M, Saeki C, Torisu Y, and Saruta M (2018). Mouse models for investigating the underlying mechanisms of nonalcoholic steatohepatitis-derived hepatocellular carcinoma. World J Gastroenterol 24, 1989–1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teufel A, Itzel T, Erhart W, Brosch M, Wang XY, Kim YO, von Schonfels W, Herrmann A, Bruckner S, Stickel F, et al. (2016). Comparison of Gene Expression Patterns Between Mouse Models of Nonalcoholic Fatty Liver Disease and Liver Tissues From Patients. Gastroenterology 151, 513–525 e510. [DOI] [PubMed] [Google Scholar]
- Tilg H, and Moschen AR (2006). Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nat Rev Immunol 6, 772–783. [DOI] [PubMed] [Google Scholar]
- Tsuchida T, Lee YA, Fujiwara N, Ybanez M, Allen B, Martins S, Fiel MI, Goossens N, Chou HI, Hoshida Y, et al. (2018). A simple diet- and chemical-induced murine NASH model with rapid progression of steatohepatitis, fibrosis and liver cancer. J Hepatol 69, 385–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umemura A, He F, Taniguchi K, Nakagawa H, Yamachika S, Font-Burgada J, Zhong Z, Subramaniam S, Raghunandan S, Duran A, et al. (2016). p62, Upregulated during Preneoplasia, Induces Hepatocellular Carcinogenesis by Maintaining Survival of Stressed HCC-Initiating Cells. Cancer Cell 29, 935–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbeek J, Lannoo M, Pirinen E, Ryu D, Spincemaille P, Vander Elst I, Windmolders P, Thevissen K, Cammue BP, van Pelt J, et al. (2015). Roux-en-y gastric bypass attenuates hepatic mitochondrial dysfunction in mice with non-alcoholic steatohepatitis. Gut 64, 673–683. [DOI] [PubMed] [Google Scholar]
- Wolf MJ, Adili A, Piotrowitz K, Abdullah Z, Boege Y, Stemmer K, Ringelhan M, Simonavicius N, Egger M, Wohlleber D, et al. (2014). Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell 26, 549–564. [DOI] [PubMed] [Google Scholar]
- Yatsuji S, Hashimoto E, Tobari M, Taniai M, Tokushige K, and Shiratori K (2009). Clinical features and outcomes of cirrhosis due to non-alcoholic steatohepatitis compared with cirrhosis caused by chronic hepatitis C. J Gastroenterol Hepatol 24, 248–254. [DOI] [PubMed] [Google Scholar]
- Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, Iwakura Y, Oshima K, Morita H, Hattori M, et al. (2013). Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 499, 97–101. [DOI] [PubMed] [Google Scholar]
- Yu LX, and Schwabe RF (2017). The gut microbiome and liver cancer: mechanisms and clinical translation. Nature reviews. Gastroenterology & hepatology 14, 527–539. [DOI] [PMC free article] [PubMed] [Google Scholar]