

Summary
Successful metastasis requires the co-evolution of stromal and cancer cells. We used stable isotope labeling of amino acids in cell culture (SILAC) coupled with quantitative, label-free phosphoproteomics to study the bidirectional signaling in ovarian cancer cells and human-derived, cancer-associated fibroblasts (CAFs) after co-culture. In cancer cells, the interaction with CAFs supported glycogenolysis under normoxic conditions and induced phosphorylation and activation of phosphoglucomutase 1 (PGM1), an enzyme involved in glycogen metabolism. Glycogen was funneled into glycolysis, leading to increased proliferation, invasion, and metastasis of cancer cells co-cultured with human CAFs. Glycogen mobilization in cancer cells was dependent on p38α MAPK activation in CAFs. In vivo, deletion of p38α in CAFs and glycogen phosphorylase inhibition in cancer cells reduced metastasis, suggesting that glycogen is an energy source used by cancer cells to facilitate metastatic tumor growth.
Keywords: glycogen, metastasis, metabolism, cancer-associated fibroblast, phosphoproteomics
eTOC Blurb
et al show that ovarian cancer cells can mobilize glycogen as an energy source, leading to increased proliferation, invasion and metastasis, following their interaction with cancer-associated fibroblasts (CAFs). This process is dependent on p38α MAPK activation in CAFs and inhibition of the pathway reduced metastatic tumor growth in vivo.
Graphical Abstract
Introduction
Cancer associated fibroblasts (CAFs), which are a major component of the tumor stroma, promote several tumor cell characteristics associated with increased aggressiveness (Bhowmick et al., 2004; Orimo et al., 2005). The interplay between CAFs and tumor cells promotes motility, invasion, and metastasis through direct contact and the exchange of soluble mediators that further activate the surrounding stroma and promote epithelial mesenchymal transition in the cancer cells (Hanahan and Coussens, 2012; Ohlund et al., 2014). While the mechanisms by which CAFs support cancer cells are only partially understood, the current perception is that successful tumorigenesis requires the co-evolution of stroma and cancer cells. Recently, CAFs were shown to participate in tumor cell metabolism by providing metabolites used by cancer cells for energy production and biosynthetic processes through the tricarboxylic acid (TCA) cycle, known as the ‘reverse Warburg effect’ (Pavlides et al., 2009; Romero et al., 2015; Sousa et al., 2016).
Given the co-evolution of cancer cells and CAFs within a tumor, it is important to understand the reciprocal signaling between them and how this signaling influences the metastatic capability of the cancer cells. While several studies have investigated changes of specific proteins in either cancer cells or CAFs, a systematic unbiased study of the bidirectional signaling pathways initiated by the interaction of human cancer cells with human-derived CAFs has not been performed. A thorough understanding of these signaling pathways could have clinical implications, since many abdominally metastasizing cancers, including ovarian cancer (OvCa), have prominent interactions between metastatic tumor cells and CAFs.
Results
Metabolic signaling is activated in OvCa by CAFs
Co-culture of an OvCa cell line with primary human CAFs isolated from the omental metastases of patients with high grade serous OvCa (Figure S1A) strongly induced the invasion of cancer cells (Figure S1B) in a manner consistent with the known tumor-promoting role of the stroma (Bhowmick et al., 2004; Orimo et al., 2005). To understand the bidirectional signaling that occurs between cancer cells and primary CAFs, we utilized stable isotope labeling of amino acids in cell culture (SILAC), allowing us to systematically analyze signaling networks in both CAFs and cancer cells simultaneously. The OvCa cell line, SKOV3ip1, was labeled with ‘heavy’ arginine and lysine (C13N15) and cultured with primary human CAFs labeled with ‘light’ arginine an d lysine (C12N14) for 4 hours, allowing communication between the two cell types. Tyrosine-phosphorylated peptides were immunoprecipitated and analyzed by nano-LC/MS on an Orbitrap-Velos high performance mass spectrometer. As depicted in Figure 1A, the differential labeling of the two cell types yields two distinct proteomes by producing a shift in the molecular weights of the phospho-peptides in the OvCa cells. In an analysis of the proteomes, we identified 236 non-redundant, unique tyrosine phospho-peptides (quantified at 1% FDR), representing 233 phospho-peptides in SKOV3ip1 cells and 226 in CAFs (Figure 1B, Table S1 and S2).After removing peptides with redundant phophosites, and using a cut-off of +/− 1 log2-fold change, 59 phosphosites, representing 45 unique proteins, were found to be significantly changed in SKOV3ip1 cells and 20 phosphosites, representing 18 unique proteins in CAFs (Table S3). Pathway analysis of the altered tyrosine phosphorylation events identified alterations of key signaling pathways upon the interaction of the two cell types (Figure 1C and Figure S1C). CAFs showed increased phosphorylation of several adaptor proteins that mediate integrin signaling (TLN1, PXN, BCAR1), as well as the EPHA2 receptor, its adaptor protein, NEDD9, and the p38-MAPK signaling pathway, a well-known mediator of mitogenic signaling and inflammation (Hickson et al., 2006; Igea and Nebreda, 2015). The siRNA-mediated inhibition of BCAR1, NEDD9 and p38α in CAFs reduced OvCa invasion, suggesting that phospho-proteins identified in CAFs are of functional significance (Figure S1D). In the OvCa cell line, phosphorylation of integrin adaptor proteins (paxillin and talin) was upregulated. Interestingly, the phosphorylation of several metabolic enzymes was induced in the cancer cells, most notably phosphoglucomutase-1 (PGM1), a central member of the glycogen metabolism pathway (Bae et al., 2014; Gururaj et al., 2004) (Figure 1C, Figure S2A, Table S3).
Figure 1. Bidirectional signaling between cancer cells and CAFs mapped with quantitative phosphoproteomics.
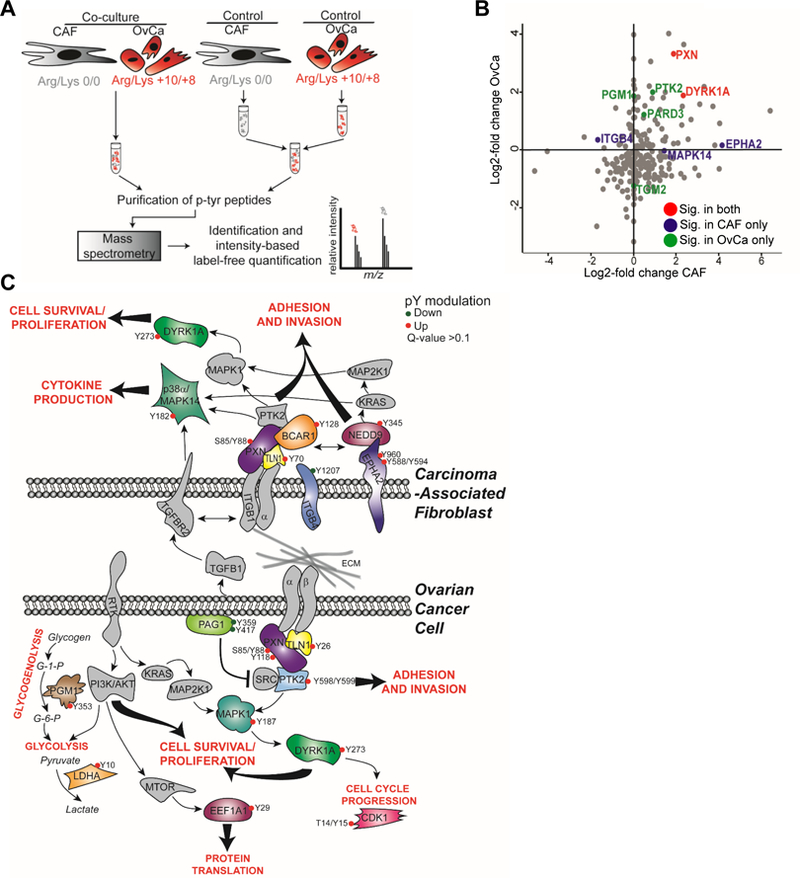
(A) Experimental design. The OvCa cell line SKOV3ip1 was SILAC labeled with heavy arginine and lysine while CAFs were labeled with light Arg and Lys. Cells were co-cultured or cultured independently (control) for 4 hr prior to lysate collection (n=5/group). Control lysates were mixed together and then samples were digested with trypsin and phosphotyrosine containing peptides extracted using immunoprecipitation and identified by nano-LC/MS. For each identified peptide, the isotopic label was used to distinguish which cell population the peptide originated from. Intensity-based label-free quantification was used to determine relative levels of phosphorylated peptides in both cell types. (B) Scatter plot showing distribution of identified p-tyr proteins and their relative fold-change in both cancer cells and CAFs. Selection of significantly (Sig.) changed proteins from Table S3 are highlighted in either OvCa (green), CAF (blue), or in both cell types (red). (C) Schematic of selected significantly modified proteins highlighting key regulatory pathways. Phosphoproteins changed by co-culture are shown in color. Phosphotyrosine sites in red are increased and in green are decreased upon co-culture.
Recent findings suggest that stromal cells in the tumor microenvironment regulate metabolism in adjacent cancer cells (Hanahan and Coussens, 2012; Romero et al., 2015). Therefore, we focused on the finding that PGM1 was phosphorylated in cancer cells upon interaction with CAFs. The PGM1 enzyme catalyzes the inter-conversion between glucose 1-phosphate and glucose 6-phosphate, acting as a gatekeeper in the metabolic pathway between glucose and glycogen (Figure S2A) (Bae et al., 2014; Gururaj et al., 2004; Roach et al., 2012; Zois et al., 2014). Upon interaction with human primary CAFs, PGM1 was phosphorylated in cancer cells on tyrosine 353, a previously uncharacterized and evolutionarily conserved phosphorylation site in eukaryotes (Figure S2B, C). A search of the PhosphositePlus (Hornbeck et al., 2015) database showed that this tyrosine phosphorylation site was the most common protein modification (#200 records) identified across PGM1. Because this phosphosite has never been functionally analyzed, we raised an antibody against phospho-Y353 PGM1, which recognized WT PGM1 protein immunoprecipitated from OvCa cells (Figure S2D) but did not detect PGM1 protein with tyrosine-353 mutated to phenylalanine (Y353F) (Figure S2E). To confirm the phosphoproteomic findings, we incubated primary human CAFs, separated by a transwell insert, with the SKOV3ip1 cell line (probably reflecting clear cell OvCa (Coscia et al., 2016; Domcke et al., 2013), a histologic subtype known for high glycogen content) or with TYK-nu cells (representing high grade serous OvCa (Coscia et al., 2016; Domcke et al., 2013)). The co-culture induced PGM1-Y353 phosphorylation in both cancer cell lines (Figure 2A). In cells expressing the Y353F mutant, PGM1 activity was decreased 4-fold as compared to the WT protein (Figure 2B, Figure S2F), suggesting that phosphorylation of this residue is critical for enzyme activity. To determine whether the Y353F mutation may have altered the stability of the PGM1 protein, a cellular thermal shift assay (CETSA) was performed and showed minimal differences in thermal stability as compared to WT PGM1 (Figure S2G). In addition, co-culture between TYK-nu cells and CAFs in a transwell insert stimulated PGM1 activity (Figure 2C). Since PGM1 is not the rate-limiting enzyme of glycogenolysis, we next tested the effect of co-culture with CAFs on the regulatory enzyme of glycogenolysis, glycogen phosphorylase (Roach et al., 2012; Zois et al., 2014) (Figure S2A). Analyzing data from our recently published proteomic analysis of ovarian cancer cell lines (Coscia et al., 2016) both the liver (PYGL) and brain (PYGB) isoforms of glycogen phosphorylase are expressed in SKOV3ip1 and TYK-nu cell lines (Figure S2H). Similar to PGM1, glycogen phophorylase activity in TYK-nu cells was also increased after co-culture in a transwell insert with CAFs (Figure 2D), while total expression of glycogen phosphorylase was unchanged (Figure S2I).
Figure 2. CAFs promote mobilization of glycogen stores in cancer cells.
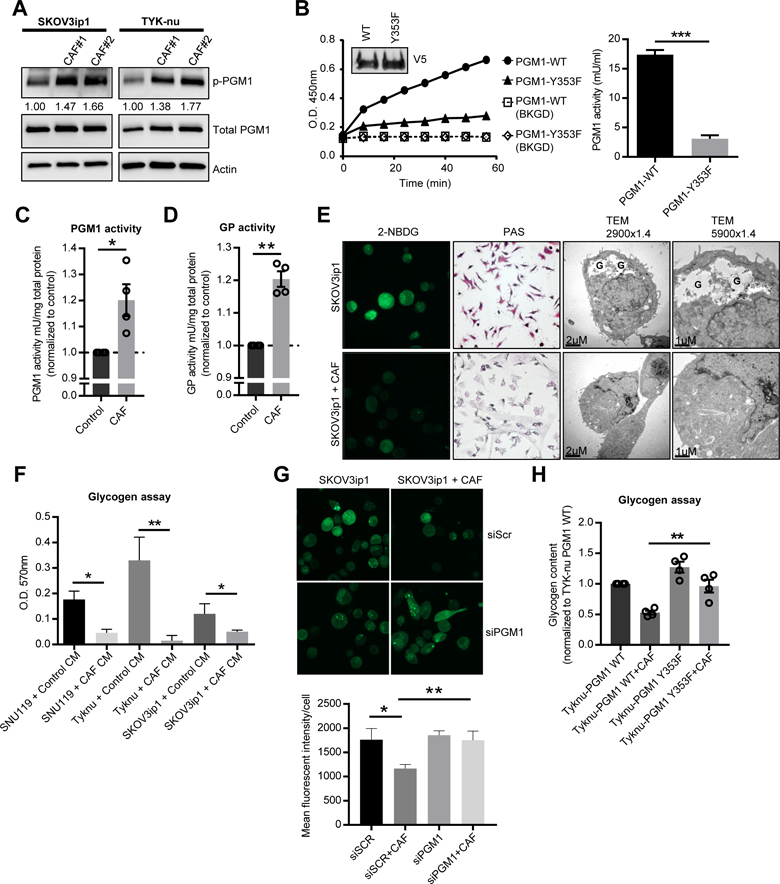
(A) Western blot detecting phosphorylated phosphoglucomutase 1 (PGM1) at Y353 in SKOV3ip1 and TYK-nu cells co-cultured with primary human CAFs separated by a transwell cell culture insert. Values below p-PGM1 indicate the relative band intensity normalized to total PGM1 and fold change compared to the control. Images are representative of three biological repeats. (B) PGM1 enzyme activity assay of PGM1 immunoprecipitated from TYK-nu cells stably expressing the WT or Y353F mutant constructs (inset) and analyzed for PGM1 activity using glucose-1-phosphate as the substrate. Right panel, activity of PGM1 (***p< 0.001). Background (BKGD). (C) PGM1 enzyme activity assay and (D) glycogen phosphorylase (GP) activity in TYK-nu cells co-cultured with primary human CAFs (3 hr) separated by a transwell cell culture insert. Values are normalized to enzyme activity in TYK-nu cells cultured alone as indicated by the dotted line. Values are mean + SEM from 4 independent experiments (n=6/group/experiment) *p<0.05, **p< 0.01. (E) Glycogen stores were visualized by 2-NBDG fluorescence, PAS staining and transmission electron microscopy (TEM) in SKOV3ip1 cells co-cultured with or without CAFs (4 hr). In the TEM images, glycogen pools are identified by a “G”. (F) Glycogen assay on SKOV3ip1, TYK-nu, and SNU119 cells with and without CAF conditioned media (CM) after 4 hr. *p<0.05, **p< 0.01. Values are mean + SEM n=3. Data is representative of two biological repeats with CAF CM from two different patient-derived CAFs. (G) Glycogen content in SKOV3ip1 cells transfected with PGM1 siRNA or scramble control (siScr) visualized by 2-NBDG fluorescence. Values are mean + SEM from 3 independent experiments (n=50 cells/group). (H) Glycogen assay on TYK-nu cells expressing either wild-type (WT) PGM1 or the Y353F PGM1 mutant and co-cultured with primary human CAFs in a transwell cell culture insert. Values are mean + SEM from 3 independent experiments. **p<0.01.
Glycogen mobilization is stimulated by CAFs in cancer cell lines
Given that PGM1 and glycogen phosphorylase are central members of the glycogen metabolism pathway, we evaluated the effect of CAFs on glycogen content in OvCa cells. After four hours, human-derived primary CAFs induced a reduction of glycogen stores in OvCa cells as shown by PAS staining, transmission electron microscopy, and by the conversion of a fluorescent glucose analog (2-NBDG) into glycogen granules (Figure 2E). Glycogen levels were consistently very low in CAFs and remained unchanged by the co-culture (Figure S2J). In addition, the CAF-induced depletion of glycogen was confirmed in five out of six additional cancer cell lines studied, including two high grade serous OvCa cell lines, another clear cell OvCa (ES-2) cell line, a breast cancer cell line, and a prostate cancer cell line (Figure S3A). The colon cancer cell line, RKO, which has a constitutively low glycogen level, was not affected by CAF co-culture. OvCa cells cultured in direct contact with CAFs and then sorted by FACS (Figure S3B) or treated with CAF conditioned media showed significantly reduced glycogen content (Figure 2F). In addition, knock-down of PGM1 in SKOV3ip1 and TYK-nu OvCa cells impaired the ability of CAFs to mobilize glycogen stores in the cancer cells (Figure 2G and Figure S3C, D) while it had no effect on the activation of glycogen phosphorylase in cancer cells stimulated by CAFs (Figure S3E). Furthermore, glycogen mobilization stimulated by CAF co-culture was inhibited in cells expressing the PGM1 Y353F mutant as compared to cells expressing wild-type PGM1 (Figure 2H), confirming that PGM1 plays an important role in the utilization of glycogen. Taken together, these data strongly suggest that fibroblasts can induce a rapid mobilization of glycogen in cancer cells that is mediated through the exchange of soluble factors.
Glycogen fuels glycolysis in cancer cells
Next, we examined the metabolic and functional consequences of fibroblast-induced glycogen mobilization in cancer cells. Reduction of glycogen by CAF conditioned media in OvCa cells was paralleled by an increase in cancer cell glycolysis, glycolytic capacity, and glycolytic reserve (Figure 3A, Figure S4A), with no changes in oxidative phosphorylation (Figure S4B). Glucose was readily taken up by both CAFs and TYK-nu cells in co-culture as assessed by the uptake of 2-NBDG (Figure S4C). In addition, lactate production was dramatically increased both in cancer cells stimulated with CAF conditioned media (CM) and in CAFs stimulated with cancer cell CM (Figure S4D). Consistent with the CAF-induced increase in glycolysis, there was also an increase in the energy potential of the cell as shown by an increase in the ATP/ADP ratio in TYK-nu and SKOV3ip1 cells (Figure 3B, Figure S4E). The CAF-mediated increase in glycolysis and in the ATP/ADP ratio was blocked by CP-91149 (Figure 3A, B and Figure S4A, E), an inhibitor of glycogen phosphorylase (Henke and Sparks, 2006) (Figure S2A), which prevented CAF-mediated glycogen mobilization (Figure S4F). The inhibitor, however, had no effect on the control treated cancer cells. Given that CAFs promoted glycogenolysis in cancer cells, we asked whether glycogenesis would be inhibited in parallel. Indeed, treating OvCa cells with conditioned media collected from human-derived primary CAFs reduced expression of glycogen synthase, the regulatory enzyme of glycogenesis (Figure S2A, Figure S4G). These data suggest that CAFs initiate a switch in OvCa cells that shifts glycogen metabolism from glycogen storage to glycogen utilization.
Figure 3. Glycogen fuels glycolysis to promote proliferation and invasion.
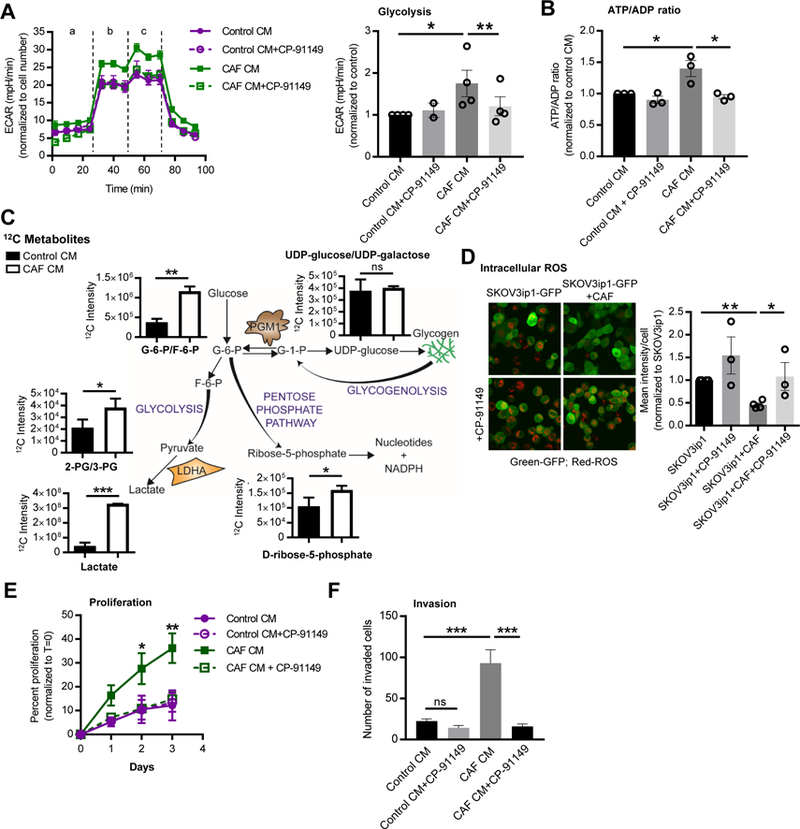
(A) TYK-nu cells were pretreated (1 hr) with the glycogen phosphorylase inhibitor CP-91149 followed by stimulation with conditioned media (CM) from primary human CAFs for 16 hr. Glycolysis was measured using the Seahorse SF96 Extracellular Flux Analyzer. Left, extracellular acidification rates (ECAR) are shown normalized to cell number. ‘a’, ba sal glycolysis (no glucose); ‘b’, glycolysis (stimulated with glucose); ‘c’, glycolytic capacity (stimulated with oligomycin). Right panel shows the average of the 3 glycolysis time points for each group. Values are from 4 independent experiments (n=15/group). Comparisons were made to CAF CM using a paired, two-tailed t test (*p<0.05, **p<0.01). (B) ATP/ADP ratios in TYK-nu cells treated with CAF or control CM pretreated with or without CP-91149, an inhibitor of glycogen phosphorylase. Values are mean + SEM from 3 independent experiments (n=4/group). Comparisons were made to CAF CM using a paired, two-tailed t test (*p<0.05). (C) Steady state 12C metabolites in TYK-nu cells stimulated with control CM or CAF CM for 6 hr (n=3/group). Key metabolites are shown along with a schematic depicting the central pathways (* p<0.05, **p<0.01, ***p<0.001). (D) ROS level in GFP-labeled SKOV3ip1 cancer cells with or without CAF co-culture and CP-91149. Left, representative images of ROS staining (red). Right, quantification of 3 independent experiments. Values are mean + SEM. Samples were compared to OvCa-GFP+CAF using an unpaired, two-tailed t test (* p<0.05, **p<0.01). (E) Proliferation of TYK-nu cells exposed to CAF CM with or without the glycogen phosphorylase inhibitor CP-91149. Values are mean + SEM from 4 independent experiments (n=6/group). Comparisons were made to CAF CM using a Two-way ANOVA (* p<0.05, **p<0.01). (F) Invasion of TYK-nu cells exposed to CAF CM with or without the glycogen phosphorylase inhibitor CP-91149. Data is representative of 3 independent experiments. Values are mean + SEM (n=3/group). ***p<0.001.
In order to understand further the metabolic changes induced by CAFs, we performed targeted metabolic profiling of OvCa cells stimulated with control or CAF conditioned media using high-performance liquid chromatography (HPLC) coupled to high-resolution mass spectrometry. Consistent with our previous data, we observed after 6 hr increased glycolysis in cancer cells, indicated by increased glucose-6-phosphate/fructose-6-phosphate, 2/3-phospho-glycerate, and lactate production with CAF conditioned media (Figure 3C). The levels of UDP-glucose/UDP-galactose, intermediates for glycogen synthesis, remained constant. A higher level of ribose-5-phosphate was also detected in the cancer cells stimulated with CAF conditioned media (Figure 3C) suggesting an increase in the pentose phosphate pathway. As shown by a quantitative assay, glucose-1-phosphate was also increased in OvCa cells following stimulation with CAF CM, which is consistent with an increased breakdown of glycogen (Figure S4H). Since the pentose phosphate pathway contributes to both increased ribose-5-phosphate and an increased availability of reducing equivalents, we next asked if CAF co-culture affects reactive oxygen species (ROS) levels in cancer cells. Indeed, CAFs decreased the production of ROS in the cancer cells, an effect that was rescued by treatment with the glycogen phosphorylase inhibitor (Figure 3D). This is consistent with a report showing that glycogen phosphorylase inhibition induces ROS under conditions of hypoxia (Favaro et al., 2012). Increased glycolysis was paralleled by increased proliferation and invasion in TYK-nu and SKOV3ip1 cell lines following stimulation by CAF conditioned media. Treatment with the glycogen phosphorylase inhibitor, CP-91149, blocked the increase in both proliferation and invasion (Figure 3E, F and Figure S5A, B). Direct CAF co-culture showed similar effects on proliferation in SKOV3ip1 and in one primary OvCa cell clone (Figure S5C). Of note, treatment with CP-91149 had no effect on the basal level of glycolysis, proliferation or invasion.
We next asked if hydrolyzed glycogen contributes directly to the metabolite pool in cancer cells. To accomplish this, we devised a new glycogen labeling strategy in which cancer cells were fed U-13C-glucose for 48hr to label glycogen (Figure 4A) and then media was changed to 12C-glucose for 1 hour prior to the experiment. This would leave glycogen as the singular source for 13C-carbon in the cell. 13C-labeling patterns were analyzed following culture either with or without human-derived primary CAFs in a transwell insert. U-13C-glycogen was hydrolyzed and contributed to the levels of glycolytic metabolites, including glucose-6-phosphate/fructose-6-phosphate (m+6), dihydroxyacetone phosphate (DHAP) (m+3), and 2/3-phospho-glycerate (m+3), all of which were increased upon CAF co-culture (Figure 4B). Glycogen also contributed to increases in citrate/isocitrate (m+2) and α-ketoglutarate (m+2), while there was no increase in additional TCA cycle intermediates such as fumarate and malate. These data suggest that CAFs induce cancer cells to metabolize glycogen through glycolysis, which increases energy production and tumor aggressiveness.
Figure 4. Glycogen-derived metabolites feed glycolysis.
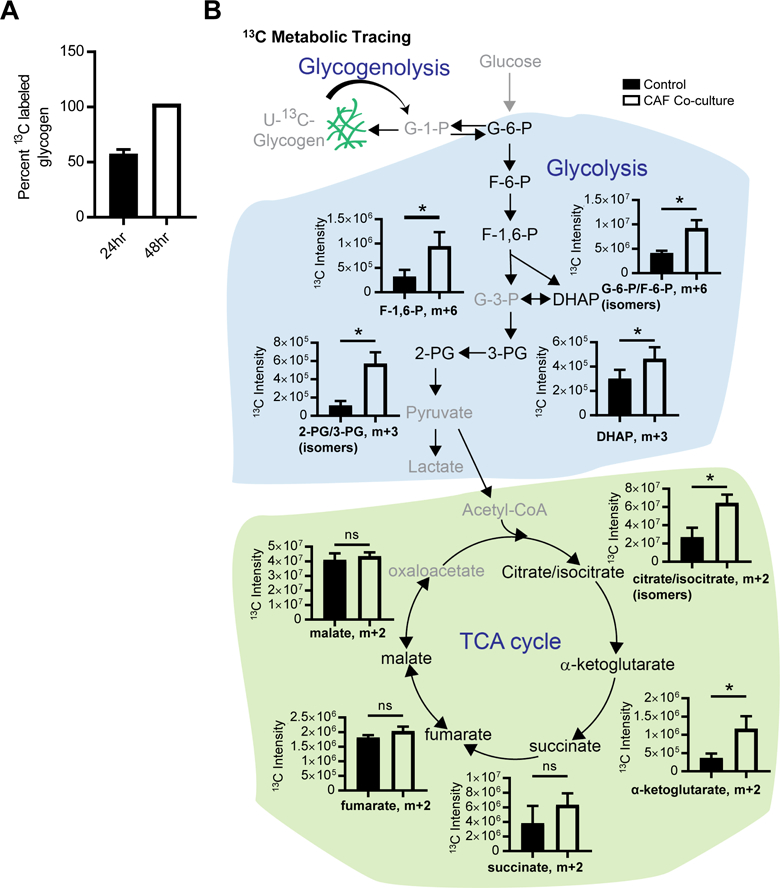
(A) Percent of glycogen labeled with U-13C-glucose tracer at 24 and 48hr. 13C-glucose (m+6) was measured using mass spectrometry following hydrolysis of U-13C-glycogen to glucose. TYK-nu cells were cultured in U-13C-glucose containing DMEM for 24hr or 48hr to synthesize U-13C-glycogen. (B) Glycogen tracing analysis. 13C metabolite tracing analysis of TYK-nu cells cultured with or without CAFs in a transwell insert for 30 min (n=4/group). Prior to the experiment, TYK-nu cells were cultured for 48hr in media containing 13C-glucose (to label glycogen), followed by culture in 12C-glucose for 1 hr to label pathway intermediates 12C, leaving U-13C-glycogen as the only 13C source for glycolysis. Peak intensities of the 13C metabolites are shown, along with a schematic depicting the pathways. (* p<0.05). G-1-P, glucose-1-phosphate; G-6-P, glucose-6-phosphate; F-6-P, fructose-6-phosphate; F-1,6-P, fructose-1,6-bisphosphate; DHAP, dihydroxyacetone phosphate; G-3-P, glyceraldehyde-3-phosphate; 3-PG, 3-phosphoglycerate; 2-PG, 2-phosphoglycerate.
Glycogen mobilization is dependent on p38-MAPK signaling in CAFs.
A key signaling pathway activated in fibroblasts upon co-culture with OvCa cells, as determined using SILAC (Figure 1C), was p38α MAPK. We and others have shown that cancer cells secrete TGF-β1 (Guido et al., 2012; Kenny et al., 2014; Schauer et al., 2011), which is used in heterotypic signaling and is known to regulate p38α MAPK signaling (Igea and Nebreda, 2015). Knocking down TGF-β1 in the OvCa cells or the receptor for TGF-β1, TGFβRII, in the CAFs, downregulated the phosphorylation of p38α in CAFs following stimulation with OvCa conditioned media (Figure 5A), suggesting that p38α activation in CAFs is induced, at least in part, through the production of TGF-β1 by OvCa cells. Neither primary human CAFs with stable p38α knockdown (Figure 5B, C), nor CAFs treated with a chemical inhibitor of p38α MAPK (Figure 5D, E), were able to induce glycogen mobilization in SKOV3ip1 cells. Consistent with these results, conditioned media from primary CAFs expressing a p38α shRNA was unable to stimulate phosphorylation of PGM1 (Figure 5F) and the CAFs were less efficient in promoting in vitro proliferation (Figure 5G), invasion (Figure 5H), and subcutaneous tumor growth in mice (Figure 5I) compared to CAFs expressing a control shRNA. Tumors containing CAFs expressing a p38α shRNA, contained higher levels of glycogen, as assessed by PAS staining, when compared the tumors with control shRNA CAFs (Figure 5J).
Figure 5. Glycogen utilization is dependent on p38α MAPK activity in cancer associated fibroblasts.
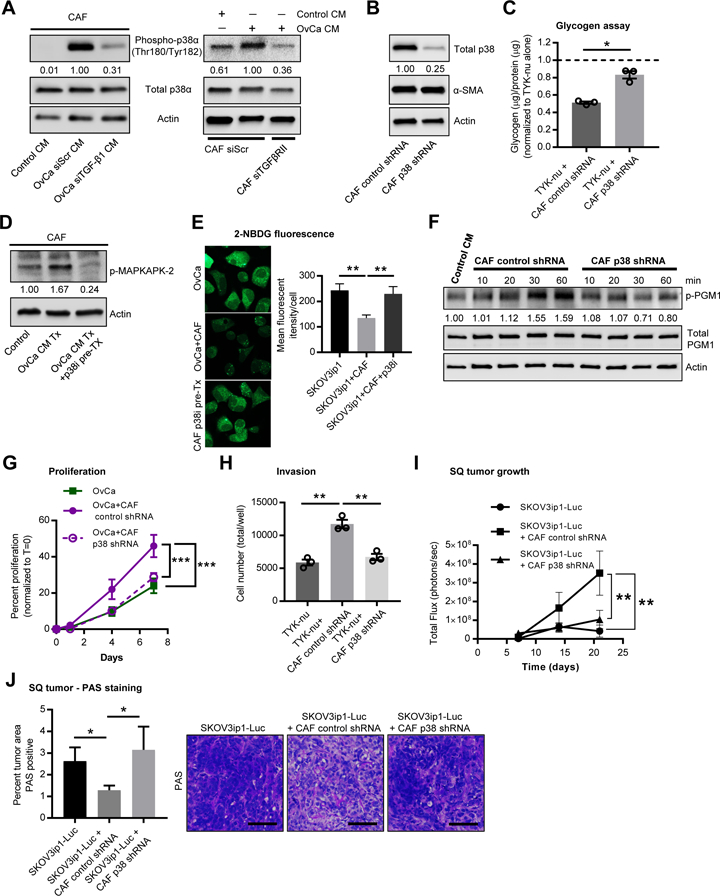
(A) Western blot for p38α phosphorylation in CAFs. Left, CAFs treated with conditioned media (CM) from SKOV3ip1 cells transfected with either TGF-β1 or scrambled (siScr) siRNA. Right, CAFs transfected with either TGF-βRII or scrambled (siScr) siRNA were stimulated with control or SKOV3ip1 CM for 4 hr. Values represent relative band intensity of p-p38 expression normalized to total p38 and fold change relative to the OvCa stimulated sample. Images are representative of three biological repeats. (B) Validation of p38 knockdown in primary CAFs. α-SMA (smooth muscle actin) is unchanged with p38 knockdown. Values represent relative band intensity of total p38 expression normalized to actin and fold change relative to the control shRNA sample. Images are representative of three biological repeats with primary CAFs from three patients. (C) Glycogen assay on TYK-nu cells stimulated with CM collected from CAFs expressing a control shRNA or a p38α shRNA. Values are normalized to TYK-nu alone shown by the dotted line and represent 3 biological repeats. Values are mean + SEM (*p< 0.05). (D) Western blot for phospho-MAPKAPK-2, a downstream effector of the p38 MAPK pathway, in CAFs treated with and without PH-797804, a p38α MAPK inhibitor. Values represent relative band intensity of p-p38 normalized to actin. (E) Glycogen storage (left) was visualized by 2-NBDG fluorescence in SKOV3ip1 cancer cells after 4 hr of co-culture with CAFs. CAFs were pretreated with 2 µM of PH-797804 for 1 hr prior to co-culture. Right, quantification of 2-NBDG fluorescence. (F) Western blot for p-PGM1 at Y353, total PGM1 and actin in TYK-nu cells following stimulation by CM collected from CAFs expressing a control shRNA or a p38α shRNA for the indicated time. Values represent relative band intensity of p-PGM1 normalized to total PGM1 and fold change relative to the control CM sample. Images are representative of 4 biological replicates. (G) Proliferation of TYK-nu cells exposed to CM from primary CAFs expressing a control shRNA versus CAFs expressing a p38α shRNA. Data is representative of 2 independent experiments. Values are mean + SEM (n=6/group), ***p<0.001. (H) Boyden chamber invasion assay of TYK-nu treated with CM from either primary CAFs expressing a control shRNA or p38α shRNA. Values are mean + SEM from 3 independent experiments (n=3/group/experiment), **p< 0.01. (I) In vivo tumor growth of SKOV3ip1-Luc cells injected alone (n=6) or co-mixed with either primary CAFs expressing a control shRNA (n=8) or a p38α shRNA (n=8) and then injected subcutaneously into the flanks of female athymic nude mice. Bioluminescence imaging is shown over time. Comparisons were made using a repeated measure Two-way ANOVA (**p<0.01 for matching across groups) and Tukey’s multiple comparisons post-test was used to compare each group at day 21, **p< 0.01. (J) PAS staining of sub-cutaneous (SQ) tumors from experiments in Figure 5I. Left, quantification of percent of tumor area staining positive for PAS with background staining (distase insensitive areas, see methods) removed. Values are mean + SEM (n=6–8/group), *p< 0.05. Right, representative images of PAS staining in tumors from each group. Scale bar = 100µm.
Because glycogen mobilization was stimulated by both direct co-culture and CAF conditioned media and given that cytokine and chemokine production in senescent stromal fibroblasts is mediated through p38α activation (Alspach et al., 2014), we sought to determine whether cytokines and chemokines are regulated by p38α in OvCa cell stimulated CAFs. We compared the conditioned media from human-derived primary CAFs with conditioned media from CAFs that were previously stimulated by OvCa cell conditioned media with or without pretreatment with a p38α MAPK inhibitor (Figure 6A, schematic). The OvCa cell conditioned media induced CAFs to secrete IL-6, IL-8 and CXCL10, which were all repressed by the p38α MAPK inhibition (Figure 6A). CCL5 was produced at high levels following OvCa conditioned media stimulation, as previously reported by us (Mitra et al., 2012), but was not significantly reduced with p38α MAPK inhibition. Consistent with a dependence on the TGF-β1/p38α signaling axis, treatment with recombinant TGF-β1 or OvCa cell conditioned media stimulated upregulation of CXCL10, IL-6, and IL-8 mRNA levels, which was abrogated by knockdown of p38α using an siRNA in CAFs (Figure S5D). Moreover, the conditioned media from CAFs pretreated with OvCa cell conditioned media, as described in Figure 6A, abolished glycogen synthase expression in cancer cells (Figure 6B) and depleted cancer cells of their glycogen content (Figure 6C). These findings were paralleled by the stimulation of cancer cell migration (Figure 6D) and a marked increase in in vivo metastasis to the omentum (Figure 6E) by CAF conditioned media. The omentum, a large fat pad covering the intestines, is the primary site of metastasis for intra-abdominally metastasizing tumors, including serous ovarian, pancreatic, and colon cancer (Lengyel, 2010). The CAF-mediated increase in migration and metastasis was reversed when p38α MAPK was inhibited in the CAFs (Figure 6D, E) as shown in the schematic in Figure 6A. Consistent with these findings linking p38α MAPK regulation in CAFs and glycogenolysis in cancer cells, treatment with the glycogen phosphorylase inhibitor reduced CAF-stimulated metastasis (Figure 6F).
Figure 6. p38α MAPK activity in CAFs regulates cytokines that induce ovarian cancer glycogen release and stimulate metastasis.
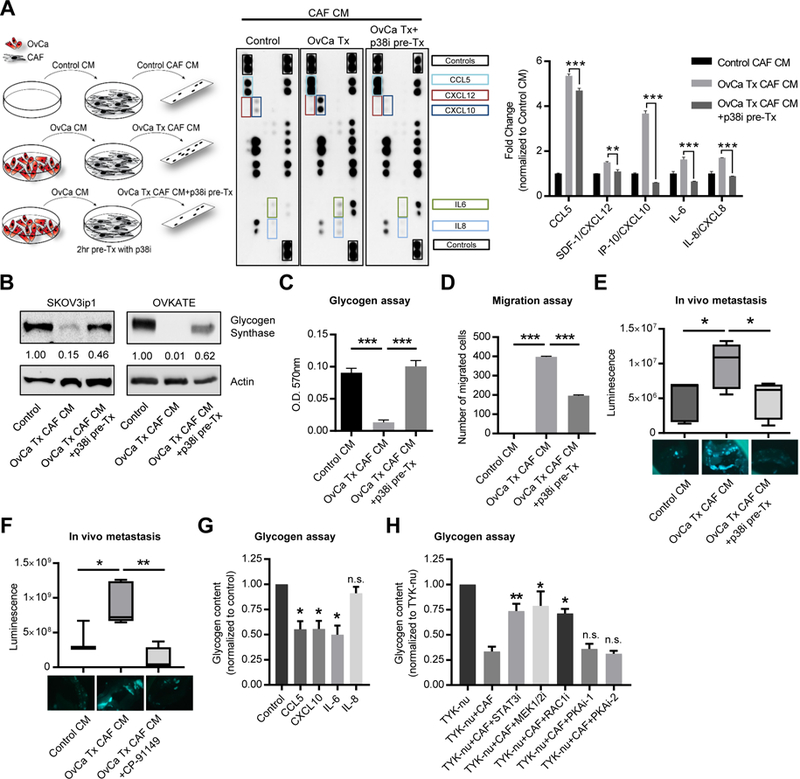
(A) Left, schematic of treatments for cytokine and chemokine array analysis (center) of conditioned media (CM) from CAFs stimulated with control CM (Control), or TYK-nu CM (OvCa Tx CM) either with or without pre-treatment with 2μM of the p38 M APK inhibitor PH-797804 (OvCa Tx+p38i pre-Tx). Following pre-stimulation, media was removed and replaced with serum-free media. CAF CM was collected 48 hr later. Right, Quantification of selected cytokine signals normalized to control CM. Values represent the average of the two technical replicates on the same membrane (**p< 0.01, **p< 0.001). Membrane images are representative images of two biological replicates with primary CAFs from two patients. (B) Western blot of glycogen synthase. SKOV3ip1 and OVKATE ovarian cancer cells were stimulated with CAF conditioned media (OvCa Tx CAF CM) or CM from CAFs pre-treated with the p38α inhibitor PH-797804 (OvCa Tx CAF CM +p38i pre-Tx). CM was generated as shown in Figure 6A and OvCa cells were incubated for 4 h. Values below glycogen synthase represent relative band intensity normalized to actin and fold change relative to the control sample. (C) Glycogen assay of SKOV3ip1 cells treated with CAF CM obtained as shown in the schematic in Figure 6A. ***p<0.001. Result is representative of 3 biological repeats. (D) Migration of SKOV3ip1 cells towards CAF CM generated as shown in Figure 6A. **p<0.01. Result is representative of 3 biological repeats. (E, F). Short term in vivo metastasis assay to the omentum. CM was generated as shown in the schematic in Figure 6A. Images below show GFP fluorescent SKOV3ip1 cells attached to the omentum in PBS. (E) SKOV3ip1-GFP-Luc OvCa cells were treated with Control CM, with CAF CM (OvCa Tx CAF CM), or CAF CM from CAFs pretreated with a p38 inhibitor (OvCa Tx CAF CM + p38 pre Tx). n=5/group. Luminescence was quantified by a luciferase assay on the isolated omentum. (F) SKOV3ip1-GFP-Luc OvCa cells were treated with Control CM (n=3), with CAF CM (OvCa Tx CAF CM) (n=5), or CAF CM plus a glycogen phosphorylase inhibitor (OvCa Tx CAF CM + CP-91149) (n=5). Luminescence was quantified by live animal imaging using the Xenogen IVIS Spectrum Imaging System after 24hr. Comparisons between control CM and OvCa Tx CAF CM + CP-91149 were not significant. *p< 0.05, **p<0.01. (G) Glycogen assay of TYK-nu cells stimulated with recombinant cytokines CCL5 (CXCL10 (50ng/ml), ng/ml), IL-6 (100ng/ml), or IL-8 (100ng/ml) after 6hr. Values are normalized to TYK-nu alone and represent 3 biological replicates. Values are mean + SEM (*p< 0.05). (H) Glycogen assay of TYK-nu cells with CAF co-culture and the indicated inhibitors (i): STAT3 inhibitor (Stattic, 10μM), MEK1/2 inhibitor (Tramet inib, 10μM), RAC1 inhibitor (NSC23766, 50μM), PKA inhibitor-1 (H-89, 10μM), or PKA inhibitor-2 (14–22 amide, 10μM) after 1hr. Cancer cells were pretreated with the compounds or vehicle control for 30 min and then co-cultured with CAFs for 1hr prior to the assay. Values are normalized to TYK-nu alone and represent 3–6 biological replicates. Values are mean + SEM (*p< 0.05, **p< 0.01).
In order to assess whether chemo/cytokine signaling could directly induce glycogen mobilization in the absence of CAFs, we stimulated cancer cells with recombinant chemo/cytokines that were upregulated in CAFs following exposure to cancer cell conditioned media. Stimulation of cancer cells with recombinant CCL5, CXCL10 and IL-6 induced glycogen mobilization, while IL-8 had no effect (Figure 6G). We next sought to test whether glycogen utilization could be prevented by blocking chemo/cytokine receptor downstream signaling. Despite the well-known role of protein kinase A (PKA) in the regulation of glycogenolysis through the activation of phosphorylase kinase (Roach et al., 2012), two different PKA inhibitors had no effect on CAF-mediated glycogen mobilization in cancer cells (Figure 6H). However, inhibition of the key effectors of cytokine signaling (STAT3, MEK1/2, RAC1) in cancer cells, blocked the utilization of glycogen stimulated by co-culture with CAFs (Figure 6H). These data suggest that glycogen mobilization in cancer cells is stimulated by chemo/cytokines secreted by CAFs following co-culture with cancer cells and that glycogen mobilization can be inhibited by blocking the downstream signaling of cytokine receptor.
Glycogen is stored in early human metastases
The in vitro and in vivo results collectively suggest a prominent role for glycogen in early metastasis. Therefore, we evaluated glycogen content in human epithelial high-grade serous ovarian cancers (HGSOC), comparing early microscopic metastatic foci (FIGO stage IIIA) with large metastatic nodules of at least 2 cm size (FIGO stage IIIC) from patients who underwent primary surgical debulking for high-grade serous ovarian cancer. All 11 early metastatic tumors stained positive for glycogen, while the advanced tumors (n=12), which were characterized by the transformation of the entire omentum by cancer with heavy fibroblast involvement, showed little to no glycogen staining (Figure 7A). Consistent with this finding, immunohistochemical p-p38α MAPK staining of human HGSOC omental tumors and normal omentum demonstrated that p38α was phosphorylated in human omental CAFs from invasive tumors, but not in fibroblasts in normal human omentum (Figure S6A). To answer the question of whether CAFs from other tumor sites within the abdominal cavity also modulate glycogen content, we isolated primary CAFs from peritoneal, endometrial, colorectal, and pancreatic cancers. CAFs from other primary tumor sites were able to reproduce the reduction in cancer cell glycogen content found with CAFs isolated from omental OvCa metastasis to varying extents (Figure S6B), suggesting that the phenomenon is not limited to omental-derived CAFs. Notably, normal omental fibroblasts and CAFs from primary endometrial cancers were unable to reduce glycogen to the same extent as CAFs from omental metastases (Figure S6B). Lastly, other tumors known to metastasize to the omentum (colon, gastric, pancreatic, serous endometrial cancer) were analyzed for glycogen content at the metastatic site. Cancer cells that were part of an early, small metastatic implant (Figure S6C) contained significantly more glycogen than tumor cells that were part of a large metastatic omental tumor with CAF infiltration (Figure 7B).
Figure 7. Glycogen utilization fuels early metastasis.
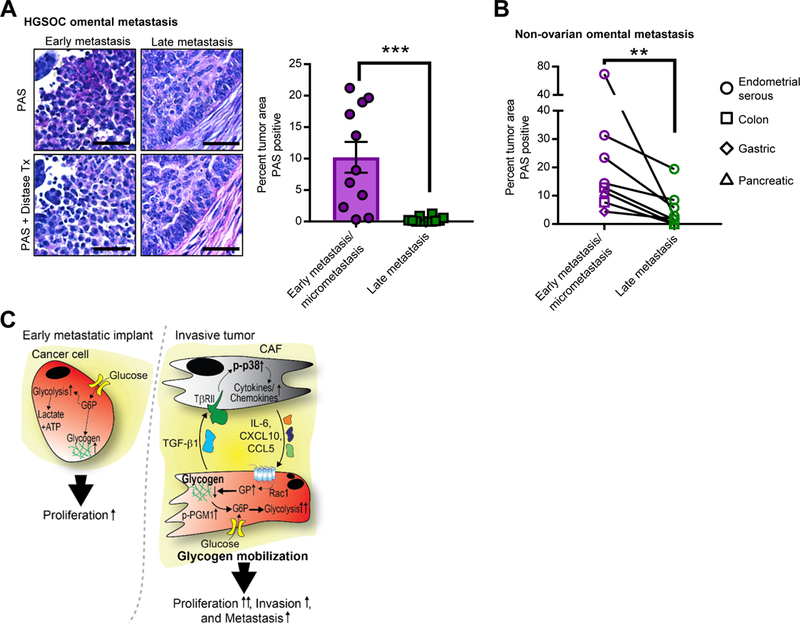
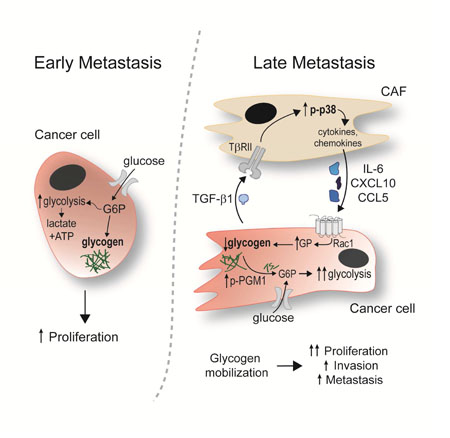
(A) PAS Staining of human OvCa tissue Left, representative images of PAS staining indicating glycogen storage (deep purple stain) in early, microscopic omental implants (FIGO IIIA disease) and late, advanced metastasis (FIG IIIC) of high grade serous OvCa. Distase treatment (Tx) was used to specifically digest glycogen from the tissue prior to PAS staining. PAS positive, distase sensitive areas of tumor were quantified in early implants (n=11) and invasive tumor (n=12). Scale bar = 50μm. Right, the average percent PAS positive tumor area from 5 fields. Comparisons were made using an unpaired, one-way, Mann Whitney test, ***p< 0.001. (B) Glycogen content in early and late metastasis in colon, gastric, pancreatic, and serous endometrial cancer. Quantification of PAS staining comparing paired samples of early metastasis/micrometastases to large metastatic lesions (late metastasis) in the omentum from the same patient (n=7). Samples quantified as in Fig. 7A. Comparisons were made using a Wilcoxon matched-pairs signed rank test, **p< 0.01. (C) Model of CAF-mediated glycogen hydrolysis in cancer cells. OvCa cells store glycogen when excess glucose is available. During early metastasis, as the tumor takes hold, stromal changes occur, including the generation and recruitment of CAFs through TGF-β1. The tumor cells and CAFs then engage in bidirectional signaling: p38α MAPK is activated in CAFs resulting in CAF production of chemokines and cytokines that initiate the mobilization of glycogen stores in cancer cells through activation of glycogen phosphorylase. This burst of additional energy allows the cancer cells to begin high energy tasks such as migration, invasion and further metastasis.
Discussion
Ovarian cancer is an aggressive tumor that metastasizes early to the adipocyte-rich omentum within the abdominal cavity (Lengyel, 2010). At this site, cancer cells use lipids released by adipocytes to fuel proliferation (Nieman et al., 2011). However, once the lipids supplied by the adipocytes are depleted, CAFs and tumor cells dominate the microenvironment. Using primary human CAFs isolated from over 100 serous ovarian cancer patients, we show that CAFs accelerate tumor metastasis by inducing changes in glycogen metabolism in cancer cells. Using quantitative phosphoproteomics, which allowed the simultaneous characterization of the bidirectional signaling between human primary CAFs and cancer cells, we discovered that co-culture leads to phosphorylation of p38α MAPK in CAFs and phosphorylation of a functionally unique site on PGM1 in cancer cells. This led us to investigate the role glycogen plays in the metabolism of ovarian tumor cells. We show that cancer cells produce TGF-β1, which activates p38-MAPK signaling in CAFs, and in response, the CAFs secrete cytokines and chemokines that trigger mobilization of glycogen in cancer cells in a glycogen phosphorylase dependent manner. The CAF-mediated glycogen mobilization then results in the enhanced proliferation, invasion, and metastasis of the cancer cells (Figure 7C).
Cellular glycogen storage is conserved evolutionarily in all organisms and has been observed in several types of cancer cells (Rousset et al., 1981), but little is known about the functional and metabolic consequences of its utilization in cancer. In times of hypoxia, glycogen stores in cancer cells increase and are available for energy production during nutrient deprivation (Favaro et al., 2012; Pelletier et al., 2012; Pescador et al., 2010). Hypoxic regions and a scarcity of nutrients characterize the tumor microenvironment of most solid tumors. However, the unique tumor microenvironment of the omentum is highly vascularized and is rich in nutrients including glucose and fatty acids. In our study, glycogen was produced under normoxic conditions, which is consistent with recent findings that yeast cells produce glycogen during aerobic glycolysis as a way to maintain glycolytic intermediates and ATP in homeostasis (Shulman and Rothman, 2015) and a study showing that dendritic cells use glycogen metabolism to modulate the immune response (Thwe et al., 2017).
Using 13C-labeled glucose to label glycogen, we were able to trace the fate of glycogen into glycolysis, which provides a boost of glycolytic metabolites to the cancer cell following stimulation by CAFs allowing for the enhanced proliferation and invasion of the cancer cells during early metastasis. Our study also demonstrated that, following co-culture with CAFs, glycogen led to the increased generation of citrate/isocitrate and α-ketoglutarate, central metabolites of the tricarboxylic acid (TCA) cycle, while other metabolites were unchanged, including fumarate and malate. No increase in the oxygen consumption rate was observed in cancer cells stimulated by CAFs, suggesting that the cancer cells may be using TCA cycle-derived metabolites to fuel other processes within the cell. In dendritic cells, glycogen-derived citrate can be shuttled to the cytosol for use in fatty acid, amino acid, and NADPH synthesis. Availability of these metabolic building blocks leads to ER and Golgi membrane expansion contributing to dendritic cell activation (Thwe et al., 2017). In cancer cells, these building blocks are likely to be essential for fueling the large increase in proliferation stimulated by CAF co-culture.
Paralleling an increase in PGM1 activity, the activity of glycogen phosphorylase, the first enzyme in glycogen degradation, was also increased upon co-culture with CAFs. The activity of glycogen phosphorylase is known to be directly regulated by phosphorylase kinase, which is activated by protein kinase A. However, our study shows that PKA inhibition had no effect on glycogen mobilization in cancer cells. Rather, inhibition of downstream signaling molecules (STAT3, MEK1/2, or RAC1) involved in cytokine signaling pathways were able to block the utilization of glycogen stimulated by CAFs. This finding is in line with a previous study, which identified RAC1 as a key mediator of glycogen phosphorylase activity following stimulation by IL-2 (Arrizabalaga et al., 2012). We identified several chemo/cytokines that were secreted by CAFs following stimulation by cancer cell conditioned media. Stimulation of cancer cells with recombinant CCL5, IL-6, or CXCL10 was able to induce glycogen mobilization. Several studies have established a role for CCL5 in the promotion of metastasis (; Mitra et al., 2012; Sicoli et al., 2014) and while CCL5 secretion was not p38-dependent in our study, CCL5 still plays a role in CAF-mediated glycogen mobilization. It is likely that the unique combination of chemo/cytokines produced by CAFs is required for full stimulation of glycogen mobilization and subsequent metastasis.
PGM1 is the second enzyme in the glycogen degradation pathway, responsible for the interconversion of glucose-1-phosphate to glucose-6-phosphate (Figure S2A). We identified a functionally novel phosphorylation site on PGM1 which, when mutated, blocked its enzyme activity and prevented glycogen mobilization stimulated by CAFs. Based on an analysis of the PGM1 crystal structure, tyrosine 353 is an accessible substrate for a kinase; however, a candidate kinase has not yet been identified. A previous study found that the kinase, PAK1, controls PGM1 activity through phosphorylation of a threonine residue (Gururaj et al., 2004), which suggests that the regulatory phosphorylation of PGM1 could be a common mechanism and explains how mitogenic signaling connects to glycogen metabolism and triggers the mobilization of energy in cancer cells.
We have previously shown that the initial homing of cancer cells to the omentum is mediated by visceral adipocytes through the secretion of cytokines (Nieman et al., 2011). Once the cancer cells colonize the omentum, they stimulate the release of lipids from adipocytes and use them as an energy source in β-oxidation. As the tumor cells begin to proliferate and invade, they remodel the microenvironment by activating resident fibroblasts, which eventually replace the adipocyte-rich stroma with CAFs (Nieman et al., 2013). Here, we show that the bi-directional signaling between cancer cells and stromal CAFs triggers a metabolic switch in the cancer cells, turning off glycogen synthesis and prompting glycogen utilization. Therefore, blocking glycogen mobilization in cancer cells with glycogen phosphorylase (Henke and Sparks, 2006; Rines et al., 2016; Ritterson Lew et al., 2015) or PGM1 inhibitors might be a therapeutic strategy for the reduction of tumor dissemination in abdominally metastasizing cancers (e.g. ovarian, gastric, pancreatic, and colon cancer) after optimal cytoreduction.
Limitations of Study
While the role of glycogen in cancer metastasis had not been investigated until our study, a few limitations of our work do exist. Much of the previous work on glycogen metabolism in cancer cells has focused on the influence of hypoxia. Our study relied primarily on in vitro culture of primary human fibroblasts with human cell lines under normoxic conditions. In vivo, a hypoxic microenvironment may alter the influence of fibroblasts on glycogen metabolism in cancer cells even more. The use of genetic mouse models would provide a more comprehensive analysis of the fate of glycogen in vivo. In addition, we found that glycogen metabolism was dependent on RAC1/STAT3 signaling in the cancer cells rather than the well described mechanism of PKA activation. Further investigation is required to understand the full signaling mechanism.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to the Lead Contact, Ernst Lengyel (elengyel@uchicago.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
Cultivation of primary human fibroblasts and cancer cells
Carcinoma-associated fibroblasts (CAF) were isolated from tumor samples as previously described (Kenny et al., 2007). Tumors were removed from patients undergoing primary surgery by a Gynecologic oncologist (SDY, EL) at the University of Chicago Hospital. Informed consent was obtained before surgery and the study was approved by the IRB of the University of Chicago. Unless otherwise designated, all tumors came from chemo naïve, omental tumors from female patients with high grade serous ovarian cancer (HGSOC). Isolated CAFs were cultured in DMEM (4.5g/L glucose) with 10% FBS and 1% penicillin/streptomycin. Primary CAFs at passages between 2 and 6 after isolation were used for experiments. To generate conditioned media, CAFs were seeded into 15cm dishes and allowed to reach 90% confluence. Media was replaced with DMEM (4.5g/L glucose) supplemented with pen/strep and 0.1% BSA and cells were incubated for 72hr. Media was collected, filtered with a 0.8μm filter to remove cell debris and then frozen at −80°C. Control CM was generated by incubating DMEM supplemented with pen/strep and 0.1% BSA for 72hr in a dish without cells. For all experiments, media underwent only one freeze/thaw. All experiments were repeated with at least 3 different preparations of primary CAFs from individual patients. All co-culture experiments were performed in DMEM media containing 4.5g/L glucose.
Primary human cancer cells were isolated from ascites. Briefly, spheroids larger than 40 μm were collected, digested with trypsin for 10 min, and seeded directly for the assay. Cancer cells were characterized by the absence of α-smooth muscle actin and the presence of PAX-8 and cytokeratin.
SKOV3ip1, HeyA8, ES2, MDA-MB-231, and HEK293T were cultured in DMEM with 10% FBS and 1% penicillin/streptomycin. RKO, TYK-nu, OVKATE, Caco-2 were cultured in Eagle’s MEM with 10% FBS and 1% penicillin/streptomycin. SNU-119 was cultured in RPMI-1640 and PC-3 in F12K medium supplemented with 10% FBS and 1% penicillin/streptomycin. All cell lines were cultured in an incubator at 37°C with 5% CO2.
Mouse models
For the subcutaneous model, 2×105 primary CAFs expressing a control shRNA (n=8) or a p38α shRNA (n=8) were co-mixed with 1×105 SKOV3ip1-Luc cells and then injected subcutaneously into the flanks of 6 week old female athymic nude mice (Harlan; Hsd: Athymic Nude-Foxn1nu). SKOV3ip1-Luc cells were injected alone as a control (n=6). Tumor growth was imaged over time every week starting on day 4 for 21 days using the Xenogen IVIS Spectrum Imaging System (Caliper Life Sciences, Hopkinton, MA) and tumor size was quantified using Living Image software (Caliper Life Sciences).
For the in vivo metastasis assay with conditioned media, 5×105 SKOV3ip1-Luc-GFP cells were injected intraperitoneally in 6 weeks old athymic nude female mice (n=5/group) in 500 µl of conditione d media from 3 different conditions as described in Fig. 6A. 24 hr later mice were sacrificed and the omentum was removed. Fluorescent images were taken of the omentum in PBS prior to incubation for 1hr in Passive Lysis Buffer (Promega). Lysates from the omentum were assayed for luciferase activity using LAR II substrate (Promega) and quantified using a luminometer. Alternatively, cells were treated with 30µM CP-91149 for 1hr in 10% FBS, DMEM, then t rypsinized and cells resuspended in indicated media followed by injection intraperitoneally in nude mice. 24hr following injection mice were imaged using IVIS Spectrum Imaging System (Caliper Life Sciences, Hopkinton, MA) and tumor size was quantified using Living Image software (Caliper Life Sciences). After 48 hr, mice were sacrificed and the omentum was removed. Fluorescent images were taken of the omentum in PBS.
METHOD DETAILS
Kinetic cell monitoring
96-well black wall plates (NUNC) were coated with a mixture of 2.5 μg/μl fibronectin and 2.5 μg/μl collagen in PBS for 30 minutes at 37 °C. After one wash with PBS, Oris Cell Seeding Stoppers (Platypus Technologies, Fitchburg, WI) were placed into the wells. CAFs were then seeded around the plugs. 24 hr later, GFP expressing cancer cells were plated on top of the CAFs. After an overnight incubation the Oris Cell Seeding Stoppers were carefully removed from the wells leaving an empty space in the middle of the well. Growth-factor reduced Matrigel was mixed 1:1 with serum-free, phenol-free media and was placed on top of the cells. The gel was allowed to harden for 30 minutes at 37°C and then media containing FBS was placed on top of the gel. Plates were placed in the IncuCyte® ZOOM (Essen BioScience, Ann Arbor, MI) and imaged every 4 hr for 72 hr. Invasion of cancer cells into the center of the well was tracked using GFP fluorescence. Images at time zero were used as a baseline to calculate the change in percent confluence in the center of the well. For siRNA studies, cells were transfected with 30nM siGENOME SMARTpool siRNA (Dharmacon) 48 hr prior to the beginning of the invasion assay. Samples were analyzed with 6 technical replicates. Data is representative of 2 independent experiments.
SILAC labeling and co-culture
SKOV3ip1 cells were grown for 8 passages in SILAC DMEM (4.5g/L glucose) (Thermo Scientific) supplemented with ‘heavy’ arginine and lysine (C13N15) (Cambridge Isotope Laboratories, Tewksbury, MA) and 10% dialyzed FBS (Gibco). Labeling efficiency was evaluated by analyzing peptides from labeled and unlabeled lysates of SKOV3ip1 cells. Overall, ~98% metabolic incorporation of heavy arginine and lysine was achieved.
Primary CAFs isolated from an omental metastasis were immortalized using hTERT as previously described (Orimo et al., 2005). CAFs were seeded in regular growth media with ‘light’ arginine and lysine (C12N14) at 2.5e6/15cm dish x 2 dishes/replicate. After 24 hr, ‘heavy’ labeled SKOV3ip1 cells were collected by trypsinization and resuspended in SILAC DMEM lacking Arg/Lys with 0.1% BSA. CAFs were washed one time with SILAC DMEM lacking Arg/Lys. SKOV3ip1
(7.5e6/dish ×2 dishes/replicate) cells were then plated with CAFs or on plastic and cultured for 4 hr. CAFs alone were left in SILAC DMEM lacking Arg/Lys for 4 hr. Samples were analyzed with 5 technical replicates/group. One biological replicate was analyzed by phosphoproteomics.
Cell lysis, protein reduction, alkylation, digestion, and peptide immunoprecipitation
To halt the culture, media was removed from the cells and lysis buffer (9 M urea, 1 mM sodium orthovanadate, 20 mM HEPES, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, pH 8.0) was added to the cells. Lysates were prepared from the co-culture of CAF and SKOV3ip1 for 4 hours or the control cells with lysates of CAF alone and SKOV3ip1 alone mixed together. Lysates were sonicated at a 30 watt output with 2 bursts of 30 seconds each and cleared at 20,000×g for 15 minutes at 4°C. Lysates were reduced with 45 mM DTT for 20 minutes at 60°C, and alkylated with 100 mM iodoacetamide for 15 minutes at room temperature in the dark. Lysates were then diluted 4-fold with 20 mM HEPES buffer, pH 8.0 and digested with TPCK-treated trypsin (Worthington, Lakewood, NJ) in a 1:1 (w/w) trypsin:protein ratio overnight at room temperature. Tryptic peptides were acidified to pH 2.0 with 20% trifluoroacetic acid (TFA), cleared at 1,800×g for 5 minutes at room temperature, and desalted using C18 Sep-Pak plus cartridges (Waters, Milford, MA). Eluents containing peptides were lyophilized for 48 hours to dryness. Peptide immunoprecipitation was performed using p-Tyr-100 phosphotyrosine antibody beads (Cell Signaling Technology) as previously described (Helou et al., 2013).
Automated nano-LC/MS
Phosphopeptides were eluted into a Linear Trap Quadropole (LTQ) Orbitrap Velos mass spectrometer (Thermo Fisher Scientific) through a PicoFrit analytical column (360 µm outer diameter 75 μm inner diameter-fused silica with 12 cm of 3-μM Monitor C18 particles; New Objective, Woburn, MA) with a reversed-phase gradient (0–70% 0.1 M ace tic acid in acetonitrile in 90 minutes). Spectra were collected in positive ion mode and in cycles of one full MS scan in the Orbitrap (m/z 400–1800). This was followed by data-dependent tandem mass spectrometry (MS/MS) scans in the LTQ Velos, sequentially, of the ten most abundant ions in each MS scan with charge state screening for +1, +2, +3 ions and dynamic exclusion time of 30 seconds. The automatic gain control was 1,000,000 for the Orbitrap scan and 10,000 for the LTQ scans. The maximum ion time was 100 milliseconds for the LTQ scan and 500 milliseconds for the Orbitrap full scan. Orbitrap resolution was set at 60,000.
Database analysis
MS/MS spectra were searched against the human UNIPROT non-redundant “complete proteome set” protein database using Mascot (Matrix Science) (Perkins et al., 1999). Peak lists were generated using extract_msn.exe (1/10/11) using a mass range of 600–4500. The UNIPROT human database contained 88,832 forward and an equal number of reverse protein sequence entries. The Mascot database search was performed with the following parameters: trypsin enzyme specificity, 2 possible missed cleavages, 7 ppm mass tolerance for precursor ions, and 0.5 Da mass tolerance for fragment ions. Search parameters specified a differential modification of phosphorylation (+79.9663 Da) on serine, threonine, and tyrosine residues and methionine oxidation (+15.9949 Da) as well as a static modification of carbamidomethylation (+57.0215 Da) on cysteine. Search parameters also included a differential modification for arginine (+10.00827 Da) and lysine (+8.01420 Da) amino acids. To provide high confidence phosphopeptide sequence assignments, Mascot results were first filtered by Mowse score (>20) and precursor mass error (< 2 ppm), and the resulting peptide assignments were filtered down to 1% false discovery rate (FDR) by a logistic spectral score (Yu et al., 2009). FDR was estimated with the decoy database approach after final assembly of non-redundant data into heatmaps (Elias and Gygi, 2007). To validate the position of the phosphorylation sites, the Ascore algorithm (Beausoleil et al., 2006) was applied to all data, and the reported phosphorylation site position reflected the top Ascore prediction. The mass spectrometry proteomics data sets have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomeexchange.org) via the PRIDE partner repository with the dataset identifier PXD003574.
Quantitation of relative phosphopeptide abundance
Relative quantitation of phosphopeptide abundance was performed via calculation of select ion chromatogram (SIC) peak areas for heavy and light SILAC-labeled phosphopeptides identified from 5 biological replicate experiments. Peak areas were calculated by inspection of SICs using software programmed in Microsoft Visual Basic 6.0 based on Xcalibur Development kit 2.0 SR2 (Thermo Fisher Scientific). Quantitative data was calculated automatically for every assigned phosphopeptide using the ICIS algorithm available in the Xcalibur XDK. Retention time alignment of individual replicate analyses was performed as described previously (Demirkan et al., 2011). SIC peak areas were determined for every phosphopeptide that was identified by MSMS. In the case of a missing MSMS for a particular peptide, in a particular replicate, peak areas were calculated according to the peptide’s isolated mass and the retention time calculated from retention time alignment. A minimum SIC peak area equivalent to the typical spectral noise level of 300 was required of all data reported for label-free quantitation.
For label free quantitation of phosphopeptide abundance, individual SICs were normalized to an exogenously spiked standard phosphopeptide LIEDAEpYTAK peak area. The LIEDAEpYTAK phosphopeptide was added at the same amount in each cell condition and replicate sample and accompanied cellular phosphopeptides through peptide immunoprecipitation and reversed-phase elution into the mass spectrometer. A q-value is defined as the measure of the minimum FDR at which a test can be called significant (Storey, 2002). For each time point, q values for multiple hypothesis tests were calculated based on the determined p-values using the R package QVALUE as previously described (Storey, 2003; Storey and Tibshirani, 2003). The www.Phosphosite.org website was used to search for PGM1 phosphorylation sites.
2-NBDG labeling to visualize glycogen
Cancer cells were seeded in 24-well plates and primary CAFs were seeded on poly-D-lysine coated cover glass. The next day media was removed from cancer cells and replaced with glucose-free DMEM (Life Technologies). Glycogen was visualized using a previously described method with modifications (Louzao et al., 2008). After 30 min 2-NBDG (Caymen Chemical) was added at 500 μM. Cells were incubated for 2 hr with 2-NBDG, then all media was removed and the cells were collected via trypsinization. CAFs were washed one time with serum-free DMEM and then the 2-NBDG labeled cancer cells were plated with CAFs or alone on poly-D-lysine coated coverslips. After 4 hours the live cells were imaged using a Zeiss 510 confocal microscope. Images are representative of 2 technical replicates from 3 independent experiments.
Transmission electron microscopy
SKOV3ip1 cells in co-culture with CAFs or on plastic were fixed in 2% glutaraldehyde and 4% paraformaldehyde in 0.1 M sodium cacodylate buffer for 5 min. Cells were then collected and pelleted to be processed for electron microscopy. The samples were postfixed in osmium tetroxide, dehydrated in ethanol, treated with propylene oxide, and embedded in Spurr’s epoxy resin. 90 nm sections were stained with uranyl acetate and lead citrate prior to examination under 300KV using a FEI Tecnai F30 electron microscope equipped with a Gatan CCD digital camera. Images were taken at 4060× and 8260× magnification (Favaro et al., 2012; Nieman et al., 2011). Three different fields were acquired from each sample. Representative images are shown.
Periodic acid Schiff (PAS) staining
For cell lines, after 4 hours of co-culture, cells were fixed with 4% paraformaldehyde for 10 min. Representative data from 3 independent experiments is shown. For tumor tissue, slides were deparaffinized and rehydrated to deionized water. Adjacent sections of tumor were treated with a 0.01 g/ml amylase (Sigma-Aldrich) solution for 15 min at 37°C to serve as a negative control for glycogen staining. Samples were then incubated in 1% periodic acid (Sigma-Aldrich) for 5 min, rinsed in water, and placed in Schiff’s reagent (Sigma-Aldrich) for 15 min. Finally, samples were washed under running water for 5 min, counterstained with Hematoxylin solution and allowed to air-dry. Representative images are shown. Tumors were identified as either early or late metastatic by a gynecologic pathologist (AM, RL). To quantify the percent of the tumor that was PAS positive, 5 images were taken of the PAS stained slides along with identical fields of view with distase staining. PAS positivity, which was removed by distase treatment was classified as glycogen. ImageJ was use to quantify the full area of the tumor and the area which was PAS positive for each field.
Glycogen content assay
Glycogen content was measured using either a Glycogen Assay Kit (BioVision, Milpitas, CA) or using the anthrone reagent as previously described with slight modifications for a multi-well plate (Schnier et al., 2003). The assays were performed with three experimental variations. GFP-labeled
cancer cells were co-cultured with primary CAFs for 4 hours, and then sorted via FACS to remove the CAFs prior to the assay. Alternatively, cancer cells were cultured in control or CAF CM for 4 hours before being collected for the assay. In order to test many fibroblasts simultaneously, we plated CAFs beneath a 6-well transwell insert with a 0.4μm poly ester membrane (Corning). TYK-nu cells were plated on the top of the inserts in serum-free media. After 6hr, media was removed, cells were washed 1× with PBS prior to being collected for the assay. In the Glycogen Assay Kit glucoamylase hydrolyzes glycogen to glucose, which is oxidized. The product reacts with OxiRed and generates color (570 nm).
In the anthrone method, cells were collected in 0.2 ml of 30% KOH and incubated for 15 min at 95 °C to extract glycogen. Afterward, 50μl of the s upernatant was mixed with 25μl 2% NaSO4 in a microwell of a 96-well plate. Glycogen was precipitated by addition of 150μl ethanol. Following centrifugation, the precipitate was resuspended in 200μl H2O/H2SO4 (3:7.6 ratio) containing 0.15% anthrone and heated for 15 min at 95 °C. Glycogen content was determined by measuring absorbance at 620 nm and the amount of glycogen was calculated using a standard curve of commercial glycogen. Glycogen content was normalized to protein content. Experiments were done with 3–5 technical replicates.
PGM1 mutagenesis
A lentiviral plasmid was generated containing full-length PGM1 by recombining the entry vector, pDONR221-PGM1, with the destination vector pLX304 using the Gateway LR Clonase II Enzyme (Life Technologies). To generate the Y353F mutant PGM1, the following primers were used to produce a phenylalanine substitution for tyrosine 353 with the Q5 Site-Directed Mutagenesis Kit (New England Biolabs, Ipswich, MA): Fwd – GATTGCTTTGttcG AGACCCCAAC, Rev – TTTGTAGCACTAGCCACC. Plasmid sequences were validated using Sanger sequencing. To generate stable cell lines expressing wild-type or mutant PGM1, cancer cells were infected with virus generated by the lentiviral vectors pLX304-PGM1 or pLX304-PGM1-Y353F respectively.
Phosphoglucomutase activity assay
PGM1 activity was measured using the Phosphoglucomutase Colorimetric Assay Kit (BioVision, Milpitas, CA). The principle of the assay is that PGM1 in the lysate converts glucose-1-phosphate to glucose-6-phosphate, which is then oxidized by glucose-6-phosphate dehydrogenase to form NADH, which reduces a probe that can be detected using a fluorometer at 450 nm (Figure S2F). Briefly, lysates from pLX304-PGM1 WT or Y353F mutant cell lines were collected in RIPA buffer. Anti-V5 antibody was pre-conjugated to Protein A/G PLUS-Agarose beads (Santa Cruz, Dallas, TX) for 1 hr at 4°C. 500 μg of total protein was pre-cleared using 20 μl of Protein A/G PLUS-Agarose then incubated with the anti-V5-conjugated beads for 2 hr at 4°C. Beads were washed extensively, resuspended in PGM1 assay buffer and then processed according to the manufacturer’s protocol. Samples were analyzed with 3 technical replicates. Data is representative of 2 independent experiments for each cell line.
For co-culture with CAFs, CAFs were plated beneath a 6-well transwell insert with a 0.4µm polyester membrane (Corning). TYK-nu cells were plated on the top of the inserts in serum-free media. After 3hr, media was removed from the cells and the TYK-nu cells were collected for the PGM1 activity assay in 200μl TES Buffer (20 mM Tris, pH 7.4, 1 mM EDTA, 225 mM sucrose). PGM1 activity was assayed in the direction of glycogenolysis (Figure S2A) using a previously described protocol (Bae et al., 2014). Briefly, samples were sonicated and cleared by centrifugation (13,500 rpm, 4C). Then 20μg total protein was mixed with assay buffer (50 mM K2H2PO4, pH 7.5, 10 mM MgCl2, 5 mM EDTA pH containing 0.5 mM NADP+, 1.5 units/ml glucose-6-phosphate dehydrogenase, 5mM Glucose-1-phosphate, 0.02mM glucose 1,6-bisphosphate to a total volume of 150μl. Reagents were from Sigma. Absorbance at 340 nm was measured kinetically to assess the amount of NADPH produced using a SpectraMax i3 (Molecular Devices, Sunnyvale, CA).
Cellular thermal shift assay (CETSA)
The assay was performed as previously described (Jafari et al., 2014). Briefly, cells were collected in PBS and exposed to increasing temperatures. Following lysis, protein aggregates were removed by centrifugation and the supernatant was loaded on an SDS-PAGE gel and immunoblotting for the V5 tagged PGM1 was performed. The percentage of non-denatured protein was quantified by measuring the relative intensities of the protein.
Glycogen phosphorylase activity assay
CAFs were plated beneath a 6-well transwell insert with a 0.4μm polyester membrane (Corning). TYK-nu cells were plated on the top of the inserts in serum-free media. After 3hr, media was removed from the cells and the TYK-nu cells were collected for the glycogen phosphorylase (GP) activity assay in 200μl TES Buffer (20 mM Tris, pH 7.4, 1 mM EDTA, 225 mM sucrose). The GP activity assay was performed in the direction of glycogenolysis as previously described (Arrizabalaga et al., 2012). Briefly, samples were sonicated and cleared by centrifugation (13,500 rpm, 4°C). Then 100μg total protein in 100μl was mixed with 100μl assay buffer (50 mM K2H2 PO4, pH 7.5, 10 mM MgCl2, 5 mM EDTA pH containing 0.5 mM NADP+, 1.5 units/ml glucose-6-phosphate dehydrogenase, 1 unit/ml phosphoglucomutase, 0.1 mg/ml glycogen) for a total volume of 200μl. Reagents were from Sigma. Absorbance at 340 nm was measured kinetically to assess the amount of NADPH produced using a SpectraMax i3 (Molecular Devices, Sunnyvale, CA).
Bioenergetic assays: Glycolysis and oxidative phosphorylation
Glycolysis and oxidative phosphorylation were measured with the Seahorse Extracellular Flux XF-96) analyzer (Seahorse Bioscience, North Billerica, MA) (Ladanyi et al., 2018). Cells were seeded in Seahorse XF-96 plates at a density of 10,000 cells per well and cultured for 24hr. The next day media was replaced with either control or CAF conditioned media. For glycolysis, pretreatment with CP-91149 was done at 20 μM where indicated. After 24 hr, cel ls were changed to unbuffered DMEM in the absence of glucose. Sequential injections were performed with D-glucose (10 mM), oligomycin (1 μM), and 2-deoxyglucose (100 mmol/L). The extracellular acidification rates (ECAR) after the injection of D-glucose was a measure of glycolysis, and the ECAR after the injection of oligomycin represented maximal glycolytic capacity. Non-glycolytic activity was quantified by the measure of ECAR after the injection of 2-deoxyglucose. Samples were analyzed with 10 technical replicates. Data is representative of 4 independent experiments.
For oxidative phosphorylation, 24 hr after cell seeding, media was changed to unbuffered DMEM containing 2 mM glutamine, 1 mM pyruvate, and 10 mM glucose. Sequential injections were performed with oligomycin (2 μM), FCCP (1 μM), and antimycin/rotenone (1 μM). The oxygen consumption rate (OCR) after the injection of oligomycin was a measure of ATP-linked respiration and the OCR after the injection of FCCP represented maximal respiratory capacity. Basal respiration was quantified by the measure of OCR prior to the injection of oligomycin. Samples were analyzed with 10 technical replicates. Data is representative of 2 independent experiments.
ATP/ADP ratio assay
The ratio of ATP to ADP was analyzed in TYK-nu and SKOV3ip1 cells exposed to control or CAF CM for 16hr. Cells were pretreated with CP-91149 for 1hr prior to stimulation with CM. The ratio was determined using the ATP/ADP ratio kit (Sigma) following the manufacturers protocol. Luminescence was detected using the SpectraMax i3 (Molecular Devices, Sunnyvale, CA). Samples were analyzed with 4 technical replicates and the experiment was performed 3 times with CM from independent patients.
Glucose-1-phosphate assay
To quantify G-1-P in the cell extract, we used an enzymatic assay with minor modifications (Zhu et al., 2011). Briefly, cell pellets were resuspended in 200 μl cold PBS and sonicated. The same volume of perchloric acid (6%) was added and 2.5 N K2CO3 was added dropwise to neutralize the reaction. The sample was cleared by centrifugation at 13,000 × g for 10 min. Ten microliter of supernatant was mixed with 90 μl of the complete assay mixture containing 130 mM glycine (pH7.4), 5 mM MgCl2, 600 mM NADP, 10 mM Resazurin, 1 U/ml G-6-P dehydrogenase, 0.2 U/ml of diaphorase and incubated for 10 min at room temperature. Fluorescence at 590 nm was measured using excitation at 530 nm using a SpectraMax i3 (Molecular Devices, Sunnyvale, CA). Background fluorescence was corrected by subtracting the value of the buffer control from all sample readings. For quantification of G-1-P, 4U/ml of recombinant PGM1 was added to convert G-1-P to G-6-P followed by quantifying total G-6-P. G-1-P amount was calculated by the subtraction of the G-6-P amount resulting from the reaction without exogenous PGM1.
Glucose uptake assay
TYK-nu cells and CAFs were seeded either alone or in a co-culture in a 8-well glass chamber slide. After incubation overnight, media was changed to glucose-free DMEM with 500µM 2-NBDG. Cells were incubated for 30min, media was removed, and cells were washed 2 times with phenol-free DMEM. Glucose uptake was assessed by confocal microscopy.
Lactate assay
TYK-nu cells and CAFs were seeded in a 24-well plate and incubated for 24hr. Media was changed and replaced with control CM, TYK-nu CM, or CAF CM. Cells were then incubated for 24hr, media was collected and lactate was measured using the EnzyChrom L-Lactate Assay Kit (BioAssay Systems) according to the manufacturer’s instructions. Baseline values of lactate in the CM were subtracted from the TYK-nu and CAF stimulated lactate concentrations and values were normalized to cell number.
Proliferation assay
Primary CAFs were seeded in 96-well plates and incubated for 24 hr. Then GFP-labeled SKOV3ip1, HeyA8 cells (Kenny et al., 2014) or a CMFDA labeled primary ovarian cancer cell clone were added to the wells. Cancer cells were pretreated with inhibitors as indicated and media was changed to 5% FBS containing media. Alternatively, SKOV3ip1 and TYK-nu cells were plated in 96-well plates and cultured with control or CAF CM with or without 1 hr pre-treatment of 30µM CP-91149. Proliferation of the cancer cells over three - seven days was tracked by imaging the wells with a SpectraMax i3 (Molecular Devices, Sunnyvale, CA) every 24hr. Samples were analyzed with 6 technical replicates. Data is representative of 2 independent experiments for each cell line.
Invasion assay
Boyden chamber invasion assays were performed as previously described (Zillhardt et al., 2011) with a few modifications. Control or CAF CM was placed on the bottom of the transwell inserts and cancer cells TYK-nu were plated on top of the insert in serum-free media with or without 30μM CP-91149. After 48hr, the cells were fixed, wiped from the top of the insert and stained by crystal violet. Crystal violet was extracted with 10% acetic acid and the number of invaded cells was quantified by measuring absorbance at 570nm and fitting the value to a standard curve of known cell numbers. Alternatively, SKOV3ip1-GFP cells were quantified in five 100X magnification fields using CellProfiler and averaged for each well by fluorescent microscopy on a Zeiss AxioObserver A.1 microscope equipped with a digital camera. Samples were run in triplicate and the experiment was performed 3 times with CM from independent patients.
Metabolomics
TYK-nu cells were stimulated with control CM (n=3) or CAF CM (n=3) for 6 hr then metabolites were extracted using 80% methanol/water and analyzed on a liquid chromatography-Q Exactive plus mass spectrometer (Thermo Scientific). Metabolite peak area (intensity) was used to represent relative metabolite level. A detailed method description was published previously (Liu et al., 2014).
ROS detection
ROS levels were measured using CellRox Deep Red Reagent (Molecular Probes) as an indicator of intracellular ROS. For microscopic analysis, GFP labeled SKOV3ip1 cells were cultured with CAFs or alone on poly-D-lysine coated cover glass for 4 hr. Cells were then loaded with 5 μM CellRox Deep Red for 30 min, washed with PBS and imaged live using a Zeiss 510 confocal microscope. Quantification from 3 independent experiments is shown. Representative images are shown.
U-13C-glycogen labeling
TYK-nu cells were cultured in glucose-free, DMEM (Thermo Fisher Scientific) supplemented with 4.5g/L U- 13C-glucose (Cambridge Isotope Laboratories, Andover, MA) and 10% FBS for 24 or 48 hrs. The cells were then washed two times with glucose-free DMEM and the media was replaced with DMEM containing 4.5g/L 12C-glucose (Corning) with 10% FBS and cultured for 1hr to label glycolytic and pentose phosphate pathway intermediates with 12C, leaving glycogen as the only 13C source for glycolysis. Cells were collected and homogenized in 250μl of ice-cold water. Samples were boiled for 15min and cleared by centrifugation for 10 min at 16,000 rcf. The supernatant was split in half, with half treated with Glycogen Hydrolysis Enzyme (Caymen Chemical, Ann Arbor, MI) in 50 mM ammonium acetate buffer, pH 4.5 for 30 min. After 30 min, the enzymatic reaction was quenched by adding 4 volumes of ice-cold methanol and vortexing for 30 seconds. Samples were dried in a speed vacuum (Thermo Fisher Scientific) and analyzed on a liquid chromatography-Q Exactive plus mass spectrometer (Thermo Scientific). The peak area of 12C6-glucose and 13C6-glucose was measured in samples that were treated with or without the hydrolysis enzyme. To calculate the percent of U-13C-glycogen labeling, the amount of 12C-glucose in samples without hydrolysis was subtracted from the amount of 12C-glucose in samples treated with the hydrolysis enzyme and then compared to the amount of detected 13C-glucose in the hydrolyzed samples.
U-13C-glycogen tracing assay
Primary CAFs were plated on the bottom surface of a transwell insert with a polyester membrane and 0.4 μm pores (Corning). After 16hr, inserts with CAFs were turned over, placed in DMEM containing 4.5g/L 12C-glucose (Corning) with 10% FBS, and cultured for 48hr. TYK-nu cells were cultured in glucose-free, DMEM (Thermo Fisher Scientific) supplemented with U-13C-glucose (Cambridge Isotope Laboratories, Andover, MA) and 10% FBS for 48hr. TYK-nu cells were then washed two times with glucose-free DMEM and the media was replaced with DMEM containing 4.5g/L 12C-glucose (Corning) with 10% FBS and cultured for 1hr to label glycolytic and pentose phosphate pathway intermediates with 12C, leaving glycogen as the only 13C source for glycolysis. Media was removed from CAFs and after two washes with glucose-free DMEM, the media was replaced with 3ml glucose-free DMEM. TYK-nu cells containing U-13C-glycogen were trypsinized, washed two times with glucose-free DMEM and then plated in serum-free/glucose-free DMEM on the top surface of the transwell insert either with or without CAFs on the bottom surface. After 30min of culture, cancer cell metabolites were extracted using 80% methanol/water from the top surface of the membrane and analyzed on a liquid chromatography-Q Exactive plus mass spectrometer (Thermo Scientific). Metabolite peak area (intensity) was used to represent relative metabolite level. A detailed method description was published previously (Liu et al., 2014).
Cytokine and Chemokine array
Primary CAFs were pretreated with the p38 MAPK inhibitor PH-797804 (Selleck Chemicals, Houston, TX) at 2 μM for 1 hr and then stimulated with conditioned media from TYK-nu cells or control media. After 4 hr, media and drug was removed and fresh serum-free DMEM with 0.1% BSA was placed on cells. Cells were cultured for 48 hr then conditioned media from the CAFs was collected. The Human Cytokine Array, Panel A (R&D Systems, Minneapolis, MN) was used to analyze 42 different cytokines and chemokines in each sample according to the manufacturer’s recommendations. Expression levels were analyzed using Gilles Carpentier’s Protein Array Analyzer macro (available online: http://rsb.info.nih.gov/ij/macros/toolsets/Protein Array Analyzer.txt with ImageJ software. The experiment was performed with two different primary CAF cultures.
Migration assay
Migration was performed as previously described (Kenny et al., 2008). Briefly, 30,000 SKOV3ip1 cells were plated in boyden chambers with conditioned media from 3 different conditions in the bottom well as described in Fig. 6A. Five technical replicates were used per condition. Data is representative of 2 biological replicates.
Immunohistochemistry
Slides from human ovarian tumors were deparaffinized and re-hydrated. After washing with water for 5 min, endogenous peroxidase activity was blocked for 15 min at RT in peroxidase blocking buffer. Thereafter slides were washed in tap water and antigen unmasking was performed. After antigen retrieval, slides were washed with PBS and unspecific background was blocked with blocking buffer (10% normal goat serum and 0.3% Triton x-100 in PBS). Slides were stained with phospho-p38 MAPK antibody (1:50, overnight 4°C) from Cell Signaling Technology (#4631). After washing, HRP conjugated secondary antibody (anti-rabbit IgG ImmunoLogic DPVR110HRP; 45 min at RT) was added to the samples and then signals were visualized with DAB (3,3-diaminobenzidine), using hematoxylin as a counterstain. Tissue was mounted with DPX mounting medium after washing with PBS and dehydrated in a descending series of ethanol solutions, cleared in xylol.
Phospho-p38 immunohistochemistry quantification
Quantification and localization of nuclear phospho-P38 protein expression was performed using Aperio ImageScope and Spectrum software on 50 samples of human omental tumor from patients with HGSOC. Patients were selected by a gynecologic pathologist. The intensity and % positive nuclei of DAB staining was determined in the stroma specific regions of the tumor cores using the nuclear localization algorithm. Results were confirmed by the pathologist.
Ten samples of omentum from patients undergoing surgery for benign conditions were also analyzed by a pathologist for regions of fibroblasts.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analyses were performed using GraphPad Prism (GraphPad, La Jolla, CA). Sample sizes were determined based on previous experience with the individual experiment except for animal studies where power calculations were used. The mean and the standard error of the mean (s.e.m) indicating variance are reported for all graphs. For experiments making one comparison, data was analyzed using a two-tailed Mann Whitney U test to account for non-normal distribution of the data. For experiments with more than one comparison, One-Way ANOVA with Tukey’s multiple comparisons post-test was used. Before applying ANOVA, we first tested whether the variation was similar among the groups using the Bartlett’s test. Where the standard deviations were significantly different, a log2 transformation was applied to the data before analysis. Differences were considered significant if p<0.05.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Total glycogen synthase | Cell Signaling Technology |
Cat#3886; RRID:AB_2116392 |
| p-p38 (thr180/tyr182) | Cell Signaling Technology |
Cat#9215; RRID:AB_331762 |
| p-p38 (thr180/tyr182) | Cell Signaling Technology |
Cat#4631; RRID:AB_331765 |
| Total p38 | Cell Signaling Technology |
Cat#9212; RRID:AB_330713 |
| p-MAPKAPK-2 (thr334) | Cell Signaling Technology |
Cat#3007; RRID:AB_490936 |
| anti-rabbit IgG-HRP | Cell Signaling Technology |
Cat#7074; RRID:AB_2099233 |
| anti-mouse IgG-HRP | Cell Signaling Technology |
Cat#7076; RRID:AB_330924 |
| Actin | Sigma-Aldrich | Cat#A5441; RRID:AB_476744 |
| V5 | Life Technologies | Cat#MA5–15253; RRID:AB_10977225 |
| p-PGM1(Y353) | GenScript | Custom order |
| Total PGM1 | Santa Cruz Biotechnology |
Cat#sc-373796; RRID:AB_10918014 |
| Total PGM1 | Proteintech | Cat#15161–1-AP; RRID:AB_2161415 |
| PYGL | Proteintech | Cat#15851–1-AP; RRID:AB_2175014 |
| Bacterial and Virus Strains | ||
| NEB® 5-alpha Competent E. coli (High Efficiency) | New England BioLabs | Cat#C2987H |
| Biological Samples | ||
| Human carcinoma-associated fibroblasts | This paper | |
| Human normal fibroblasts | This paper | |
| Chemicals, Peptides, and Recombinant Proteins | ||
| CP-91149 | Sigma-Aldrich | Cat#PZ0104 |
| PH-797804 (p38α inhibitor) | Selleck Chemical | Cat# S2726 |
| C13N15 Arginine | Cambridge Isotope Laboratories | Cat#CNLM-539-PK |
| C13N15 Lysine | Cambridge Isotope Laboratories |
Cat#CNLM-291-H- |
| Collagenase type 3 | Worthington Biochemical Cooperation |
Cat#LS004208 |
| Hyaluronidase | Worthington Biochemical Cooperation |
Cat#LS002592 |
| 2-NBDG | Caymen Chemical | Cat#11046 |
| U-13C-glucose | Cambridge Isotope Laboratories |
Cat#CLM-1396-PK |
| Glycogen Hydrolysis Enzyme | Caymen Chemical | Cat#700483 |
| CellRox Deep Red Reagent | Invitrogen | Cat#C10422 |
| Matrigel | BD Biosciences | Cat#356231 |
| Gateway LR Clonase II Enzyme | Invitrogen | Cat#11791–020 |
| phosphoglucomutase | Sigma-Aldrich | Cat#P3397 |
| glycogen | Sigma-Aldrich | Cat#G0885 |
| NADP+ | Sigma-Aldrich | Cat#N0505 |
| IL-6 | PeproTech | Cat#200–06 |
| IL-8 | PeproTech | Cat#200–08 |
| CXCL10 | PeproTech | Cat#300–12 |
| CCL5 | PeproTech | Cat#300–06 |
| Stattic | MedChem Express | Cat#HY-13838 |
| Trametinib | MedChem Express | Cat#HY-10999 |
| NSC23766 | Sigma-Aldrich | Cat#SML0952 |
| H-89 | Tocris Bioscience | Cat#2910 |
| 14–22 amide | Tocris Bioscience | Cat#2546 |
| Resazurin | Sigma-Aldrich | Cat#R7017 |
| Diaphorase | Sigma-Aldrich | Cat#D5540 |
| Glucose-6-phosphate dehydrogenase | Sigma-Aldrich | Cat#G7877 |
| Critical Commercial Assays | ||
| Glycogen Assay Kit | BioVision | Cat#K646–100 |
| Q5 Site-Directed Mutagenesis Kit | New England Biolabs | Cat#E0554S |
| Phosphoglucomutase Colorimetric Assay Kit | BioVision | Cat#K774–100 |
| ATP/ADP ratio kit | Sigma-Aldrich | Cat#MAK135–1KT |
| The Human Cytokine Array, Panel A | R&D Systems | Cat#ARY005 |
| EnzyChrom L-Lactate Assay Kit | BioAssay Systems | Cat#ECLC-100 |
| Deposited Data | ||
| Phosphoproteomics | ProteomeXchange | PXD003574 |
| Experimental Models: Cell Lines | ||
| SKOV3ip1 | Gordon Mills, M.D. Anderson Cancer Center, Houston, TX |
RRID:CVCL_0C84 |
| HeyA8 | Gordon Mills, M.D. Anderson Cancer Center, Houston, TX |
RRID:CVCL_8878 |
| ES2 | American Type Tissue Collection |
ATCC Cat# CRL- 1978, RRID:CVCL_3509 |
| MDA-MB-231 | American Type Tissue Collection |
ATCC Cat# HTB-26, RRID:CVCL_0062 |
| RKO | American Type Tissue Collection |
ATCC Cat# CRL- 2577, RRID:CVCL_0504 |
| HeyA8-cGFP-Luc | (Mitra et al., 2012) | N/A |
| SKOV3ip1-cGFP-Luc | (Mitra et al., 2012) | N/A |
| TYK-nu | Dr. Gottfried Koneczny, UCLA |
RRID:CVCL_1776 |
| OVKATE | Dr. Gottfried Koneczny, UCLA |
RRID:CVCL_3110 |
| Caco-2 | American Type Tissue Collection |
ATCC Cat# HTB-37, RRID:CVCL_0025 |
| SNU-119 | Korean Cell Line Bank | KCLB Cat# 00119, RRID:CVCL_5014 |
| PC-3 | American Type Tissue Collection |
ATCC Cat# CRL- 1435, RRID:CVCL_0035 |
| HEK293T | Lucy Godley, University of Chicago |
RRID:CVCL_0063 |
| Tyknu-V5-PGM1-WT | this paper | |
| Tyknu-V5-PGM1-Y353F | this paper | |
| SKOV3ip1-V5-PGM1-WT | this paper | |
| SKOV3ip1-V5-PGM1-Y353F | this paper | |
| Experimental Models: Organisms/Strains | ||
| Athymic nude mice (Hsd: Athymic Nude-Foxn1nu) | Envigo (Harlan) | |
| Oligonucleotides | ||
| PGM1 siGENOME SMARTpool siRNA | Dharmacon, GE Healthcare |
Cat#M-009569–02- 0005 |
| PGM1 mutagenesis forward primer GATTGCTTTGttcGAGACCCCAAC |
Integrated DNA Technologies |
custom |
| PGM1 mutagenesis reverse primer TTTGTAGCACTAGCCACC |
Integrated DNA Technologies |
custom |
| MAPK14 siGENOME SMARTpool siRNA | Dharmacon, GE Healthcare |
Cat# M-003512–06- 0005 |
| Recombinant DNA | ||
| pLX304 | Addgene | Cat#25890 |
| pDONR221-PGM1 | DNASU Plasmid Repository |
clone HsCD00039472 |
| pLX304-V5-PGM1 WT | this paper | |
| pLX304-V5-PGM1-Y353F | this paper | |
| pLKO.1 TRC control | Addgene | Cat#10879 |
| pLKO-shp38 | Addgene | Cat#52920 |
| Software and Algorithms | ||
| Prism software | GraphPad | RRID: SCR_002798 |
| Incucyte Zoom software | Essen Bioscience | |
| Microsoft Visual Basic 6.0 | Thermo Fisher Scientific |
|
| R package - QVALUE | Bioconductor | RRID:SCR_001073 |
| ImageJ | https://imagej.nih.gov/ij/ | RRID:SCR_003070 |
| Living Image Software | Caliper Life Sciences | RRID:SCR_014247 |
Highlights.
Cancer cells activate p38-signaling in neighboring fibroblasts
Fibroblast-derived p38 regulated cytokines mobilize glycogen in cancer cells
Glycogen is used by cancer cells to fuel glycolysis promoting invasion and metastasis
Inhibition of glycogen phosphorylase blocks CAF-stimulated tumor metastasis
Acknowledgments
We are very grateful to Phoebe Rice, PhD Dept. of Biochemistry, University of Chicago for in silico modeling of the PGM1 protein, Elisabet Llonch, IRB Barcelona, for phospho-p38 immunohistochemistry. We are very thankful to Gail Isenberg, University of Chicago for editing the manuscript.
This work was supported by the Cancer Research Foundation Fletcher Scholars Program, Bears Care, the charitable beneficiary of the Chicago Bears Football Club, and the National Cancer Institute grant R01 CA169604 (E. Lengyel), R01 CA193256 (J.W. Locasale), R00CA168997 (J.W. Locasale), and R21CA201963 (J.W. Locasale). A.R. Nebreda’s research group was supported by the European Commission grant ERC 294665. We acknowledge the University of Chicago Human Tissue Resource Center for PAS staining funded by the Cancer Center Support Grant (P30CA014599) as well as the University of Chicago Advanced Electron Microscopy Core Facility for transmission electron microscopy.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare no competing interests.
DATA AND SOFTWARE AVAILABILITY
The mass spectrometry proteomics datasets have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomeexchange.org) via the PRIDE partner repository with the dataset identifier PXD003574.
References:
- Alspach E, Flanagan KC, Luo X, Ruhland MK, Huang H, Pazolli E, Donlin MJ, Marsh T, Piwnica-Worms D, Monahan J , et al. (2014). p38MAPK plays a crucial role in stromal-mediated tumorigenesis. Cancer Discov 4, 716–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrizabalaga O, Lacerda HM, Zubiaga AM, and Zugaza JL (2012). Rac1 protein regulates glycogen phosphorylase activation and controls interleukin (IL)-2-dependent T cell proliferation. J Biol Chem 287, 11878–11890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae E, Kim HE, Koh E, and Kim KS (2014). Phosphoglucomutase1 is necessary for sustained cell growth under repetitive glucose depletion. FEBS Lett 588, 3074–3080. [DOI] [PubMed] [Google Scholar]
- Beausoleil SA, Villen J, Gerber SA, Rush J, and Gygi SP (2006). A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat Biotechnol 24, 1285–1292. [DOI] [PubMed] [Google Scholar]
- Bhowmick NA, Neilson EG, and Moses HL (2004). Stromal fibroblasts in cancer initiation and progression. Nature 432, 332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coscia F, Watters KM, Curtis M, Eckert MA, Chiang CY, Tyanova S, Montag A, Lastra RR, Lengyel E, and Mann M (2016). Integrative proteomic profiling of ovarian cancer cell lines reveals precursor cell associated proteins and functional status. Nat Commun 7, 12645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demirkan G, Yu K, Boylan JM, Salomon AR, and Gruppuso PA (2011). Phosphoproteomic profiling of in vivo signaling in liver by the mammalian target of rapamycin complex 1 (mTORC1). PLoS One 6, e21729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domcke S, Sinha R, Levine DA, Sander C, and Schultz N (2013). Evaluating cell lines as tumour models by comparison of genomic profiles. Nat Commun 4, 2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias JE, and Gygi SP (2007). Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods 4, 207–214. [DOI] [PubMed] [Google Scholar]
- Favaro E, Bensaad K, Chong MG, Tennant DA, Ferguson DJ, Snell C, Steers G, Turley H, Li JL, Gunther UL , et al. (2012). Glucose utilization via glycogen phosphorylase sustains proliferation and prevents premature senescence in cancer cells. Cell Metab 16, 751–764. [DOI] [PubMed] [Google Scholar]
- Guido C, Whitaker-Menezes D, Capparelli C, Balliet R, Lin Z, Pestell RG, Howell A, Aquila S, Andò S, Martinez-Outschoorn U , et al. (2012). Metabolic reprogramming of cancer-associated fibroblasts by TGF-β drives tumor growth: Connecting TGF-β signaling with “Warburg-like” cancer metabolism and L-lactate production. Cell cycle 11, 3019–3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gururaj A, Barnes CJ, Vadlamudi RK, and Kumar R (2004). Regulation of phosphoglucomutase 1 phosphorylation and activity by a signaling kinase. Oncogene 23, 8118–8127. [DOI] [PubMed] [Google Scholar]
- Hanahan D, and Coussens LM (2012). Accessories to the crime: Functions of cell recruited to the tumor microenvironment. Cancer Cell 21, 309–322. [DOI] [PubMed] [Google Scholar]
- Helou YA, Nguyen V, Beik SP, and Salomon AR (2013). ERK positive feedback regulates a widespread network of tyrosine phosphorylation sites across canonical T cell signaling and actin cytoskeletal proteins in Jurkat T cells. PLoS One 8, e69641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henke BR, and Sparks SM (2006). Glycogen phosphorylase inhibitors. Mini Rev Med Chem 6, 845–857. [DOI] [PubMed] [Google Scholar]
- Hickson JA, Huo D, Vander Griend DJ, Lin A, Rinker-Schaeffer CW, and Yamada SD (2006). The p38 kinases MKK4 and MKK6 supress metastatic colonization in human ovarian carcinoma. Cancer Res 66, 2264–2270. [DOI] [PubMed] [Google Scholar]
- Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, and Skrzypek E (2015). PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res 43, D512–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igea A, and Nebreda AR (2015). The Stress Kinase p38alpha as a Target for Cancer Therapy. Cancer Res 75, 3997–4002. [DOI] [PubMed] [Google Scholar]
- Jafari R, Almqvist H, Axelsson H, Ignatushchenko M, Lundback T, Nordlund P, and Martinez Molina D (2014). The cellular thermal shift assay for evaluating drug target interactions in cells. Nat Protoc 9, 2100–2122. [DOI] [PubMed] [Google Scholar]
- Karnoub A, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW, Richardson AL, Polyak K, Tubo R, and Weinberg RA (2007). Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 449, 557–563. [DOI] [PubMed] [Google Scholar]
- Kenny HA, Chiang CY, White EA, Schryver EM, Habis M, Romero IL, Ladanyi A, Penicka CV, George J, Matlin K , et al. (2014). Mesothelial cells promote early ovarian cancer metastasis through fibronectin secretion. J Clin Invest 124, 4614–4628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny HA, Kaur S, Coussens LM, and Lengyel E (2008). The initial steps of ovarian cancer cell metastasis are mediated by MMP-2 cleavage of vitronectin and fibronectin. J Clin Invest 118, 1367–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny HA, Krausz T, Yamada SD, and Lengyel E (2007). Use of a novel 3D culture model to elucidate the role of mesothelial cells, fibroblasts and extra-cellular matrices on adhesion and invasion of ovarian cancer cells to the omentum. Int J Cancer 121, 1463–1472. [DOI] [PubMed] [Google Scholar]
- Ladanyi A, Mukherjee A, Kenny HA, Johnson A, Mitra AK, Sundaresan S, Nieman KM, Pascual G, Benitah SA, Montag A , et al. (2018). Adipocyte-induced CD36 expression drives ovarian cancer progression and metastasis. Oncogene [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengyel E (2010). Ovarian cancer development and metastasis. Am J Pathol 177, 1053–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Ser Z, and Locasale JW (2014). Development and Quantitative Evaluation of a High-Resolution Metabolomics Technology. Anal Chem 86, 2175–2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louzao MC, Espina B, Vieytes MR, Vega FV, Rubiolo JA, Baba O, Terashima T, and Botana LM (2008). “Fluorescent glycogen” formation with sensibility for in vivo and in vitro detection. Glycoconj J 25, 503–510. [DOI] [PubMed] [Google Scholar]
- Mitra AK, Zillhardt M, Hua YJ, Tiwari P, Murmann AE, Peter ME, and Lengyel E (2012). MicroRNAs reprogram normal fibroblasts into cancer-associated fibroblasts in ovarian cancer. Cancer Discov 2, 1100–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR, Romero IL, Carey MS, Mills GB, Hotamisligil GS , et al. (2011). Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med 17, 1498–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieman KM, Romero IL, Van Houten B, and Lengyel E (2013). Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochim Biophys Acta 1831, 1533–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlund D, Elyada E, and Tuveson D (2014). Fibroblast heterogeneity in the cancer wound. J Exp Med 211, 1503–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orimo A, Gupta P, Sgroi D, Arenzana-Seisdedos F, Delaunay T, Naeem R, Carey V, Richardson A, and Weinberg RA (2005). Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL 12 secretion. Cell 121, 335–348. [DOI] [PubMed] [Google Scholar]
- Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, Casimiro MC, Wang C, Fortina P, Addya S , et al. (2009). The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell cycle 8, 3984–4001. [DOI] [PubMed] [Google Scholar]
- Pelletier J, Bellot G, Gounon P, Lacas-Gervais S, Pouyssegur J, and Mazure NM (2012). Glycogen synthesis is induced in hypoxia by the Hypoxia-Inducible Factor and promotes cancer cell survival. Front Oncol 2, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins DN, Pappin DJ, Creasy DM, and Cottrell JS (1999). Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567. [DOI] [PubMed] [Google Scholar]
- Pescador N, Villar D, Cifuentes D, Garcia-Rocha M, Ortiz-Barahona A, Vazquez S, Ordonez A, Cuevas Y, Saez-Morales D, Garcia-Bermejo ML , et al. (2010). Hypoxia promotes glycogen accumulation through hypoxia inducible factor (HIF)-mediated induction of glycogen synthase 1. PLoS One 5, e9644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rines AK, Sharabi K, Tavares CD, and Puigserver P (2016). Targeting hepatic glucose metabolism in the treatment of type 2 diabetes. Nat Rev Drug Discov 15, 786–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritterson Lew C, Guin S, and Theodorescu D (2015). Targeting glycogen metabolism in bladder cancer. Nat Rev Urol 12, 383–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roach PJ, Depaoli-Roach AA, Hurley TD, and Tagliabracci VS (2012). Glycogen and its metabolism: Some new developments and old themes. Biochem J 441, 763–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero IL, Mukherjee A, Kenny HA, Litchfield LM, and Lengyel E (2015). Molecular Pathways: Trafficking of metabolic resources in the tumor microenvironment. Clin Cancer Res 21, 680–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousset M, Zweibaum A, and Fogh J (1981). Presence of glycogen and growth-related variations in 58 cultured human tumor cell lines of various tissue origins. Cancer Res 41, 1165–1170. [PubMed] [Google Scholar]
- Schauer IG, Sood AK, Mok S, and Liu J (2011). Cancer-associated fibroblasts and their putative role in potentiating the initiation and development of epithelial ovarian cancer. Neoplasia 13, 393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnier JB, Nishi K, Monks A, Gorin FA, and Bradbury EM (2003). Inhibition of glycogen phosphorylase (GP) by CP-91,149 induces growth inhibition correlating with brain GP expression. Biochem Biophys Res Commun 309, 126–134. [DOI] [PubMed] [Google Scholar]
- Shulman RG, and Rothman DL (2015). Homeostasis and the glycogen shunt explains aerobic ethanol production in yeast. Proc Natl Acad Sci U S A [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sicoli D, Jiao X, Ju X, Velasco-Velazquez M, Ertel A, Addya S, Li Z, Ando S, Fatatis A, Paudyal B, et al. (2014). CCR5 receptor antagonists block metastasis to bone of v-Src-oncogene-transformed metastatic prostate cancer cell lines. Cancer Res 74, 7103–7114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa CM, Biancur DE, Wang X, Halbrook CJ, Sherman MH, Zhang L, Kremer D, Hwang RF, Witkiewicz AK, Ying H, et al. (2016). Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 536, 479–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey JD (2002). A direct approach to false discovery rates. Journal of the Royal Statistical Society Series B-Statistical Methodology 64, 4479–4498. [Google Scholar]
- Storey JD (2003). The positie false discoery rate: A Bayesian interpretation and the q-value. The Annals of Statistics 31, 2013–2035. [Google Scholar]
- Storey JD, and Tibshirani R (2003). Statistical significance for genomewide studies. Proceedings of the National Academy of Sciences 100, 9440–9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thwe PM, Pelgrom L, Cooper R, Beauchamp S, Reisz JA, D’Alessandro A, Everts B, and Amiel E (2017). Cell-Intrinsic glycogen metabolism supports early glycolytic reprogramming required for dendritic cell immune responses. Cell Metab 26, 558–567 e555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K, Sabelli A, DeKeukelaere L, Park R, Sindi S, Gatsonis CA, and Salomon A (2009). Integrated platform for manual and high-throughput statistical validation of tandem mass spectra., Proteomics 9, 3115–3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu A, Romero R, and Petty HR (2011). An enzymatic colorimetric assay for glucose-6-phosphate. Anal Biochem 419, 266–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zillhardt M, Park SM, Romero IL, Sawada K, Montag A, Krausz T, Yamada SD, Peter ME, and Lengyel E (2011). Foretinib (GSK1363089), an orally available multikinase inhibitor of c-Met and VEGFR-2, blocks proliferation, induces anoikis, and impairs ovarian cancer metastasis. Clin Cancer Res 17, 4042–4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zois CE, Favaro E, and Harris AL (2014). Glycogen metabolism in cancer. Biochem Pharmacol 92, 3–11. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.